

Abstract
Background
The ketone body 3‐hydroxybutyrate (3‐OHB) increases cardiac output (CO) in patients with heart failure through unknown mechanisms. 3‐OHB activates the hydroxycarboxylic acid receptor 2 (HCA2), which increases prostaglandins and suppresses circulating free fatty acids. We investigated whether the cardiovascular effects of 3‐OHB involved HCA2 activation and if the potent HCA2‐stimulator niacin may increase CO.
Methods and Results
Twelve patients with heart failure with reduced ejection fraction were included in a randomized crossover study and examined by right heart catheterization, echocardiography, and blood sampling on 2 separate days. On study day 1, patients received aspirin to block the HCA2 downstream cyclooxygenase enzyme, followed by 3‐OHB and placebo infusions in random order. We compared the results with those of a previous study in which patients received no aspirin. On study day 2, patients received niacin and placebo. The primary end point was CO. 3‐OHB increased CO (2.3 L/min, P<0.01), stroke volume (19 mL, P<0.01), heart rate (10 bpm, P<0.01), and mixed venous saturation (5%, P<0.01) with preceding aspirin. 3‐OHB did not change prostaglandin levels, neither in the ketone/placebo group receiving aspirin nor the previous study cohort. Aspirin did not block 3‐OHB‐induced changes in CO (P=0.43). 3‐OHB decreased free fatty acids by 58% (P=0.01). Niacin increased prostaglandin D2 levels by 330% (P<0.02) and reduced free fatty acids by 75% (P<0.01) but did not affect CO.
Conclusions
The acute increase in CO during 3‐OHB infusion was not modified by aspirin, and niacin had no hemodynamic effects. These findings show that HCA2 receptor‐mediated effects were not involved in the hemodynamic response to 3‐OHB.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: NCT04703361.
Keywords: HCA2 receptor, heart failure, hemodynamics, ketone body, metabolic
Subject Categories: Heart Failure
Nonstandard Abbreviations and Acronyms
- 3‐OHB
3‐hydroxybutyrate
- CO
cardiac output
- FFA
free fatty acid
- HCA2
hydroxycarboxylic acid receptor 2
- HFrEF
heart failure with reduced ejection fraction
- PAP
mean pulmonary artery pressure
- PCWP
pulmonary capillary wedge pressure
- PGD2
prostaglandin D2
- PGE2
prostaglandin E2
- PGI2
prostaglandin I2
Clinical Perspective.
What Is New?
In patients with heart failure with reduced ejection fraction, infusion of the ketone body 3‐hydroxybutyrate (3‐OHB) increased cardiac output by 2.3 L/min and reduced circulating free fatty acids by 58%.
The increase in cardiac output during 3‐OHB infusion was not blocked when the HCA2 downstream enzyme cyclooxygenase was inhibited with aspirin, and circulating prostaglandin levels did not change during 3‐OHB infusion compared with placebo.
Stimulation of HCA2 with the potent niacin (B3 vitamin) suppressed free fatty acids to a similar extent as 3‐OHB and increased circulating prostaglandins but did not change cardiac output.
What Are the Clinical Implications?
3‐OHB may be a beneficial treatment for patients with heart failure with reduced ejection fraction because it increases cardiac output. The hemodynamic changes were not mediated through HCA2 activation, prostaglandin release, or circulating free fatty acid suppression.
The ketone body 3‐hydroxybutyrate (3‐OHB) is produced in the liver during ketogenic dieting, fasting, and stress 1 and is oxidized in vital organs such as brain, heart, and skeletal muscle. 2 We recently demonstrated that 3‐OHB infusion increases myocardial blood flow by 75% in healthy participants 3 and cardiac output (CO) by 40% in patients with chronic heart failure with reduced ejection fraction (HFrEF). 4 Animal studies have shown that 3‐OHB has cardioprotective effects in heart failure 5 ; however, the exact underlying mechanism is not fully understood. In patients with heart failure, myocardial genes coding for enzymes involved in 3‐OHB oxidation are upregulated. 6 Experimental studies have reported that heart failure is accompanied by a cardiac metabolic remodeling process that shifts the substrate utilization from free fatty acids (FFAs) toward 3‐OHB. 7 , 8 Enzymes involved in 3‐OHB oxidation are upregulated in cardiomyocytes from patients with HFrEF, 6 and studies show that the failing heart increases utilization of 3‐OHB 9 without compromising glucose or FFA consumption. 10 , 11 Thus, current data support that 3‐OHB is an important adjunct fuel in the failing heart, potentially exerting its beneficial effects through enhanced ATP production. 12 , 13
In addition to its metabolic effects, 3‐OHB has pleiotropic effects. The metabolite 3‐OHB is the only physiologically important endogenous ligand of the G‐protein‐coupled receptor hydroxycarboxylic acid receptor 2 (HCA2). 14 , 15 Physiological 3‐OHB concentrations activate HCA2 16 on immune cells and adipocytes. 15 In macrophages, HCA2 activation has downstream effects resulting in production and release of prostaglandin D2 (PGD2) and prostaglandin E2 (PGE2) through cyclooxygenase enzymes. 15 , 17 Aspirin blocks the cyclooxygenase enzymes and thereby prostaglandin production. 18 In adipocytes, HCA2 activation by 3‐OHB decreases FFA release by inhibition of the hormone‐sensitive lipase and adipocyte triglyceride lipase. 15 , 19 This is known to constitute a negative feedback loop, whereby hepatic ketogenesis from circulating FFAs during fasting prevents excessive FFA release from adipose tissue 15 (Figure 1). Our previous study showed that circulating FFAs are suppressed during 3‐OHB infusion. 4 Therefore, it is possible that the observed favorable hemodynamic effects of 3‐OHB infusion were due to both reduced plasma FFA concentrations and altered myocardial substrate metabolism. However, it is not fully understood whether the hemodynamic effect of 3‐OHB in patients with HFrEF involves a HCA2‐mediated decrease in circulating FFAs.
Figure 1. HCA2.
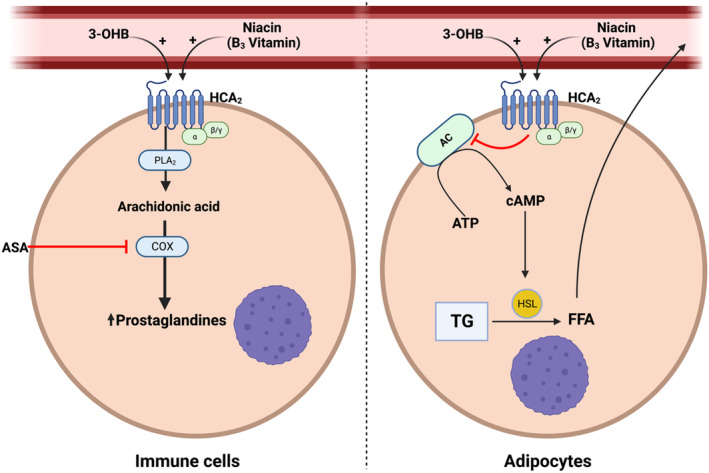
Modulation of prostaglandin and free fatty acid production through HCA2 receptor activation. 3‐OHB indicates 3‐hydroxybutyrate; AC, adenylyl cyclase; ASA, aspirin; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; COX, cyclooxygenase; FFA, free fatty acid; HCA2, hydroxycarboxylic acid 2 receptor; HSL, hormone‐sensitive lipase; PLA2, phospholipase A2; and TG, triglyceride. Created with BioRender.com
The hemodynamic effects of 3‐OHB in patients with HFrEF 4 involve a marked reduction in systemic vascular resistance (SVR) that could play a major role in the increased CO observed. Therefore, in the present study, we addressed the potential effects of 3‐OHB‐mediated activation of the HCA2 receptor and release of vasodilatory prostaglandins (PGD2, PGE2, and prostaglandin I2). Prostaglandins stimulate the receptors DP1 and EP2/4 on blood vessels, which activate the process of cAMP‐mediated vasodilation. 17 It is unclear whether the observed increase in CO during 3‐OHB infusion is a consequence of increased left ventricular contractility, direct vasodilatory effect on vessels, or a combination thereof.
