

Abstract
Background
Identifying new therapeutic targets for preventing the myocardial ischemia–reperfusion injury would have profound implications in cardiovascular medicine. Myocardial ischemia–reperfusion injury remains a major clinical burden in patients with coronary artery disease.
Methods and Results
We studied several key mechanistic pathways known to mediate cardioprotection in myocardial ischemia–reperfusion in 2 independent genetic models with reduced cardiac phosphoinositide 3‐kinase‐α (PI3Kα) activity. P3Kα‐deficient genetic models (PI3KαDN and PI3Kα‐Mer‐Cre‐Mer) showed profound resistance to myocardial ischemia–reperfusion injury. In an ex vivo reperfusion protocol, PI3Kα‐deficient hearts had an 80% recovery of function compared with ≈10% recovery in the wild‐type. Using an in vivo reperfusion protocol, PI3Kα‐deficient hearts showed a 40% reduction in infarct size compared with wild‐type hearts. Lack of PI3Kα increased late Na+ current, generating an influx of Na+, facilitating the lowering of mitochondrial Ca2+, thereby maintaining mitochondrial membrane potential and oxidative phosphorylation. Consistent with these functional differences, mitochondrial structure in PI3Kα‐deficient hearts was preserved following ischemia–reperfusion injury. Computer modeling predicted that PIP3, the product of PI3Kα action, can interact with the murine and human NaV1.5 channels binding to the hydrophobic pocket below the selectivity filter and occluding the channel.
Conclusions
Loss of PI3Kα protects from global ischemic–reperfusion injury linked to improved mitochondrial structure and function associated with increased late Na+ current. Our results strongly support enhancement of mitochondrial function as a therapeutic strategy to minimize ischemia–reperfusion injury.
Keywords: ischemia–reperfusion, mitochondria, NaV1.5, PI3Kα
Subject Categories: Computational Biology, Contractile function, Calcium Cycling/Excitation-Contraction Coupling, Ischemia, Metabolism
Nonstandard Abbreviations and Acronyms
- Akt
protein kinase B
- DN
dominant negative
- IR
ischemia–reperfusion
- OCR
oxygen consumption rate
- PI3Kα
phosphoinositide 3‐kinase‐α
- PIP3
phosphatidylinositol (3,4,5)‐trisphosphate
- RCR
respiratory control ratio
- RIP
phospho‐receptor‐interacting serine/threonine‐protein kinase
- WT
wild type
Research Perspective.
What Is New?
Genetic loss of phosphoinositide 3‐kinase‐α markedly protects the heart from global ischemic–reperfusion injury.
Phosphoinositide 3‐kinase‐α–deficient hearts display preserved mitochondrial function and maintained ATP production.
Phosphoinositide 3‐kinase‐α–deficient hearts have increased Na+ influx via NaV1.5 channels, possibly due to the lack of blocking effect of phosphatidylinositol (3,4,5)‐trisphosphate) on NaV1.5 channels.
What Question Should Be Addressed Next?
Protecting mitochondrial structure and function to reduce myocardial ischemia–reperfusion damage.
Stimulating the mitochondrial sodium‐calcium exchanger to maintain mitochondrial ATP production in the stressed myocardium.
Coronary artery disease is the leading cause of death worldwide, and myocardial ischemia–reperfusion (IR) injury associated with restoring blood supply to the ischemic myocardium limits therapeutic effects. 1 , 2 , 3 Although the cause of IR injury seems rather complex, mitochondrial function is generally considered the predominant determinant for cardiomyocyte viability following IR injury. 4 , 5 , 6 In addition to sustaining the high energy demand of the myocardium, mitochondria can also trigger cell death and are a major source of reactive oxygen species. In this context, the functional state of mitochondria in cardiomyocytes during IR injury is critical to maintaining mitochondrial and cellular homeostasis. 3 , 7
The chronic loss of phosphoinositide 3‐kinase alpha (PI3Kα) signaling is known to increase the susceptibility to myocardial infarction 8 , 9 and pressure‐induced 10 heart failure. Surprisingly, PI3Kα‐dominant negative hearts (p110α‐DN) are resistant to IR injury. However, the mechanism of this resistance remains elusive. 11 , 12 PI3Kα signaling regulates the reperfusion injury salvage kinase pathway, 8 , 11 , 12 ischemia‐induced necroptosis, 12 , 13 and the late sodium current, 13 , 14 which in turn can affect mitochondrial Ca2+ handling and homeostasis, thereby potentially allowing all these pathways to exert a protective effect in IR injury. 4 , 6 Using 2 genetic models of PI3Kα‐deficiency, p110α‐dominant negative (DN) (constituent) 8 , 11 , 12 and p110α‐Mer (inducible; αMHC‐Cre‐ER), we demonstrated that loss of PI3Kα signaling results in less IR injury and reperfusion arrhythmias. We linked these changes to improved mitochondrial structure and function. Increased late sodium current in PI3Kα‐deficient cardiomyocytes is supported by our modeling data demonstrating the ability of phosphatidylinositol (3,4,5)‐trisphosphate (PIP3), the product of PI3Kα, to block the NaV1.5 channels.
METHODS
The data supporting findings of this study are available from the corresponding author upon reasonable request.
Experimental Animal Protocols
C57Bl/6J littermate wild‐type controls, p110α‐DN (C57Bl/6J background), and p110α‐Cre (C57Bl/6J background) were used in this study. We investigated 2 genetic models of PI3Kα‐deficiency: p110α‐DN (constituent) 12 and p110α‐Mer (inducible; αMHC‐Cre‐ER), whereby deletion of p110α was achieved after 2 days of tamoxifen treatment (40 mg/kg; oral gavage). 8 , 14 , 15 , 16 Mice were housed on a 12‐hour light/12‐hour dark cycle with ad libitum access to chow diet (#5001 from Lab Diet, St. Louis, MO, with 13.5% kcal from fat) and water. All experiments were performed in accordance with the University of Alberta institutional guidelines, which conformed to guidelines published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised 2011).
Ischemia Reperfusion in Ex Vivo Perfused Hearts
Mice were heparinized and anesthetized with 2% isoflurane. Hearts were excised, mounted on the Langendorff system, and perfused in retrograde at 80 mm Hg and 37 °C with modified Krebs–Henseleit solution (in mmol/L: 116 NaCl, 3.2 KCl, 2.0 CaCl2, 1.2 MgSO4, 25 NaHCO3, 1.2 KH2PO4, 11 glucose, 0.5 EDTA, and 2 pyruvate), which was continuously oxygenated with 95% O2/5% CO2 to maintain a pH of 7.4. 11 , 12 A water‐filled balloon connected to a pressure transducer was inserted into the left ventricular chamber, and the pressure changes were recorded by a PowerLab system (AD Instruments, USA). After stabilization and 10 minutes baseline recording, global ischemia was induced by interrupting the perfusion for 30 minutes followed by 40 minutes reperfusion. The coronary effluents were collected at baseline and initial reperfusion to determine creatine kinase activity using a colorimetric commercial kit (BioAssay). Following reperfusion, ventricles were snap‐frozen in liquid N2 and stored at −80 °C for further processing.
In Situ Imaging of Mitochondrial Membrane Potential in Cardiac Tissue
Hearts were perfused as described in the previous section (ischemia reperfusion in ex vivo perfused hearts). Florescent probes were added to the perfusion buffer. Tetramethylrhodamine, ethyl ester (100 nmoL/L) and Hoechst (0.5 μmoL/L) were added at the beginning of reperfusion, and Sytox green (1 μmoL/L) was added at 20 minutes of the reperfusion phase. At 30 minutes of the reperfusion phase, the fluorescent probes were washed out for 10 minutes. At the end of the reperfusion phase, a longitudinal slice of the left ventricle free wall was cut for immediate confocal imaging.
Ischemia Reperfusion of the In Vivo Hearts
Twelve‐week‐old mice were anesthetized with 2.5% isoflurane, intubated, and underwent left thoracotomy in the fourth intercostal space. The left anterior descending coronary artery was encircled and ligated with a 6‐0 silk suture for 30 minutes; the reperfusion was achieved by releasing the suture. 17 Three hours later, the mice were euthanized. Hearts were quickly excised, sectioned in 7 slices from apex to base (base slice has been discarded), incubated in 1% triphenyl tetrazolium chloride (Sigma‐Aldrich, Canada) at 37 °C for 20 minutes, and placed into 10% formalin. The heart slices were imaged 3 to 4 days later using Infinity1 camera (Teledyne Lumenera, Canada) mounted on the Olympus SZ61 microscope (Microscopy Technologies, Evident Corporation, Japan). The infarct areas were identified by a blinded observer and traced using ImageJ 1.54c (National Institutes of Health, USA).
