

Abstract
Background
Data mining of electronic health records to identify patients suspected of familial hypercholesterolemia (FH) has been limited by absence of both phenotypic and genomic data in the same cohort.
Methods and Results
Using the Geisinger MyCode Community Health Initiative cohort (n=130 257), we ran 2 screening algorithms (Mayo Clinic [Mayo] and flag, identify, network, deliver [FIND] FH) to determine FH genetic and phenotypic diagnostic yields. With 29 243 excluded by Mayo (for secondary causes of hypercholesterolemia, no lipid value in electronic health records), 52 034 excluded by FIND FH (insufficient data to run the model), and 187 excluded for prior FH diagnosis, a final cohort of 59 729 participants was created. Genetic diagnosis was based on presence of a pathogenic or likely pathogenic variant in FH genes. Charts from 180 variant‐negative participants (60 controls, 120 identified by FIND FH and Mayo) were reviewed to calculate Dutch Lipid Clinic Network scores; a score ≥5 defined probable phenotypic FH. Mayo flagged 10 415 subjects; 194 (1.9%) had a pathogenic or likely pathogenic FH variant. FIND FH flagged 573; 34 (5.9%) had a pathogenic or likely pathogenic variant, giving a net yield from both of 197 out of 280 (70%). Confirmation of a phenotypic diagnosis was constrained by lack of electronic health record data on physical findings or family history. Phenotypic FH by chart review was present by Mayo and/or FIND FH in 13 out of 120 versus 2 out of 60 not flagged by either (P<0.09).
Conclusions
Applying 2 recognized FH screening algorithms to the Geisinger MyCode Community Health Initiative identified 70% of those with a pathogenic or likely pathogenic FH variant. Phenotypic diagnosis was rarely achievable due to missing data.
Keywords: electronic health records, familial hypercholesterolemia, genetic testing, machine learning
Subject Categories: Genetics
Nonstandard Abbreviations and Acronyms
- DLCN
Dutch Lipid Clinics Network
- FH
familial hypercholesterolemia
Clinical Perspective.
What Is New?
Using information technology tools applied to an electronic health record database, about 1 out of 6 of patients are eligible for medical evaluation for familial hypercholesterolemia (FH).
Screening just these individuals, about 70% of those in the entire cohort with a genetic variant causing FH will be found.
Phenotypic FH cannot be reliably diagnosed from the electronic health records.
What Are the Clinical Implications?
Electronic health record screening identifies a sufficiently high percentage of those with an FH genetic variant to suggest that health care systems that combine such strategies with patient evaluation, genetic testing of at‐risk patients, and cascade screening can identify a majority of patients with FH in their catchment area.
Familial hypercholesterolemia (FH) is underdiagnosed worldwide, despite high known risk for premature atherosclerotic cardiovascular disease and the known ability to prevent adverse outcomes with lipid‐lowering treatment. 1 To improve identification of those likely to have an FH diagnosis, many studies have used strategies, based on clinical characteristics (eg, low‐density lipoprotein cholesterol [LDL‐C] levels, premature heart attacks) or more complex machine learning algorithms (eg, flag, identify, network, deliver [FIND] FH) to search electronic health records (EHRs) to find at‐risk individuals. 2 , 3 , 4 , 5 , 6 , 7 However, other than studies in the UK Biobank, these efforts have generally not been performed in cohorts with both phenotypic EHR data and genomic information on the entire cohort, including those with and without FH.
The purpose of this study was to apply 2 recognized EHR screening algorithms (Mayo Clinic and FIND FH) to the Geisinger MyCode Community Health Initiative (MyCode) data set, to determine the yields for (1) a genetic diagnosis of FH based on the presence of a pathogenic or likely pathogenic variant identified via genomic screening, or (2) by phenotypic criteria, using the Dutch Lipid Clinic Network (DLCN) score. 2 , 3 , 8 Additional analyses looked at clinical characteristics of those identified by each algorithm, the relationship of variants of unknown significance on study covariates, and the relationship of the gene involved (eg, LDLR or APOB) to results.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Geisinger MyCode Community Health Initiative
In 2007, Geisinger launched the MyCode Community Health Initiative, creating a biobank of serum, blood, and DNA samples for health discovery research. 8 , 9 , 10 , 11 , 12 Research related to MyCode, and the analyses reported in this article were approved by the Geisinger institutional review board. All Geisinger patients are eligible to participate, irrespective of health status, and provide written informed consented in person when they present for routine care or via the patient portal in the EHR. Through the MyCode Genomic Screening and Counseling program, clinically actionable genomic risk results are identified, confirmed, and disclosed to patient participants and their clinicians. 13 When a pathogenic or likely pathogenic (P/LP) variant is identified in the MyCode exome sequencing data in a gene returned through the Genomic Screening and Counseling program, the variant is clinically confirmed in a clinical laboratory improvement amendments‐certified genetics laboratory before disclosure. 14 For this study, an adult (>18 years old) cohort comprising all those with completed exome sequencing data by October 2019 were included. After exclusion based on current consent status and age to exclude pediatric participants, a cohort of 130 257 eligible adult participants was created. FIND FH and the Mayo algorithms were run on this cohort.
Variants in the 3 main FH genes (LDLR, APOB, PCSK9) were annotated with variant effect predictor version 100, 15 SpliceAI version 106, 16 , 17 gnomAD version 2.1.1, 18 Rare Exome Variant Ensemble Learner (REVEL, 19 and ClinVar 18 , 20 (April 16, 2022) and filtered 14 based on minor allele frequency <0.01 and high impact by Clinical Laboratory Improvement Amendmentsor 2* P/LP in ClinVar. Filtered variants were manually reviewed based on elements from the current LDLR variant interpretation guideline. 20 Participants were considered as variant positive if the P/LP variant was clinical laboratory improvement amendments‐confirmed orthogonally or quality metrics met conservative thresholds (allele balance ≥0.35, genotype quality ≥90, and depth ≥20). For the expanded analysis into burden of LDLR variants, all LDLR variants, including ±15 bp into intronic regions and untranslated regions, were identified from the cohort of eligible participants and required to meet the same quality metrics above. Those not considered P/LP were considered variants of uncertain significance (VUS), unless the minor allele frequency based on gnomAD version 2.1.1 PopMax was ≥0.002, which were considered as likely to be benign. VUS with a REVEL score of ≥0.75, missense variants in exon 4 or that remove a conserved cysteine residue were considered suspicious VUS. Participants were then categorized for analysis as having a P/LP variant (excluded from further analysis), suspicious VUS, other VUS, or those with comparatively more common variants (minor allele frequency >0.002, rest of MyCode). Participants with multiple variants were categorized according to the variant with the highest‐ranking category. Copy number variants were called from the whole exome sequence data using the copy number estimation using the Lattice‐Aligned Mixture Models algorithm. 21 Copy number variants in the LDLR gene considered to be P/LP included (1) duplications overlapping with a previously reported tandem duplication (exon 13 to exon 17) known to be P/LP or (2) deletions spanning at least 2 exons located upstream of the penultimate exon and LDLR was the only gene within the breakpoints.