Niacin (vitamin B3) is a well‐described potent HCA2 receptor stimulator. 20 Niacin stimulation of HCA2 has an antilipolytic effect and has therefore been used in dyslipidemia treatment. 19 , 21 However, the hemodynamic effects of niacin have never been investigated.
The aims of this study were to investigate, first, if the hemodynamic effect of 3‐OHB in patients with HFrEF involves HCA2 activation; second, to explore the hemodynamic effects of the exogenous and even stronger HCA2‐agonist niacin; third, to investigate the release of prostaglandins by HCA2 activation; and finally, to examine the interaction between HCA2 stimulation, suppressed FFA release from adipose tissue, and hemodynamic changes in patients with HFrEF.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Participants
We included patients with HFrEF with left ventricular ejection fraction ≤40% who were in New York Heart Association class II to III and able to give informed consent. Exclusion criteria were significant cardiac valve disease (symptomatic cardiac valve disease or disease requiring intervention according to guidelines), history of myocardial infarction within 1 month, insulin treatment, or significant liver disease. All patients were recruited from the Heart Failure Clinic, Department of Cardiology, Aarhus University Hospital, Denmark.
Design
This was a randomized, controlled, single‐blinded crossover study (Figure 2). Twelve patients were studied on 2 days separated by a minimum 7‐day washout period. All participants fasted overnight and avoided heavy exercise before each visit. On study days, a pulmonary artery catheter was placed, and a peripheral venous catheter was inserted in the cubital vein. Patients were then left to rest for at least 45 minutes before baseline measurements were made.
Figure 2. Study flowchart.
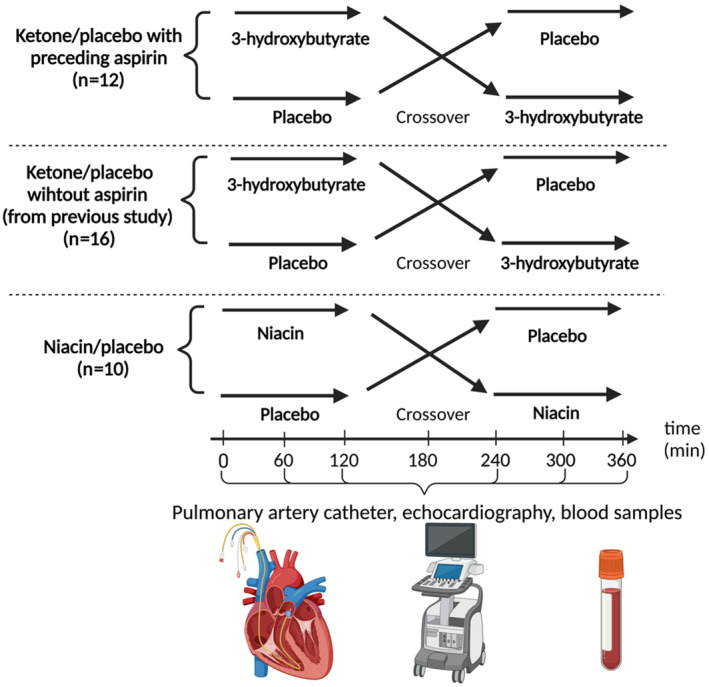
Overview of the study design for all 3 study groups: Ketone/placebo with preceding aspirin, Ketone/placebo without aspirin (from previous study), Niacin/placebo. The interventions were initiated after baseline measurements (0 minutes) and were crossed over to the other intervention at 180 minutes. Invasive measurements using pulmonary artery catheter, echocardiography, and blood samples were conducted hourly during the study period.
On study day 1 (Ketone/placebo with preceding aspirin), all participants received a 650 mg bolus of intravenous aspirin (Bayer, Germany), which is known to irreversibly block the cyclooxygenase enzyme. 18 Thirty minutes later, the patients received a continuous 3‐OHB infusion (0.18 g/kg per hour) and isovolumetric placebo (isotonic saline) for 3 hours, administered in random order (Figure 2).
Sixteen control patients with HFrEF (ketone/placebo without aspirin) were derived from a previous study by our group. 4 These patients also received the infusions and investigations described above for study day 1 but received no intravenous aspirin (Figure 2).
On study day 2 (Niacin/placebo), all participants received tablets of 1000 mg niacin orally (Natur Drogeriet, Denmark) and placebo tablets (lactose monohydrate, the Hospital Pharmacy Aarhus University Hospital, Aarhus, Denmark) in random order (Figure 2). The purpose of utilizing niacin in this study was to serve as a positive control for the HCA2 receptor, as it is a well‐known strong agonist of this receptor. Measurements were made for 3 hours after intake of each study drug. The niacin dose was reduced to 500 mg halfway through the study because 4 patients experienced flushing and discomfort.
Solutions and Infusions
The 3‐OHB solution was prepared as a 7.5% concentration of 3‐OHB by the local pharmacy. Potassium chloride (60 mmol/L) was added to the 3‐OHB solution. Infusion was initiated following baseline measurements. Infusion was changed at crossover after 3 hours. The patients received an oral bolus of 60 mmol KCl when randomized to initiate with 3‐OHB and 20 mmol when randomized to initiate with placebo. To maintain euglycemia and minimize endogenous ketogenesis, all patients received a low‐dose insulinemic euglycemic clamp (0.3 mU insulin/kg per min) with glucose (20% solution, 60 mmol/L KCl) throughout the 6‐hour study period.
End Points and Measurements
All patients were monitored with pulmonary artery catheter measurements, echocardiography, and blood samples at baseline and hourly for a 6‐hour period on each study day. The primary end point was difference between CO at the end of each infusion period. Secondary end points were mixed venous saturation, pulmonary capillary wedge pressure, mean pulmonary artery pressure (PAP), pulmonary vascular resistance, systemic vascular resistance (SVR), left ventricular ejection fraction, left ventricular global longitudinal strain, FFAs, and circulating prostaglandin levels.
Noninvasive Hemodynamic Measurements
Blood pressure and heart rate were measured with Phillips IntelliVue X3 (Phillips, Netherlands) monitoring system. Mean arterial pressure was calculated as one‐third systolic blood pressure + two‐thirds diastolic blood pressure.
