

Abstract
Background
Although the critical role of pericytes in maintaining vascular integrity has been extensively demonstrated in the brain and the retina, little is known about their role in the heart. We aim to investigate structural and functional consequences of partial pericyte depletion (≈60%) in the heart of adult mice.
Methods and Results
To deplete pericytes in adult mice, we used platelet‐derived growth factor receptor β–Cre/ERT2; RosaDTA mice and compared their phenotype with that of control mice (RosaDTA) chosen among their littermates. Cardiac function was assessed via echocardiography and left ventricular catheterization 1 month after the first tamoxifen injection. We found mice depleted with pericytes had a reduced left ventricular ejection fraction and an increased end‐diastolic pressure, demonstrating both systolic and diastolic dysfunction. Consistently, mice depleted with pericytes presented a decreased left ventricular contractility and an increased left ventricular relaxation time (dP/dtmin). At the tissue level, mice depleted of pericytes displayed increased coronary endothelium leakage and activation, which was associated with increased CD45+ cell infiltration. Consistent with systolic dysfunction, pericyte depletion was associated with an increased expression of myosin heavy chain 7 and decreased expression of ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 and connexin 43. More important, coculture assays demonstrated, for the first time, that the decreased expression of connexin 43 is likely attributable to a direct effect of pericytes on cardiomyocytes. Besides, this study reveals that cardiac pericytes may undergo strong remodeling on injury.
Conclusions
Cardiac pericyte depletion induces both systolic and diastolic dysfunction, suggesting that pericyte dysfunction may contribute to the occurrence of cardiac diseases.
Keywords: cardiac function, coronary microvasculature, heart, pericytes
Subject Categories: Heart Failure, Vascular Disease
Nonstandard Abbreviations and Acronyms
- ATP2A2
ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2
- CX43
connexin 43
- DTA
diphtheria toxin A
- EC
endothelial cell
- GFP
green fluorescent protein
- HMVEC‐C
human cardiac microvascular endothelial cell
- ICAM1
intercellular adhesion molecule 1
- NG2
chondroitin sulfate proteoglycan 4
- Notch3
Notch receptor 3
- PDGFRB
platelet‐derived growth factor receptor β
- SMA
α‐smooth muscle actin
- SMC
smooth muscle cell
- VCAM1
vascular cell adhesion molecule 1
Research Perspective.
What Is New?
The present study demonstrates that cardiac pericytes are critical to maintain proper cardiac function.
Cardiac pericytes modulate the properties of cardiomyocytes directly, notably connexin 43 expression level.
Cardiac pericytes prevent cardiac inflammation by downregulating interleukin‐6 in both endothelial cells and cardiomyocytes.
What Question Should Be Addressed Next?
Do cardiac pericytes participate in the pathophysiological features of cardiac diseases, notably heart failure?
How is the phenotype of cardiac pericyte modified by cardiovascular risk factors?
Pericytes are cells that reside in the microvasculature and share a basement membrane with the underlying endothelial cells (ECs). Pericyte density varies between different organs. In the brain, the organ with the highest density of pericytes, the EC/pericyte ratio varies between 1:1 and 3:1. 1 Accordingly, the brain is also the organ in which the role of pericytes has been the most investigated. Notably, in this organ, pericytes are thought to play a role in regulating permeability across the blood‐brain barrier. 2 , 3 Indeed, mice deficient in pericytes or exhibiting decreased pericyte function have increased vascular permeability. 2 , 3 Notably, pericyte function and maintenance depends on platelet‐derived growth factor subunit B expression by quiescent adult microvascular brain endothelium. 4
The pericyte content of the cardiac microvasculature is thought to be closer to the one of the cerebral vasculature, with an endothelial/pericyte ratio of 2:1 to 3:1. 5 It is also likely that pericytes contribute to the maintenance of microvascular function in the heart given that the loss of pericytes has been associated with increased vascular permeability, 6 , 7 endothelial activation, 7 and altered vasomotricity. 6 Notably, in mice treated with PDGF receptor inhibitors (sunitinib and CP‐673451), decreased pericyte coverage of cardiac capillaries was associated with increased permeability, impaired endothelium‐dependent vasodilation, and decreased coronary flow reserve. 6 Besides, the expression of a mutant form of pro–nerve growth factor, unable to be matured and which may target pericytes, was shown to induce endothelial activation and vascular leakage. 7 Finally, pericyte loss observed in both Sirtuin 3 8 and Notch receptor 3 (Notch3) 9 deficient mice was associated with decreased coronary flow reserve. More important, in each of these studies, impaired microvascular integrity and function has been also associated with cardiac dysfunction. Notably, administration of the PDGF receptor–specific inhibitor CP‐673451 was shown to decrease left ventricular ejection fraction (LVEF), 6 expression of the mutant form of pro–nerve growth factor–induced dilated cardiomyopathy, cardiac fibrosis, and contractile dysfunction. 7 Sirtuin 3–deficient mice showed decreased ejection fraction and fractional shortening after myocardial infarction, 8 whereas Notch3‐deficient mice were shown to exhibit cardiac hypertrophy. 9 Altogether, these studies suggest that pericytes may be beneficial to the coronary vasculature, which may have a direct or indirect impact on cardiac function. However, in the heart, pericytes were also proposed to have deleterious effects. Notably, pericytes may become myofibroblasts and contribute to fibrosis. 10 Also, capillary pericytes are suggested to be responsible for coronary no reflow after myocardial ischemia by inducing capillary constriction 11 via a mechanism involving G‐protein–coupled receptor 39. 12
In pathophysiological conditions, cardiac pericytes have been shown to either decrease or increase in number. Loss of pericytes has been shown to constitute a remarkable feature of aging in several organs, including the heart in both mice and humans. 13 Also, high‐fat diet was shown to induce pericyte loss in the heart of mice. 14 On the contrary, cardiac pericytes were shown to be disorganized and more numerous in Zucker fatty/spontaneously hypertensive heart failure F1 hybrid rats. 15 In other conditions, such as SARS‐CoV‐2 infection, the phenotype of pericytes is proposed to be modified by the acquisition of a more contractile phenotype. 16 Finally, pericyte dedifferentiation leading to increased endothelial permeability is proposed to be induced by the downregulation of hypoxia‐induced endoplasmic reticulum stress regulating long noncoding RNA in human heart failure. 17
In conclusion, cardiac pericytes may participate in the pathophysiological features of cardiac diseases through a wide range of mechanisms. However, studies performed to date are mainly descriptive, and their conclusions are based on associations. The purpose of the present study is to investigate the specific consequences of pericyte depletion on cardiac structure and function.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files. Also, we used the Animal Research: Reporting of In Vivo Experiments reporting guidelines. 18
Mice
B6.Cg‐Tg(platelet‐derived growth factor receptor β [Pdgfrb]–cre/ERT2)6096Rha/J (strain number 029684), Gt(ROSA)26Sortm4(ACTB‐tdTomato, ‐EGFP)Luo/J (strain number 007576) (named RosamTmG in the article), and B6.129P2‐Gt(ROSA)26Sortm1(DTA)Lky/J (strain number 009669) (named RosaDTA in the article) were obtained from the Jackson Laboratory.
Pdgfrb‐cre/ERT2 mice were crossed with RosaDTA/DTA mice to obtain Pdgfrb‐CreERT2; RosaDTA/+ (Pdgfrb–diphtheria toxin A [DTA]) mice and control RosaDTA/+ mice.
