

Abstract
Background and Objectives
LAMA2-related muscular dystrophy (LAMA2-MD) is a rare neuromuscular disease characterized by proximal and axial muscle weakness, rigidity of the spine, scoliosis, and respiratory impairment. No curative treatment options exist, yet promising preclinical studies are ongoing. Currently, there is a paucity on natural history data, and appropriate clinical and functional outcome measures are needed. We aim for deep clinical phenotyping, establishment of a well-characterized baseline cohort for prospective follow-up and recruitment for future clinical trials, improvement of clinical care, and selection of outcome measures for reaching trial readiness.
Methods
We performed a cross-sectional, single-center, observational study. This study included neurologic examination and functional measurements among others the Motor Function Measure 20/32 (MFM-20/32) as primary outcome measure, accelerometry, questionnaires, muscle ultrasound, respiratory function tests, electrocardiography and echocardiography, and dual-energy X-ray absorptiometry.
Results
Twenty-seven patients with genetically confirmed LAMA2-MD were included (21 ± 13 years; M = 9; ambulant = 7). Axial and proximal muscle weakness was most pronounced. The mean MFM-20/32 score was 42.0% ± 29.4%, with domain 1 (standing and transfers) being severely affected and domain 3 (distal muscle function) relatively spared. Physical activity as measured through accelerometry showed very strong correlations to MFM-20/32 (Pearson correlation, −0.928, p < 0.01). Muscle ultrasound showed symmetrically increased echogenicity, with the sternocleidomastoid muscle most affected. Respiratory function was impaired in 85% of patients without prominent diaphragm dysfunction and was independent of age. Ten patients (37%) needed (non)invasive ventilatory support. Cardiac assessment revealed QRS fragmentation in 62%, abnormal left ventricular global longitudinal strain in 25%, and decreased left ventricular ejection fraction in 14% of patients. Decreased bone quality leading to fragility fractures was seen in most of the patients.
Discussion
LAMA2-MD has a widely variable phenotype. Based on the results of this cross-sectional study and current standards of care for congenital muscular dystrophies, we advise routine cardiorespiratory follow-up and optimization of bone quality. We propose MFM-20/32, accelerometry, and muscle ultrasound for assessing disease severity and progression. For definitive clinical recommendations and outcome measures, natural history data are needed.
Clinical Trials Registration
This study was registered at clinicaltrials.gov (NCT04478981, 21 July 2020). The first patient was enrolled in September 2020.
Introduction
Laminin α2–related muscular dystrophy (LAMA2-MD) is a congenital muscular dystrophy with a clinical phenotype ranging from a severe congenital muscular dystrophy type 1A (MDC1A) (complete merosin deficiency) to a milder, late-onset LAMA2-MD (partial merosin deficiency).1 Key features include axial and proximal muscle weakness, delayed motor milestones, early-onset spinal rigidity, joint contractures, scoliosis, and respiratory insufficiency.1 In addition, patients may experience epileptic seizures and show characteristic diffuse brain white matter lesions on MRI.2 Manifesting symptoms of patients with a severe form of LAMA2-MD include postnatal hypotonia, a weak cry, and reduced spontaneous movements.1 Facial weakness, macroglossia, protruding tongue, and progressive limitation of extraocular movements are common later in life. Contrarily, most patients with a mild form of LAMA2-MD present during (early) childhood with delayed motor milestones.1 The clinical diagnosis of LAMA2-MD is confirmed by 2 recessive (likely) pathogenic variants in the LAMA2 gene (OMIM-number 156225). Currently, no curative treatments exist, but preclinical studies are ongoing.3,4 To pave the way toward clinical trials, it is essential to identify and characterize patients clinically and genetically and to select sensitive, patient-friendly, and sustainable clinical and functional outcome measures. Previous clinical studies have been performed to characterize the natural history of patients diagnosed with LAMA2-MD, showing a significant rate of change in Motor Function Measurement 32 (MFM-32) score in nonambulant patients (aged 5–17 years), a linear annual decline in forced vital capacity (FVC) (percentage predicted) in patients with complete merosin deficiency, cardiac ventricular dysfunction and rhythm disturbances, and pneumonia as the main cause of death.5,6 However, these studies had several major limitations, including a limited subset of outcome measures, an absence