Right Heart Catheterization
The pulmonary artery catheter was placed with fluoroscopy guidance under sterile conditions through the internal jugular vein. Right arterial pressure, PAP, PCWP, mixed venous saturation, and CO were measured at baseline and hourly after the intervention was initiated. CO was measured by the thermodilution technique, and the mean of 3 measurements was used. SVR was calculated as (mean arterial pressure–right arterial pressure)/CO. Pulmonary vascular resistance was calculated as (PAP–PCWP)/CO.
Echocardiography
All echocardiographic acquisitions were made using GE Vivid E95 or GE Vivid E9 (GE Healthcare, USA). EchoPAC software (GE‐Vingmed Ultrasound, Norway) was used for analysis.
Blood Samples
Blood samples were drawn from the cubital vein via a peripheral venous catheter. pH, potassium, sodium, calcium, and lactate were analyzed immediately after sampling using YSI STAT 2100 (YSI Inc., Netherlands). Glucose was analyzed using Contour Xt Glucometer (Bayer, Germany). D‐3‐OHB was measured with FreeStyle Precision Neo (Abbott, USA). All other samples were analyzed as a batch at the end of the study. Total 3‐OHB was measured with hydrophilic interaction liquid chromatography tandem mass spectrometry. Prostaglandin I2 (PGI2), PGE2, and PGD2 were analyzed with the 6‐keto Prostaglandin F1alpha ELISA kit (Cayman Chemical, USA), Prostaglandin E Metabolite ELISA kit (Cayman Chemical, USA), and the Prostaglandin D2‐MOX Express ELISA Kit (Cayman Chemical, USA), respectively.
Statistical Analysis
The SD of CO (primary end point) is 0.8 L/min. 4 By enrolling 12 patients, a relative difference in CO of 20% can be detected with a power of 90% and a 2‐sided significance level of 5%. For the niacin part of the study, 10 patients completed the study. Post‐hoc power for this part of the study was 88.5%, based on the given parameters of 10 participants, a 20% difference in CO, and an SD of 0.8 L/min. Randomization was made in STATA 17 (StataCorp, USA) by the investigator. Participants were blinded to the randomization order. Data were inspected for normal distribution as required. Baseline data (Table 1) are presented as mean±SD if normally distributed or median (interquartile range) if not normally distributed. Paired 2‐tailed t test was used to compare the effect of the interventions with that of placebo. A 2‐sample t test was used to compare the means of continuous variables between the 2 groups: ketone/placebo with the preceding aspirin and ketone/placebo without aspirin, whereas Fisher exact test was utilized to compare the categorical variables between the 2 groups. Differences are presented as mean with 95% CI. A P‐value <0.05 was considered statistically significant. Data analyses were performed with STATA 17 (StataCorp, USA). All figures were created with Prism 8 (Graphpad Software, LLC).
Table 1.
Baseline Characteristics
| Ketone/placebo with preceding aspirin and niacin/placebo (n=12) | Ketone/placebo without aspirin (n=16) | P value | |
|---|---|---|---|
| Sex (male/female) | 11/1 | 14/2 | 1.00 |
| Age, y | 64±11 | 60±12 | 0.38 |
| BMI, kg/m2 | 30±5 | 26±5 | 0.07 |
| IHD, n (%) | 6 (50%) | 12 (75%) | 0.17 |
| AFIB, n (%) | 3 (25%) | 1 (6%) | 0.28 |
| Heart rate, beats/min | 72 (67–84) | 67 (58–72) | 0.07 |
| Systolic BP, mm Hg | 120±14 | 130±18 | 0.15 |
| Diastolic BP, mm Hg | 76±13 | 77±10 | 0.78 |
| NT‐proBNP, ng/L | 360 (206–1113) | 617 (418–1306) | 0.15 |
| HbA1c, mmol/mol | 41±4 | 39±4 | 0.24 |
| eGFR, mL | 85 (71–90) | 90 (66–90) | 0.82 |
| NYHA class | 2 (2–3) | 2 (2–2) | 0.06 |
| LVEF, (%) | 33±5 | 36±4 | 0.16 |
| GLS, (%) | −9.3±4 | −10±3 | 0.60 |
| ACE‐I/ARB, n (%) | 8 (67%) | 15 (94%) | 0.13 |
| Platelet inhibitors, n (%) | 4 (33%) | 12 (75%) | 0.05 |
| β‐blockers, n (%) | 12 (100%) | 16 (100%) | 1.00 |
| Diuretics, n (%) | 7 (58%) | 8 (50%) | 0.66 |
| MRA, n (%) | 11 (92%) | 10 (63%) | 0.08 |
| Statin, n (%) | 8 (67%) | 14 (88%) | 0.35 |
| SGLT2 inhibitor | 3 (25%) | 0 (0%) | 0.07 |
| ICD/CRT/CRT‐D, n (%) | 7 (58%) | 12 (75%) | 0.43 |
Data are mean±SD or median (interquartile range). ACE‐I indicates angiotensin‐converting enzyme inhibitors; AFIB, atrial fibrillation; ARB, angiotensin‐2 receptor blockers; BMI, body mass index; BP, blood pressure; CRT, cardiac resynchronization therapy; CRT‐D, implantable cardioverter defibrillators with cardiac resynchronization capabilities; eGFR, estimated glomerular filtration rate; GLS, global longitudinal strain; HbA1c, hemoglobin A1c; ICD, implantable cardioverter‐defibrillator; IHD, ischemic heart disease; LVEF, left ventricular ejection fraction; MRA, mineralocorticoid receptor antagonists; NT‐proBNP, N‐terminal pro brain‐natriuretic‐peptide; NYHA, New York Heart Association; and SGLT2, sodium‐glucose cotransporter‐2.
Ethics
The local Ethical Committee of the Central Denmark Region approved the study. All patients provided written informed consent for participation. The study was registered with the Danish Data Protection Agency and clinicaltrials.gov (NCT04703361). NG had full access to all study data and takes responsibility for its integrity and data analysis.
Results
Patients
Sixteen patients were eligible for inclusion. Three patients declined to participate, and 1 patient did not meet the inclusion criteria. Thus, 12 patients were included in the study. They all completed study day 1 (ketone/placebo with preceding aspirin), but 2 did not complete study day 2 (the niacin/placebo day). One declined to participate; the other had a vasovagal syncope on the day of examination. All patients received treatment in the intended randomly assigned order. The patient studies were conducted from February 2021 to September 2021, and blood sample analyses were finalized in September 2022. The baseline characteristics are shown in Table 1. Patient characteristics for patients from our previous study 4 (ketone/placebo without aspirin) are also presented in Table 1.
Hemodynamic Effects of 3‐OHB With Preceding Aspirin
Intravenous aspirin was administrated 42±1 minutes before interventions were initiated. Circulating 3‐OHB levels increased by 4.1 mmol/L (95% CI, 3.5– 4.6 mmol/L) during the ketone infusion period compared with the placebo period. CO increased by 2.3 L/min (95% CI, 1.6–2.9 L/min) after 3 hours of 3‐OHB infusion compared with 3 hours of placebo infusion (P<0.01). The increase in CO was driven by a 19 mL increase in stroke volume (SV) (95% CI, 14–24 mL) and an increase in heart rate of 10 beats per minute (95% CI, 7–13 beats per minute). Mixed venous saturation increased by 5 percentage points (95% CI, from 2%–8%) after 3‐OHB infusion compared with placebo (P<0.01). SVR was reduced by 5 Wood units compared with placebo (95% CI, −6 to −4 Wood units). PCWP, PAP, and pulmonary vascular resistance did not change significantly (Table 2).