Ex Vivo Epicardial Optical Mapping of Voltage and Ca2+
Mice were heparinized and anesthetized with 2% isoflurane. Hearts were excised, mounted on the Langendorff system, perfused in retrograde at 80 mm Hg and 37 °C with modified Normal Tyrode's solution (in mmol/L: 135 NaCl, 5.4 KCl, 1.2 CaCl2, 1.0 MgCl2, 1.0 NaH2PO4, 10 HEPES, 10 taurine, and 10 glucose) containing 1 g/L albumin and 10 μmoL/L (−)‐blebbistatin, and oxygenated with 100% O2. 14 After initial perfusion for 5 to 10 minutes, hearts were loaded with Ca2+‐sensitive dye Rhod‐2AM (ThermoFisher Scientific, Canada; 80 μL per heart of 1 g/L solution) for 15 minutes followed by loading with voltage‐sensitive dye RH237 (ThermoFisher Scientific; 12 μL per heart of 1 g/L solution) for 6 minutes. MiCAM Ultima (Brainvision Inc., Japan) was used to record and process optical signals from the hearts. Images were recorded at a frame rate of 1 kHz. Hearts were paced at 12 Hz for 1.5 seconds (applied to the left ventricle) and then allowed to excite autonomously. Baseline measurements were taken 5 minutes after loading the voltage dye. Action potential durations are reported as averages for the heart. Global ischemia was induced by interrupting the perfusion for 30 minutes, followed by reperfusion.
Measurements of Mitochondrial Respiration
Mitochondrial oxygen consumption was measured in permeabilized mouse cardiac fibers following IR injury using an Oxygraph 2k (OROBOROS Instruments, Innsbruck, Austria). At the end of reperfusion, fresh cardiac fibers were isolated from the left ventricles of the perfused hearts as previously described. 18 , 19 Briefly, ventricular tissues were dissected under a dissecting microscope in ice‐cold isolation buffer (2.77 mmol/L CaK2EGTA, 7.23 mmol/L K2EGTA, 20 mmol/L imidazole, 20 mmol/L taurine, 49 mmol/L K‐MES, 3 mmol/L K2HPO4, 9.5 mmol/L MgCl2, 5.7 mmol/L ATP, 1 μmol/L leupeptin, and 15 mmol/L phosphocreatine). A 3–5 mm strip of the anterior left ventricle was isolated and cleaned from the remaining fats and vessels. Myocardial strips were disassembled into bundles (5–6 fibers each, 1 mm wide, 3–4 mm long). Fibers were permeabilized in isolation buffer containing saponin (100 μg/mL) for 20 minutes followed by washing 3 times for 5 minutes in ice‐cold respiration buffer (0.5 mmol/L EGTA, 3 mmol/L MgCl2, 20 mmol/L taurine, 10 mmol/L KH2PO4, 20 mmol/L HEPES, 1 g/L fatty‐acid‐free BSA, 60 mmol/L potassium lactobionate, 110 mmol/L D‐sucrose). Fibers were then immediately added to the respiration chamber containing 2 mL respiration buffer. To stimulate basal respiration, glutamate (10 mmol/L) and malate (2 mmol/L) or succinate (10 mmol/L) in the presence of rotenone (2 μmol/L) were added as substrates for complex I or complex II, respectively. ADP (0.5 mmol/L) was added to stimulate ATP‐dependent respiration, which was subsequently inhibited using the ATP synthase inhibitor, oligomycin (0.5 μmol/L). Maximal mitochondrial respiration was recorded after adding step‐wise titrations of 0.1 μmol/L carbonyl cyanide p‐trifluoro‐methoxyphenyl hydrazone (up to 1 μmol/L) as a mitochondrial oxidative phosphorylation uncoupler. Finally, rotenone (2 μmol/L) or antimycin A (5 μmol/L) were used to inhibit complex I or II‐mediated respiration, respectively. Following each step, the oxygen consumption rate (OCR) was measured and normalized to the dry weight of the fiber. The respiratory control ratio (RCR) was calculated as the ratio between ATP‐dependent and basal respiration rates to estimate mitochondrial respiratory efficiency.
Isolation of Cardiac Myocytes
The mouse was anesthetized with 2% inhaled isoflurane. The heart was removed and promptly perfused with a collagenase solution, and cardiomyocytes were isolated as previously described. 10 , 14 , 20 2,3‐Butanedione monoxime was replaced by (−)‐blebbistatin (25 μmol/L; Toronto Research Chemicals) in stopping buffer to inhibit cardiomyocyte contraction after isolation.
Measurements of Late Na+ Current
Myocytes were placed in a bath mounted on top of an inverted microscope (Olympus IX71; Olympus, Canada), and rod‐shaped quiescent myocytes were selected for the study. Myocytes were superfused with modified Tyrode's solution at 35 to 36 °C. Pipettes with a resistance of 1.5–2.5 MOhm filled with Cs+ solution (in mmol/L: 25 CsCl, 5 NaCl, 110 CsOH, 110 aspartic acid, 5 MgATP, 5 EGTA, and 10 HEPES) were zeroed in the solution, then used to form a tight seal, and after that, the membrane under the pipette was ruptured using the zap function of the amplifier and gentle suction. Current and membrane potential was measured using a Multiclamp 700B amplifier (Molecular Devices, USA) in the voltage‐clamp mode. The measured signal was digitized at 10 kHz by 16‐bit analog‐digital board DigiData 1440A (Molecular Devices) under the control of pClamp 10 software (Molecular Devices, USA) and stored for offline analysis. Late Na+ current was measured in nominally K+/Ca2+‐free modified Tyrode's solution as superfusate with 1 μmoL/L nisoldipine (Sigma Aldrich, Canada). The current was elicited by depolarizations from −120 (pre‐pulse) to −40 mV. Tetrodotoxin in citrate buffer (5 μmoL/L tetrodotoxin; Abcam, USA) was used to record background and isolate INa‐L. 14
Measurement of Mitochondrial Ca2+ in Isolated Cardiac Myocytes
An aliquot of myocytes was placed in a 35‐mm culture dish. Fluo‐4‐AM (Invitrogen; 5 μg/mL) and Pluronic F‐127 (Invitrogen; 0.005%) in DMEM were added. The myocytes were incubated for 45 minutes at room temperature (21 °C) to promote compartmentalization of the Fluo‐4‐AM to mitochondria. 21 After that, the solution was changed 3 times to DMEM to wash off the excess of Fluo‐4‐AM, and the myocytes were left to equilibrate at room temperature for 10 minutes. The myocytes were imaged by a Zeiss Axio Observer Z1 inverted epifluorescence microscope using Zeiss Zen software with 40×/63× oil objective lens and maintained at 37°C throughout the experiment at a baseline with 21% O2 and after 30 minutes of hypoxia (<1% O2).
Transmission Electron Microscopy and Quantification of the Images
Small blocks (≈1 mm3) of cardiac tissue were fixed with 2.5% glutaraldehyde (EM grade) saline, contrasted osmium tetroxide, incorporated into resin blocks, cut into 200‐nm slices, and the slices were placed on copper mesh for imaging. Images were obtained on Hitachi H‐7650 TEM. Mitochondria in images were graded by a blinded observer on a scale from 0 to 4, with 0 being “empty” (lacking any cristae) and 4 being normal healthy mitochondria. At least 4 fields of view were graded per biological replicate. The average grade was reported per biological replicate.
Western Blot Analysis
Heart tissue was homogenized using a TissueLyser II in CelLytic M Cell Lysis Reagent (Sigma) with cOmplete and PhosSTOP inhibitors (Roche). Protein concentrations were standardized using a Bradford assay (Bio‐Rad); samples were boiled in a denaturing Laemmli load dye (Bio‐Rad). Samples were separated using SDS‐PAGE to Immobilon polyvinylidene difluoride membranes (Millipore). Polyvinylidene difluoride membranes were stained for total protein as a loading control using MemCode (Thermo). Membranes were probed using the following list of antibodies at 1:1000–1:2000 concentration in Tris‐buffered saline solution with 1.5% BSA, followed by HRP conjugated secondary antibodies (Cell Signaling) at 1:5000 concentration and imaged with ECL (GE Amersham) chemiluminescence using an ImageQuant TL (GE). The proteins were detected using the following antibodies: protein kinase B total (Cell Signaling, 9272), P‐Akt Ser 473 (Cell Signaling, 9271), P‐Akt Thr 308 (Cell Signaling, 9275), ERK 1/2 total (Cell Signaling, 4695), P‐ERK 1/2 (Cell Signaling, 4377), Caspase 3 (Cell Signaling, 9662), Caspase 8 (Cell Signaling, 4927), phospho‐receptor‐interacting serine/threonine‐protein kinase 1 (RIP‐1) (Cell Signaling, 3493), RIP‐3 (Cell Signaling, 75 702), P‐calmodulin‐dependent protein kinase II Thr 286 (Cell Signaling, 12 716), bcl‐2‐like protein 4 (Cell Signaling, 2772), Bcl‐2 homologous antagonist/killer (Cell Signaling, 12 105), Cox4 (Cell Signaling, 4850), mitochondrial calcium uniporter (Cell Signaling, 14 997), and mitochondrial Na+/Ca2+ exchanger (GeneTex, GTX87452), voltage‐dependent anion channel (Abcam, ab14734).