of convenient muscle visualizing techniques, an absence of bone quality assessment, and a limited age range. Prospective cross-sectional and natural history studies including a large subset of clinical, functional, and imaging outcome measures in a cohort with variable and large age range and disease severity are missing. In this study, we present a cross-sectional study on LAMA2-MD, which fills in this gap. Hereby, we aim for deep clinical phenotyping and to establish a well-characterized baseline cohort of patients with LAMA2-MD for prospective follow-up and recruitment for future clinical trials. Moreover, we want to validate the use of a large subset of outcome measures in LAMA2-MD. We finally aim to contribute to optimal clinical management for patients with LAMA2-MD across the life span. The primary outcome measure is the Motor Function Measure 20/32 (MFM-20/32) because it evaluates fine and gross motor ability and is generally accepted as a reliable and valid method to measure disease severity and progression in patients with neuromuscular diseases.7
Methods
Study Design
This cross-sectional study in adult and pediatric patients with LAMA2-MD is part of the LAST STRONG Study: a study on LAMA2-MD and SELENON-RM To Study Trial Readiness, Outcome measures and Natural history, a prospective, single-center, observational study with repeated measurements. An overview on the LAST STRONG study protocol can be found elsewhere.8
Study Population
Patients were recruited nonselectively and consecutively from September 2020 to June 2022. Inclusion criteria include a genetic confirmation of LAMA2-MD by 2 recessive (likely) pathologic variants in the LAMA2 gene or typical clinical and histologic alterations combined with genetic confirmation in a first-degree relative. Exclusion criteria were an insufficient understanding of the Dutch language or lack of informed consent. If patients did not wish or were not able to visit our center, they were offered to participate in this study through home visits.
Neurologic Examination and Functional Measurements
Patients underwent a standard neurologic examination by 2 assessors (K.B. and C.E. or N.V.). Rigid spine was defined by a decreased flexibility in the neck or lower back. Furthermore, muscle strength was measured using handheld dynamometry (Citec, CT3002) in muscles with an MRC score of ≥4 and was compared with age-specific and sex-specific reference values.9,10 Muscle strength was decreased if below the fifth percentile (P5). The passive range of motion of the elbow, hip, knee, and ankle joints was assessed by goniometry. Functional measurements include MFM-20/32, Hammersmith Functional Motor Scale (HFMS), Pediatric Balance Scale (PBS), Mini Balance Evaluation Systems Test (miniBEST), and Graded and Timed Function Tests (GTFTs, time to rise from the floor, 30 seconds sit to stand, 10-m Walk Test, 6-Minute Walk Test, and Timed Up and Go Test). All functional measurements were adapted to the patient's age and functional abilities. The MFM-20/32 was the primary outcome measure.
Accelerometry
An accelerometer (GENEActiv Original, Activinsights Ltd, 87.5 Hz sampling) on the nondominant wrist measured the daily activity in the home situation for 7 days. The gravity-subtracted sum of vector magnitudes (SVMgs) was used as the activity measure. All data were subsequently analyzed using MATLAB R2018a Update 4 (9.4.0.902940) for windows. Activity was expressed as counts per day and the percentage of sedentary, light, moderate, or vigorous activity corrected for age.11-13
Questionnaires
Patients and/or their parent(s) completed age-adapted questionnaires, including Pediatric Quality of Life Inventory (PedsQL) Generic Core Scale, Neuromuscular Module (NMM) and Multidimensional Fatigue Scale (MFS), Research and Development-36 (RAND-36), Individualized Neuromuscular Quality of Life (INQoL), McGill Pain Questionnaire, Wong-Baker Faces Pain rating scale, Checklist Individual Strength (CIS), ACTIVLIM, Impact on Participation and Autonomy (IPA), and Egen Klassifikation version 2 (EK2).
Muscle Ultrasound
Muscle thickness and echogenicity (quantitative grayscale analysis and visual grading using the Heckmatt score) of a set of bilateral muscles were assessed by muscle ultrasound, using an Esaote MyLabTwice ultrasound scanner (Esaote SpA, Genoa, Italy) with a 3–13 MHz broadband linear transducer and a 53-mm footprint. The mean grayscale level was determined within a manually selected region of interest in the ultrasound image using an in-house developed MATLAB software package (version 2013b, Mathworks, Natick, MA).8 The muscle thickness and echogenicity were compared with reference values and represented by their z score.