Table 2.
Ketone/Placebo With Preceding Aspirin and Without Aspirin
| Ketone/placebo with preceding aspirin | Ketone/placebo without aspirin | Ketone/placebo with preceding aspirin vs without aspirin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (saline) (n=12) | 3‐OHB (n=12) | Difference | P value | Placebo (saline) (n=16) | 3‐OHB (n=16) | Difference | P value | Difference of difference | P value | |
| Hemodynamic parameters | ||||||||||
| CO, L/min | 5.1±1.5 | 7.4±2.0 | 2.3 [1.6, 2.9] | <0.01* | 4.8±0.6 | 6.8±1.0 | 2.0 [1.6–2.4] | <0.01* | 0.3 [−0.4, 0.9] | 0.43 |
| SV, mL | 80±15 | 100±16 | 19 [14– 24] | <0.01* | 75±16 | 95±18 | 20 [15–24] | <0.01* | 0 [7–6] | 0.90 |
| RAP, mm Hg | 5±2 | 5±2 | 0 | 0.83 | 4.4±2.7 | 3.7±3.0 | −0.7 [−1.4, −0.02] | 0.05 | 1 [0–1.6] | 0.23 |
| PAP, mm Hg | 19±6 | 19±5 | 0 [−2, 2] | 0.91 | 19.8±10.6 | 19.6±8.7 | −0.2 [−2.0, 1.7] | 0.80 | 0 [−2, 3] | 0.82 |
| PCWP, mm Hg | 9±2 | 8±2 | −1 [0–1] | 0.11 | 10±5 | 9±5 | −1 [−2, 0] | 0.03* | 1 [−1, 2] | 0.37 |
| MAP, mm Hg | 81±16 | 77±12 | −4 [−9, 0.8] | 0.09 | 92±13 | 91±10 | −1 [−5, 2] | 0.48 | −3 [−9, 3] | 0.32 |
| SVR, WU | 15±3 | 10±3 | −5 [−6, −4] | <0.01* | 19±3 | 13±2 | −5 [−4, −2] | <0.01* | 0 [−1, 2] | 0.60 |
| Heart rate, bpm | 63±9 | 73±11 | 10 [7–13] | <0.01* | 66 ± 16 | 73±14 | 7 [4–11] | <0.01* | 3 [−2, 7] | 0.21 |
| SVO2, % | 70±7 | 75±5 | 5 [2–8] | <0.01* | 72±3 | 79±4 | 6 [5–8] | <0.01* | −2 [− 4, 1] | 0.28 |
| Echocardiography | ||||||||||
| LVOT VTI, cm | 22.4±3.8 | 23.3±3.9 | 0.9 [−1.7, 3.5] | 0.46 | 18.1±3.5 | 22.1±4.3 | 4.0 [2.8–5.2] | <0.01* | −3.1 [−5.6, −0.6] | <0.02* |
| GLS, % | 10.3±3.9 | 12.3±4.2 | 2.1 [1.2–3.0] | <0.01* | 9.7±3.2 | 11.7±3.6 | 2.0 [1.3–2.7] | <0.01* | 0.1 [−0.9, 1–1] | 0.83 |
| S′, cm/s | 4.4±1.1 | 5.6±1.9 | 1.2 [0.5– 1.8] | <0.01* | 3.7±0.8 | 4.5±1.3 | 0.8 [0.3–1.3] | <0.01* | 0.4 [−0.4, 1–1] | 0.32 |
| LVEF, % | 40±8.6 | 45±7 | 6 [3–9] | <0.01* | 35±7 | 43±9 | 8 [5–11] | <0.01* | −2 [−6, 1] | 0.21 |
| LVEDV, mL | 143±41 | 127±40 | −16 [−29, 3] | 0.02* | 174±48 | 165±47 | −9 [−20, 2] | 0.09 | −7 [−23, 9] | 0.38 |
| LVESV, mL | 97±46 | 72±30 | −25 [−44, −6] | 0.02* | 115±40 | 97±41 | −17 [−24, 10] | <0.01* | −8 [−25, 9] | 0.35 |
| TAPSE, cm | 1.8±0.4 | 2.0±0.5 | 0.2 [0.0, 0.4] | 0.07 | 1.9±0.5 | 2.1±0.5 | 0.2 [0.1–0.3] | <0.01* | 0.0 [−0.2, 0.2] | 0.96 |
| Substrates and hormones | ||||||||||
| 3‐OHB, mM | 0.4±0.4 | 4.4±0.9 | 4.1 [3.5–4.6] | <0.01* | 0.3±0.3 | 3.3±0.4 | 2.9 [2.7–3.1] | <0.01* | 1.1 [0.6–1.6] | <0.01* |
| D‐3‐OHB, mM | 0.1±0.1 | 1.7±0.6 | 1.6 [1.2, 1.9] | <0.01* | NA | NA | NA | NA | – | – |
| Lactate, mmol/L | 0.9±0.2 | 1.3±0.3 | 0.4 [0.2–0.60.0] | <0.01* | 1.1±0.5 | 1.5±0.7 | 0.4 [0.3–0.6] | <0.01* | 0.0 [−0.2, 0.2] | 0.84 |
| Glucose, mmol/L | 7.6±0.6 | 7.7±0.8 | 0.0 [−0.2, 0.3] | 0.72 | 5.7±0.7 | 5.7±0.6 | 0.0 [−0.4, 0.4] | 0.99 | −0.1 [−0.8, 0.3] | 0.30 |
| FFA, mmol/L | 0.17±0.13 | 0.07±0.08 | −0.10 [−0.17, − 0.03] | 0.01* | 0.17±0.18 | 0.09±0.12 | ‐ 0.08 [−0.14, −0.01] | 0.02* | −0.02 [−0.12, 0.07] | 0.60 |
| Insulin, pM | 225.4±95.0 | 236.6±135.4 | 11.2 [−33.7, 56.0] | 0.59 | 175.3±72.1 | 183.4±48.4 | 8.2 [−20.4, 36.8] | 0.55 | 53.16 [−21.8, 128.1] | 0.16 |
| Prostaglandins | ||||||||||
| PGE2, pg/mL | 126.1±137.2 | 78.0±27.2 | −48.0 [−125.6, 29.5] | 0.20 | 157.3±87.2 | 108.3±50.8 | ‐ 49.0 [−107.9, 9.8] | 0.10 | 1.0 [−89.6, 91.7] | 0.98 |
| PGI2, pg/mL | 669.3±526.9 | 788.7±560.9 | 119.4 [−299.9, 538.7] | 0.54 | 554.6±1236.7 | 581.4±660 | 26.8 [−815.4, 869.0] | 0.95 | 92.6 [−851.5, 1036.7] | 0.84 |
| PGD2, pg/mL | 60.2±30.1 | 57.3±25.6 | −2.8 [−10.0, 4.3] | 0.40 | 117.7±44.9 | 109.4±40.1 | −8.3 [−21.2, 4.6] | 0.19 | 5.5 [−10.1, 21.0] | 0.48 |
The data are shown as mean±SD. The measures were performed at the end of each intervention period. P values refer to paired t tests. bpm indicates beats per minute; 3‐OHB, 3‐hydroxybutyrate; CO, cardiac output; D‐3‐OHB, D‐3‐hydroxybutyrate; FFA, free fatty acids; GLS, numeric global longitudinal strain; LVEDV, left ventricular end‐diastolic volume; LVEF, left ventricular ejection fraction; LVESV, left ventricular end‐systolic volume; LVOT VTI, left ventricular outflow tract velocity time integral; MAP, mean arterial pressure; NA, not available; PAP, mean pulmonal artery pressure; PCWP, pulmonary capillary wedge pressure; PGD2, prostaglandin D2; PGE2, prostaglandin E2; PGI2, prostaglandin I2; RAP, right arterial pressure; S′, systolic mitral plane peak excursion velocity; SV, stroke volume; SVO2, mixed venous saturation; SVR, systemic vascular resistance;TAPSE, tricuspid annular peak systolic excursion; and WU, Wood units.