Find FH
FIND FH is a proprietary machine learning algorithm developed by the Family Heart Foundation. 2 , 7 Two years of health care encounter history, including a patient's diagnoses, procedures, medications, and laboratory results, are used to score the patient's relative likelihood of having yet‐to‐be‐diagnosed FH. The algorithm scores only individuals with sufficient data and at least 1 cardiac comorbidity or primary prevention condition recorded in their history. 2 There were 17 426 unique individuals excluded with insufficient data, and 49 389 were excluded for not having any history relevant to cardiac conditions. An additional 187 were excluded for a previous FH diagnosis. Some patients had >1 exclusion, resulting in a total of 52 034 unique patients excluded. Patients with FIND FH scores ≥0.23 were identified and subjected to further postprocessing filter rules, including removing those individuals with diagnosed nephrotic syndrome, triglycerides laboratory values >400 mg/dL, and age >85 years. The threshold of 0.23 was chosen, based on precision and recall calculated when the algorithm was applied to the holdout data set after training. 2
Mayo Clinic Algorithm
The algorithm currently in use at the Mayo Clinic for identifying patients with LDL‐C ≥190 mg/dL was adapted for use in the Geisinger EHR. 3 To use this algorithm, LDL‐C must be present in the EHR. Patients with laboratory values consistent with proteinuria (urine protein >3000 mg/24 h), severe liver disease (alkaline phosphatase >200 IU/L), triglycerides >400 mg/dL, or uncontrolled hypothyroidism (thyroid stimulating hormone >10 mIU/L) are excluded. The highest LDL‐C in the record is used for assignment in the presence of multiple values. If statin use is present, the highest on‐treatment value is divided by 0.7 to estimate pretreatment LDL‐C. These criteria led to 29 243 exclusions.
Based on the entry criteria for each algorithm, a final cohort for analysis of 59 729 subjects was created for whom both algorithms could be run, on whom exome sequencing data were available, and on whom a prior diagnosis of FH based on International Classification of Diseases, Tenth Revision (ICD‐10) codes had not been established. The Consolidated Standards of Reporting Trials (CONSORT) diagram describes the assembly of the final cohort (Figure 1).
Figure 1. CONSORT diagram showing creation of the cohort.
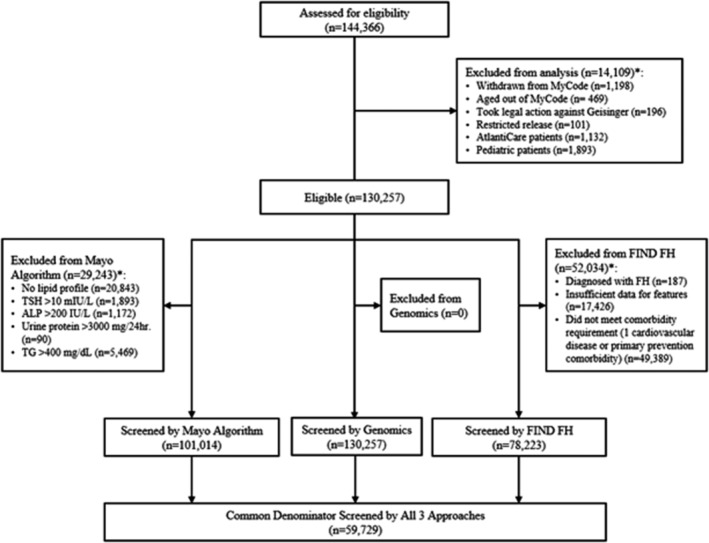
The diagram depicts the construction of the final cohort for this study. *Patients may fall into >1 exclusion category. ALP indicates alkaline phosphatase; CONSORT, Consolidated Standards of Reporting Trials; FIND, Flag, Identify, Network, Deliver; FH, familial hypercholesterolemia; TG, triglycerides; and TSH, thyroid stimulating hormone.
Chart Reviews
Chart reviews from the EHR were conducted manually on a total of 420 subjects; 240 out of 280 with a positive FH variant, 60 without an FH variant and not identified by either algorithm (controls), and 120 identified by either algorithm (40 Mayo only, 40 FIND FH only, and 40 identified by both). Subjects were chosen randomly from the specific subgroups. Study data were collected and managed using REDCap electronic data capture tools hosted at Geisinger. 22 , 23 Information needed to calculate the DLCN score was extracted from both structured and unstructured EHR data by following predetermined instructions that included defined search terms related to cholesterol, FH, family history, atherosclerotic cardiovascular disease (ASCVD), and physical findings. 24 For LDL‐C values, the highest untreated value was used whenever possible; if untreated LDL‐C was unavailable, an untreated value was calculated from the subject's first treated LDL‐C and associated medication regimen using a web‐based tool. 25 LDL‐C percentiles were calculated using a web‐based lipid reference value tool. 25 , 26 To satisfy components of each respective criteria, relevant responses of yes, no, or unknown were indicated. A response of unknown was designated for elements that could not be ascertained from the EHR. When discrepancies occurred, charts were reviewed by a second reviewer, and discordant findings adjudicated. Mayo algorithm performance was evaluated for accuracy using manual chart review.
Statistical Analysis
Data were summarized using median and interquartile ranges for continuous variables, and frequency and percentage for categorial variables. Comparisons between groups of interest was accomplished using the Wilcoxon rank sum, Kruskal‐Wallis, and the Pearson χ2 tests. Post hoc pairwise comparisons used the Sidak multiple comparison criteria for determining significance. 27 A Venn diagram was used to graphically show the overlap of positive FH variant and identification by the FIND FH and Mayo algorithms. Test characteristics (sensitivity, specificity, positive predictive value, negative predictive value) for prediction of the presence of a genetic variant were calculated. SAS version 9.4 (SAS Institute, Cary, NC) and R version 4.2.2 (The R Foundation for Statistical Computing) were used for all analyses.
Results
Results of the primary analysis, yield of the presence of a genetic variant for FH in those identified by either FIND FH or the Mayo algorithm, are shown in the Venn diagram (Figure 2). Overall, 280 out of 59 729 (0.45%) had a P/LP FH variant; the same percentage of variants was identified in the excluded cohort. Of these, 144 were in LDLR and 96 were in APOB. Those with a P/LP variant are included in the gray circle, those positive for FIND FH are in the orange circle, and those identified by Mayo are in the blue circle. FIND FH flagged 573 subjects; 34 (5.9%) had a P/LP FH variant. Mayo flagged 10 415 subjects; 195 (1.9%) had a P/LP FH variant. Of the 280 with a P/LP variant, 197 (70%) were identified by at least 1 algorithm; 103 were in LDLR and 64 were in APOB. No P/LP variants were identified in PCSK9. Test characteristics are presented for identifying an FH genetic variant in Table 1.
Figure 2. Venn diagram showing yield for an FH genetic variant of the FIND FH and Mayo algorithms.