Modeling of NaV1.5 and Its Interaction With 3,4,5‐Phosphatidylinositol Triphosphate
The protein sequences of human (Homo sapiens) and murine (Mus musculus) Na+ channel proteins (NaV1.5) were obtained from the UniProtKB sequence database (https://www.uniprot.org/help/uniprotkb). Sequence homology search in BLAST 22 for human and murine NaV1.5 showed 97% and 100% homology with rat (Rattus norvegicus) NaV1.5, which was used as a template for homology‐based modeling. The template's crystal structure (PDB ID: 6UZ0) 23 was retrieved from the RCSB database (https://www.rcsb.org/). The homology modeling was performed on Swiss‐model server 24 using the user‐specified modeling approach for the uploaded template structure. The binding grid of the flecainide inside the Na+ channel 23 was used to dock PIP3 within human and murine NaV1.5 protein. The complexes of PIP3 with the human and murine NaV1.5 proteins have been evaluated using PROCHECK, ERRAT, and VERIFY3D. 25 , 26 Molecular interaction between protein and ligand was determined by the PLIP server. 27 Normal mode analysis of the PIP3 unbound and bound structure of both the human and murine NaV1.5 protein has been performed in WEBnm@ server, 28 and the residual atomic displacement between the 2 conformers (PIP3 unbound and bound) of both human and murine NaV1.5 protein was calculated. The correlation matrix of different conformers of these 2 proteins was calculated to display the correlation between the motions of all the Cα atoms in the protein structures. Multiple normal modes were compared for the protein structures with the transition from 1 conformation to another of the same protein. The binding energy between the human and murine NaV1.5 proteins and PIP3 was determined using the DINC server. 29 The grid center and dimensions used for docking purposes have also been taken as input for the protein‐ligand energy calculations.
Statistical Analysis
Statistical analyses were carried out using SPSS Statistics 24 software, and statistical significance was defined as P<0.05 (2‐sided). Data were presented in scatterplots with mean±SEM. The differences between the 2 independent groups were evaluated using an independent t test after normality examination. For multiple comparisons, a 1‐way ANOVA test was performed after normality examination, and the Tukey post‐test was used to determine the significance of pairwise comparisons.
RESULTS
PI3Kα and Ischemia Reperfusion Ex Vivo and In Vivo
Two arms of the reperfusion–injury salvage kinase pathway are known to be involved in cardioprotection (Figure 1A). In this study, 2 genetic models of PI3Kα‐deficiency (p110α‐DN—constituent 12 and p110α‐Mer—inducible, Figure 1B) demonstrated marked protection from global IR injury in response to 30‐ and 60‐minute ischemic periods and 40‐minute reperfusion protocol (Figure 1C and 1D). In the case of 60‐minute ischemia, both genetic models partially recovered after ischemia and maintained normal appearance, whereas wild‐type (WT) hearts were noncontractile and appeared pale (Figure 1D inset). However, neither MEK‐1/2‐specific inhibitor (MEK‐162) nor protein kinase B‐specific inhibitor (MK‐2206) affected recovery from IR (Figure 1E and 1F), suggesting that neither arm of the reperfusion–injury salvage kinase pathway is responsible for the enhanced recovery from IR in PI3Kα‐deficient hearts. To confirm that both MEK‐162 and MK‐2206 are inhibiting their respective targets, we performed Western blot analysis of tissue lysates of WT and p110α‐DN hearts following IR and found (1) marked reduction in phosphorylation levels of mitogen‐activated protein kinase encoded by the MAPK3 and MAPK1 in response to application of MEK‐162, and (2) marked reduction in Ser473‐protein kinase B and Thr308‐protein kinase B levels in response to application MK‐2206 (Figure 1G). PI3Kα‐deficient (p110α‐DN) and WT mice were subjected to the in vivo reperfusion by ligating the left anterior descending artery for 30 minutes. Three hours later, the hearts were excised, sectioned, and stained with triphenyl tetrazolium chloride. The p110α‐DN hearts had significantly smaller infarcted areas than WT hearts (Figure 1H and 1I), confirming that the marked protection of PI3Kα‐deficient genotype from IR injury is extended to the in vivo condition.
Figure 1. Risk pathway, PI3Kα, and ischemia–reperfusion injury.
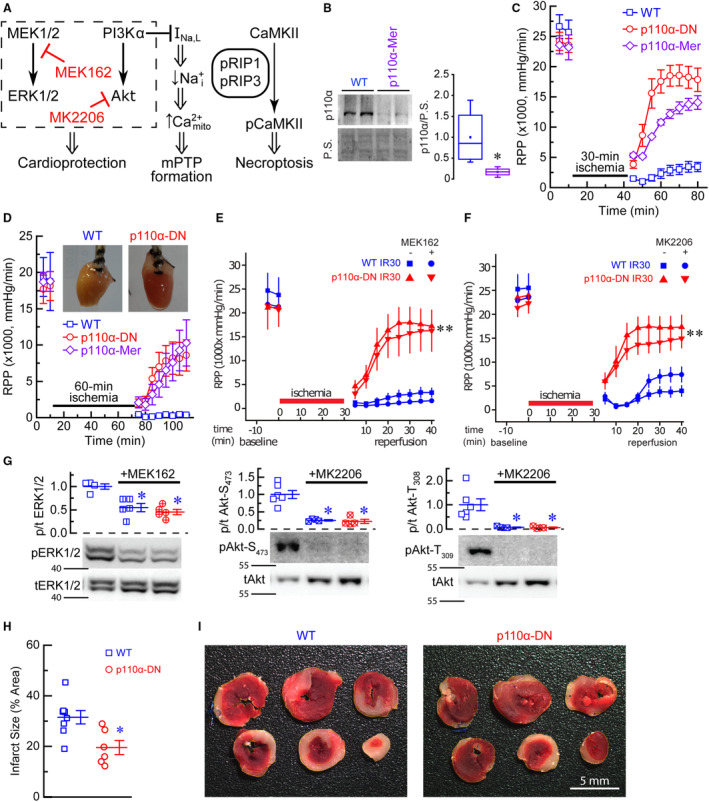
A, Schematic of the risk pathway, a possible link between PI3Kα and mPTP formation, and necroptosis pathways. B, Deletion of p110α after 2 days of tamoxifen treatment (40 mg/kg) in p110α‐Mer compared with non‐Mer (WT) littermate controls (n=4). C, RPP in response to 30‐minute ischemia in WT, constituent PI3Kα‐deficient (p110α‐DN), and inducible PI3Kα‐deficient (p110α‐Mer) excised hearts (n=6–8). D, Rate pressure product (RPP) in response to 60‐minute ischemia in WT (open squares), constituent PI3Kα‐deficient (p110α‐DN, open circles), and inducible PI3Kα‐deficient (p110α‐Mer, open rhombi) excised hearts (n=5–6). Inset: pictures of WT (left) and p110α‐DN (right) hearts. E, Effect of MEK162 on RPP in response to 30‐minute ischemia in WT and constituent PI3Kα‐deficient (p110α‐DN) excised hearts (n=4–6). IR30, 30‐minute ischemia followed by reperfusion. F, Effect of MK2206 on RPP in response to 30‐minute ischemia in WT and constituent PI3Kα‐deficient (p110α‐DN) excised hearts (n=4–6). G, Phosphorylation levels of ERK1/2 in cell lysates from WT, WT+MEK162, and p110α‐DN+MEK162 hearts; phosphorylation levels of pAkt‐S473 in cell lysates from WT, WT+MK2206, and p110α‐DN+MK2206 hearts; phosphorylation levels of pAkt‐T309 in cell lysates from WT, WT+MK2206, and p110α‐DN+MK2206 hearts. H, Infarcted areas as % of total area for WT (blue squares) and p110α‐DN (red circles). I, Representative images of heart sections for WT and p110α‐DN hearts. Data are presented as mean±SEM; statistical significance is calculated using Student t test in (B, H) and 1‐way ANOVA in (E through G); **P<0.05 vs WT after 30‐minute reperfusion (WT IR30) no MEK162 (E), **P<0.05 vs WT after 30‐minute reperfusion (WT IR30) no MK2206 (F), *P<0.05 vs WT (B, G, H). Akt indicates protein kinase B; Ca2+mito, mitochondrial Ca2+; CaMKII, calmodulin‐dependent protein kinase II; ERK1/2, a mitogen‐activated protein kinase encoded by the MAPK3 and MAPK1; INa,L, late Na+ current; MEK1/2, mitogen‐activated protein kinase 1/2; MEK162, MEK1/2 inhibitor; MK2206, Akt inhibitor; mPTP, mitochondrial permeability transition pore; Na+i, intracellular sodium; P.S., protein stain; p110α, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K), catalytic subunit α; PI3Kα, phosphoinositide 3‐kinase α; RIP, phospho‐receptor‐interacting serine/threonine‐protein kinase; and RPP, rate pressure product.