Pdgfrb‐cre/ERT2 mice were crossed with RosamTmG/mTmG mice to obtain Pdgfrb‐CreERT2; RosamTmG/+ mice.
The Cre recombinase in Pdgfrb‐Cre/ERT2 mice was activated by intraperitoneal injection of 50 mg/kg per day tamoxifen (Sigma‐Aldrich) for 5 consecutive days at 8 weeks of age and eventually repeated 2 weeks later at 10 weeks of age.
Animals were house in a conventional animal facility. Animal experiments were performed in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and approved by the local Animal Care and Use Committee of Bordeaux University. The appearance, weight, clinical signs, and behavior of the mice were monitored every day to prevent animal pain and distress. Both male and female mice were used. The number of animals per group necessary to have a 90% chance to find a significant difference between 2 groups was calculated using G power 3.1 software. The calculation was made for the measurement of cardiac function (notably LVEF). Mice were euthanized by either cervical dislocation or exsanguination under deep anesthesia (ketamine, 100 mg/kg, and xylazine, 20 mg/kg; intraperitoneally). A total of 60 mice were used in the study. No animals were excluded from the study.
Echocardiography
LVEF and left ventricular (LV) dimension were measured on a high‐resolution echocardiographic system equipped with a 30‐MHz mechanical transducer (VEVO 2100; VisualSonics Inc), as previously described. 19 , 20 Mice were anchored to a warming platform in a supine position, limbs were taped to the echocardiographic electrodes, and chests were shaved and cleaned with a chemical hair remover to minimize ultrasound attenuation. UNI'GEL ECG (Asept Inmed), from which all air bubbles had been expelled, was applied to the thorax to optimize the visibility of the cardiac chambers. LVEF was evaluated by planimetry as recommended. 21 Two‐dimensional, parasternal long‐axis and short‐axis views were acquired, and the endocardial area of each frame was calculated by tracing the endocardial limits in the long‐axis view, then the minimal and maximal areas were used to determine the left ventricular end‐systolic volume and end‐diastolic volume, respectively. The system software uses a formula based on a cylindrical‐hemiellipsoid model (volume=8×area2/3π×length). 22 The LVEF was derived from the following formula: (end‐diastolic volume–end‐systolic volume)/end‐diastolic volume×100. The cardiac wall thickness, LV posterior wall, interventricular septum, and LV internal diameter were calculated by tracing wall limits in both the long‐axis and short‐axis views.
LV Pressure/Systolic Blood Pressure Measurement
LV diastolic pressure measurement was assessed via an invasive catheterization technique. Mice were anesthetized with isoflurane. A Scisense pressure catheter (Transonic) was inserted into the LV through the common carotid artery. Pressure was recorded using LabChart software. End‐diastolic pressure, dP/dt minimum and maximum, Tau, and heart rate were automatically calculated by a curve fit through end‐systolic and end‐diastolic points on the pressure plot.
Blood Sampling for Biochemical Marker Analysis/Complete Blood Count
Blood samples were collected by the heparin retro‐orbital bleeding method at euthanasia. Blood cell counts were determined using an automated counter (scil Vet abc Plus+). Plasma was separated by a 10‐minute centrifugation at 2500g and then stored at −80 °C. Concentrations of the following biomarkers were measured using an Architect CI8200 analyzer (Abbott Diagnostics, North Chicago, IL): triglycerides, using the lipoprotein‐lipase/glycerol kinase/oxidase/peroxidase method; total cholesterol, using the esterase/oxidase/peroxidase method; and high‐density lipoprotein cholesterol, using the accelerator/selective detergent/esterase/oxidase/peroxidase method. Low‐density lipoprotein cholesterol was then estimated using the Friedewald formula [low‐density lipoprotein cholesterol (mmol/L)=total cholesterol– high‐density lipoprotein cholesterol–(triglycerides/2.2), or low‐density lipoprotein cholesterol (mg/dL)=total cholesterol– high‐density lipoprotein cholesterol–(triglycerides/5)].
Tissue Staining/Immunostaining
ECs were identified using anti‐CD31 or anti–podocalyxin‐like antibodies or biotin‐conjugated isolectin B4 (Vector). Muscularized vessels were identified using anti–α‐smooth muscle actin (SMA) antibodies. Pericyte were identified using anti–chondroitin sulfate proteoglycan 4 (CSPG4; also known as NG2) or anti‐PDGFRB antibodies. Leucocyte, macrophage, T‐cell, and B‐cell infiltrations were quantified using anti‐CD45, anti‐CD68, anti‐CD3, and anti‐B220 antibodies, respectively. Endothelial adherens junction was characterized using anti–cadherin‐5 antibodies. Edema was measured after fibrinogen B chain staining of muscle sections and quantified as the mean fibrinogen B chain–positive areas (see Table S1 for antibody references).
Cardiomyocyte mean surface area was measured using ImageJ software after membrane staining with wheat germ agglutinin, Alexa Fluor 488 Conjugate (Invitrogen). Fibrosis was assessed after Sirius red staining of heart sections by quantifying the percentage of red stained area.
Quantifications done on images were acquired by using an Axiozoom V16 or axioscope A1 (Zeiss) under ×200 magnification. They were conducted on 10 randomly selected images, performed using ImageJ/Fiji v2.0.0‐rc‐59 software (National Institutes of Health) by an investigator blinded to genotype. More precisely, each sample received a unique number. At the end of the experiment, the genotype/treatment for each sample was revealed to enable data analysis.
For immunohistochemical analyses, primary antibodies were sequentially coupled with biotin‐conjugated secondary antibodies and streptavidin–horseradish peroxidase complex (Amersham). Staining was then revealed with a diaminobenzidine substrate kit (Vector Laboratories), and tissue sections were counterstained with hematoxylin (see Table S2 for secondary antibody references).
For immunofluorescence analyses, primary antibodies were resolved with Alexa‐Fluor–conjugated secondary polyclonal antibodies (see Table S2). Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole.
For both immunohistochemical and immunofluorescence analyses, negative control experiments to check for antibody specificity were performed using secondary antibodies.
Cell Culture
Human cardiac microvascular ECs (HMVEC‐Cs) (Lonza) were cultured in endothelial basal medium‐2 supplemented with endothelial growth medium (EGM)‐2 BulletKits (Lonza). Cells at passage 3 were used.
Mouse pericytes were isolated from 10‐day‐old pups. Briefly, hearts were dissociated using 2 mg/mL type IV collagenase (Gibco, ThermoFisher) for 1 hour at 37 °C, and the resulting dissociated cells were filtrated on a 30‐μm strainer. Pericytes were labeled with rat anti‐mouse CD146 microbeads (Miltenyi Biotec). Labeled cells were then isolated magnetically, plated, and cultured in pericyte Growth Medium 2 (PromoCell). Cells at passages 1 and 2 were used.
HMVEC‐Cs and mouse pericytes were cocultured in 0.4 μm–pore Transwells in 6‐well plates (Corning). HMVEC‐Cs were cultured together or not with pericytes for 24 hours.