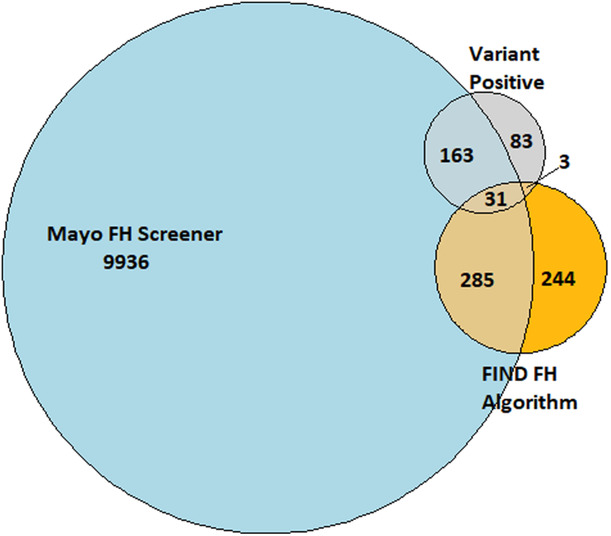
The Venn diagram shows the overlap among those who were identified by Flag, Identify, Network, Deliver (FIND) FH (orange) and the Mayo algorithm (blue), and those who had a pathogenic or likely pathogenic familial hypercholesterolemia (FH) variant (gray).
Table 1.
Test Characteristics for Prediction of an FH Genetic Variant for Each Algorithm and the Algorithms Combined
| Mayo algorithm | FIND FH algorithm | Mayo or FIND FH | |
|---|---|---|---|
| Sensitivity | 69.3% | 12.1% | 70.4% |
| Specificity | 82.8% | 99.1% | 82.4% |
| Positive predictive value | 1.9% | 5.9% | 1.8% |
| Negative predictive value | 99.8% | 98.6% | 99.8% |
FIND indicates Flag, Identify, Network, Deliver; and FH, familial hypercholesterolemia.
For the expanded analysis based on variants within LDLR, the distribution of LDL‐C values by variant category are shown in Table 2. Those with a P/LP FH variant had the highest mean LDL‐C values, followed by those with suspicious VUS. Average LDL‐C for those with an LDLR variant versus an APOB variant was 256.34 (SD, 99.18) versus 217.71 (SD, 62.75), respectively (P=0.0028). Those with a P/LP FH variant also had the highest prevalence of ASCVD, family history of ASCVD, and use of lipid‐lowering therapy. Conversely, they also had the lowest triglycerides, body mass index, and blood pressure. Those with suspicious VUS had intermediate values for LDL‐C, use of lipid‐lowering medications, atherosclerotic vascular disease, family history of atherosclerotic vascular disease, and identification by 1 of the algorithms as compared with those with P/LP FH variants and the rest of MyCode (those without an LDLR VUS). Important for screening, the prevalence of hypothyroidism, diabetes, and chronic kidney disease did not differ across variant groups. Black subjects were less likely to have a P/LP FH variant but more likely to have a VUS or benign/likely benign variant (Table 3).
Table 2.
Summary Statistics by FH Variant Groups
| P/LP (N=280) | Suspicious VUS (N=142) | VUS (N=1555) | LB/B (N=2875) | No variants (N=54 877) | P value | |
|---|---|---|---|---|---|---|
| Age, y | ||||||
| Mean (SD) | 58.2 (13.97) | 61.5 (13.16) | 60.0 (13.99) | 59.9 (13.85) | 60.6 (13.70) | 0.0011* |
| Median (IQR) | 60.0 (48.0–68.0) | 62.0 (52.0–71.0) | 62.0 (51.0–71.0) | 61.0 (51.0–71.0) | 62.0 (52.0–71.0) | |
| Female sex, n (%) | 160 (57.1%) | 72 (50.7%) | 967 (62.2%) | 1745 (60.7%) | 32 483 (59.2%) | 0.0121 |
| Body mass index | ||||||
| N (missing) | 277 (3) | 142 (0) | 1548 (7) | 2870 (5) | 54 730 (147) | 0.0141* |
| Mean (SD) | 31.1 (7.66) | 33.1 (7.64) | 32.2 (8.25) | 32.5 (7.94) | 33.6 (75.76) | |
| Median (IQR) | 31 (26–35) | 32 (27–38) | 31 (27–36) | 31 (27–37) | 31 (27–37) | |
| Lipid‐lowering therapy, n (%) | 90 (32.1%) | 35 (24.6%) | 302 (19.4%) | 520 (18.1%) | 9847 (17.9%) | <0.0001* , ¶ |
| Highest LDL‐C | ||||||
| Mean (SD) | 240.8 (87.44) | 182.4 (74.09) | 153.7 (51.03) | 147.9 (50.92) | 151.7 (57.60) | <0.0001* , † , § , || , ¶ , # |
| Median (IQR) | 226 (176–286) | 164 (133–217) | 150 (119–179) | 141 (115–171) | 146 (119–175) | |
| HDL‐C | ||||||
| N (missing) | 260 (20) | 128 (14) | 1398 (157) | 2578 (297) | 49 089 (5788) | 0.0238† , # |
| Mean (SD) | 51.7 (14.82) | 47.3 (12.47) | 52.2 (14.71) | 52.1 (14.83) | 52.0 (15.24) | |
| Median (IQR) | 49 (41–62) | 47 (39–55) | 50 (42–60) | 50 (42–60) | 49 (41–60) | |
| Total cholesterol | ||||||
| N (missing) | 261 (19) | 128 (14) | 1411 (144) | 2595 (280) | 49 266 (5611) | |
| Mean (SD) | 292.3 (72.65) | 243.1 (58.13) | 222.4 (55.18) | 215.3 (45.01) | 219.6 (44.50) | |
| Median (IQR) | 281 (243–332) | 241 (202–269) | 220 (190–252) | 213 (185–245) | 219 (189–248) | |
| Triglycerides | ||||||
| N (missing) | 258 (22) | 124 (18) | 1374 (181) | 2516 (359) | 48 047 (6830) | 0.0164 |
| Mean (SD) | 133.6 (67.52) | 151.9 (69.85) | 140.8 (64.12) | 139.0 (63.56) | 141.3 (64.39) | |
| Median (IQR) | 116 (84–169) | 140 (97–193) | 127 (95–175) | 128 (92–175) | 129 (94–176) | |
| Systolic blood pressure | ||||||
| N (missing) | 278 (2) | 142 (0) | 1551 (4) | 2874 (1) | 54 797 (80) | 0.0227* |
| Mean (SD) | 124.3 (15.15) | 127.9 (17.13) | 126.9 (16.14) | 127.1 (15.13) | 127.2 (15.81) | |
| Median (IQR) | 122 (112–132) | 126 (118–138) | 126 (116–136) | 126 (118–137) | 126 (118–136) | |
| Diastolic blood pressure | ||||||
| N (missing) | 278 (2) | 142 (0) | 1551 (4) | 2874 (1) | 54 797 (80) | 0.0354 |
| Mean (SD) | 72.8 (9.95) | 73.9 (10.01) | 74.2 (10.14) | 74.7 (10.11) | 74.3 (9.94) | |
| Median (IQR) | 72 (66–80) | 74 (68–80) | 74 (68–80) | 74 (68–80) | 74 (68–80) | |
| Family history of heart disease, n (%) | 26 (9.3%) | 11 (7.7%) | 96 (6.2%) | 148 (5.1%) | 3330 (6.1%) | 0.0392 |
| ASCVD, n (%) | 101 (36.1%) | 38 (26.8%) | 346 (22.3%) | 600 (20.9%) | 11 950 (21.8%) | <0.0001* , ¶ |
| Diabetes, n (%) | 72 (25.7%) | 46 (32.4%) | 419 (26.9%) | 810 (28.2%) | 15 008 (27.3%) | 0.5133 |
| CKD, n (%) | 38 (13.6%) | 25 (17.6%) | 265 (17.0%) | 480 (16.7%) | 9918 (18.1%) | 0.0859 |
| Smoking status, n (%) | ||||||
| Current | 46 (16.4%) | 26 (18.3%) | 258 (16.6%) | 477 (16.6%) | 8250 (15.0%) | 0.4067 |
| Former | 94 (33.6%) | 47 (33.1%) | 543 (34.9%) | 1039 (36.1%) | 20 248 (36.9%) | |
| Never | 140 (50.0%) | 69 (48.6%) | 753 (48.5%) | 1359 (47.3%) | 26 342 (48.0%) | |
| Unknown | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 9 (0.0%) | |
| Missing | 0 | 0 | 1 | 0 | 28 | |
| Hypothyroidism, n (%) | 44 (15.7%) | 30 (21.1%) | 318 (20.5%) | 600 (20.9%) | 11 445 (20.9%) | 0.3296 |
| Mayo algorithm screen positive | 194 (62.3%) | 48 (33.8%) | 299 (19.2%) | 450 (15.6%) | 9424 (17.2%) | <0.0001* , † , || , ¶ , # |
| FIND FH algorithm screen positive | 34 (12.1%) | 10 (7.0%) | 12 (0.8%) | 19 (0.7%) | 498 (0.9%) | <0.0001* , † , ¶ , # |
ASCVD indicates atherosclerotic cardiovascular disease; CKD, chronic kidney disease; FIND, Flag, Identify, Network, Deliver; FH, familial hypercholesterolemia; HDL‐C, high‐density lipoprotein cholesterol; IQR, interquartile range; LB/B, likely benign or benign; LDL‐C, low‐density lipoprotein cholesterol; P/LP, pathogenic or likely pathogenic; and VUS, variants of uncertain significance.