Apoptosis and Necrosis in Ex Vivo Ischemia Reperfusion
Neither initiator caspase 8 (Figure 2A) nor executioner caspase 3 (Figure 2B) were cleaved after reperfusion in both WT and p110α‐DN hearts, but significantly greater levels of caspase 3 were detected in p110α‐DN hearts compared with WT in post‐IR heart tissue (Figure 2B). Terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling staining found no apoptotic nuclei in post IR hearts (Figure 2C). Bcl‐2 family members bcl‐2‐like protein 4 and Bcl‐2 homologous antagonist/killer (commonly associated with apoptosis induction, localized to mitochondrial membranes, and regulated by PI3K signaling) 30 had overall levels unchanged or higher in nonperfused p110α‐DN hearts compared with nonperfused WT hearts (Figure 2D). A comparison of mitochondrial fractions of bcl‐2‐like protein 4 and Bcl‐2 homologous antagonist/killer in post‐IR hearts showed that bcl‐2‐like protein 4 levels were significantly higher in p110α‐DN hearts, but Bcl‐2 homologous antagonist/killer levels were the same for both genotypes (Figure 2E). We further explored the role of the necroptosis cell death pathway in mediating this protection from myocardial IR injury. 13 Mediators of regulated necrosis (RIP‐1 and RIP‐3) were increased in p110α‐DN hearts compared with WT in nonperfused hearts and were minimally detectable in IR hearts (Figure 2F). In WT hearts, RIP‐1 is predominantly lost by the end of ischemia, while RIP‐3 is lost within 10 minutes of reperfusion with an increase in phosphorylated calmodulin‐dependent protein kinase II, a known substrate of RIP3, increased in p110α‐DN hearts compared with WT after IR (Figure 2F). Collectively, these results fail to implicate a dominant role of primary cell death pathways in the resistance to IR injury seen in hearts with reduced PI3Kα activity.
Figure 2. Apoptosis and necrosis in ex vivo ischemia reperfusion.
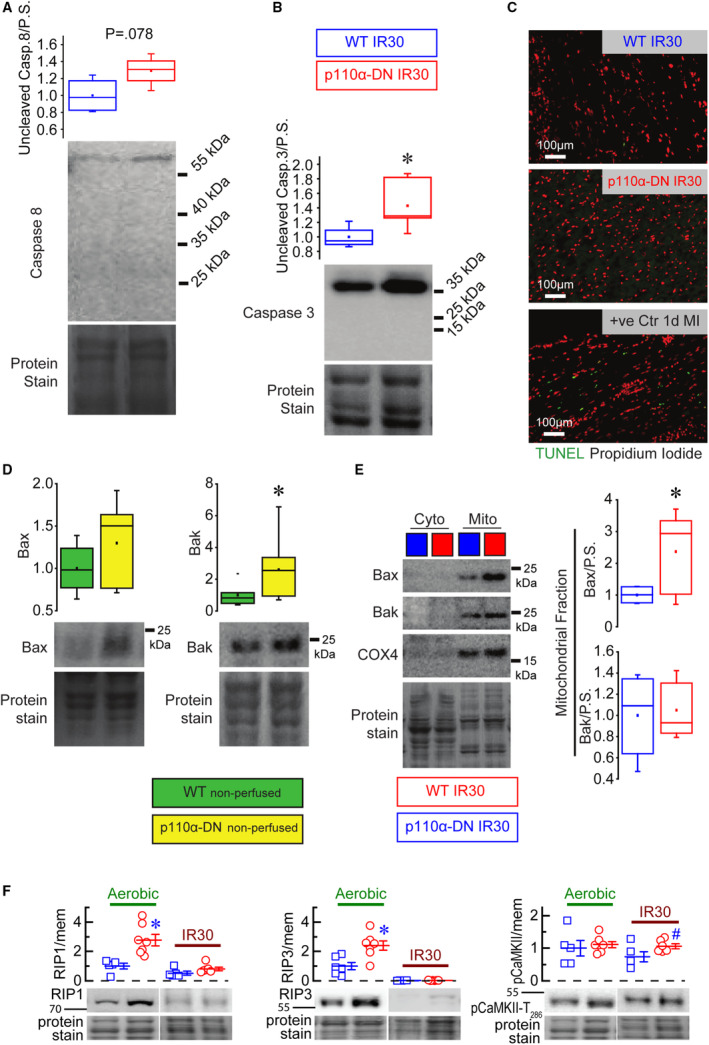
A, Uncleaved caspase (Casp) 8 in lysates from control WT (wild‐type) 30‐minute ischemia after reperfusion (IR30) and PI13Kα‐dominant negative hearts (p110α‐DN) IR30 hearts (n=4). B, Uncleaved caspase 3 in lysates from WT IR30 (30‐minute ischemia followed by reperfusion) and p110α‐DN (constituent PI3Kα‐deficient) IR30 hearts (n=4). C, Terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) staining of the apoptotic nuclei (green) and propidium iodide staining of nuclei (red). D, Bax (bcl‐2‐like protein 4) and Bak (Bcl‐2 homologous antagonist/killer) protein levels in the lysates from nonperfused WT and p110α‐DN hearts (n=6). E, Bax and Bak protein levels in the cytoplasmic (cyto) and mitochondrial (mito) fractions of the lysates from perfused WT IR30 and p110α‐DN IR30 hearts (n=6). F, RIP1 (receptor‐interacting serine/threonine‐protein kinase 1), RIP3 (receptor‐interacting serine/threonine‐protein kinase 3), and pCaMKII (calmodulin‐dependent protein kinase II) protein levels in aerobically perfused WT, aerobically perfused p110α‐DN, 30‐minute ischemia reperfusion WT, and 30‐minute ischemia reperfusion p110α‐DN. Data are presented as mean±SEM; statistical significance is calculated using a 2‐sided t test with Welsch correction in (A, B, D, and E) as well as 1‐way ANOVA in F; *P<0.05 vs WT IR30 (B), *P<0.05 vs WT nonperfused (D), *P<0.05 vs mito WT IR30 (E), *P<0.05 vs aerobic WT (F), # P<0.05 vs WT IR30 (F).
Mitochondrial Function and Respiration in Response to Ischemia Reperfusion
In response to IR, WT hearts lost their hyperpolarized mitochondrial potential, whereas PI3Kα‐deficient hearts (p110α‐DN and p110α‐Mer) preserved it, resulting in WT hearts displaying considerable cellular damage as indicated by sytox green staining (Figure 3A and 3B). Similarly, in response to IR, mitochondrial structure from p110α‐DN hearts had less cristae degradation following IR injury (Figure 3C and 3D). Subjecting isolated cardiomyocytes with preferential stain for mitochondrial Ca2+ to 30‐minute hypoxia showed that p110α‐DN myocytes had lower mitochondrial Ca2+ levels during hypoxia (Figure 3E), and their viability was preserved, unlike WT cardiomyocytes (Figure 3F). Mitochondrial proteins that regulate Ca2+ and Na+ movement across the inner mitochondrial membrane, such as mitochondrial calcium uniporter, mitochondrial Na+/Ca2+ exchanger, and voltage‐dependent anion channel, were not different in the hearts subjected to aerobic perfusion or 30‐minute IR in both genotypes (Figure 3G).
Figure 3. Mitochondrial function and structure in ischemia reperfusion.
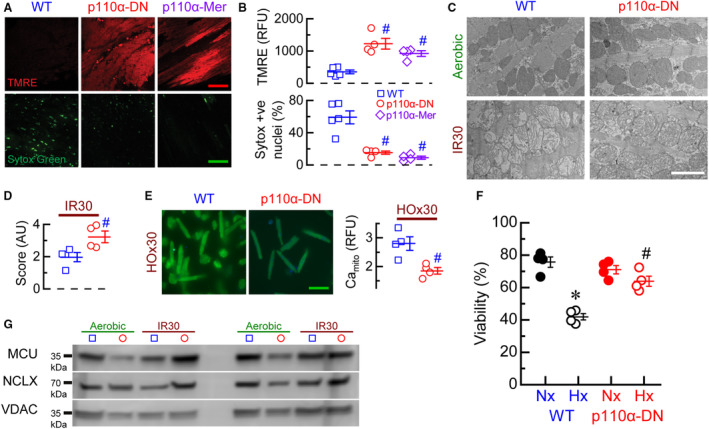
A, Representative images of tetramethylrhodamine ethyl (TMRE, red) and sytox green stainings of in situ heart sections after 30‐minute ischemia and reperfusion for WT (wild‐type), p110α‐DN (constituent PI3Kα‐deficient), and p110α‐Mer (inducible PI3Kα‐deficient) hearts; calibration bars 50=μm. B, Quantification of in situ TMRE and sytox stainings of heart after 30‐minute ischemia and reperfusion for WT, p110α‐DN, and p110α‐Mer hearts. C, Representative images of transmission electron microscopy of sections of aerobically perfused WT, aerobically perfused p110α‐DN, 30‐minute ischemia–reperfusion (IR30) WT, and IR30 p110α‐DN hearts; calibration bar 2=μm. D, Quantification of mitochondria structure of transmission electron microscopy images of sections of aerobically perfused WT, aerobically perfused p110α‐DN, 30‐minute ischemia–reperfusion (IR30) WT, and IR30 p110α‐DN hearts (score: from 0 being “empty” [lacking any cristae] to 4 being normal healthy mitochondria). E, Left: representative images of mitochondrial Ca2+ staining for WT and p110α‐DN myocytes after 30 minutes hypoxia (HO×30); calibration bar 100=μm. Right: quantification of mitochondrial Ca2+ staining (Camito) for WT (blue) and p110α‐DN (red) myocytes after 30‐minute hypoxia. F, Cell viability for WT and p110α‐DN myocytes after 30‐minute normoxia (Nx) or 30‐minute hypoxia (Hx). G, Representative blots for mitochondrial calcium uniporter (MCU), mitochondrial Na+/Ca2+ exchanger (NCLX), and voltage‐dependent anion channel (VDAC) proteins from lysates of aerobically perfused WT, aerobically perfused p110α‐DN, 30‐minute ischemia–reperfusion (IR30) WT, and IR30 p110α‐DN hearts. Data are presented as mean±SEM; statistical significance is calculated using 1‐way ANOVA in (B and F); 2‐sided t test with Welsch correction in (D, E); # P<0.05 vs WT (B), # P<0.05 vs WT IR30 (D), # P<0.05 vs WT HO×30 (E), *P<0.05 vs WT Nx and # P<0.05 vs p110α‐DN Nx (F).