Mouse adult cardiomyocytes were isolated: hearts were dissociated using 300 U/mL Collagenase type 2 (Worthington) via aortic catheterization and a 5‐minute perfusion at 37 °C. The resulting dissociated cells were then filtrated on a 400‐μm mesh and centrifuged at 40g for 5 minutes to separate cardiomyocytes. Centrifuged cardiomyocytes were then incubated in increasing CaCl2 concentrations (from 12.5 to 900 μmol/L in the presence of 2 mmol/L ATP; Sigma). Cells were centrifuged again at 40g for 5 minutes, resuspended in minimum essential medium (Gibco) containing 5% fetal bovine serum, 1.26 mmol/L CaCl2, 10 mmol/L 2,3‐butanedione monoxime (Sigma), and 2 mmol/L ATP and seeded in cell culture wells coated with 10 μg/mL Laminine (Sigma). After a 2‐hour incubation at 37 °C, culture medium was replaced by minimum essential medium containing 0.2% bovine serum albumin (Sigma), 1.26 mmol/L CaCl2, 1× insulin/transferrin/selenium (Gibco), and 25 μmol/L blebbistatin (Sigma).
Mouse cardiac pericytes were added or not to the cardiomyocytes at the same time as the change of the adhesion medium into culture medium. Cardiomyocytes were cultured together or not with pericytes for 24 hours.
Quantitative Polymerase Chain Reaction
RNA was isolated using Tri Reagent (Molecular Research Center Inc) as instructed by the manufacturer, from heart tissue that had been snap frozen in liquid nitrogen and homogenized. For quantitative polymerase chain reaction analyses, total RNA was reverse transcribed with M‐MLV reverse transcriptase (Promega), and amplification was performed on an AriaMx Real Time PCR system (Agilent Technologies) using GoTaq quantitative polymerase chain reaction master mix (Promega). Primer sequences are reported in Table S3.
The relative expression of each mRNA was calculated by the comparative threshold cycle method and normalized to 18S rRNA or beta‐actin mRNA expression.
Western Blot Analysis
Intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) protein levels were evaluated by SDS‐PAGE using goat anti‐ICAM1 (R&D Systems; catalog number AF‐796) and rabbit anti‐VCAM1 (Abcam; catalog number ab134047) antibodies, respectively. ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (ATP2A2) protein level was evaluated by SDS‐PAGE using rabbit anti‐ATP2A2 antibodies (Badrilla; catalog number A010‐80). Phospholamban phosphorylation at serine 16 and threonine 17 was evaluated by SDS‐PAGE using rabbit anti–phosphorylated phospholamban serine 16 (Badrilla; catalog number A010‐12), rabbit anti–phosphorylated phospholamban threonine 17 (Badrilla; catalog number A010‐13), and mouse anti–total phospholamban (Badrilla; catalog number A010‐14). Ryanodine receptor 2 phosphorylation was evaluated by SDS‐PAGE using rabbit anti–phosphorylated ryanodine receptor 2 antibodies (Badrilla; catalog number A010‐31).
Human cadherin‐5 and ICAM1 protein levels were evaluated by SDS‐PAGE using rabbit anti–cadherin‐5 (Cell Signaling Technology; catalog number 2500) and mouse anti‐ICAM1 (Santa Cruz Biotechnology; catalog number sc‐8439) antibodies, respectively.
Protein loading quantity was controlled using mouse monoclonal anti–α‐tubulin antibodies (Sigma; catalog number T5168).
Statistical Analysis
Results are reported as mean±SEM. Comparisons between groups were analyzed for significance with the nonparametric Mann‐Whitney test using GraphPad Prism v8.0.2 (GraphPad Inc, San Diego, CA). The normality and variance were not tested. Differences between groups were considered significant when P≤0.05.
Results
Cardiac Capillaries Are Covered by Smooth Muscle Myosin Heavy Chain–Negative, NG2 + Pericytes
In the heart, <2% of vessels, essentially arterioles, are covered by smooth muscle myosin heavy chain–positive smooth muscle cells. Indeed, the vast majorities of capillaries (ie, 95% of them) are covered by NG2‐positive, SMA‐negative pericytes (Figure 1A and 1C). Cardiac pericytes are stellate‐shaped cells with cytoplasmic processes contacting several capillaries (Figure 1D and 1F). More important, contrary to the brain, which contains a significant amount of ensheathing and mesh pericytes, 23 >95% of cardiac pericytes are thin‐stranded pericytes (Figure S1) that do not express smooth muscle myosin heavy chain (Figure S2).
Figure 1. A total of 95% of the cardiac vasculature is covered by pericytes.
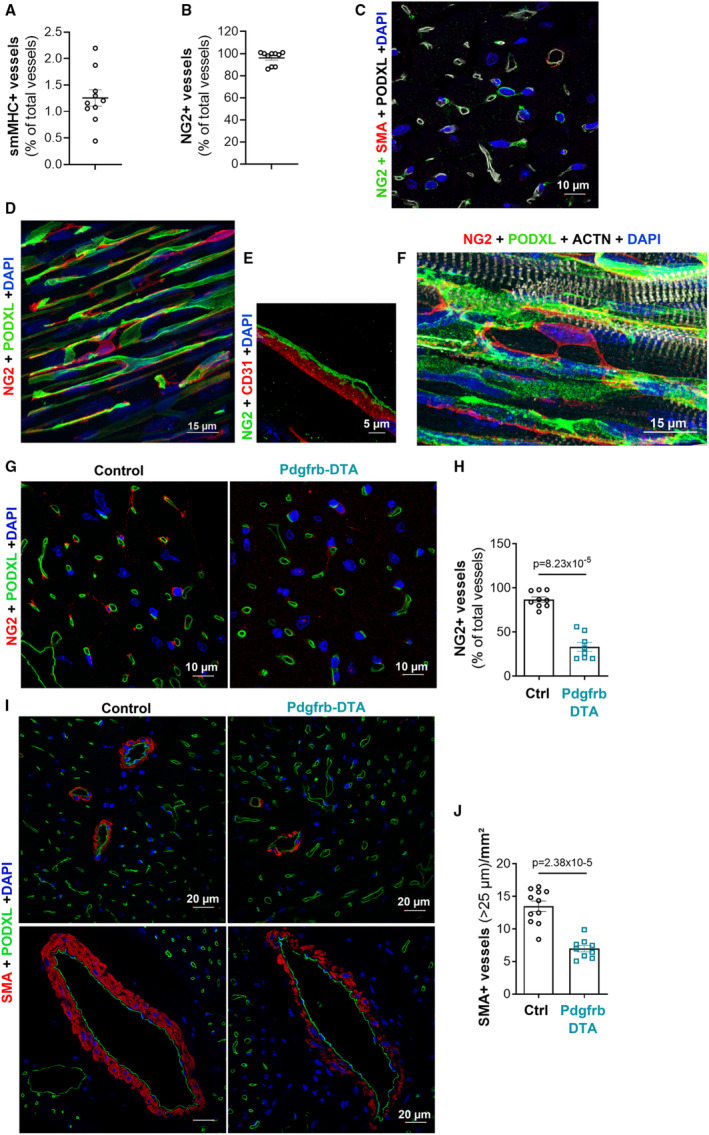
A, Heart cross‐sections were coimmunostained with anti–smooth muscle myosin heavy chain (smMHC) and anti–podocalyxin‐like (PODXL) antibodies to identify smooth muscle cells (SMCs) and endothelial cells (ECs), respectively. The percentage of smMHC+ vessels/total PODXL+ vessels was calculated. B, Heart cross‐sections were coimmunostained with anti–neuronal/glial antigen 2 (NG2) and anti‐PODXL antibodies to identify pericytes and ECs, respectively. The percentage of NG2+ vessels/total PODXL+ vessels was calculated. C, Heart cross‐sections were coimmunostained with anti–smooth muscle actin (SMA) (red), anti‐NG2 (green), and anti‐PODXL (white) antibodies to identify SMCs, pericytes, and ECs, respectively. D, Heart thick sections were coimmunostained with anti‐NG2 (in red) and anti‐PODXL (in green) antibodies to identify pericytes and ECs, respectively. E, Heart thick sections were coimmunostained with anti‐NG2 (green) and anti‐CD31 (red) antibodies to identify pericytes and ECs, respectively. F, Heart thick sections were coimmunostained with anti‐NG2 (red), anti‐PODXL (green), and anti–actinin α 2 (ACTN2) (white) antibodies to identify pericytes, ECs, and cardiomyocytes, respectively. G through J, Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. G, Heart cross‐sections were coimmunostained with anti‐NG2 (red) and anti‐PODXL (green) antibodies to identify pericytes and ECs, respectively. H, The percentage of NG2+ vessels/total PODXL+ vessels was calculated (n=8 and 9 mice/group). I, Heart cross‐sections were coimmunostained with anti‐SMA (red) and anti‐PODXL (green) antibodies to identify SMCs and ECs, respectively. J, The number of SMA+ vessels per mm2 was counted (n=9 and 11 mice/group). Mann‐Whitney tests were used. DAPI indicates 4′;6‐diamidino‐2‐phenylindole.