Respiratory Function
Patients (age 5 years or older) underwent spirometry in upright and supine positions (SpiroUSB, Vyaire Medical connected to PC Spirometry software, Spida CareFusion 2.3.0.10 for Windows 7). In addition, maximum expiratory pressure (MEP), maximum inspiratory pressure (MIP), and sniff nasal inspiratory pressure (SNIP) were measured in upright position (Micro RPM, Micro Medical, CareFusion, United Kingdom). Diaphragm ultrasound (MyLabTwice, Esaote SpA, Genoa, Italy) was performed to assess bilateral diaphragm thickness (end-expiratory and maximum inspiratory thicknesses) and thickening ratio.14
Cardiac Assessment
Heart axis (QRS axis between −30° and +90°), morphology of the P, QRS, and T waves, ST segment morphology (elevation or depression), the presence of repolarization disorders (poor R-wave progression), and QRS fragmentation (additional R wave or notching in the nadir of the R wave or S wave or more than 1 R′ without a typical bundle branch block) were assessed.15 Furthermore, conventional transthoracic echocardiography (TTE) with speckle tracking and tissue Doppler imaging (TDI) was performed by certified sonographers using an Affinity70 General (Philips Health care, Best, the Netherlands) for adults and a Vivid E9 or Vivid E95 ultrasound scanner (GE Health care Ultrasound, Horten, Norway) for pediatric patients according to the EACVI recommendations for cardiac chamber quantification. Left ventricular ejection fraction (LVEF) was decreased if <52% in male individuals, <54% in female individuals, and <55% in the pediatric population.16,17 Global longitudinal strain was abnormal if above −18% in adults and above −19.5% in children.18,19
Bone Quality
A dual-energy X-ray absorptiometry (DEXA) scan of the right femoral neck and lumbar spine, including a vertebral fracture assessment, was performed using the Hologic Discovery A Horizon DXA System (S/N 303053M). For adults, osteopenia and osteoporosis were defined by a t score between −1 and −2.5 SD, and < −2.5 SD, respectively.20 In children, low bone quality is defined as a z score ≤ −2.0 on DEXA scan without a clinically significant fracture history. Osteoporosis is defined as low bone mineral density (z score ≤ −2.0) combined with a clinically significant fracture history (≥2 fragility long bone fractures before the age of 10 years, ≥3 fragility long bone fractures before the age of 19 years, or the presence of 1 or more vertebral compression fractures without major trauma or local disease).21
Statistical Analysis