To assess the effect of 30‐minute IR on mitochondrial respiration, 4 groups of hearts were compared: aerobic WT, aerobic p110α‐DN, 30‐minute ischemia followed by 40‐minute reperfusion (IR30) WT, and IR30 p110α‐DN. Isolated fibers from those hearts were subjected to the respiration protocol (Figure 4A) to measure the basal OCR, ATP‐dependent OCR, ATP‐independent OCR, maximal electron‐transport capacity, and nonmitochondrial OCR for complex I (Figure 4B) and complex II (Figure 4C). In aerobic conditions, there was no difference between WT and p110α‐DN genotypes for complex‐I parameters (Figure 4B). However, maximal electron‐transport capacity (state 3) for complex II was significantly higher in p110α‐DN fibers (Figure 4C). After 30‐minute IR (IR30), for complex I, ATP‐dependent OCR and maximal electron‐transport capacity were reduced in WT but not in p110α‐DN fibers (Figure 4B). For complex II, only maximal electron‐transport capacity was reduced in both genotypes (Figure 4C). There were no differences in basal or ATP‐independent (“leak”) respiration for either group in both complex I (Figure 4B) and complex II (Figure 4C). Calculation of RCR for complex I (Figure 4B) and complex II (Figure 4C) showed that RCR was preserved for p110α‐DN fibers for complex I (Figure 4B). In complex II, p110α‐DN fibers showed significantly higher RCR in aerobic conditions, but the RCR was reduced in both genotypes (Figure 4C).
Figure 4. Mitochondrial respiration in isolated fibers.
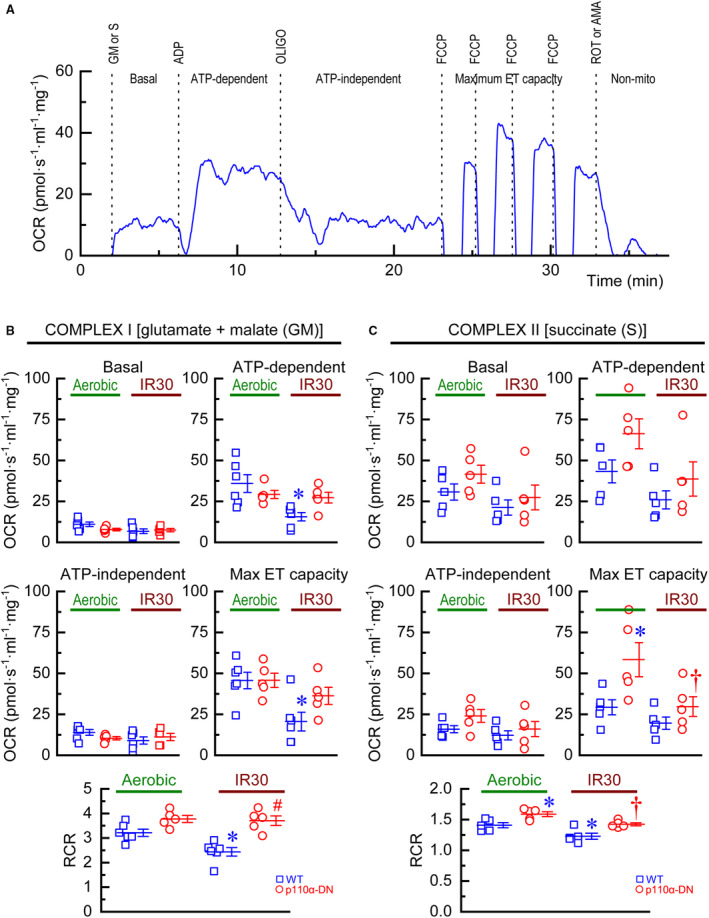
A, Representative time course of oxygen consumption rate (OCR) of wild‐type (WT) fiber showing applications of glutamate+malate (GM) or succinate (S), ADP, oligomycin (OLIGO), carbonyl cyanide p‐trifluoro‐methoxyphenyl hydrazone (FCCP), and rotenone (ROT) or antimycin A (AMA); non‐mito, non‐mitochondrial respiration. B, Complex I: measurements of basal OCR, ATP‐dependent OCR, ATP‐independent OCR, maximal electron‐transport (ET) capacity, and respiratory control ratio (RCR) for WT and p110α‐DN fibers under aerobic conditions (Aerobic) or after 30‐minute ischemia followed by 40‐minute reperfusion (IR30). C, Complex II: measurements of basal OCR, ATP‐dependent OCR, ATP‐independent OCR, maximal electron‐transport (ET) capacity, and respiratory control ratio (RCR) for WT and p110α‐DN fibers under aerobic conditions (Aerobic) or after 30‐minute ischemia followed by 40‐minute reperfusion (IR30). Data are presented as mean±SEM; statistical significance is calculated using 1‐way ANOVA in (B and C); *P<0.05 vs WT Aerobic (B, C), # P<0.05 vs WT IR30, † P<0.05 vs p110α‐DN Aerobic.
Late Na+ Current and Reperfusion Arrhythmias in Response to IR Injury
PI3Kα is known to suppress late Na+ current via the production of PIP3. 14 Late Na+ current (tetrodotoxin‐sensitive current) was markedly larger in cardiomyocytes from PI3Kα‐deficient hearts (p110α‐DN and p110α‐Mer) compared with WT cardiomyocytes (Figure 5A). To investigate reperfusion arrhythmia, we recorded the electrical activity of the heart at 1 to 4 minutes after 30‐minute ischemia in response to 12‐Hz burst stimulation. WT hearts had reperfusion arrhythmias in 4 out of 8 hearts, whereas no reperfusion arrhythmias have been registered in p110α‐DN hearts (Figure 5B). Furthermore, spontaneous frequency measured after 1 minute of reperfusion (when reperfusion arrhythmias are detected) was significantly lower in p110α‐DN hearts compared with littermate WT hearts (Figure 5C). Illustrative examples of the optimal mapping of the reperfusion arrhythmias recorded from 4 distinct locations (Figure 5D) supported by video recordings (Videos S1 through S4) reflect the marked suppression of ventricular arrhythmias in p110α‐DN hearts.
Figure 5. Late Na+ current and reperfusion arrhythmias in PI3Kα mutant hearts.
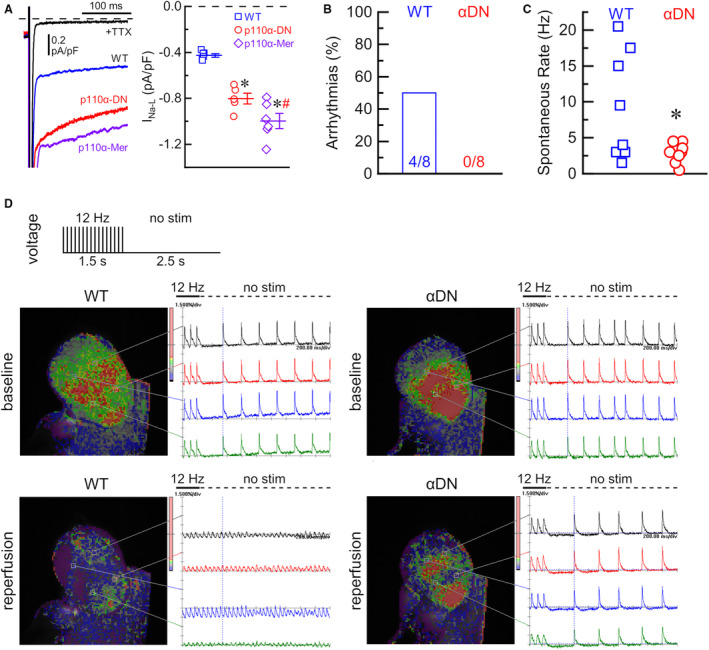
A, Left: representative late Na+ currents (INa‐L) in response to step depolarization from −120 to −40 mV in wild type (WT, blue), constituent PI3Kα‐deficient (p110α‐DN, red), and inducible PI3Kα‐deficient (p110α‐Mer, purple) myocytes, and background current after application of specific Na+ channel blocker, tetrodotoxin (TTX) in black; Right: average current densities of INa‐L (TTX‐sensitive current) for WT (blue), p110α‐DN (red), and p110α‐Mer (purple) myocytes. B, Fraction of arrhythmias at reperfusion in wild‐type (WT) and p110α‐DN (αDN) excised hearts. C, Spontaneous heart rate (no stimulation) after reperfusion in WT and p110α‐DN (αDN) excised hearts. D, Representative images and voltage traces from WT and p110α‐DN (αDN) excised hearts at baseline and reperfusion; no stim, no stimulation. Data are presented as mean±SEM; statistical significance is calculated using 1‐way ANOVA in (A) and 2‐sided t test with Welsch correction in (C); *P<0.05 vs WT (A, C), # P<0.05 vs p110α‐DN (A).