Pdgfrb‐DTA Mice Display a 60% Pericyte Depletion in the Heart
To investigate the role of cardiac pericytes, we have chosen to measure the pathophysiological consequences of partial pericyte depletion on cardiac microvasculature, cardiomyocytes, and heart function. To do so, we specifically induced DTA expression under the Pdgfrb promoter in adult mice. At first, we verified that the Pdgfrb promoter was active in cardiac pericytes in adult mice, by crossing Pdgfrb‐Cre/ERT2 mice with RosamTmG mice. As shown in Figure S3A and S3B, GFP (green fluorescent protein) expression in the heart was detected in most NG2+ pericytes but also, as expected, by SMA+ smooth muscle cells.
In the meantime, we have examined GFP expression in several other organs, notably the aorta, in which GFP expression was detected in a few smooth muscle cells of the media, in the brain, in which GFP was detected in perivascular cells but also unexpectedly in glial cells; in the kidney, in which GFP expression was detected in some SMA+ cells of arterioles; but not in NG2+ cells within the glomerulus. Also, GFP expression was detected in capillaries within the lacteals but not in smooth muscle cells (SMCs) surrounding the intestine. In the lung, GFP expression was detected in both arterial and capillary mural cells; however, mural cells covering lung capillaries did not express NG2. Finally, in the skeletal muscle, similarly to what we found in the heart, GFP expression was detected in every pericyte and a few SMCs (Figure S4).
Pericyte depletion was then induced by tamoxifen injections in 8‐week‐old Pdgfrb‐Cre/ERT2; Rosa‐DTA mice. Notably, to maintain constant pericyte depletion in the heart over time, we had to repeat tamoxifen administrations every 2 weeks. Indeed, in mice administered with tamoxifen once during 5 consecutive days, although the percentage of NG2‐positive vessels was ≈20% 7 days after the first tamoxifen injection, it was back to 90% after 28 days (Figure S5A and S5B), suggesting cardiac pericytes can be renewed.
After 2 series of tamoxifen injections (ie, 1 month after the first one), the percentage of cardiac capillaries covered by pericytes was reduced by ≈60% in Cre‐positive mice compared with Cre‐negative mice (Figure 1G and 1H), and the number of arterioles covered by SMCs was reduced by ≈50% (Figure 1I and 1J); SMC coverage of coronary arteries was sparser (Figure 1I).
In parallel, we have examined the effectiveness of mural cell depletion in other organs. The most severe phenotypes were observed in the aorta, in which the media was strongly decellularized; in the intestine lacteals; and in the skeletal muscle, in which NG2+ pericytes have been fully depleted (Figure S6). These “side” depletions induced significant weight loss (Figure S7A and S7B) associated with circulating markers of malnutrition, including decreased hemoglobin, triglyceride, cholesterol, and protein levels together with decreased aspartate aminotransferase and alkaline phosphatase levels (Figure S7C–S7K). As expected, the SMC loss in the aorta decreased diastolic blood pressure. Systolic blood pressure and heart rate were not modified (Figure S7L–S7O). Finally, consistent with pericyte loss in the skeletal muscle, mice were severely intolerant to exercise (Figure S7P).
Altogether, these results demonstrate that Pdgfrb‐DTA mice display successful pericyte depletion in the heart. However, considering the absence of pericyte‐specific marker and/or the absence of cardiac mural cell–specific markers allowing pericyte depletion in the heart only, the interpretation of the data presented in this article may have some limitations.
Cardiac Pericytes Are Renewed
While performing a thorough histological analysis of the heart of Pdgfrb‐DTA mice, we were surprised to see that the number of small vessels (mainly capillaries with a diameter <10 μm) covered by SMA+ cells was significantly increased in the heart of Pdgfrb‐DTA mice compared with control mice (Figure 2A and 2B). These SMA+ cells were actually NG2 positive, suggesting that the remaining pericytes in these mice display an activated phenotype characterized by SMA expression (Figure S8A). Besides, the heart of Pdgfrb‐DTA mice displayed an increased number of vimentin‐positive mesenchymal cells (Figure S8B and S8C). Notably, among vimentin‐positive cells, the number of PDGFRB‐negative cells (fibroblasts) did not increase (Figure S8D and S8E), whereas the number of PDGFRB‐positive cells significantly increased (Figure S8F). These results suggest that (1) the number of cardiac fibroblasts did not change and (2) some pericytes expressed PDGFRB but not NG2, which we verified by doing a PDGFRB, NG2 double immunostaining (Figure 2C). Indeed, we found that, although all NG2+ pericytes expressed PDGFRB in both Pdgfrb‐DTA and control mice (Figure 2D), all PDPGFRB+ pericytes did not express NG2, especially in Pdgfrb‐DTA mice (Figure 2E). Considering that we found that cardiac pericytes are renewed (Figure S5), and that mice were euthanized 10 to 13 days after the last tamoxifen injection, we believe that PDGFRB+NG2 cells are pericytes being renewed.
Figure 2. Cardiac pericytes may undergo strong remodeling.

Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. A, Heart cross‐sections were coimmunostained with anti–α‐smooth muscle actin (SMA) (red) and anti–podocalyxin like (PODXL) (green) antibodies to identify smooth muscle cells and endothelial cells, respectively. B, The number of SMA+ vessels with a diameter ≤25 μm per mm2 was counted (n=9 and 11 mice/group). C, Heart cross‐sections were coimmunostained with anti‐PDGFRB (green) and anti–neuronal/glial antigen 2 (NG2) (red) antibodies. D, The percentage of PDGFRB+ cells among NG2+ cells was calculated (n=7 and 10 mice/group). E, The percentage of NG2+ cells among PDGFRB+ cells was calculated (n=7 and 10 mice/group). Mann‐Whitney tests were used. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Consistent with the fact that the number of cardiac fibroblasts did not change, interstitial fibrosis was not significantly modulated in Pdgfrb‐DTA mice; only perivascular fibrosis was modulated (Figure S9A–S9C). Moreover, the expression of several markers of fibrosis, including collagen type I α 1 chain, collagen type III α 1 chain, transforming growth factor β 1, and connective tissue growth factor mRNA, was not significantly different between Pdgfrb‐DTA mice and control mice (Figure S9D–S9G).