Descriptive statistics were used to summarize our data in IBM SPSS Statistics 25.0.0.1 for Windows (SPSS, Inc., Chicago, IL). Values are mean ± SD, unless otherwise stated. The Pearson correlation was considered moderate (r = 0.40–0.59), strong (r = 0.60–0.80), or very strong (r = 0.80–1.0). A paired sample t test was used to compare the mean echogenicity of the right and left sides. An independent sample t test was performed to compare echogenicity and bone mineral density of the femoral neck and lumbar spine between ambulant and nonambulant patients. An independent sample t test was performed to compare MFM-20/32 and accelerometry between patients with partial and complete merosin deficiency and to assess difference in MFM-20/32 between genotypes. A nonparametric test for independent samples (Mann–Whitney U test) was performed to compare the Heckmatt score between ambulant and nonambulant patients. If reference values are available, overlapping confidence intervals were checked. In case of missing data, patients were excluded from specific subanalysis. Differences were considered significant if p < 0.05. The statistical analysis plan was published.8
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the medical ethical reviewing committee of Region Arnhem-Nijmegen (NL-number NL64269.091.17, dossier number 2017-3911; 8 July 2020) and was registered at clinicaltrials.gov (NCT4478981). From all patients, or in case of children their parents or legal guardian, informed consent was obtained for participating in our study. A consent-to-disclose form was signed by any identifiable person.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Study Population
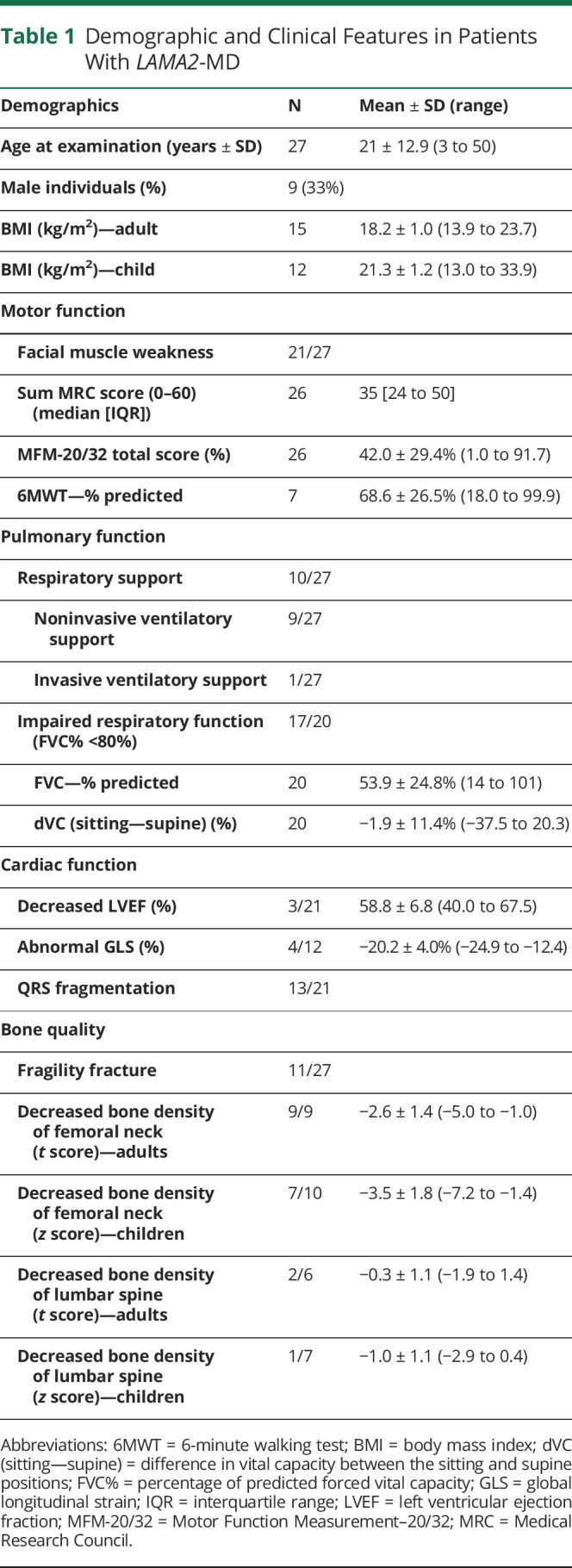
We were able to contact 31 patients with LAMA2-MD, of whom 27 patients were included (mean age: 21 ± 13 years (range 3–50 years), F = 18, M = 9). One Belgium patient could not participate because of COVID-19 pandemic rules, and 3 patients did not want to participate in scientific research because of variable reasons, without being more or less severely affected than the participants. Twenty-five patients were of Dutch origin and 2 from the Dutch-speaking part of Belgium (Flanders). Five patients participated through home visits and thus underwent only part of the protocol, which is summarized in eTable 1 (links.lww.com/NXG/A619). Key clinical characteristics are summarized in Table 1.
Table 1.
Demographic and Clinical Features in Patients With LAMA2-MD
Genetic Characteristics
All patients had genetic confirmation before enrollment in the study (Table 2).2,22-30 The Dutch founder mutation (homozygous and compound heterozygous) was most prevalent (c.5562+5G>C splice site mutation in exon 38). No individuals were excluded from this study because of variants of unknown significance.
Table 2.