P/LP vs no variants.
Suspicious VUS vs no variants.
VUS vs no variants.
LB/B vs no variants.
P/LP vs suspicious VUS.
P/LP vs VUS.
Suspicious VUS vs VUS.
Table 3.
Variant Group by Race Categories
| FH variant group | White (n=58 121) | Black (n=1133) | Other (n=475)* | P value |
|---|---|---|---|---|
| P/LP | 279 (0.5%) | 1 (0.1%) | 0 (0%) | 0.1384 |
| Suspicious VUS | 134 (0.2%) | 7 (0.6%) | 1 (0.2%) | 0.0063 |
| VUS | 1492 (2.6%) | 47 (4.2%) | 16 (3.4%) | <0.0001 |
| LB/B | 2603 (4.5%) | 248 (21.9%) | 24 (5.0%) | <0.0001 |
| No variants | 56 613 (92.2%) | 830 (73.3%) | 434 (91.4%) | … |
The distribution of the variant groups was significantly different between Black and White biobank participants (P<0.0001).
FH indicates familial hypercholesterolemia; LB/B, likely benign or benign; P/LP, pathogenic or likely pathogenic; and VUS, variants of uncertain significance. *Other includes 38% Asian, 24% Unknown, 20% American Indian or Alaska Native, and 18% Native Hawaiian or Other Pacific Islander.
Comparison of those with P/LP FH variants identified by either algorithm with those not identified are presented in Table 4. Those not identified by Mayo or FIND FH were more likely to have elevated LDL‐C and total cholesterol and be on lipid‐lowering therapy. They were slightly more likely to be women.
Table 4.
Participants With a P/LP FH Variant: Comparison of Those Identified by Algorithm Versus Those Not Identified
| Variable | Participants with P/LP FH variants identified by either FIND FH or Mayo | P value | |
|---|---|---|---|
| No (N=83) | Yes (N=197) | ||
| FH gene | |||
| APOB | 32 (43.8%) | 64 (38.3%) | 0.4226 |
| LDLR | 41 (56.2%) | 103 (61.7%) | |
| Age, y | |||
| Mean (SD) | 59.4 (13.97) | 57.7 (13.97) | 0.2165 |
| Median (IQR) | 63.0 (50.0–70.0) | 59.0 (47.0–67.0) | |
| Female sex, n (%) | 40 (48.2%) | 120 (60.9%) | 0.0495 |
| Race, n (%) | |||
| Black | 0 (0.0%) | 1 (0.5%) | 0.5155 |
| White | 83 (100.0%) | 196 (99.5%) | |
| Body mass index | |||
| Mean (SD) | 30.1 (8.34) | 31.5 (7.35) | 0.0919 |
| Median (IQR) | 29 (24–34) | 31 (26–35) | |
| Lipid‐lowering therapy, n (%) | 9 (10.8%) | 81 (41.1%) | <0.0001 |
| LDL‐C | |||
| Mean (SD) | 153.7 (26.59) | 277.6 (77.56) | <0.0001 |
| Median (IQR) | 160 (134–174) | 259 (221–313) | |
| HDL‐C | |||
| Mean (SD) | 49.8 (13.40) | 52.4 (15.33) | 0.2472 |
| Median (IQR) | 47 (40–62) | 50 (42–62) | |
| Total cholesterol | |||
| Mean (SD) | 222.6 (32.10) | 319.9 (65.41) | <0.0001 |
| Median (IQR) | 230 (200–249) | 305 (274–350) | |
| Triglycerides | |||
| N (missing) | 75 (8) | 183 (14) | 0.858 |
| Mean (SD) | 120.9 (57.69) | 138.8 (70.65) | |
| Median (IQR) | 103 (77–155) | 119 (86–178) | |
| Systolic blood pressure | |||
| Mean (SD) | 125.1 (17.37) | 123.9 (14.17) | 0.9633 |
| Median (IQR) | 122 (112–134) | 122 (113–132) | |
| Diastolic blood pressure | |||
| Mean (SD) | 72.9 (9.45) | 72.8 (10.17) | 0.8473 |
| Median (IQR) | 74 (64–80) | 72 (66–80) | |
| Family history of heart disease | 9 (10.8%) | 17 (8.6%) | 0.5599 |
| ASCVD, n (%) | 32 (38.6%) | 69 (35.0%) | 0.5744 |
| Diabetes, n (%) | 24 (28.9%) | 48 (24.4%) | 0.4263 |
| CKD, n (%) | 11 (13.3%) | 27 (13.7%) | 0.9196 |
| Smoking status | |||
| Current | 16 (19.3%) | 30 (15.2%) | 0.0919 |
| Former | 20 (24.1%) | 74 (37.6%) | |
| Never | 47 (56.6%) | 93 (47.2%) | |
| Hypothyroidism, n (%) | 11 (13.3%) | 33 (16.8%) | 0.4626 |
ASCVD indicates atherosclerotic cardiovascular disease; CKD, chronic kidney disease; FIND, Flag, Identify, Network, Deliver; FH, familial hypercholesterolemia; HDL‐C, high‐density lipoprotein cholesterol; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; and P/LP, pathogenic or likely pathogenic.