Altogether, these results suggest that partial pericyte depletion induces activation and renewal of the remaining pericytes. Pericytes undergoing remodeling may at some points express SMA and/or PDGFRB, but not NG2.
Pericyte Depletion Modifies the Phenotype of Cardiac Capillary
Then, we measured the consequences of mural cell depletion on the cardiac microvasculature. As mentioned above, this was done 1 month after the induction of pericyte depletion. As shown in Figure 3A and 3B, pericyte depletion did not induce microvessel rarefaction, because capillary density was equivalent in both Pdgfrb‐DTA and control mice. However, pericyte depletion led to an increased capillary diameter (Figure 3C), possibly reflecting capillary disorganization, and endothelial activation characterized by a significant increase in ICAM1 and VCAM1 expression. This phenotype was attested via immunostaining (Figure 3D and 3E) and confirmed by Western blot analyses of total heart extracts (Figure 3F and 3H). Also, pericyte depletion led to altered endothelial intercellular junctions because we observed that cadherin‐5 staining was discontinuous in pericyte‐depleted mice (Figure 3I and 3J). We confirmed that endothelial intercellular junction integrity was altered in Pdgfrb‐DTA mice because fibrinogen extravasation was significantly increased in Pdgfrb‐DTA mice compared with control mice (Figure 3K and 3L), revealing that capillary permeability is abnormally increased in pericyte‐depleted mice.
Figure 3. Pericytes are necessary to maintain cardiac capillary integrity.

Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. A, Heart cross‐sections were immunostained with anti– podocalyxin like (PODXL) antibodies to identify endothelial cells. B, The number of PODXL+ capillaries per mm2 was counted (n=14 and 11 mice/group). C, The mean capillary diameter was measured (n=10 mice in each group). D, Heart cross‐sections were coimmunostained with anti–intercellular adhesion molecule‐1 (ICAM‐1) (green) and anti‐CD31 (red) antibodies. E, Heart cross‐sections were coimmunostained with anti– vascular cell adhesion molecule‐1 (VCAM‐1) (green) and anti‐PODXL (red) antibodies. F, ICAM‐1 and VCAM‐1 protein expression was evaluated in total heart extract by Western blot analyses. ICAM‐1 (G) and VCAM‐1 (H) protein was quantified using ImageJ software and normalized to α‐tubulin (n=8 mice/group). I and J, Heart cross‐sections were immunostained with anti–cadherin‐5 (CDH5) antibodies to identify adherens junction. K, Heart cross‐sections were coimmunostained with anti–fibrinogen B chain (FGB) and anti‐PODXL antibodies. L, FGB+ surface area was measured using ImageJ software (n=10 and 8 mice/group). Mann‐Whitney tests were used. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
To make sure these phenotypes were not attributable to the “unwanted” features of Pdgfrb‐DTA mice, such as malnutrition or decreased diastolic pressure, we confirmed the critical role of cardiac pericytes in maintaining endothelial integrity and immune quiescence in vitro. To do so, we cocultured mouse cardiac pericytes with HMVEC‐Cs (Figure S10A). HMVECs cocultured with pericytes expressed significantly higher cadherin‐5 protein levels, whereas cadherin‐2 mRNA was downregulated (Figure S10B–S10D), signaling increased endothelial differentiation. Consistent with in vivo data, pericytes prevented HMVEC‐C activation by decreasing ICAM1, VCAM1, and interleukin‐6 levels (Figure S10E–S10I).
Overall, these results confirm association studies and demonstrate that pericytes are necessary for cardiac capillary integrity and immune quiescence, just as they are in the brain.
Notably, modification of the coronary vessel phenotype did not lead to cardiac hypoxia, because hypoxia‐inducible factor 1 subunit α levels were not different in the heart of Pdgfrb‐DTA and control mice (Figure S11A). However, consistent with endothelial activation, Pdgfrb‐DTA mice display cardiac inflammation with significantly increased interleukin‐6 mRNA level (Figure S11B) and increased CD45+ leukocyte infiltration (Figure S11C and S11D) in the heart. Notably, neither CD68+ macrophage nor CD3+ T‐cell infiltration was increased (Figure S11E–S11H), whereas B220+ B‐cell infiltration was significantly increased (Figure S11I and S11J).
Pericyte Depletion Modifies the Phenotype of Cardiomyocytes
To assess the phenotype of cardiomyocytes, we first performed wheat germ agglutinin staining on heart cross‐sections and found that cardiomyocytes did not show hypertrophy (Figure 4A and 4B). On the contrary, Pdgfrb‐DTA mice even show slight cardiac atrophy. Indeed, the heart weight/tibia length ratio tended to decrease (Figure 4C), which was confirmed by measuring the LV mass via echocardiography (Figure 4D). Consistently, cardiac wall thickness (ie, interventricular septum and LV posterior wall) measured via echocardiography was thinner in Pdgfrb‐DTA mice (Figure 4E and 4F). The LV internal diameter in diastole was not modified (Figure 4G).
Figure 4. Pericyte depletion induces cardiomyocyte dedifferentiation but not cardiomyocyte hypertrophy.

Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. A, Heart cross‐sections were costained with wheat germ agglutinin (WGA) (green) and anti–actinin α 2 (ACTN2) (red) antibodies. B, Cardiomyocyte cross‐sectional area was measured using ImageJ software (n=11 and 9 mice/group). C, The heart weight/tibia length was measured (n=10 and 8 mice/group). D, The left ventricular (LV) mass corrected was measured via echocardiography. Interventricular septum (IVS) thickness (E), LV posterior wall (LVPW) thickness (F), and LV internal diameter (LVID) (G) were measured by echocardiography in diastole. H, Myosin heavy chain 7 (Myh7) mRNA expression was quantified by quantitative polymerase chain reaction in total heart extract and normalized to 18S rRNA (n=9 and 10 mice/group). I, Heart cross‐sections were coimmunostained with anti–connexin 43 (CX43) (green) and anti‐ACTN2 (red) antibodies. CX43 protein expression was analyzed by Western blot (J) and quantified using ImageJ software (K) (n=7 and 8 mice/group). Mann‐Whitney tests were used. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; IVSd, IVS diastole; LVIDd, LVID diastole; and LVPWd, LVPW diastole.
Notably, wheat germ agglutinin staining revealed that Pdgfrb‐DTA had expanded cardiac interstitial space (Figure 4A), which is consistent with the increased vascular leakage.
More important, cardiomyocytes showed a modified phenotype with increased myosin heavy chain 7 mRNA expression (Figure 4H) and decreased connexin 43 (CX43) levels (Figure 4I and 4K). To test whether cardiomyocyte contractility could be affected by pericyte depletion, we measured expression and/or phosphorylation of proteins regulating calcium homeostasis. We first measured ATP2A2 expression and found it significantly downregulated at both mRNA and protein levels in Pdgfrb‐DTA mice compared with control mice (Figure 5A and 5C). This was associated with diminished phospholamban phosphorylation at both serine 16 and threonine 17 (Figure 5D and 5F). On the contrary, ryanodine receptor 2 phosphorylation at serine 2814 was not significantly different in both groups (Figure 5G and 5H). To assess cardiomyocyte stiffness, we quantified the mRNA expression of each Titin isoform. Expression of stiff Titin isoform N2B was significantly diminished, whereas expression of Titin N2BA isoform tended to increase. Accordingly, the N2BA/N2B ratio was significantly increased (Figure 5I and 5K).
Figure 5. Pericyte depletion modifies ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (ATP2A2) level and Titin (Ttn) splicing in cardiomyocytes.