Clinical and Genetic Features of Patients With LAMA2-MD

Neurologic Examination
Facial muscle weakness was observed in 21/27 patients (Figure 1 and Table 3). All patients had skeletal muscle weakness, most pronounced in axial and proximal muscles. The median sum MRC was 35/60 [IQR 24–50] (Figure 1 and Table 3). Elbow, hip, knee, and ankle contractures were frequent and limited muscle strength scoring. Biceps, triceps, knee, and Achilles heel deep tendon reflexes were absent or reduced in 25/27 patients. Sensation was normal in all patients. Seven of 27 patients had a macroglossia, a high arched palate was found in 17/27 patients, while 13/27 patients had an elongated face. Rigid spine was seen in all, and scapular winging including scapular dyskinesia was observed in 17/24 patients. We were unable to assess scapular winging in some patients because of the inability to actively and/or passively move the arms (muscle weakness, contractures) combined with spinal or chest deformities. Both pseudohypertrophy and hypotrophy, especially of the calf musculature, were seen. Nineteen out of 27 patients had a spinal deformity (scoliosis and/or lordosis) or had undergone surgical correction.
Figure 1. Facial Muscle Weakness and Dysmorphic Features in Patients With LAMA2-MD.

(A) neutral position, showing bilateral ptosis. (B) orbicularis oculi weakness. (C) inadequate puckering movements, indicating orbicularis ori weakness. (D) inadequate blowing movements, indicating orbicularis ori weakness. (E) risorius weakness. (F) macroglossia and elongated face. (G) inability of neck flexion indicating rigid spine. (H) scapular winging. (I) spinal and thorax deformities. (J) pseudohypertrophy and hypotrophy of calve muscles, respectively. All identifiable patients provided consent for publication of their pictures. (K) Median muscle strength according to the MRC grading scale in patients with LAMA2-MD. The median muscle strength (MRC) in neck flexor, neck extensor, deltoid, biceps brachii, triceps brachii, wrist flexor, wrist extensor, finger flexor, finger extensor, iliopsoas, gluteus, quadriceps, hamstrings, foot flexor, foot extensor, and toe flexor and extensor hallucis longus muscles.
Table 3.
Neurologic Examination, Functional Measurements, and Accelerometry in Patients With LAMA2-MD

Functional Measurements
A detailed overview is listed in Table 3.
Motor Function Measurement 20/32
Domain 1 (standing and transfers) of MFM-20/32 was most severely affected, while domain 3 (distal muscle function) was relatively spared. MFM-20/32 was higher in patients with partial (52.1% ± 18.8%) than complete (22.5% ± 8.5%) merosin deficiency (p = 0.002) (Figure 2). Patients with the compound homozygous Dutch founder mutation were less affected (90.4% ± 1.1%) than patients with the c.3799_3821del allele (13.9% ± 8.4%) (p < 0.001). There was no correlation with age. MFM-20/32 correlated moderately with PedsQL NMM filled in by mothers (0.589, p < 0.05) and muscle weakness subscale of INQoL (−0.585, p < 0.05).
Figure 2. Scatter Plot.

Total MFM-20/32 score (%) against age (years) in patients with decreased merosin expression, absent merosin expression, or an unknown level of merosin expression.
Mini-Balance Evaluation Systems Test
Four of 15 adult patients performed the MiniBEST. Anticipatory movements and reactive postural control were most severely affected, while sensory orientation and dynamic gait were relatively spared.
Pediatric Balance Scale
Three of 12 children performed the PBS. One child achieved the maximum score. “Standing unsupported with 1 foot in front” and “standing on one leg” were most severely affected.
Hammersmith Functional Motor Scale
The HFMS was performed in 19/27 patients. The items “lifting head from supine,” “propping on extended arms,” and “stepping” were most severely affected. “Sitting on a plinth or chair” and “putting 1 hand on top of the head” were less severely affected.
Graded and Timed Function Tests
Seven of 27 patients performed the 6MWT (percentage predicted: 68.6% ± 26.5%). Two patients had a walked distance above the lower limit of normal: 558 m (female, age 32 years) and 460 m (female, age 7 years).31,32 Seven patients performed the Timed Up and Go Test (TUG), with a delay >10% in 3 patients in the cognitive task version. Four patients were able to perform the 30-second sit-to-stand test (mean number of 11.8 ± 4.1 stands). Seven patients were able to climb and descend 4 steps, of whom 4 and 5 patients were able to do with alternating steps and without the use of their hands, respectively. Nine patients were able to independently raise from the ground to a standing position, of whom 5 needed a nearby object for support and 2 needed the support of their hands on the ground.