Subjects with a P/LP FH variant were then compared with those identified by either algorithm, those identified by FIND FH alone, those identified by Mayo alone, and the remainder of the cohort (Table 5). Those not identified by either algorithm had slightly higher body mass index, were much less likely to be on lipid‐lowering therapy, had lower LDL‐C (by definition), and the lowest prevalence of family history of ASCVD. The highest LDL‐C was in those identified by the Mayo algorithm. Those identified by FIND FH alone had the lowest LDL‐C levels, and intermediate risk factor levels between those not identified by either algorithm and either those identified by the Mayo algorithm, both algorithms, or those with P/LP FH variants. The lower LDL‐C levels in those with a P/LP FH variant were explained by the presence of lower LDL‐C in those with APOB variants.
Table 5.
Comparison of Those With a P/LP FH Variant With Those Identified by Algorithms and the Remainder of the Cohort
| Variable | P/LP (N=280) | Mayo+FIND FH (N=285) | FIND FH only (N=254) | Mayo only (N=9936) | Remainder of cohort* (N=48 974) | P value |
|---|---|---|---|---|---|---|
| Age, y | ||||||
| Mean (SD) | 58.2 (13.97) | 60.8 (12.87) | 63.5 (12.42) | 63.8 (11.44) | 59.9 (14.05) | <0.0001† , ‡ , § , ¶ , # |
| Median (IQR) | 60.0 (48.0–68.0) | 62.0 (53.0–71.0) | 65.0 (58.0–71.0) | 65.0 (56.0–72.0) | 62.0 (51.0–71.0) | |
| Female sex, n (%) | 160 (57.1%) | 199 (69.8%) | 148 (58.3%) | 6246 (62.9%) | 28 674 (58.5%) | <0.0001§ , ** , †† |
| Race, n (%) | ||||||
| Black | 1 (0.4%) | 7 (2.5%) | 5 (2.0%) | 157 (1.6%) | 963 (2.0%) | 0.0441 |
| Other§§ | 0 (0.0%) | 1 (0.4%) | 2 (0.8%) | 68 (0.7%) | 404 (0.8%) | |
| White | 279 (99.6%) | 277 (97.2%) | 247 (97.2%) | 9711 (97.7%) | 47 607 (97.2%) | |
| Body mass index | ||||||
| N (missing) | 277 (3) | 285 (0) | 254 (0) | 9926 (10) | 48 825 (149) | <0.0001† , § , †† , ‡‡ |
| Mean (SD) | 31.1 (7.66) | 30.4 (6.94) | 31.4 (7.47) | 32.5 (52.11) | 33.8 (76.72) | |
| Median (IQR) | 31 (26–35) | 29 (26–33) | 30 (26–35) | 31 (27–36) | 31 (27–37) | |
| Lipid‐lowering therapy, n (%) | 90 (32.1%) | 160 (56.1%) | 57 (22.4%) | 5624 (56.6%) | 4863 (9.9%) | <0.0001 |
| Highest LDL‐C | ||||||
| Mean (SD) | 240.8 (87.44) | 288.0 (410.04) | 149.1 (29.67) | 231.8 (40.75) | 134.6 (30.88) | <0.0001* , † , § , || , ¶ , # , ** , †† , ‡‡ |
| Median (IQR) | 226 (176–286) | 254 (215–301) | 155 (131–172) | 219 (201–251) | 137 (113–159) | |
| HDL‐C | ||||||
| N (missing) | 260 (20) | 256 (29) | 236 (18) | 8740 (1196) | 43 961 (5013) | 0.0070 |
| Mean (SD) | 51.7 (14.82) | 54.3 (15.57) | 54.5 (15.44) | 51.6 (13.91) | 52.0 (15.44) | |
| Median (IQR) | 49 (41–62) | 52 (43–63) | 53 (43–64) | 50 (42–59) | 49 (41–60) | |
| Total cholesterol | ||||||
| N (missing) | 261 (19) | 257 (28) | 237 (17) | 8768 (1168) | 44 138 (4836) | |
| Mean (SD) | 292.3 (72.65) | 299.8 (54.39) | 218.6 (38.14) | 271.1 (39.54) | 208.8 (37.72) | |
| Median (IQR) | 281 (243–332) | 296 (267–332) | 223 (189–246) | 274 (246–295) | 210 (183–236) | |
| Triglycerides | ||||||
| N (missing) | 258 (22) | 255 (30) | 236 (18) | 8517 (1419) | 43 053 (5921) | <0.0001* , ‡ , § , || , ** , †† |
| Mean (SD) | 133.6 (67.52) | 163.1 (72.20) | 130.7 (56.89) | 166.4 (66.05) | 136.1 (62.79) | |
| Median (IQR) | 116 (84–169) | 145 (111–203) | 121 (89–159) | 156 (117–205) | 124 (90–170) | |
| Systolic blood pressure | ||||||
| N (missing) | 278 (2) | 285 (0) | 254 (0) | 9932 (4) | 48 893 (81) | <0.0001‡ , § , ‡‡ |
| Mean (SD) | 124.3 (15.15) | 127.5 (16.99) | 126.1 (13.67) | 128.0 (16.11) | 127.0 (15.72) | |
| Median (IQR) | 122 (112–132) | 126 (116–138) | 126 (118–134) | 126 (118–138) | 126 (118–136) | |
| Diastolic blood pressure | ||||||
| N (missing) | 278 (2) | 285 (0) | 254 (0) | 9932 (4) | 48 893 (81) | 0.0025 |
| Mean (SD) | 72.8 (9.95) | 74.4 (9.88) | 73.1 (9.40) | 74.1 (9.90) | 74.4 (9.97) | |
| Median (IQR) | 72 (66–80) | 76 (70–80) | 72 (68–80) | 74 (68–80) | 74 (68–80) | |
| Family history of heart disease, n (%) | 26 (9.3%) | 34 (11.9%) | 11 (4.3%) | 670 (6.7%) | 2870 (5.9%) | <0.0001* , § , || , †† |
| ASCVD, n (%) | 101 (36.1%) | 98 (34.4%) | 52 (20.5%) | 2870 (28.9%) | 9914 (20.2%) | <0.0001* , § , || , ¶ , †† , ‡‡ |
| Diabetes, n (%) | 72 (25.7%) | 79 (27.7%) | 64 (25.2%) | 3197 (32.2%) | 12 943 (26.4%) | <0.0001§ |
| CKD, n (%) | 38 (13.6%) | 59 (20.7%) | 41 (16.1%) | 2336 (23.5%) | 2336 (23.5%) | <0.0001‡ , § |
| Smoking status, n (%) | ||||||
| Current | 46 (16.4%) | 43 (15.1%) | 31 (12.3%) | 1647 (16.6%) | 7290 (14.9%) | 0.0040§ |
| Former | 94 (33.6%) | 102 (35.8%) | 100 (39.5%) | 3692 (37.2%) | 17 983 (36.7%) | |
| Never | 140 (50.0%) | 140 (49.1%) | 122 (48.2%) | 4593 (46.2%) | 23 668 (48.4%) | |
| Unknown | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 9 (0.0%) | |
| Missing | 0 | 0 | 1 | 4 | 24 | |
| Hypothyroidism, n (%) | 44 (15.7%) | 70 (24.6%) | 57 (22.4%) | 2450 (24.7%) | 9816 (20.0%) | <0.0001‡ , § |
ASCVD indicates atherosclerotic cardiovascular disease; CKD, chronic kidney disease; FIND, Flag, Identify, Network, Deliver; FH, familial hypercholesterolemia; HDL‐C, high‐density lipoprotein cholesterol; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; and P/LP, pathogenic or likely pathogenic.