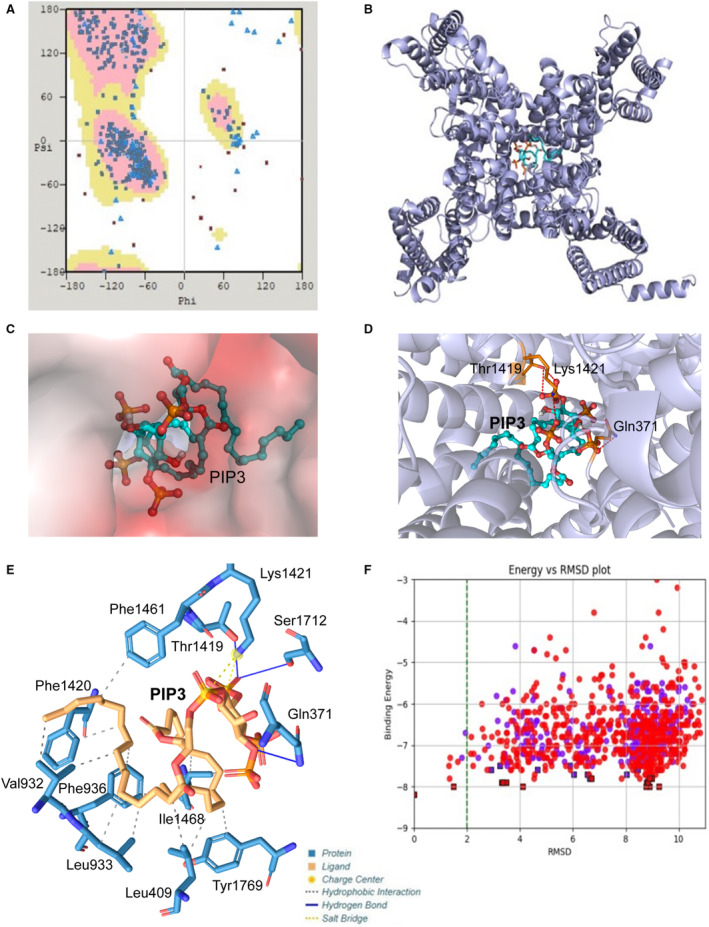
Modeling of the Interaction of PIP3 With Murine and Human NaV1.5
Modeled structure of murine NaV1.5 contains 24 transmembrane helices. Ramachandran statistics show that modeled structure has 95.1% residues in the preferred region (Figure 6A), implying good model accuracy. Docking PIP3 to the modeled protein showed that in the preferred position, PIP3 blocks opening of the channel (Figure 6B, Figure S1); see also the zoomed‐in position (top view) of PIP3 in the cavity (Figure 6C) and relative position of PIP3 to some selected amino acids (Figure 6D). Plotting out the residues that interact with PIP3 indicated that the phospholipid interacts with Lys1421 (part of the selectivity filter) and Thr1419, suggesting that PIP3 binds just below the selectivity filter (from the intracellular side; Figure 6E) in the region similar to where lidocaine, flecainide, and ranolazine bind. 31 Binding energy calculation revealed that murine NaV1.5 has a PIP3 binding energy of −8.20 kcal/mol (Figure 6F). Human NaV1.5 had a structure very similar to murine protein (Figure S2), with 95.4% of residues in the preferred region (Figure 7A). Docking PIP3 to the human NaV1.5 also showed PIP3 in the blocking position (Figure 7B, Figure S3A, Figure S3B; see also the zoomed‐in position [top view] of PIP3 in the cavity (Figure 7C) and relative position of PIP3 to loop 4 and helix 18 [Figure 7D]) similar involvement of equivalent residues Lys1419 (part of the selectivity filter) and Thr1417, similar location of the binding site for PIP3 (intracellular side near selectivity filter; Figure 7E, Figure S3C), and similar binding energy (Figure 7F). Both murine and human NaV1.5 have equivalent residues interacting with PIP3 with similar distances between ligand and residue (Table).
Figure 6. Modeling of the interaction of phosphatidylinositol‐3, 4, 5‐triphosphate (PIP3) with murine NaV1.5.
A, Ramachandran plot of the distribution of residues between preferred regions (pink, 1094, 95.13%), allowed regions (yellow, 36, 3.13%), and outliers (20, 1.74%). B, Top (extracellular) view of the murine NaV1.5 model with PIP3 inside the channel. C, Electrostatic potential surface of the cavity in the murine NaV1.5 binding PIP3. D, Depiction of the region of the murine NaV1.5 interacting with PIP3. E, Structure of PIP3 binding site of NaV1.5 channel. F, Plot of binding energy vs root‐mean‐square deviation (RMSD) for PIP3 and murine NaV1.5 channel. Gln indicates glutamine; Ile, isoleucine; Leu, leucine; Lys, lysine; Phe, phenylalanine; Ser, serine; Thr, threonine; Tyr, tyrosine; and Val, valine.
Figure 7. Modeling of the interaction of phosphatidylinositol‐3, 4, 5‐triphosphate (PIP3) with human NaV1.5.
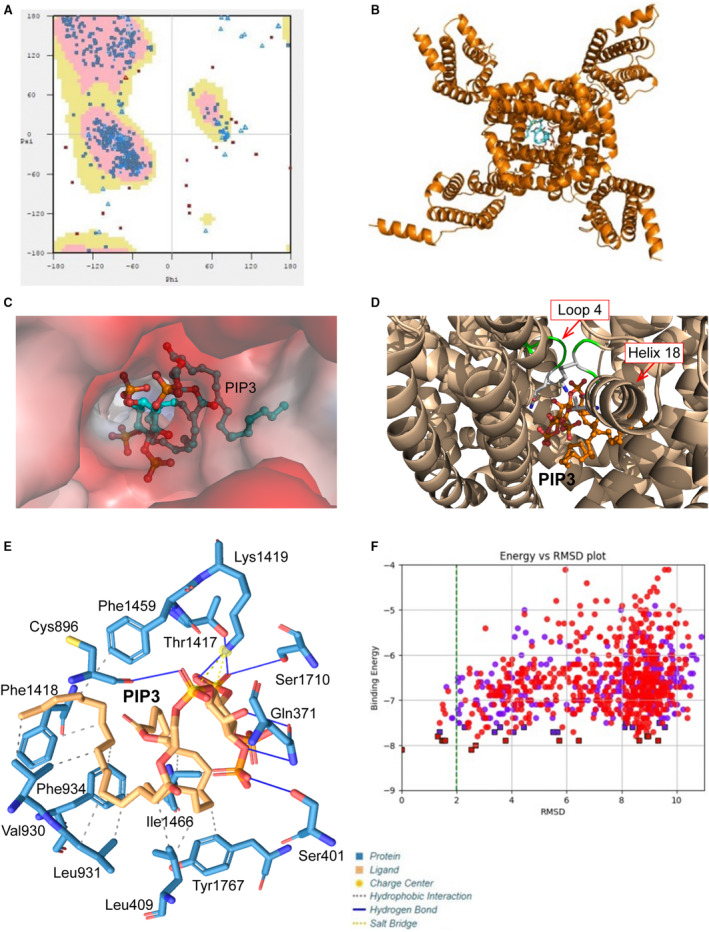
A, Ramachandran plot of the distribution of residues between preferred regions (pink, 1089, 95.36%), allowed regions (yellow, 33, 2.89%), and outliers (20, 1.75%). B, Top (extracellular) view of NaV1.5 model with PIP3 inside the channel. C, Electrostatic potential surface of the cavity in the human NaV1.5 binding PIP3. D, Depiction of the region of the human NaV1.5 interacting with PIP3. E, Structure of PIP3 binding site of NaV1.5 channel. F, Plot of binding energy vs root‐mean‐square deviation (RMSD) for PIP3 and human NaV1.5 channel. Cys indicates cysteine; Gln, glutamine; Ile, isoleucine; Leu, leucine; Lys, lysine; Phe, phenylalanine; Ser, serine; Thr, threonine; Tyr, tyrosine; and Val, valine.
Table 1.