Handheld Dynamometry
The foot flexor muscles had the highest percentage of the predicted strength (right: 75% ± 42%; left: 68% ± 45%), with 3 patients having bilateral strength above the lower limit of normal (Table 3).
Accelerometry
The average activity (counts per day) was correlated with the MFM-20/32 total score (Pearson correlation, 0.889, p < 0.01). MFM-20/32 total score was correlated with sedentary (Pearson correlation, −0.928, p < 0.01), light (Pearson's correlation, 0.901, p < 0.01), moderate (Pearson correlation, 0.887, p < 0.01), and vigorous activity (Pearson correlation, 0.530, p < 0.01). The counts per day and percentage sedentary, light, and moderate activities differed between patients with partial (212,825 and 93.39%, 4.71%, and 1.69%, respectively) and complete (126,908 and 98.49%, 1.19%, and 0.16%, respectively) merosin deficiency (p = 0.005, p = 0.005, p = 0.007, and p = 0.007, respectively).
Questionnaires
A detailed overview is listed in eTable 2 (links.lww.com/NXG/A620).
RAND-36
The subdomain physical functioning and general health were significantly lower than those for healthy controls (p < 0.05). The subdomains emotional well-being and pain were found significantly higher than those for healthy controls (p < 0.05). There was no difference in all other domains compared with that in healthy controls.
Individualized Neuromuscular Quality of Life
Weakness (60.4% ± 22.1%) and independence (60.0% ± 31.1%) were most severely affected. Patients experienced minimal problems of muscle locking, pain, and emotions.
Checklist Individual Strength
Ten of 15 patients reported problematic fatigue (>76 of 140 points), i.e., negatively influencing the person's performance in the occupational and home setting. The subjective fatigue subdomain score was increased in 12 patients, of whom 6 patients experienced severe subjective fatigue (>35 of 56 points).
Pediatric Quality of Life Inventory
The Generic Core score was significantly lower compared with that in healthy controls (p < 0.05).33 The NMM score was not altered compared with patients with spinal muscular atrophy. PedsQL MFS score was not changed compared with that in healthy controls, while it was decreased in the parent proxy reports (p < 0.05).
ACTIVLIM
A mean of 11 ± 7 from a total of 18 activities were reported to be impossible, 2 ± 2 difficult, and 5 ± 6 easy to perform. Eleven of 25 patients reported that it is impossible to perform any of the listed activities.
Impact on Participation and Autonomy
Seven patients experienced no problems, 6 patients experienced minor problems, and 1 patient experienced major problems in participation and autonomy. The work and education subscale was most severely affected.
McGill Pain Questionnaire
Nine of 19 patients had pain in daily life, with pain in the back and/or neck region being the most prevalent. Two had problems with normal movements or adopting normal postures or hobbies, and 1 patient could not sleep because of the pain. Four patients indicated analgesic use, including paracetamol, non-steroidal anti-inflammatory drugs, morphine, and/or cannabidiol oil.
Egen Klassifikation Version 2
The mean EK2, a scale to measure physical function over time, was 23 ± 15 of 51 points. A score of zero indicates the highest physical function and a score of 51 the lowest.
Wong-Baker Faces Pain Rating Scale
Sixteen of 27 patients indicated to have had no pain during examination or during the previous week, while 4 patients had pain every day.
Muscle Ultrasound
Echogenicity and Heckmatt Score
We found no difference between left and right muscles, except for the Heckmatt score of the biceps femoris muscles. No scanned muscle groups were spared (Figure 3 and Table 4). The echogenicity of the sternocleidomastoid and the biceps brachii muscles were most severely affected, and the soleus muscle was least affected. The Heckmatt scoring system showed that the tibialis anterior muscle was most severely affected, and the soleus muscle was least severely affected. The echogenicity and Heckmatt score of a subset of muscles and the MFM-20/32 total score, MRC scale, and/or age were correlated (Table 4). Furthermore, nonambulant patients showed significantly higher echogenicity and Heckmatt score in a subset of muscles compared with those in ambulant patients.
Figure 3. Overview on Echogenicity z Score (Mean) and Heckmatt Score (Median) per Muscle in Patients With LAMA2-MD.

The z score of the echogenicity (A) and the median Heckmatt score (B) in the temporalis, sternocleidomastoid, biceps brachii, flexor carpi radialis, rectus abdominis, rectus femoris, vastus lateralis, tibialis anterior, biceps femoris, gastrocnemius medial head, and soleus muscles are shown.