P<0.05.
Echocardiographic systolic parameters including global longitudinal strain, left ventricular ejection fraction, and systolic myocardial velocity at mitral annulus (s') all increased during 3‐OHB infusion compared with placebo (Table 2).
Hemodynamic Effects of 3‐OHB With and Without Preceding Aspirin Treatment
Hemodynamic data for ketone/placebo without aspirin treatment have previously been published by our group, 4 and the results are included in Table 2. These data showed an increase in CO of 2.0 L/min (95% CI, 1.6–2.4 L/min) after 3 hours of 3‐OHB infusion as compared with placebo.
In the present study, where aspirin treatment preceded infusions, we observed an increase in CO of 2.3 L/min (95% CI, 1.6–2.9 L/min) after 3 hours of 3‐OHB infusion as compared with placebo. Thus, no significant difference was observed between the increase in CO between patients with HFrEF with and without preceding aspirin treatment (0.3 L/min; 95% CI, −0.4 to 0.9 L/min). Likewise, no differences in other hemodynamic parameters were observed between patients with HFrEF with and without preceding aspirin treatment (Table 2).
Hemodynamic Effects of Niacin
Plasma niacin levels increased by 8.1 mg/mL (95% CI, 3.6–12.6 mg/mL) after niacin administration compared with placebo. CO did not change (0 L/min; 95% CI –0.4 to 0.4 L/min) after niacin administration compared with placebo. Likewise, no differences were observed in mixed venous saturation, SVR, PCWP, PAP, and pulmonary vascular resistance (Table 3). No changes were observed in echocardiographic parameters (Table 3).
Table 3.
Study Day 2 (Niacin/Placebo Day)
| Placebo (n=10) | Niacin (n=10) | Difference | P value | |
|---|---|---|---|---|
| Hemodynamic parameters | ||||
| CO, L/min | 4.5±0.9 | 4.5±0.6 | 0 [−0.4, 0.4] | 0.98 |
| SV, mL | 72±12 | 70±11 | ‐ 3 [−9, 3] | 0.35 |
| RAP, mm Hg | 5±1 | 6±1 | 1 [0.1–1.7] | 0.03† |
| PAP, mm Hg | 18±8 | 18±7 | 0 [−2, 3] | 0.76 |
| PCWP, mm Hg | 10±6 | 9±6 | −1 [−2, 0] | 0.06 |
| TPG, mm Hg | 8.9±4.3 | 9.3±3.7 | 0.4 [−2.6–3.36] | 0.77 |
| PVR, WU | 2.0±1.0 | 2.0±0.8 | 0.0 [−0.7–1.0] | 0.91 |
| MAP, mm Hg | 86±13 | 84±14 | ‐ 2 [−12, 8] | 0.72 |
| SVR, WU | 18.3±2.6 | 17.5±2.3 | −0.8 [−3.6, 2.0] | 0.55 |
| Heart rate, bpm | 62±8 | 64±59 | 3 [−1, 6] | 0.18 |
| SVO2, % | 65±7 | 63±6 | −2 [−6, 2] | 0.33 |
| Echocardiography | ||||
| LVOT VTI, cm | 18.3±3.6 | 17.2±2.6 | −1.0 [−3.0, 0.9] | 0.25 |
| GLS, % | 9.1±2.4 | 10.3±6.9 | 1.1 [−0.4, 2.6] | 0.12 |
| S′, cm/s | 4.1±1.4 | 4.2±1.6 | 0.1 [−0.7, 0.9] | 0.75 |
| LVEF, % | 38.8±7.5 | 38.4±6.8 | −0.4 [−2.3, 1.6] | 0.66 |
| LVEDV, mL | 135.4±57.9 | 134±59.1 | −1.4 [−30.5, 27.7] | 0.91 |
| LVESV, mL | 84.3±42.6 | 84.4±45.4 | 0.1 [−18.7, 19.0] | 0.99 |
| TAPSE, cm | 16.3±3.5 | 17.6±4.6 | 1.2 [−2.5, 4.9] | 0.47 |
| Blood samples | ||||
| Lactate, mmol/L | 1.0±0.2 | 1.1±0.2 | 0.1 [−0.1, 0.3] | 0.25 |
| Glucose, mmol/L | 6.0±0.8 | 5.5±0.8 | −0.5 [−1.3, 0.3] | 0.18 |
| FFA*, mmol/L | 0.3±0.1 | 0.2±0.1 | −0.2 [−0.3, −0.7] | <0.01† |
| pH | 7.4±0.0 | 7.4±0.0 | 0.0 [0.0–0.0] | 0.22 |
| Niacin, μg/mL | 1.6±1.0 | 9.7±7.2 | 8.1 [3.6–12.6] | <0.01† |
| Prostaglandins | ||||
| PGE2, pg/mL | 138.8±97.7 | 125.6±69.5 | −13.2 [−102.9, 76.5] | 0.74 |
| PGI2, pg/mL | 634.1±352.8 | 1308.4±1658 | 674.2 [−543.2, 1891.7] | 0.24 |
| PGD2, pg/mL | 95.6±54.3 | 150.8±97.7 | 55.3 [0.9–109.6] | 0.047† |
The data are shown as mean±SD. The measures were performed at the end of each intervention period. P values refer to paired t test. bpm indicates beats per minute; CO, cardiac output; FFA, free fatty acids, GLS, numeric global longitudinal strain; LVEDV, left ventricular end‐diastolic volume; LVEF, left ventricular ejection fraction; LVESV, left ventricular end‐systolic volume; LVOT VTI, left ventricular outflow tract velocity time integral; MAP, mean arterial pressure; PAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PGD2, prostaglandin D2; PGE2, prostaglandin E2; PGI2, prostaglandin I2; PVR, pulmonary vascular resistance; RAP, right arterial pressure; S′, systolic mitral plane peak excursion velocity; SV, stroke volume; SVO2, mixed venous saturation; SVR, systemic vascular resistance; TAPSE, tricuspid annular peak systolic excursion; TPG, transpulmonary pressure gradient; and WU, Wood units.