No variants identified through genomic screening and not flagged by either FIND FH or Mayo algorithms. Mayo only vs FIND FH only.
Mayo only vs Mayo + FIND FH.
Mayo only vs P/LP.
Mayo only vs none.
FIND FH only vs Mayo + FIND FH.
FIND FH only vs P/LP.
FIND FH only vs none.
Mayo + FIND FH vs P/LP.
Mayo + FIND FH vs none.
P/LP vs none.
Other includes 38% Asian, 24% Unknown, 20% American Indian or Alaska Native, and 18% Native Hawaiian or Other Pacific Islander.
Phenotypic diagnosis was severely constrained by absence of data in the EHR on physical findings associated with FH and family history data for either premature ASCVD (as opposed to any family history), presence of an FH genetic variant, or high cholesterol as specified by DLCN criteria, as presented in Table 6, after data searches performed by trained observers. Chart reviews of those who were variant negative showed phenotypic FH, as defined by a DLCN score of >5, was present in 13 out of 120 identified by either algorithm versus 2 out of 60 identified by neither (P<0.09). Note that 13 out of 240 of those who were variant positive had probable versus definite FH by DLCN criteria, because a genetic variant contributes 8 points to the score, and 9 points are needed for a definite diagnosis.
Table 6.
Chart Review DLCN of Diagnosis and Unknown Subcategory
| Diagnosis* | All variant positive (n=240) | All variant negative (n=180) | |||
|---|---|---|---|---|---|
| Mayo only (n=40) | FIND FH only (n=40) | Mayo + FIND FH only (n=40) | Unflagged (n=60) | ||
| DLCN | |||||
| Definite (n=230) | 227 | 1 | 0 | 2 | 0 |
| Probable (n=25) | 13 | 2 | 1 | 7 | 2 |
| Possible (n=62) | 0 | 26 | 5 | 24 | 7 |
| Unlikely (n=103) | 0 | 11 | 34 | 7 | 51 |
| Prevalence of missing data by DLCN category | |||||
| DLCN subcategory unknowns† , ‡ | |||||
| First‐degree relative with known premature CHD or first‐degree relative with known LDL‐C level >95th percentile (n=305) | 157 | 32 | 35 | 29 | 52 |
| First‐degree relative with tendon xanthoma and/or corneal arcus or child(ren) <18 years with LDL‐C level >95th percentile (n=417) | 237 | 40 | 40 | 40 | 60 |
| Subject has premature CHD (n=74) | 54 | 1 | 7 | 4 | 8 |
| Subject has premature cerebral or peripheral vascular disease (n=92) | 63 | 4 | 6 | 7 | 12 |
| Tendon xanthoma (n=378) | 207 | 39 | 39 | 35 | 58 |
| Corneal arcus in person <45 years (n=388) | 213 | 40 | 38 | 37 | 60 |
| Causative mutation shown in an FH gene (n=178) | 0 | 39 | 40 | 39 | 60 |
CHD indicates coronary heart disease; DLCN, Dutch Lipid Clinics Network; FIND, Flag, Identify, Network, Deliver; FH, familial hypercholesterolemia; and LDL‐C, low‐density lipoprotein cholesterol.
DLCN diagnoses: definite >8; probable = 6–8; possible = 3–5; unlikely <3.
Numbers represent charts where the subcategory was marked as unknown.
In cases of true missingness of data in the electronic health records (ie, complete absence or no explicit documentation of a positive or negative finding in the patient's chart), the corresponding subcategory has been marked as unknown. An unknown in any category would result ultimately in an inconclusive status for DLCN, regardless of DLCN diagnosis.
Discussion
We were able to find about 70% of adult patients with a P/LP FH variant not previously diagnosed with FH using 2 information technology tools, the first based on identifying those with high LDL‐C (Mayo) and the second based on machine learning technology (FIND FH). 2 , 3 Those with a P/LP LDLR variant were more likely to be identified than those with a P/LP APOB variant, because the latter have lower rates of ASCVD and lower LDL‐C. Collectively, the algorithms identified about 17% of the entire cohort at risk for having FH. It has been estimated that to identify virtually all people in a given population with FH using a combination of index cases and cascade testing, about 50% to 70% of index cases must be known. 28 Therefore, the combination of using information technology in combination with cascade screening to achieve FH recognition is possible, but limited by the relatively large number of people identified at risk who do not actually have FH. A second limitation is lack of EHR information necessary to help make an FH diagnosis. A substantial percentage of the full MyCode cohort was excluded from the Mayo analysis because of the absence of a lipid profile (the FIND FH model can be run even without a lipid panel in the structured EHR data).
As in other studies of genetically defined FH, including those from this cohort, participants with a P/LP FH variant had the highest likelihood of ASCVD, high LDL‐C, higher likelihood of family history of ASCVD, and younger age. 1 , 8 The lower percentage of men likely is secondary to early death in some of those with FH, creating a selection bias toward a higher percentage of women in the genetic variant positive cohort. Black subjects were less likely to have a P/LP variant but more likely to have a VUS, consistent with the fact that FH has been studied predominantly in White and Asian populations. By definition, those identified by the Mayo algorithm had the highest LDL‐C; those with FH were less likely to get statins than those identified by the Mayo algorithm. However, given the generally high‐risk profiles of individuals identified by both algorithms, even in the absence of them having FH, flagging these individuals will likely result in identification of many people who need intensification of lipid‐lowering and other preventive treatments.
The yield of P/LP FH variants from the FIND FH machine learning model was almost 6%, slightly higher than the yield of variants from screening those with premature ASCVD as assessed in recent meta‐analyses. 29 , 30 This was true despite the fact that many participants in the cohort with a confirmed genetic diagnosis were excluded because of a prior FH diagnosis. The model was trained to identify individuals with phenotypic and/or genotypic FH. An interesting finding was that about 45% of those identified by FIND FH appeared to have relatively low overall risk profiles (Table 4); this allowed the identification of 3 subjects with LDL‐C <190 mg/dL, and confirms the algorithm relies on characteristics beyond LDL‐C alone to identify subjects at risk. Recent studies of FH screening done in population‐based cohorts show that the overlap of phenotypic and genotypic FH is not as great as previously assumed. 31 , 32 In a cohort of 79 058 adults from Iceland, only 5.2% of those with FH according to the modified DLCN score also tested positive for monogenic FH. 32 Similarly, in a cohort of 1682 individuals from Minnesota with LDL‐C >155 mg/dL, only 7% with phenotypic FH also had monogenic FH. 31 In these studies, many individuals had a polygenic cause for hypercholesterolemia rather than monogenic FH. The characterization of yield using FIND FH in the current study focuses primarily on the assessment of monogenic FH but does not assess polygenic hypercholesterolemia and underestimates the presence of phenotypic FH, due to missing family history data in the EHR.