Distances for Various Interactions Between NaV 1.5 Channel and PIP3
| Mouse | Human | |||||
|---|---|---|---|---|---|---|
| Residue | AA | Distance, Å | Residue | AA | Distance, Å | |
| Hydrophobic interactions | 409 | LEU | 3.60 | 409 | LEU | 3.59 |
| 409 | LEU | 3.77 | 409 | LEU | 3.76 | |
| 932 | VAL | 3.63 | 930 | VAL | 3.63 | |
| 932 | VAL | 3.71 | 930 | VAL | 3.71 | |
| 933 | LEU | 3.54 | 931 | LEU | 3.55 | |
| 933 | LEU | 3.78 | 931 | LEU | 3.79 | |
| 936 | PHE | 3.69 | 934 | PHE | 3.68 | |
| 936 | PHE | 3.73 | 934 | PHE | 3.73 | |
| 1420 | PHE | 3.72 | 1418 | PHE | 3.71 | |
| 1461 | PHE | 3.40 | 1459 | PHE | 3.39 | |
| 1468 | ILE | 3.57 | 1466 | ILE | 3.59 | |
| 1468 | ILE | 3.66 | 1466 | ILE | 3.65 | |
| 1769 | TYR | 3.75 | 1767 | TYR | 3.77 | |
| Hydrogen bonds* | 371 | GLN | 3.41 | 371 | GLN | 3.44 |
| – | – | – | 371 | GLN | 2.87 | |
| – | – | – | 401 | SER | 3.80 | |
| – | – | – | 896 | CYS | 2.77 | |
| 1419 | THR | 2.12 | 1417 | THR | 2.12 | |
| – | – | – | 1419 | LYS | 2.64 | |
| 1712 | SER | 3.03 | 1710 | SER | 3.05 | |
| Salt bridges | 1421 | LYS | 4.38 | 1419 | LYS | 4.33 |
| 1421 | LYS | 4.97 | 1419 | LYS | 4.99 | |
AA indicates amino acid; CYS, cysteine; GLN, glutamine; ILE, isoleucine; LEU, leucine; LYS, lysine; PHE, phenylalanine; SER, serine; THR, threonine; TYR, tyrosine; and VAL, valine.
For hydrogen bonds, the hydrogen‐acceptor distance is given.
DISCUSSION
Coronary artery disease remains a major cause of heart disease resulting in increased morbidity and mortality. 32 In acute myocardial infarction, strategies to restore blood supply have been effective; however, IR injury remains a significant residual burden with limited therapeutic progress. 1 , 2 , 3 Genetic models with altered PI3Kα signaling have provided abundant evidence for the critical role of this signaling cascade in various types of heart disease. 9 , 10 , 33 We used 2 different genetic models with loss of PI3Kα signaling and showed congruent results demonstrating a marked resistance to IR injury ex vivo and in vivo. However, the mechanism behind acute IR injury is likely distinct compared with chronic postmyocardial infarction and pressure overload remodeling. For example, in response to myocardial infarction, pressure overload, and chemotherapy, PI3Kα mutant mice demonstrate exacerbated adverse remodeling 8 , 9 , 10 , 15 , 16 but the marked protection seen in response to acute global IR injury. 12 While PI3Kα inhibition is not a feasible therapeutic approach, our results highlight the critical role of mitochondrial protection and preserving metabolic status in alleviating myocardial IR injury.
We propose that activation of late Na+ current and consequent increase in intracellular Na+ in PI3Kα‐deficient hearts lower intramitochondrial Ca2+, thereby preserving oxidative phosphorylation and mitochondrial ATP production. 4 , 6 Our current study elucidated key mechanistic pathways mediating cardioprotection in myocardial IR in genetic models with reduced cardiac PI3Kα activity. Our results suggest that increased late Na+ current provides an influx of Na+, facilitating the lowering of mitochondrial Ca2+, thereby maintaining mitochondrial membrane potential and oxidative phosphorylation. 4 , 6 Moreover, elevated cytoplasmic Na+ commonly observed during IR may also act as a preconditioning stimulus in PI3Kα‐deficient hearts, rendering them resistant to IR injury. Because late Na+ current is known to be arrhythmogenic and detrimental to long‐term Ca2+ homeostasis, 14 , 34 , 35 the current results suggest a more ambivalent role of the current: protective in the settings of the acute ischemia–reperfusion model, but physiologically detrimental in the long term. 14 , 34 , 35
Our results strongly support the enhancement of mitochondrial function as a therapeutic strategy to minimize IR injury. Preserved function of complex I is associated with improved cardiac function. 36 , 37 Maintained mitochondrial membrane potential prevents excessive production of damaging reactive oxygen species during ischemia. 38 PI3Kα‐deficient hearts had preserved complex I function and mitochondrial membrane potential, resulting in maintained ATP production and preventing excessive activation of sarcolemmal KATP channels, thereby adjusting membrane excitability to match the cellular energy demands imposed by ischemic stress resulting in overall preserved cardiac electrical stability. 39 The link between cardiac metabolic status and arrhythmias is further highlighted by the recent findings showing that the Na+‐glucose cotransporter 2 inhibitor, dapagliflozin, reduced ventricular arrhythmias and sudden death in patients with heart failure. 40 Our results are consistent with previous studies in guinea pig cardiac myocytes with pharmacological inhibition of the mitochondrial calcium uniporter, 41 studies using mice with genetic deletion of the mitochondrial calcium uniporter, 42 and the observation of accelerated mitochondrial Na+/Ca2+ exchanger–dependent mitochondrial Ca2+ extrusion (at elevated [Na+]i). 43 , 44
Our computer modeling approach was based on our previous studies examining the interaction between PIP3 and gelsolin's N and C domains. 10 Docking PIP3 within the model of NaV1.5 revealed that PIP3 binds to amino acids in the hydrophobic pocket below selectivity filter 31 including the gatekeeping lysine (Lys1419), which is part of the DEKA motif responsible for the selectivity of Na+ channel. 45 , 46 The lysine prevents the movement of Na+ ions in the reverse order (from the cytoplasm to the extracellular solution). 47 On the opposite side of the cavity, PIP3 interacts with Tyr1767, suggesting that PIP3 binds approximately in the same place as lidocaine (between Lys1419 and Tyr1767), obstructing the flow of Na+ ions. 31 Our modeling of PIP3 and NaV1.5 interaction also showed that PIP3 binding occludes the pore of the Na+ channel. This result is also supported by our previous observation that intracellular application of PIP3 (via patch pipette) blocks late Na+ current in a dose‐dependent manner. 14 Taken together, PIP3 can block the Na+ channel by binding to the hydrophobic pocket below the selectivity filter and occluding the channel. 23 Given the results of this study, the late Na+ current may have an ambivalent effect on cardioprotection. On the one hand, increased Na+ flow due to late Na+ current may contribute to cardioprotective effects during ischemia reperfusion by reducing mitochondrial Ca2+. On the other hand, late Na+ current is known to be arrhythmogenic, detrimental to long‐term Ca2+ homeostasis, and thus damaging to normal cardiac function. 14 , 34 , 35 While we documented the cardioprotective action of PI3Kα‐deficiency against acute myocardial IR ex vivo and in vivo, future studies are necessary to elucidate whether these findings translate to an improvement in response to chronic IR injury.
Sources of Funding
This work is partly supported by Canadian Institute for Health Research Project Grant (PJT‐156266) to G.Y.O., and a grant from the Canadian Institutes of Health Research (FRN 156393) to J.M.S. A.M.D. is supported by both an Alberta Innovates Graduate Studentship in Health Innovation and by the Izaak Walton Killam Memorial Scholarship.
Disclosures
None.
Supporting information
Figures S1–S3
Video S1
Video S2
Video S3
Video S4
This manuscript was sent to Ferhaan Ahmad, MD, PhD, Senior Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.022352
For Sources of Funding and Disclosures, see page 15.