Calculated from the area under the curve during the 3‐hour intervention.
P<0.05.
Effects of 3‐OHB and Niacin on Prostaglandin Release
PGI2 and PGE2 levels in plasma did not differ during 3‐OHB infusion with or without preceding aspirin treatment when compared with placebo (Table 2). No differences were observed in PGI2 levels (P=0.84) or PGE2 levels (P=0.98) during 3‐OHB infusion between patients with HFrEF with and without preceding aspirin‐induced cyclooxygenase inhibition (Table 2). Niacin treatment significantly increased PGD2 levels by 58% (55.3 pg/mL [95% CI, 0.9–109.6 pg/mL]) compared with placebo (Table 3, Figure 3). No significant change was observed in PGI2 and PGE2 levels after niacin treatment compared with placebo.
Figure 3. Changes in cardiac output, free fatty acids, and prostaglandin D2.
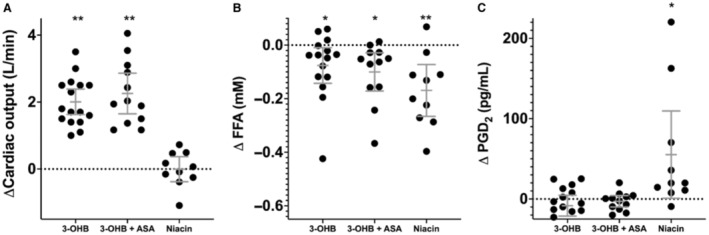
ΔCardiac output (A), ΔFFA (B), and ΔPGD2 (C) recorded during the intervention period as compared with the placebo period. 3‐OHB indicates 3‐hydroxybutyrate; ASA, aspirin; FFA, free fratty acid; and PGD2, prostaglandin D2. *P<0.05 **P<0.01. 3‐OHB: HCA2 receptors are activated; 3‐OHB+ASA: HCA2 receptors are activated, whereas the downstream cyclooxygenase enzyme is inhibited with ASA; Niacin: HCA2 receptors are activated by an even stronger HCA2 agonist than 3‐OHB.
FFA and Glucose
FFA levels were suppressed by 58% (−0.10 mmol/L; 95% CI, −0.17 to −0.03 mmol/L) during 3‐OHB infusion compared with placebo in patients with HFrEF who received preceding aspirin (Table 2). In patients without aspirin administration, FFA levels were suppressed by 47% (−0.08 mmol/L; 95% CI, −0.14 to −0.01) during 3‐OHB infusion compared with placebo (Table 2, Figure 4). No difference was recorded between the decrease in FFA levels during 3‐OHB infusion between patients with HFrEF with and without preceding aspirin treatment (P=0.60). FFA levels were suppressed by 75% during niacin treatment compared with placebo (P<0.01). Glucose levels were stable in all study arms (Tables 2 and 3).
Figure 4. FFA concentrations.
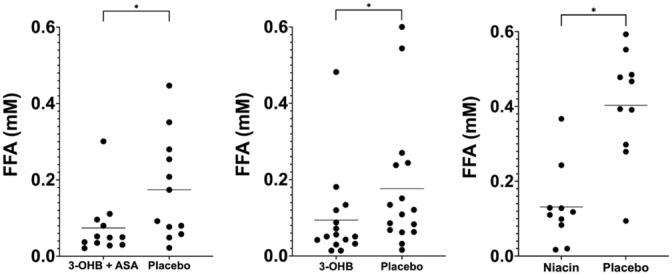
FFA levels in all 3 study groups compared with placebo. 3‐OHB indicates 3‐hydroxybutyrate; ASA, aspirin; and FFA, free fatty acid. *P<0.05.
Safety
No serious adverse events were observed during the ketone study day. However, 1 patient experienced facial heat sensation during ketone infusion. Flushing and facial heat sensations were experienced by 9 of 10 (90%) of the patients during the niacin study day. One patient experienced vasovagal syncope after niacin administration, and the study day was discontinued for this patient. Four patients experienced nausea and vomiting after niacin intake. To avoid this, the niacin dose was then reduced to 500 mg.
DISCUSSION
The present study investigated the acute effects of circulating 3‐OHB on prostaglandin release, circulating FFAs, and hemodynamics. We also tested the effects of the even stronger HCA2 stimulator niacin. The main findings of the present study were as follows: First, CO increased by 2.3 L/min and left ventricular ejection fraction by 6 percentage points during 3‐OHB infusion as compared with placebo, similar to our previous findings. 4 Second, the CO increase during 3‐OHB infusion was not affected by aspirin‐induced inhibition of the HCA2 downstream cyclooxygenase enzyme. Third, PGI2, PGE2, and PGD2 plasma levels did not change during 3‐OHB infusion compared with placebo. Fourth, 3‐OHB reduced circulating FFAs by 58%, consistent with our previous data. 4 Fifth, niacin, a high‐affinity HCA2 agonist, did not affect CO compared with placebo even though it increased prostaglandin levels. Last, niacin reduced FFA levels to the same extent as during 3‐OHB infusion but did not change CO. In summary, our findings support that acute hemodynamic changes during 3‐OHB infusion are not mediated through HCA2 stimulation, prostaglandin release, or HCA2‐induced FFA reduction.
HCA2 Stimulation by 3‐OHB, Prostaglandin Release, and Hemodynamic Effects
PGE2 levels are elevated in patients with heart failure, 22 and PGI2 treatment of patients with HFrEF increases cardiac index. 23 We hypothesized that 3‐OHB is involved in prostaglandin release and that prostaglandins mediate a beneficial hemodynamic response. HCA2 activation is known to promote production and release of PGD2 and PGE2 from macrophages, 17 and macrophages are involved in the HCA2‐mediated PGD2 and PGE2 production and cutaneous flushing during niacin treatment. 24 Activation of the G‐protein‐coupled receptor HCA2 initiates a cascade of downstream effects in the macrophages. These effects include activation of the cytosolic phospholipase A2 and release of arachidonic acid, which are converted into prostaglandins by cyclooxygenase enzymes. 17 An animal study has shown that 3‐OHB activates HCA2 and is tissue‐protective through release of prostaglandin from monocytes and macrophages. 25
3‐OHB activates HCA2 with an EC50 of ≈750 μmol/L. 26 In the present study, we reached levels of 3‐OHB that were 4 times higher. We therefore assume that the HCA2 receptor was fully stimulated. Intravenous aspirin is documented to fully suppress prostaglandin levels after 5 minutes and until 48 hours after administration. 18 In the present study, 3‐OHB infusion did not affect circulating prostaglandin levels (PGE2, PGI2, PGD2) compared with placebo (Table 2). Thus, this study showed that 3‐OHB infusion did not change HCA2‐mediated prostaglandin release and that PGI2, PGE2, and PGD2 were not involved in the acute hemodynamic effects of 3‐OHB.