The Mayo algorithm had a high yield of those needing screening for FH, about one‐sixth of the total cohort, but also a 1.9% yield for those with P/LP FH variants, thus improving the sensitivity. The algorithm likely overestimated the number of people at risk for several reasons. Our yield for at risk individuals was higher than a similarly designed study at the Mayo Clinic. 33 The median age of those identified with LDL‐C >190 mg/dL was about 60 years; because of age‐related rise in cholesterol, the FH yield will be proportionately lower than at younger ages. 34 , 35 A second reason is the inclusion of an adjustment of LDL‐C upward in those prescribed statins. This may contribute to overestimation of the number of people at risk either because of nonadherence with statin use or a lower response to treatment.
There have been several studies published on the usefulness of information technology strategies to identify patients at risk for FH. These have generally relied on making a phenotypic diagnosis of FH, often with molecular confirmation. In the United Kingdom, an algorithm based on the Simon‐Broome criteria was successful in identifying primary care patients at risk for FH. 5 At Kaiser Permanente, an algorithm based on make early diagnosis to prevent early deaths criteria, followed up by chart review identified patients with high‐risk FH. 4 At Stanford, a study that combined FIND FH with natural language processing of EHR was successful in identifying patients with FH. 7 An exercise that used the UK Biobank to identify patients at high likelihood of FH has been applied to estimate the prevalence of phenotypic and molecularly confirmed FH in the United States. 6
Results from chart reviews for identification of phenotypic FH were disappointing. Key elements of the DLCN algorithm, particularly family history and physical findings, were missing in most cases. The most frequent finding related to family history was inconclusive, defined as the absence of any information on this in the chart. Another problem was lack of specificity, including the inability to classify premature ASCVD events or family members having LDL‐C in the FH range.
It is likely that a small percentage of the suspicious VUS identified in participants are causative of FH based on the distribution of LDL‐C values in this group. However, this was too small a number to suggest that, in clinical practice, suspicious VUS meeting the selected variant characteristics in this study should be considered pathogenic. Studies combining phenotypic and broader genomic data are needed to better characterize VUS with the highest likelihood to cause disease. Studies of LDL receptor function may also help to further elucidate which VUS are likely pathogenic. 36
Strengths of the study include the combination of both FH genetic variant information and long standing EHR for the cohort. A second strength was the ability to combine 2 different algorithms to identify an at‐risk cohort.
The major limitations of this study were the older age of the cohort and either missing or inconclusive data in the EHR related to the FH phenotype. Lack of data on younger individuals, missing lipid values, and the fact that the FIND FH algorithm requires presence of at least 1 cardiac comorbidity or primary prevention condition recorded in their history led to exclusion of about half the MyCode cohort eligible for the study. There may be a bias toward identifying those currently prescribed statins, because the correction factor may overadjust for the effect of statins, and those on statins are more likely to have multiple lipid profiles in the record. The cohort was not racially diverse, so generalizability beyond White individuals is not possible. An unusual limitation was the relatively high rate of FH diagnosis in this cohort, due to the presence of returned results to many of those eligible for the study via the existing MyCode Genomic Screening and Counseling program, 9 making these individuals ineligible for our cohort. Despite this limitation, we were able to identify a high percentage of those remaining with FH variants.
Our data suggest information technology–based strategies to identify people at risk for having FH can be successful, within certain limits. That 70% of those with a P/LP FH variant in an undiagnosed cohort could be identified by combining 2 strategies based on information technologies is an important accomplishment and suggests with further refinement of these strategies, improved precision could be achieved. Research has shown that the ability to perform chart reviews, or to use natural language processing on those identified, may be able to increase the yield from those brought in for clinical evaluation after identification. 4 , 7 An important negative finding was that an absence of a meaningful difference in prevalence of hypothyroidism, diabetes, and chronic kidney disease in those with a genetic variant compared with those without suggests these conditions should not be excluded during information technology efforts to find those with FH. Enhanced data collection of the elements necessary to establish a phenotypic diagnosis of FH might also reduce the number of people identified, thus increasing precision of the models; elements that would be helpful include recording pretreatment LDL‐C, more precise family history information related to elevation of LDL‐C, age at ASCVD, relationship to the patient, and information on physical findings related to cholesterol deposition. A further limitation is the inability to identify those with an FH variant with lower LDL‐C levels, a group still at elevated risk of ASCVD. Thus, even in the presence of excellent strategies, identification of everyone with FH in a specific population will continue to require both screening of lipid profiles in younger individuals and robust procedures for cascade testing of relatives of those affected, because in many countries, yields from cascade screening are lower than in the most successful settings. 37
Sources of Funding
Research reported in this article was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL148246. This research is 100% supported by federal money in the amount of $2 837 141. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
S.S. Gidding is a consultant for Esperion. W. Howard is an employee of Atomo, Inc., which has pharmaceutical companies, medical communications agencies, and the Family Heart Foundation as clients. K. D. Myers is the CEO and founder of Atomo, Inc., which has pharmaceutical companies, medical communications agencies, and the Family Heart Foundation as clients. D. Staszak is an employee of Atomo, Inc., which has pharmaceutical companies, medical communications agencies, and the Family Heart Foundation as clients. A. C. Sturm is an employee and shareholder of 23andMe. L. K. Jones is a consultant for Novartis. The remaining authors have no disclosures to report.
This article was sent to Saket Girotra, MD, SM, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 11.