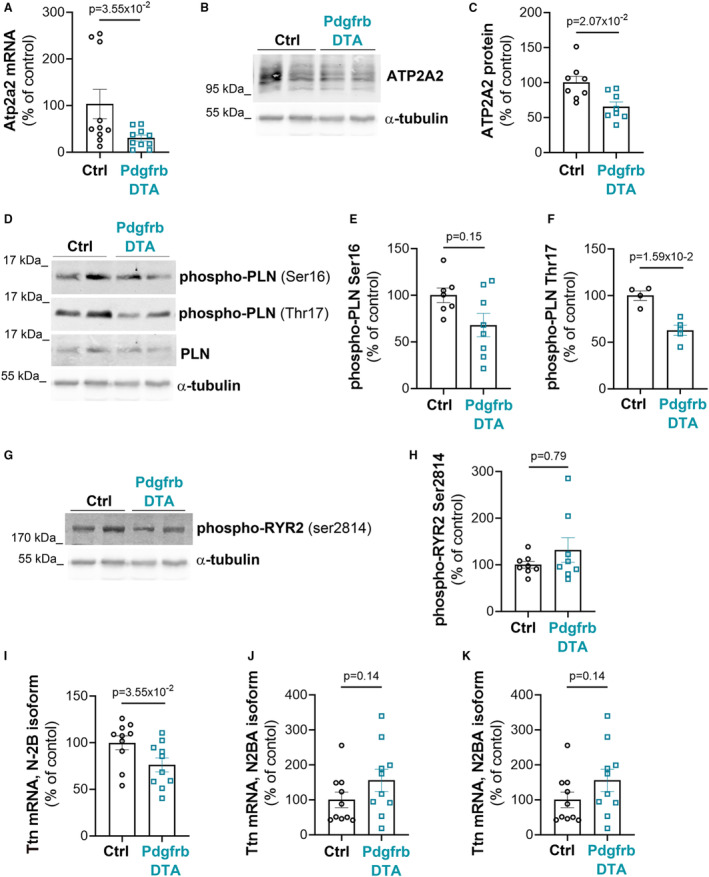
Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. A, Atp2a2 mRNA expression was quantified by reverse‐transcription quantitative polymerase chain reaction (RT‐qPCR) in total heart extract and normalized to 18S rRNA (n=10 mice in each group). ATP2A2 protein expression was analyzed by Western blot in total heart extract (B) and quantified using ImageJ software (C) (n=8 mice in each group). Phospholamban (PLN) phosphorylation was assessed by Western blot analyses (D) and quantified using ImageJ software (E and F) (n=5 to 8 mice/group). Ryanodine receptor 2 (RYR2) phosphorylation was assessed by Western blot analyses (G) and quantified using ImageJ software (H) (n=8 mice in each group). Ttn, isoform N‐2B (I), and N2BA (J) mRNA expression was quantified by RT‐qPCR in total heart extract and normalized to 18S rRNA (n=10 mice in each group). K, N2BA mRNA/N‐2B mRNA ratio was calculated. Mann‐Whitney tests were used. Ser indicates serine; and Thr, threonine.
Altogether, these results demonstrate that pericyte depletion modifies the phenotype of cardiomyocytes, which display a Titin isoform ratio consistent with heart failure in humans. 24
Once again, to make sure these changes in the phenotype of cardiomyocytes were not attributable to the “unwanted” features of Pdgfrb‐DTA mice, such as malnutrition or decreased diastolic pressure, we performed coculture assays. More specifically, mouse adult cardiomyocytes were cocultured or not with mouse cardiac pericytes (Figure S12A). Although the presence of pericytes did not modify Atp2a2 or myosin heavy chain 7 mRNA expression in cardiomyocytes (Figure S12B and S12C), it did significantly increase Cx43 mRNA expression (Figure S12D), demonstrating, for the first time, that cardiac pericytes may modify the phenotype of cardiomyocytes directly. Besides, we found that pericytes have an anti‐inflammatory effect on cardiomyocytes, like they have on ECs, by downregulating interleukin‐6 mRNA expression (Figure S12E).
To test whether cardiac inflammation (ie, interleukin‐6 overexpression) may indirectly also affect cardiomyocytes, mouse adult cardiomyocytes were treated or not with recombinant interleukin‐6. We found that interleukin‐6 did not modulate Cx43 or myosin heavy chain 7 mRNA expression. However, it significantly downregulated Atp2a2 mRNA expression.
Altogether, these results demonstrate that cardiac pericytes may affect the phenotype of cardiomyocytes directly, notably by upregulating CX43 and downregulating interleukin‐6. The increased cardiac inflammation secondary to pericyte depletion is likely responsible for the decreased Atp2a2 mRNA expression observed in the heart of Pdgfrb‐DTA mice. However, the mechanism leading to the increased myosin heavy chain 7 mRNA expression remains to be identified.
Pericyte Depletion Impairs Both Systolic and Diastolic Function
Finally, to test whether pericyte depletion and the associated cardiac remodeling result in cardiac dysfunction, we performed echocardiography and LV catheterization. As shown in Figure 6, LVEF was significantly reduced in Pdgfrb‐DTA mice, indicating systolic dysfunction (Figure 6A). LVEF was positively correlated with pericyte coverage (Figure 6B). Consistent with systolic dysfunction, cardiac contractility (attested by dP/dt maximum and contractility index) significantly decreased (Figure 6C and 6E), whereas LV end‐systolic volume significantly increased (Figure 6F and 6G) in pericyte‐depleted mice compared with control mice.
Figure 6. Pericyte depletion induces both systolic and diastolic dysfunction.

Platelet‐derived growth factor receptor β (Pdgfrb)–Cre/ERT2; RosaDTA (Pdgfrb–diphtheria toxin A [DTA]) and RosaDTA (control [Ctrl]) mice were administered with tamoxifen. Mice were euthanized 28 days after the first injection. Left ventricular (LV) ejection fraction was assessed via echocardiography (A) (n=7 and 10) and correlated with pericyte coverage (B). dP/dt max was measured by LV catheterization (C) (n=10 in each group) and correlated with pericyte coverage (D). E, The contractility index was measured by LV catheterization (n=10 mice in each group). LV volumes were measured in diastole (F) and systole (G) via echocardiography (n=7 and 10 mice/group). LV end‐diastolic pressure (LVEDP) was measured by LV catheterization (H) (n=10 mice in each group) and correlated with pericyte coverage (I). dP/dt min was measured by LV catheterization (J) (n=10 mice in each group) and correlated with pericyte coverage (K). L, Tau was measured by LV catheterization (n=10 mice in each group). Mann‐Whitney tests were used.
Mice depleted from cardiac pericytes also displayed diastolic dysfunction attested by increased end‐diastolic pressure (EDP) (Figure 6H and 6I) and decreased dP/dt minimum (Figure 6J and 6K). Relaxation constant Tau tended to increase in Pdgfrb‐DTA mice, but the difference did not reach significance (Figure 6L).
In conclusion, this study demonstrates, for the first time, that pericytes are necessary for proper cardiac function, possibly by affecting the phenotype of cardiomyocytes directly.
Discussion
The present study demonstrates, for the first time, that, just like in the brain, pericytes are necessary for endothelium integrity and immune quiescence in the heart. More important, in this organ, pericytes are not only necessary for coronary vasculature integrity but also for proper cardiac function, suggesting that modifications of pericyte properties may participate in the pathophysiological features of cardiac diseases.