Niacin Does Not Affect Hemodynamics
Niacin is an even stronger HCA2 agonist than 3‐OHB with an EC50 of 0.1 μmol/L. 26 Therefore, niacin was chosen as a positive control in the present study. We reached niacin plasma levels of 9.7 mg/mL (≈79 430 μmol/L) after niacin treatment. Niacin stimulates prostaglandin release, 27 resulting in vasodilation and cutaneous flushing. 28 , 29 The present study confirmed the increase in prostaglandin release (PGD2) during niacin treatment in patients with HFrEF. However, no hemodynamic changes were observed during niacin treatment when compared with placebo. The vasodilatory effect of niacin is predominantly described in the cutaneous vessels. 17 The lack of significant change in SVR following niacin administration may therefore be explained by isolated cutaneous vasodilation and thus is only a minor effect on SVR. Other potential explanations of no change of SVR include the possibility of a rapid and transient effect of oral niacin, 30 compensatory hemodynamic reflexes to counteract the vasodilatory effect and that most patients were on angiotensin‐converting enzyme inhibitors, and β‐blocker treatment. However, no increase in CO corroborates that prostaglandin downstream release of HCA2 stimulation does not induce any major hemodynamic changes.
HCA2 ‐Mediated Metabolic Effects
It is well known that HCA2 stimulation on adipocytes decreases FFA levels 19 , 20 (Figure 1). In the adipocyte, HCA2 stimulation inhibits adenylyl cyclase and decreases cAMP production. This results in decreased conversion of triglyceride to FFA via the hormone‐sensitive lipase and adipocyte triglyceride lipase, which decrease the level of FFA release from the adipocytes. 15 This downstream regulation is not cyclooxygenase enzyme dependent or related to prostaglandin release. 16 In the present study, 3‐OHB infusion decreased FFA levels by 58% as compared with placebo. Aspirin administration did not affect the decrease in FFA levels (Table 2 and Figure 3). The present study further confirmed that niacin decreased FFA levels (Table 3 and Figure 3). These results show that suppression of circulating FFA levels through HCA2 stimulation by 3‐OHB involves a cyclooxygenase‐independent mechanism.
In an experimental study, 3‐OHB infusion increased cardiac FFA uptake. 5 The beneficial hemodynamic response during 3‐OHB infusion could therefore be mediated through reduced plasma FFA levels and an altered myocardial substrate metabolism. 3 , 10 However, during niacin treatment, FFA levels were reduced to similar levels as during 3‐OHB infusion without significant hemodynamic changes. We did not measure myocardial FFA uptake, but it is well documented that plasma FFA levels correlate with myocardial FFA uptake. 31 , 32 Our findings indicate that the increase in CO during 3‐OHB infusion was not mediated through changes in circulating FFAs. This is further supported by other studies reporting no change in myocardial FFA uptake during 3‐OHB infusion 3 and no impact on left ventricular ejection fraction. 33
Mechanism Behind Hemodynamic Effect of 3‐OHB
3‐OHB infusion increased CO through an increase in stroke volume and heart rate and a decrease in SVR (Table 2). Further studies are needed to investigate whether the CO increase involves afterload reduction or increased contractility. Whether the changes in hemodynamic response during 3‐OHB infusion are mediated through a direct metabolic effect involving increased myocardial 3‐OHB uptake and metabolization remains unknown and warrants further studies.
Study Limitations
This study investigated the acute effects of 3‐OHB and niacin on hemodynamics and metabolism. Long‐term 3‐OHB treatment may have different effects due to HCA2 activation and prostaglandin release. Furthermore, the present study did not inhibit the HCA2 receptor directly. However, HCA2 antagonists are unavailable for human use. The infusion of 3‐OHB consisted of 50% l‐enantiomer and 50% d‐enantiomer. Only the d‐enantiomer is endogenously produced. However, HCA2 has affinity for both enanatiomers. 20
The data pertaining to patients who received ketone and placebo without aspirin were obtained from a previous study. 4 Although the study design utilized a retrospective cohort, the participants were sourced from the same medical center and were investigated by the same research team. Furthermore, all measurements were performed using the same equipment as in the current study, leading to the assumption that the 2 groups can be compared.
Circulating prostaglandin levels did not change significantly during 3‐OHB infusion compared with placebo. However, circulating prostaglandins are very unstable, so the lack of change in circulating prostaglandins during 3‐OHB infusion should be interpreted cautiously. We argue that prostaglandins were not involved in ketone‐related increase in CO, since aspirin in the given dose is documented to block prostaglandin release and production. 18
The ketone body Na‐3‐OHB dissociates into 3‐OHB− and Na+. 3‐OHB− is a weak base, which is why pH increases during 3‐OHB infusion.Alkalosis may mediate vasodilation and afterload reduction and thus increase CO. However, in most vascular beds, alkalosis either has no effect on vascular tone or causes vasoconstriction, 34 and it is unlikely that alkalosis is the only mediator of the CO increase. First, the increase in pH during 3‐OHB infusion was minimal (from 7.38 to 7.46). Second, pH did not decrease during the placebo period in the group that started with 3‐OHB, whereas CO decreased during the placebo period.
There was a statistically significant increase in the peak 3‐OHB levels in the aspirin‐receiving group when compared with the nonaspirin group. Nonetheless, we deem it improbable that the observed difference in peak circulating 3‐OHB levels can be attributed to the effect of aspirin treatment, because no effect of aspirin on ketone metabolism is known to exist. Rather, it is plausible that these fluctuations could be attributed to variations in physiological parameters or a type 1 error.
Despite active attempts to recruit female patients, we only managed to include 1. The hemodynamic findings for this 1 female participant were similar to those of the male participants.
CONCLUSIONS
In conclusion, our data demonstrated that the increase in CO during 3‐OHB infusion in patients with HFrEF is not mediated through HCA2 stimulation, prostaglandin release, or suppression of circulating FFAs.
Sources of Funding
The study was supported by Aarhus University, The Independent Research Fund Denmark (grant no. 8020‐00120A), The Novo Nordisk Foundation (grant no. NNF17OC0028230), The Lundbeck Foundation (grant no. R231‐2016‐2716), and Director Kurt Boennelycke and wife Grethe Boennelycke's Foundation.
Disclosures
Dr Wiggers has been the principal or a subinvestigator in studies involving the following pharmaceutical companies: MSD, Bayer, Daiichi‐Sankyo, Novartis, Novo Nordisk, Sanofi‐Aventis, and Pfizer. The remaining authors have no disclosures to report.
This manuscript was sent to Yen‐Hung Lin, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 12.