Table 4.
Echogenicity, Heckmatt Score, and Muscle Thickness on Muscle Ultrasound in Patients With LAMA2-MD

Muscle Thickness
Thickness of left and right muscles was equal, except for the sternocleidomastoid and vastus lateralis muscles (p < 0.05). The sternocleidomastoid muscle thickness (z score left: −1.9; right: −2.2) was decreased. Muscle echogenicity and thickness were not correlated for any of the muscles imaged.
Respiratory Function Tests
Eighty-five percent of 20 patients had a predicted FVC <80% in the upright position (53.9% ± 24.8%). The decrease in vital capacity (VC) between the upright and supine positions was <10% in all but 1 patient (−2% ± 11.4%). The peak cough flow (PCF) was decreased in all patients (194% ± 89 L/min). The MIP was decreased in 19/25 patients (63% ± 32%) and MEP in 23/25 patients (44% ± 25%). The mean SNIP was 36 ± 21 cmH2O in male patients and 46 ± 25 cmH2O in female patients. Nine of 27 patients used noninvasive ventilatory support, and 1 patient used invasive ventilatory support, all starting at pediatric age. We found no correlation between respiratory function and age.
Ultrasound of the Diaphragm
The mean end-expiratory diaphragm thickness (right: 101% ± 40% and left: 105 ± 38% of predicted), end-inspiratory diaphragm thickness (right: 84% ± 26% and left: 91% ± 29% of predicted), and thickening ratio (right: 84% ± 24% and left: 85% ± 21% of predicted) were normal.
Cardiac Assessment
Seventeen of 21 patients had ECG abnormalities, including QRS fragmentation (n = 13) and abnormal R-wave progression (n = 9). Echocardiography showed abnormal GLS in 4 patients (adult: −12.4%; −14.8%; child: −17.7%; −18,8%). LVEF was decreased in 2 of these patients (51% and 44%) and in 1 adult patient (LVEF: 40%) with a normal GLS (−18,5%), for which all 3 used cardiac medication (beta blocker, ACE inhibitor). No abnormalities of the valves, right ventricle volume/function, or atria were seen.
Bone Quality
The femoral mineral bone density was reduced in all adults (t score: −2.6 ± 1.4) and in 7 of 10 pediatric patients (z score: −3.5 ± 1.8). Mineral bone density of the lumbar spine could only be measured in 13 patients because of scoliosis surgery osteosynthesis material. The lumbar mineral bone density was reduced in 2 of 6 adults (t score: −0.3 ± 1.1) and in 1 of 7 pediatric patients (z score: −1.0 ± 1.1). The femoral neck and lumbar spine mineral bone density was lower in nonambulant (−2.0 and −4.0, respectively) than in ambulant patients (−0.3 and −2.0, respectively) (p = 0.039 and p < 0.001, respectively). Eleven patients had experienced 1 or more fragility long bone fractures. No other causes for decreased bone quality were identified.
Discussion
The key findings of our cross-sectional cohort study of 27 patients with LAMA2-MD were as follows: (1) muscle weakness uniformly presenting in a proximal to distal gradient; (2) severe muscle fatty degeneration and/or fibrosis on muscle ultrasound; (3) respiratory impairment without prominent diaphragm dysfunction; (4) cardiac abnormalities in a small subset of patients, and (5) decreased bone quality. These observations lead to a number of recommendations for clinical practice and future research, which we will discuss further.
The findings of this cross-sectional study shed light on recommendations proposed in the international consensus statements on congenital muscular dystrophies and congenital myopathies,34,35 Long-term follow-up studies are expected to further support, update, or extend these recommendations.
We advise regular respiratory function tests in (a)symptomatic patients to early detect impaired respiratory function and enable timely respiratory training, air stacking, and/or noninvasive ventilatory support. Progressive restrictive respiratory insufficiency and reduced cough capacity are common causes of morbidity and mortality in LAMA2-MD.1,36 Diaphragm ultrasound showed normal diaphragm thickness and thickening, and spirometry showed only minor decreases in VC between the sitting and supine positions. Hence, the diaphragm was relatively spared.