Besides, this study reveals that cardiac pericytes may undergo strong remodeling on injury (ie, renewal and expression of SMA). This is consistent with what has been observed in obese and hypertensive Zucker fatty/spontaneously hypertensive heart failure F1 hybrid rats, 15 in which pericytes were shown to be disorganized and more numerous. This piece of data actually suggests that, although pericytes may induce similar effects in the brain and in the heart by maintaining microvasculature integrity, pericyte turnover and biological features in these organs may be different. Indeed, in the central nervous system, pericyte impairment seems to be essentially characterized by pericyte loss. Notably, one of the earliest hallmarks of diabetic retinopathy is the loss of pericytes. The diabetic microenvironment is suggested to be particularly detrimental to pericyte survival in the retina because during the early stages of retinopathy, the pericyte/EC ratio decreases from 1:1 to 1:4. 25 In the brain, pericyte number and coverage in the cortex and hippocampus of subjects with Alzheimer disease compared with neurologically intact controls were shown to be reduced by 59% and 60%, respectively. 26 Similarly, in the spinal cord, immunostaining for PDGFRB has indicated a 54% (P<0.01) reduction in the number of pericytes in patients with amyotrophic lateral sclerosis compared with control patients. 27 However, in the heart, pericyte impairment is more likely characterized by phenotypic changes with the acquisition of an “activated” phenotype. Notably, the number of pericytes was shown to increase in the heart of mice infused with angiotensin‐2. Moreover, these pericytes express higher levels of SMA. 28 As mentioned above, cardiac pericytes were shown to be disorganized and more numerous in obese and hypertensive Zucker fatty/spontaneously hypertensive heart failure F1 hybrid rats.15 In our model, pericyte depletion induced pericyte activation and renewal.
Pericyte loss has been previously associated with reduced LVEF,6,8 dilated cardiomyopathy, cardiac fibrosis, contractile dysfunction,7 and cardiac hypertrophy.9 Our study confirms that pericytes are necessary for proper cardiac contractility and systolic function, whereas pericyte loss does not seem to promote cardiac fibrosis, hypertrophy, or dilation. In the study by Siao et al,7 cardiac fibrosis and dilation may then be attributable to the effect of the mutant form of pro–nerve growth factor on other cell types, notably cardiomyocytes and/or fibroblasts, whereas in the study by Tao et al,9 cardiac hypertrophy may be attributable to Notch3 deletion in cardiomyocytes. 29
More important, this study suggests that pericytes may modify the phenotype of cardiomyocytes directly, notably by upregulating Cx43 and downregulating interleukin‐6. Another study has also suggested that pericytes may dialogue with cardiomyocytes directly via miR‐132 transfer. 30
One of the main limitations of this study is technical because a mouse model only targeting cardiac pericytes still needs to be generated to obtain robust and specific characterization of the role of cardiac pericytes. Indeed, the Pdgfrb promoter does not only target pericytes but also SMCs, and the Pdgfrb promoter does not only target cardiac mural cells but mural cells of the entire body. However, several mouse models targeting pericytes have been used, especially in the brain, and most of these models are based on platelet‐derived growth factor subunit B–PDGFRB signaling, which is crucial for pericyte recruitment and survival. 31 At first, developmental studies were performed on Pdgfb 32 and Pdgfrb 33 deficient embryos, which are not viable. Then, mice harboring mutations within the Pdgfrb gene (the Pdgfrb redeye/redeye mutant 34 and the Pdgfrb F7/F7 mutant 35 ) were used. These mutants display progressive pericyte loss, especially in the central nervous system (brain and retina). Mouse models with inducible pericyte depletion have been developed. These models are tamoxifen inducible and use either DTA or its receptor, diphtheria toxin receptor, to induce cell ablation. 36 These last models allow the exploration of the role of pericytes in adults, excluding the potential consequences of developmental defects. The Pdgfrb‐Cre/ERT2; Rosa iDTR model is proposed to induce a less severe pericyte depletion than the Pdgfrb‐Cre/ERT2; Rosa iDTA model; however, it requires administration of both tamoxifen and diphtheria toxin. 36 Alternatively to the Pdgfrb promoter, the Cspg4 (also known as NG2) promoter has been proposed to induce pericyte depletion. However, its induction efficiency is lower than the one of Pdgfrb, and just like the Pdgfrb promoter, it induces recombination in SMCs. 37 Finally, a strategy using a double‐promoter approach with Pdgfrb and the Cspg4 has been used 38 to target pericytes more specifically, but both promoters also target SMCs, as described above. Cell culture assays also have limitations; however, they may help in demonstrating pericyte‐specific effects.
In conclusion, the present study highlights the critical role of pericytes in the heart. Just like in the brain, cardiac pericytes are necessary to maintain coronary vasculature integrity and immune quiescence. More important, cardiac pericytes may protect from cardiac inflammation, not only by acting on ECs, but also by downregulating interleukin‐6 mRNA expression in cardiomyocytes. Finally, cardiac pericytes increase Cx43 mRNA expression in cardiomyocytes, suggesting they may directly participate in the regulation of cardiac function. Altogether, these results demonstrate that the role of cardiac pericytes should be considered when investigating the pathophysiological features of cardiac diseases.
Sources of Funding
This study was supported by grants from the Fondation pour la Recherche Médicale (équipe FRM) and the Agence Nationale de la Recherche (Appel à Projet Générique). In addition, this study was cofunded by the “Institut National de la Santé et de la Recherche Médicale” and by the University of Bordeaux. Dr Cornuault received a fellowship from the Fondation pour la Recherche Médicale. Dr Hérion received a fellowship from Centre Hospitalier Universitaire de Bordeaux.
Disclosures
None.
Supporting information
Tables S1–S3
Figures S1–S12
Acknowledgments
We thank Annabel Reynaud, Sylvain Grolleau, and Maxime David for their technical help.
This article was sent to Julie K. Freed, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.029279
For Sources of Funding and Disclosures, see page 15.