REFERENCES
- 1. Cung TT, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D, Bonnefoy‐Cudraz E, Guerin P, Elbaz M, Delarche N, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. doi: 10.1056/NEJMoa1505489 [DOI] [PubMed] [Google Scholar]
- 2. Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987 [DOI] [PubMed] [Google Scholar]
- 3. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667 [DOI] [PubMed] [Google Scholar]
- 4. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22. doi: 10.1016/j.celrep.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia‐reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol. 2017;57:535–565. doi: 10.1146/annurev-pharmtox-010715-103335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017;545:93–97. doi: 10.1038/nature22082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104:292–303. doi: 10.1161/CIRCRESAHA.108.189050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen X, Zhabyeyev P, Azad AK, Vanhaesebroeck B, Grueter CE, Murray AG, Kassiri Z, Oudit GY. Pharmacological and cell‐specific genetic PI3Kalpha inhibition worsens cardiac remodeling after myocardial infarction. J Mol Cell Cardiol. 2021;157:17–30. doi: 10.1016/j.yjmcc.2021.04.004 [DOI] [PubMed] [Google Scholar]
- 9. Lin RC, Weeks KL, Gao XM, Williams RB, Bernardo BC, Kiriazis H, Matthews VB, Woodcock EA, Bouwman RD, Mollica JP, et al. PI3K(p110 alpha) protects against myocardial infarction‐induced heart failure: identification of PI3K‐regulated miRNA and mRNA. Arterioscler Thromb Vasc Biol. 2010;30:724–732. doi: 10.1161/ATVBAHA.109.201988 [DOI] [PubMed] [Google Scholar]
- 10. Patel VB, Zhabyeyev P, Chen X, Wang F, Paul M, Fan D, McLean BA, Basu R, Zhang P, Shah S, et al. PI3Kalpha‐regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat Commun. 2018;9:5390. doi: 10.1038/s41467-018-07812-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ban K, Cooper AJ, Samuel S, Bhatti A, Patel M, Izumo S, Penninger JM, Backx PH, Oudit GY, Tsushima RG. Phosphatidylinositol 3‐kinase gamma is a critical mediator of myocardial ischemic and adenosine‐mediated preconditioning. Circ Res. 2008;103:643–653. doi: 10.1161/CIRCRESAHA.108.175018 [DOI] [PubMed] [Google Scholar]
- 12. McLean BA, Kienesberger PC, Wang W, Masson G, Zhabyeyev P, Dyck JR, Oudit GY. Enhanced recovery from ischemia‐reperfusion injury in PI3Kalpha dominant negative hearts: investigating the role of alternate PI3K isoforms, increased glucose oxidation and MAPK signaling. J Mol Cell Cardiol. 2013;54:9–18. doi: 10.1016/j.yjmcc.2012.10.015 [DOI] [PubMed] [Google Scholar]
- 13. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, et al. CaMKII is a RIP3 substrate mediating ischemia‐ and oxidative stress‐induced myocardial necroptosis. Nat Med. 2016;22:175–182. doi: 10.1038/nm.4017 [DOI] [PubMed] [Google Scholar]
- 14. Zhabyeyev P, McLean B, Chen X, Vanhaesebroeck B, Oudit GY. Inhibition of PI3Kinase‐alpha is pro‐arrhythmic and associated with enhanced late Na(+) current, contractility, and Ca(2+) release in murine hearts. J Mol Cell Cardiol. 2019;132:98–109. doi: 10.1016/j.yjmcc.2019.05.008 [DOI] [PubMed] [Google Scholar]
- 15. McLean BA, Patel VB, Zhabyeyev P, Chen X, Basu R, Wang F, Shah S, Vanhaesebroeck B, Oudit GY. PI3Kalpha pathway inhibition with doxorubicin treatment results in distinct biventricular atrophy and remodeling with right ventricular dysfunction. J Am Heart Assoc. 2019;8:e010961. doi: 10.1161/JAHA.118.010961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLean BA, Zhabyeyev P, Patel VB, Basu R, Parajuli N, DesAulniers J, Murray AG, Kassiri Z, Vanhaesebroeck B, Oudit GY. PI3Kalpha is essential for the recovery from Cre/tamoxifen cardiotoxicity and in myocardial insulin signalling but is not required for normal myocardial contractility in the adult heart. Cardiovasc Res. 2015;105:292–303. doi: 10.1093/cvr/cvv016 [DOI] [PubMed] [Google Scholar]
- 17. Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, et al. Loss of apelin exacerbates myocardial infarction adverse remodeling and ischemia‐reperfusion injury: therapeutic potential of synthetic apelin analogues. J Am Heart Assoc. 2013;2:e000249. doi: 10.1161/JAHA.113.000249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61 [DOI] [PubMed] [Google Scholar]
- 19. Jamieson KL, Samokhvalov V, Akhnokh MK, Lee K, Cho WJ, Takawale A, Wang X, Kassiri Z, Seubert JM. Genetic deletion of soluble epoxide hydrolase provides cardioprotective responses following myocardial infarction in aged mice. Prostaglandins Other Lipid Mediat. 2017;132:47–58. doi: 10.1016/j.prostaglandins.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 20. O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271 [DOI] [PubMed] [Google Scholar]
- 21. McKenzie M, Lim SC, Duchen MR. Simultaneous measurement of mitochondrial calcium and mitochondrial membrane potential in live cells by fluorescent microscopy. J Vis Exp. 2017;e55166. doi: 10.3791/55166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 23. Jiang D, Shi H, Tonggu L, Gamal El‐Din TM, Lenaeus MJ, Zhao Y, Yoshioka C, Zheng N, Catterall WA. Structure of the cardiac sodium channel. Cell. 2020;180:122–134.e10. doi: 10.1016/j.cell.2019.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three‐dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0 [DOI] [PubMed] [Google Scholar]
- 26. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein‐ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tiwari SP, Fuglebakk E, Hollup SM, Skjaerven L, Cragnolini T, Grindhaug SH, Tekle KM, Reuter N. WEBnm@ v2.0: web server and services for comparing protein flexibility. BMC Bioinform. 2014;15:427. doi: 10.1186/s12859-014-0427-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Antunes DA, Moll M, Devaurs D, Jackson KR, Lizee G, Kavraki LE. DINC 2.0: a new protein‐peptide docking webserver using an incremental approach. Cancer Res. 2017;77:e55–e57. doi: 10.1158/0008-5472.CAN-17-0511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3‐kinase (PI3K)‐Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277:14040–14047. doi: 10.1074/jbc.M108975200 [DOI] [PubMed] [Google Scholar]
- 31. Nguyen PT, DeMarco KR, Vorobyov I, Clancy CE, Yarov‐Yarovoy V. Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc Natl Acad Sci USA. 2019;116:2945–2954. doi: 10.1073/pnas.1817446116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore‐Mensah Y, et al. Heart disease and stroke statistics‐2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 33. Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie‐Sasaki J, et al. Regulation of myocardial contractility and cell size by distinct PI3K‐PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8 [DOI] [PubMed] [Google Scholar]
- 34. Kaplan A, Amin G, Abidi E, Altara R, Booz GW, Zouein FA. Role of ranolazine in heart failure: from cellular to clinic perspective. Eur J Pharmacol. 2022;919:174787. doi: 10.1016/j.ejphar.2022.174787 [DOI] [PubMed] [Google Scholar]
- 35. Bersell KR, Yang T, Mosley JD, Glazer AM, Hale AT, Kryshtal DO, Kim K, Steimle JD, Brown JD, Salem JE, et al. Transcriptional dysregulation underlies both monogenic arrhythmia syndrome and common modifiers of cardiac repolarization. Circulation. 2023;147:824–840. doi: 10.1161/CIRCULATIONAHA.122.062193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti‐anginal drug ranolazine. Biochim Biophys Acta. 2012;1817:419–429. doi: 10.1016/j.bbabio.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qi B, Song L, Hu L, Guo D, Ren G, Peng T, Liu M, Fang Y, Li C, Zhang M, et al. Cardiac‐specific overexpression of Ndufs1 ameliorates cardiac dysfunction after myocardial infarction by alleviating mitochondrial dysfunction and apoptosis. Exp Mol Med. 2022;54:946–960. doi: 10.1038/s12276-022-00800-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang PT, Chen CL, Lin P, Chilian WM, Chen YR. Impairment of pH gradient and membrane potential mediates redox dysfunction in the mitochondria of the post‐ischemic heart. Basic Res Cardiol. 2017;112:36. doi: 10.1007/s00395-017-0626-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown DA, O'Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88:241–249. doi: 10.1093/cvr/cvq231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Curtain JP, Docherty KF, Jhund PS, Petrie MC, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, et al. Effect of dapagliflozin on ventricular arrhythmias, resuscitated cardiac arrest, or sudden death in DAPA‐HF. Eur Heart J. 2021;42:3727–3738. doi: 10.1093/eurheartj/ehab560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Joseph LC, Reyes MV, Homan EA, Gowen B, Avula UMR, Goulbourne CN, Wan EY, Elrod JW, Morrow JP. The mitochondrial calcium uniporter promotes arrhythmias caused by high‐fat diet. Sci Rep. 2021;11:17808. doi: 10.1038/s41598-021-97449-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation‐contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perez‐Garcia MT, Chiamvimonvat N, Ranjan R, Balser JR, Tomaselli GF, Marban E. Mechanisms of sodium/calcium selectivity in sodium channels probed by cysteine mutagenesis and sulfhydryl modification. Biophys J. 1997;72:989–996. doi: 10.1016/S0006-3495(97)78751-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hilber K, Sandtner W, Zarrabi T, Zebedin E, Kudlacek O, Fozzard HA, Todt H. Selectivity filter residues contribute unequally to pore stabilization in voltage‐gated sodium channels. Biochemistry. 2005;44:13874–13882. doi: 10.1021/bi0511944 [DOI] [PubMed] [Google Scholar]
- 47. Ahmed M, Jalily Hasani H, Ganesan A, Houghton M, Barakat K. Modeling the human Na(v)1.5 sodium channel: structural and mechanistic insights of ion permeation and drug blockade. Drug Des Devel Ther. 2017;11:2301–2324. doi: 10.2147/DDDT.S133944 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3
Video S1
Video S2
Video S3
Video S4