REFERENCES
- 1. Watts GF, Gidding SS, Mata P, Pang J, Sullivan DR, Yamashita S, Raal FJ, Santos RD, Ray KK. Familial hypercholesterolaemia: evolving knowledge for designing adaptive models of care. Nat Rev Cardiol. 2020;17:360–377. doi: 10.1038/s41569-019-0325-8 [DOI] [PubMed] [Google Scholar]
- 2. Myers KD, Knowles JW, Staszak D, Shapiro MD, Howard W, Yadava M, Zuzick D, Williamson L, Shah NH, Banda JM, et al. Precision screening for familial hypercholesterolaemia: a machine learning study applied to electronic health encounter data. Lancet Digit Health. 2019;1:e393–e402. doi: 10.1016/S2589-7500(19)30150-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Safarova MS, Liu H, Kullo IJ. Rapid identification of familial hypercholesterolemia from electronic health records: the SEARCH study. J Clin Lipidol. 2016;10:1230–1239. doi: 10.1016/j.jacl.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birnbaum RA, Horton BH, Gidding SS, Brenman LM, Macapinlac BA, Avins AL. Closing the gap: identification and management of familial hypercholesterolemia in an integrated healthcare delivery system. J Clin Lipidol. 2021;15:347–357. doi: 10.1016/j.jacl.2021.01.008 [DOI] [PubMed] [Google Scholar]
- 5. Qureshi N, Akyea RK, Dutton B, Leonardi‐Bee J, Humphries SE, Weng S, Kai J. Comparing the performance of the novel FAMCAT algorithms and established case‐finding criteria for familial hypercholesterolaemia in primary care. Open Heart. 2021;8:8. doi: 10.1136/openhrt-2021-001752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bellows BK, Khera AV, Zhang Y, Ruiz‐Negron N, Stoddard HM, Wong JB, Kazi DS, de Ferranti SD, Moran AE. Estimated yield of screening for heterozygous familial hypercholesterolemia with and without genetic testing in US adults. J Am Heart Assoc. 2022;11:e025192. doi: 10.1161/JAHA.121.025192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banda JM, Sarraju A, Abbasi F, Parizo J, Pariani M, Ison H, Briskin E, Wand H, Dubois S, Jung K, et al. Finding missed cases of familial hypercholesterolemia in health systems using machine learning. NPJ Digit Med. 2019;2:23. doi: 10.1038/s41746-019-0101-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abul‐Husn NS, Manickam K, Jones LK, Wright EA, Hartzel DN, Gonzaga‐Jauregui C, O'Dushlaine C, Leader JB, Lester Kirchner H, Lindbuchler DM, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016;354:aaf7000. doi: 10.1126/science.aaf7000 [DOI] [PubMed] [Google Scholar]
- 9. Buchanan AH, Lester Kirchner H, Schwartz MLB, Kelly MA, Schmidlen T, Jones LK, Hallquist MLG, Rocha H, Betts M, Schwiter R, et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet Med. 2020;22:1874–1882. doi: 10.1038/s41436-020-0876-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, Murray MF, Smelser DT, Gerhard GS, Ledbetter DH. The Geisinger MyCode community health initiative: an electronic health record‐linked biobank for precision medicine research. Genet Med. 2016;18:906–913. doi: 10.1038/gim.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williams MS. Early lessons from the implementation of genomic medicine programs. Annu Rev Genomics Hum Genet. 2019;20:389–411. doi: 10.1146/annurev-genom-083118-014924 [DOI] [PubMed] [Google Scholar]
- 12. Williams MS, Buchanan AH, Davis FD, Faucett WA, Hallquist MLG, Leader JB, Martin CL, McCormick CZ, Meyer MN, Murray MF, et al. Patient‐centered precision health in a learning health care system: Geisinger's genomic medicine experience. Health Aff (Millwood). 2018;37:757–764. doi: 10.1377/hlthaff.2017.1557 [DOI] [PubMed] [Google Scholar]
- 13. Schwartz MLB, McCormick CZ, Lazzeri AL, Lindbuchler DM, Hallquist MLG, Manickam K, Buchanan AH, Rahm AK, Giovanni MA, Frisbie L, et al. A model for genome‐first care: returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am J Hum Genet. 2018;103:328–337. doi: 10.1016/j.ajhg.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelly MA, Leader JB, Wain KE, Bodian D, Oetjens MT, Ledbetter DH, Martin CL, Strande NT. Leveraging population‐based exome screening to impact clinical care: the evolution of variant assessment in the Geisinger MyCode research project. Am J Med Genet C Semin Med Genet. 2021;187:83–94. doi: 10.1002/ajmg.c.31887 [DOI] [PubMed] [Google Scholar]
- 15. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 17. SpliceAi Lookup . Broad Institute Translational Genomics Group; Published May 22, 2023. Accessed June 1, 2022. https://spliceailookup.broadinstitute.org
- 18. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chora JR, Iacocca MA, Tichy L, Wand H, Kurtz CL, Zimmermann H, Leon A, Williams M, Humphries SE, Hooper AJ, et al. The Clinical Genome Rresource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel consensus guidelines for LDLR variant classification. Genet Med. 2022;24:293–306. doi: 10.1016/j.gim.2021.09.012 [DOI] [PubMed] [Google Scholar]
- 21. Packer JS, Maxwell EK, O'Dushlaine C, Lopez AE, Dewey FE, Chernomorsky R, Baras A, Overton JD, Habegger L, Reid JG. CLAMMS: a scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics. 2016;32:133–135. doi: 10.1093/bioinformatics/btv547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O'Neal L, McLeod L, Delacqua G, Delacqua F, Kirby J, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform. 2019;95:103208. doi: 10.1016/j.jbi.2019.103208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a. doi: 10.1093/eurheartj/eht273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reeskamp R, Nurmohamed NS. LDL‐C Calculator. Lipid Tools. Accessed: June 1, 2022. https://www.lipidtools.com/calculator‐pages/ldlc/
- 26. Nurmohamed NS, Collard D, Balder JW, Kuivenhoven JA, Stroes ESG, Reeskamp LF. From evidence to practice: development of web‐based Dutch lipid reference values. Neth Heart J. 2021;29:441–450. doi: 10.1007/s12471-021-01562-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sidak Z. Rectangular confidence regions for the means of multivariate normal distributions. J Am Stat Assoc. 1967;62:626–633. doi: 10.2307/2283989 [DOI] [Google Scholar]
- 28. Morris JK, Wald DS, Wald NJ. The evaluation of cascade testing for familial hypercholesterolemia. Am J Med Genet A. 2012;158a:78–84. doi: 10.1002/ajmg.a.34368 [DOI] [PubMed] [Google Scholar]
- 29. Hu P, Dharmayat KI, Stevens CAT, Sharabiani MTA, Jones RS, Watts GF, Genest J, Ray KK, Vallejo‐Vaz AJ. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta‐analysis. Circulation. 2020;141:1742–1759. doi: 10.1161/CIRCULATIONAHA.119.044795 [DOI] [PubMed] [Google Scholar]
- 30. Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia: meta‐analyses of 11 million subjects. J Am Coll Cardiol. 2020;75:2553–2566. doi: 10.1016/j.jacc.2020.03.057 [DOI] [PubMed] [Google Scholar]
- 31. Saadatagah S, Jose M, Dikilitas O, Alhalabi L, Miller AA, Fan X, Olson JE, Kochan DC, Safarova M, Kullo IJ. Genetic basis of hypercholesterolemia in adults. NPJ Genom Med. 2021;6:28. doi: 10.1038/s41525-021-00190-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bjornsson E, Thorgeirsson G, Helgadottir A, Thorleifsson G, Sveinbjornsson G, Kristmundsdottir S, Jonsson H, Jonasdottir A, Jonasdottir A, Sigurethsson A, et al. Large‐scale screening for monogenic and clinically defined familial hypercholesterolemia in Iceland. Arterioscler Thromb Vasc Biol. 2021;41:2616–2628. doi: 10.1161/ATVBAHA.120.315904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saadatagah S, Alhalabi L, Farwati M, Zordok M, Bhat A, Smith CY, Wood‐Wentz CM, Bailey KR, Kullo IJ. The burden of severe hypercholesterolemia and familial hypercholesterolemia in a population‐based setting in the US. Am J Prev Cardiol. 2022;12:100393. doi: 10.1016/j.ajpc.2022.100393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. doi: 10.1016/j.jacc.2016.03.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wald DS, Bestwick JP, Wald NJ. Child‐parent screening for familial hypercholesterolaemia: screening strategy based on a meta‐analysis. BMJ. 2007;335:599. doi: 10.1136/bmj.39300.616076.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Graca R, Alves AC, Zimon M, Pepperkok R, Bourbon M. Functional profiling of LDLR variants: important evidence for variant classification: functional profiling of LDLR variants. J Clin Lipidol. 2022;16:516–524. doi: 10.1016/j.jacl.2022.04.005 [DOI] [PubMed] [Google Scholar]
- 37. Polanski A, Wolin E, Kocher M, Zierhut H. A scoping review of interventions increasing screening and diagnosis of familial hypercholesterolemia. Genet Med. 2022;24:1791–1802. doi: 10.1016/j.gim.2022.05.012 [DOI] [PubMed] [Google Scholar]