References
- 1. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7 [DOI] [PubMed] [Google Scholar]
- 2. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- 3. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood‐brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vazquez‐Liebanas E, Nahar K, Bertuzzi G, Keller A, Betsholtz C, Mäe MA. Adult‐induced genetic ablation distinguishes PDGFB roles in blood‐brain barrier maintenance and development. J Cereb Blood Flow Metab. 2022;42:264–279. doi: 10.1177/0271678X211056395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Su H, Cantrell AC, Zeng H, Zhu S‐H, Chen J‐X. Emerging role of pericytes and their secretome in the heart. Cell. 2021;10:548. doi: 10.3390/cells10030548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, Dunner K, Pati S, Bankson JA, Pasqualini R, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate‐induced cardiotoxicity. Sci Transl Med. 2013;5:187ra69. doi: 10.1126/scitranslmed.3005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siao C‐J, Lorentz CU, Kermani P, Marinic T, Carter J, McGrath K, Padow VA, Mark W, Falcone DJ, Cohen‐Gould L, et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J Exp Med. 2012;209:2291–2305. doi: 10.1084/jem.20111749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He X, Zeng H, Chen J‐X. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int J Cardiol. 2016;215:349–357. doi: 10.1016/j.ijcard.2016.04.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tao Y‐K, Zeng H, Zhang G‐Q, Chen ST, Xie X‐J, He X, Wang S, Wen H, Chen J‐X. Notch3 deficiency impairs coronary microvascular maturation and reduces cardiac recovery after myocardial ischemia. Int J Cardiol. 2017;236:413–422. doi: 10.1016/j.ijcard.2017.01.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greenhalgh SN, Iredale JP, Henderson NC. Origins of fibrosis: pericytes take centre stage. F1000Prime Rep. 2013;5:37. doi: 10.12703/P5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Farrell FM, Mastitskaya S, Hammond‐Haley M, Freitas F, Wah WR, Attwell D. Capillary pericytes mediate coronary no‐reflow after myocardial ischaemia. Elife. 2017;6:e29280. doi: 10.7554/eLife.29280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Methner C, Cao Z, Mishra A, Kaul S. Mechanism and potential treatment of the “no reflow” phenomenon after acute myocardial infarction: role of pericytes and GPR39. Am J Physiol Heart Circ Physiol. 2021;321:H1030–H1041. doi: 10.1152/ajpheart.00312.2021 [DOI] [PubMed] [Google Scholar]
- 13. Chen J, Sivan U, Tan SL, Lippo L, De Angelis J, Labella R, Singh A, Chatzis A, Cheuk S, Medhghalchi M, et al. High‐resolution 3D imaging uncovers organ‐specific vascular control of tissue aging. Sci Adv. 2021;7:7. doi: 10.1126/sciadv.abd7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeng H, Vaka VR, He X, Booz GW, Chen J‐X. High‐fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–1856. doi: 10.1111/jcmm.12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Dijk CGM, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenço AP, et al. Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ Heart Fail. 2016;9:e002760. doi: 10.1161/CIRCHEARTFAILURE.115.002760 [DOI] [PubMed] [Google Scholar]
- 16. Robinson FA, Mihealsick RP, Wagener BM, Hanna P, Poston MD, Efimov IR, Shivkumar K, Hoover DB. Role of angiotensin‐converting enzyme 2 and pericytes in cardiac complications of COVID‐19 infection. Am J Physiol Heart Circ Physiol. 2020;319:H1059–H1068. doi: 10.1152/ajpheart.00681.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bischoff FC, Werner A, John D, Boeckel J‐N, Melissari M‐T, Grote P, Glaser SF, Demolli S, Uchida S, Michalik KM, et al. Identification and functional characterization of hypoxia‐induced endoplasmic reticulum stress regulating lncRNA (HypERlnc) in pericytes. Circ Res. 2017;121:368–375. doi: 10.1161/CIRCRESAHA.116.310531 [DOI] [PubMed] [Google Scholar]
- 18. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Br J Pharmacol. 2020;177:3617–3624. doi: 10.1111/bph.15193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Renault MA, Roncalli J, Tongers J, Misener S, Thorne T, Jujo K, Ito A, Clarke T, Fung C, Millay M, et al. The hedgehog transcription factor Gli3 modulates angiogenesis. Circ Res. 2009;105:818–826. doi: 10.1161/CIRCRESAHA.109.206706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roncalli J, Renault MA, Tongers J, Misener S, Thorne T, Kamide C, Jujo K, Tanaka T, Ii M, Klyachko E, et al. Sonic hedgehog‐induced functional recovery after myocardial infarction is enhanced by AMD3100‐mediated progenitor‐cell mobilization. J Am Coll Cardiol. 2011;57:2444–2452. doi: 10.1016/j.jacc.2010.11.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recommendations for quantitation of the left ventricle by two‐dimensional echocardiography. American Society of Echocardiography Committee on standards, subcommittee on quantitation of two‐dimensional echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/S0894-7317(89)80014-8 [DOI] [PubMed] [Google Scholar]
- 22. Mohan JC, Sethi KK, Arora R, Khalilullah M. Cross sectional echocardiographic left ventricular ejection fraction: method based variability. Indian Heart J. 1992;44:23–28. [PubMed] [Google Scholar]
- 23. Abdelazim H, Payne LB, Nolan K, Paralkar K, Bradley V, Kanodia R, Gude R, Ward R, Monavarfeshani A, Fox MA, et al. Pericyte heterogeneity identified by 3D ultrastructural analysis of the microvessel wall. Front Physiol. 2022;13:1016382. doi: 10.3389/fphys.2022.1016382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Borbély A, Falcao‐Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite‐Moreira AF, Bronzwaer JGF, Papp Z, van der Velden J, et al. Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780–786. doi: 10.1161/CIRCRESAHA.108.193326 [DOI] [PubMed] [Google Scholar]
- 25. Robison WG, Kador PF, Kinoshita JH. Early retinal microangiopathy: prevention with aldose reductase inhibitors. Diabet Med. 1985;2:196–199. doi: 10.1111/j.1464-5491.1985.tb00635.x [DOI] [PubMed] [Google Scholar]
- 26. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer's disease. Brain Pathol. 2013;23:303–310. doi: 10.1111/bpa.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood‐spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su H, Zeng H, Liu B, Chen J. Sirtuin 3 is essential for hypertension‐induced cardiac fibrosis via mediating pericyte transition. J Cell Mol Med. 2020;24:8057–8068. doi: 10.1111/jcmm.15437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Øie E, Sandberg WJ, Ahmed MS, Yndestad A, Lærum OD, Attramadal H, Aukrust P, Eiken HG. Activation of notch signaling in cardiomyocytes during post‐infarction remodeling. Scand Cardiovasc J. 2010;44:359–366. doi: 10.3109/14017431.2010.511256 [DOI] [PubMed] [Google Scholar]
- 30. Katare R, Riu F, Mitchell K, Gubernator M, Campagnolo P, Cui Y, Fortunato O, Avolio E, Cesselli D, Beltrami AP, et al. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro‐RNA‐132. Circ Res. 2011;109:894–906. doi: 10.1161/CIRCRESAHA.111.251546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–4784. doi: 10.1242/dev.00721 [DOI] [PubMed] [Google Scholar]
- 32. Levéen P, Pekny M, Gebre‐Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875 [DOI] [PubMed] [Google Scholar]
- 33. Soriano P. Abnormal kidney development and hematological disorders in PDGF beta‐receptor mutant mice. Genes Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888 [DOI] [PubMed] [Google Scholar]
- 34. Jadeja S, Mort RL, Keighren M, Hart AW, Joynson R, Wells S, Potter PK, Jackson IJ. A CNS‐specific hypomorphic Pdgfr‐beta mutant model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013;54:3569–3578. doi: 10.1167/iovs.12-11125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nikolakopoulou AM, Zhao Z, Montagne A, Zlokovic BV. Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet‐derived growth factor receptor‐β signaling. PLoS One. 2017;12:e0176225. doi: 10.1371/journal.pone.0176225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eilken HM, Diéguez‐Hurtado R, Schmidt I, Nakayama M, Jeong H‐W, Arf H, Adams S, Ferrara N, Adams RH. Pericytes regulate VEGF‐induced endothelial sprouting through VEGFR1. Nat Commun. 2017;8:1574. doi: 10.1038/s41467-017-01738-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayr D, Preishuber‐Pflügl J, Koller A, Brunner SM, Runge C, Ladek A‐M, Rivera FJ, Reitsamer HA, Trost A. Characterization of the two inducible Cre recombinase‐based mouse models NG2‐CreER™ and PDGFRb‐P2A‐CreERT2 for pericyte labeling in the retina. Curr Eye Res. 2022;47:590–596. doi: 10.1080/02713683.2021.2002910 [DOI] [PubMed] [Google Scholar]
- 38. Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, Sagare AP, Lazic D, Sweeney MD, Kong P, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22:1089–1098. doi: 10.1038/s41593-019-0434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S12