REFERENCES
- 1. Cahill GF. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258 [DOI] [PubMed] [Google Scholar]
- 2. Møller N. Ketone body, 3‐hydroxybutyrate: minor metabolite–major medical manifestations. J Clin Endocrinol Metab. 2020;105:1–9. doi: 10.1210/clinem/dgaa370 [DOI] [PubMed] [Google Scholar]
- 3. Gormsen LC, Svart M, Thomsen HH, Søndergaard E, Vendelbo MH, Christensen N, Tolbod LP, Harms HJ, Nielsen R, Wiggers H, et al. Ketone body infusion with 3‐hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study. J Am Heart Assoc. 2017;6:1–11. doi: 10.1161/JAHA.116.005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nielsen R, Møller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frøkiær J, Eiskjaer H, Jespersen NR, et al. Cardiovascular effects of treatment with the ketone body 3‐hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139:2129–2141. doi: 10.1161/CIRCULATIONAHA.118.036459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, et al. The failing heart utilizes 3‐hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4:1–19. doi: 10.1172/jci.insight.124079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bedi KC, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. doi: 10.1161/CIRCULATIONAHA.115.017545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. β‐Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl‐CoA content. Am J Physiol Heart Circ Physiol. 2003;285:1626–1631. doi: 10.1152/ajpheart.00332.2003 [DOI] [PubMed] [Google Scholar]
- 9. Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS, Arany Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science. 2020;370:364–368. doi: 10.1126/science.abc8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monzo L, Sedlacek K, Hromanikova K, Tomanova L, Borlaug BA, Jabor A, Kautzner J, Melenovsky V. Ketone body metabolism in failing heart. Metabolism. 2020;115:154452. doi: 10.1016/j.metabol.2020.154452 [DOI] [PubMed] [Google Scholar]
- 11. Ho KL, Zhang L, Wagg C, Al Batran R, Gopal K, Levasseur J, Leone T, Dyck JRB, Ussher JR, Muoio DM, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 2019;115:1606–1616. doi: 10.1093/cvr/cvz045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cahill GFJ, Veech RL. Ketoacids? Good medicine? Trans Am Clin Climatol Assoc. 2003;114:143–149. [PMC free article] [PubMed] [Google Scholar]
- 13. Abdul Kadir A, Clarke K, Evans RD. Cardiac ketone body metabolism. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165739. doi: 10.1016/j.bbadis.2020.165739 [DOI] [PubMed] [Google Scholar]
- 14. Puchalska P, Crawford PA. Multi‐dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262–284. doi: 10.1016/j.cmet.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Offermanns S. Hydroxy‐carboxylic acid receptor actions in metabolism. Trends Endocrinol Metabol. 2017;28:227–236. doi: 10.1016/j.tem.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 16. Blad CC, Ahmed K, IJzerman AP, Offermanns S. Biological and pharmacological roles of HCA receptors. Adv Pharmacol. 2011;62:219–250. [DOI] [PubMed] [Google Scholar]
- 17. Hanson J, Gille A, Offermanns S. Role of HCA2 (GPR109A) in nicotinic acid and fumaric acid ester‐induced effects on the skin. Pharmacol Ther. 2012;136:1–7. doi: 10.1016/j.pharmthera.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 18. Nagelschmitz J, Blunck M, Kraetzschmar J, Ludwig M, Wensing G, Hohlfeld T. Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers. Clin Pharmacol. 2014;6:51–59. doi: 10.2147/CPAA.S47895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lauring B, Taggart AKP, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115. doi: 10.1126/scitranslmed.3003877 [DOI] [PubMed] [Google Scholar]
- 20. Taggart AKP, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, et al. (D)‐β‐hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA‐G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200 [DOI] [PubMed] [Google Scholar]
- 21. Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta‐regression. J Am Coll Cardiol. 2013;61:440–446. doi: 10.1016/j.jacc.2012.10.030 [DOI] [PubMed] [Google Scholar]
- 22. Punzengruber C, Stanek B, Sinzinger H, Silberbauer K. Bicyclo‐prostaglandin E2 metabolite in congestive heart failure and relation to vasoconstrictor neurohumoral principles. Am J Cardiol. 1986;57:619–623. doi: 10.1016/0002-9149(86)90846-5 [DOI] [PubMed] [Google Scholar]
- 23. Califf RM, Adams KF, McKenna WJ, Gheorghiade M, Uretsky BF, McNulty SE, Darius H, Schulman K, Zannad F, Handberg‐Thurmond E, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the Flolan international randomized survival trial (FIRST). Am Heart J. 1997;134:44–54. doi: 10.1016/S0002-8703(97)70105-4 [DOI] [PubMed] [Google Scholar]
- 24. Benyó Z, Gille A, Kero J, Csiky M, Suchánková MC, Nüsing RM, Moers A, Pfeffer K, Offermanns S. GPR109A (PUMA‐G/HM74A) mediates nicotinic acid‐induced flushing. J Clin Invest. 2005;115:3634–3640. doi: 10.1172/JCI23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Müller‐Fielitz H, Pokorná B, Vollbrandt T, Stölting I, Nadrowitz R, et al. The b‐hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat Commun. 2014;5:1–11. doi: 10.1038/ncomms4944 [DOI] [PubMed] [Google Scholar]
- 26. Gille A, Bodor ET, Ahmed K, Offermanns S. Nicotinic acid: pharmacological effects and mechanisms of action. Annu Rev Pharmacol Toxicol. 2008;48:79–106. doi: 10.1146/annurev.pharmtox.48.113006.094746 [DOI] [PubMed] [Google Scholar]
- 27. Morrow JD, Parsons WG, Roberts LJ. Release of markedly increased quantities of prostaglandin D2 in vivo in humans following the administration of nicotinic acid. Prostaglandins. 1989;38:263–274. doi: 10.1016/0090-6980(89)90088-9 [DOI] [PubMed] [Google Scholar]
- 28. Eklund B, Kaijser L, Nowak J, Wennmalm A. Prostaglandins contribute to the vasodilation induced by nicotinic acid. Prostaglandins. 1979;17:821–830. doi: 10.1016/0090-6980(79)90055-8 [DOI] [PubMed] [Google Scholar]
- 29. Kaijser L, Eklund B, Olsson AG, Carlson LA. Dissociation of the effects of nicotinic acid on vasodilatation and lipolysis by a prostaglandin synthesis inhibitor, indomethacin, in man. Med Biol. 1979;57:114–117. [PubMed] [Google Scholar]
- 30. Ekström‐Jodal B, Harthon L, Häggendal E, Malmberg R, Svedmyr N. Influence of nicotinic acid and pentaerythritoltetranicotinate (Perycit®, Bofors) on the cardiac output in man. Pharmacologia Clinica. 1970;2:86–89. doi: 10.1007/BF00420712 [DOI] [Google Scholar]
- 31. Tuunanen H, Engblom E, Naum A, Scheinin M, Någren K, Airaksinen J, Nuutila P, Iozzo P, Ukkonen H, Knuuti J. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: evidence of relationship with insulin resistance and left ventricular dysfunction. J Card Fail. 2006;12:644–652. doi: 10.1016/j.cardfail.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 32. Most AS, Brachfeld N, Gorlin R, Wahren J. Free fatty acid metabolism of the human heart at rest. J Clin Invest. 1969;48:1177–1188. doi: 10.1172/JCI106082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nielsen R, Nørrelund H, Kampmann U, Kim WY, Ringgaard S, Schär M, Møller N, Bøtker HE, Wiggers H. Failing heart of patients with type 2 diabetes mellitus can adapt to extreme short‐term increases in circulating lipids and does not display features of acute myocardial lipotoxicity. Circ Heart Fail. 2013;6:845–852. doi: 10.1161/CIRCHEARTFAILURE.113.000187 [DOI] [PubMed] [Google Scholar]
- 34. Boedtkjer E, Hansen KB, Boedtkjer DMB, Aalkjaer C, Boron WF. Extracellular HCO3‐ is sensed by mouse cerebral arteries: regulation of tone by receptor protein tyrosine phosphatase γ. J Cereb Blood Flow Metab. 2016;36:965–980. doi: 10.1177/0271678X15610787 [DOI] [PMC free article] [PubMed] [Google Scholar]