We advise cardiac surveillance by regular screening of patients with ECG, Holter monitor, and echocardiography with advanced techniques for early detection and treatment of cardiac manifestations. We found decreased LVEF in a subset of patients, which is in line with our scoping review based on case reports and retrospective case series.37 QRS fragmentation is considered as a predictor of worse clinical outcomes and is related to lower LVEF and ventricular arrhythmias in Duchenne muscular dystrophy (DMD).38 GLS detects subtle left ventricular dysfunction in patients with DMD before the decline in LVEF.39 Patients with LAMA2-MD with QRS fragmentation and/or decreased GLS might thus be of an increased risk of developing cardiomyopathy and arrhythmia. QRS fragmentation and decreased GLS might help clinicians to optimize cardiac screening. Based on the low bone quality, we further recommend recurrent bone mineral density assessment and optimization of bone quality through vitamin D and calcium suppletion.
Our study provides insights into measuring disease severity and possibly disease progression for future clinical trials. The MFM-20/32 total and subdomain 2 scores are suitable (covering the clinical feature of interest) and feasible (being able to be performed by most patients) for measuring disease severity in LAMA2-MD across the life span.5 In our cohort, domain 1 and domain 3 were skewed toward minimum and maximum scores, respectively, and are unsuitable for measuring disease severity and progression in an LAMA2-MD cohort. However, domain 1 and domain 3 may be indicated for measuring disease severity and progression in very mildly and very severely affected individuals, respectively. Despite several limitations, the 6MWT is considered suitable to measure disease severity in ambulant patients with LAMA2-MD who have a near-normal score on the MFM-20/32 (>80%) without significant cardiorespiratory comorbidities.40,41 Furthermore, the time to rise from the floor is of interest due to the wide range of results, the increasing difficulty with performing this task as indicated by the patients, and its evidence in DMD.42
Accelerometry had very strong correlations with the MFM-20/32. Previous studies showed that the GENEActiv accelerometer can be used to measure physical activity and disease progression in neuromuscular disorders.43 Accelerometry also has a positive effect on physical activity through self-monitoring.44 The GENEActiv accelerometer is a small device used in the home situation, minimalizing patient burden. Especially after the COVID-19 pandemic, home measurements are of particular interest for longtime assessment of disease progression. We thus propose accelerometry as an outcome measure in LAMA2-MD.
HFMS is of limited use in LAMA2-MD because it is strongly skewed toward minimum scores in nonambulant patients. The miniBEST and PBS were considered neither suitable nor feasible for deep clinical phenotyping of an LAMA2-MD cohort. Because only a few patients were able and felt comfortable to perform these tasks, scores were skewed toward the maximum and highly influenced by muscle weakness.
Muscle ultrasound provided new insights on distribution of muscle involvement. In line with clinical observations and previous MRI studies, muscle ultrasound showed symmetrical muscle involvement in most of the studied muscles.45-48 Surprisingly, the most severely affected muscles using muscle ultrasound, the sternocleidomastoid and the biceps brachii muscles, were relatively spared in previous studies using MRI.45-47 Muscles with an increased echogenicity and normal appearance on MRI are consistent with IM fibrosis, especially in the absence of fatty infiltration.49 Contrarily, the soleus muscle was one of the least affected muscles on muscle ultrasound, while muscle MRI in other cohorts showed severe fatty infiltration.45-48 This was previously seen in facioscapulohumeral dystrophy.49 Our next step will include muscle fattening assessment on whole body MRI and correlation to muscle ultrasound. Based on the present results and available studies on MRI, we currently advise muscle ultrasound of the rectus abdominis, vastus lateralis and gastrocnemius muscles as an outcome measure for muscle imaging.
We propose RAND-36 to describe quality of life, CIS to monitor fatigue, and McGill Pain Questionnaire to assess pain in adults with LAMA2-MD. We advise PedsQL Generic Core scale, NMM, and MFS in children. Contrary to other neuromuscular diseases, quality of life related to emotional well-being and pain in RAND-36 was significantly increased in patients with LAMA2-MD compared with that in healthy controls. This might be explained by good health care for patients with neuromuscular disorders in The Netherlands and by participation in a study in this rare disease.50 As in other neuromuscular diseases, adult patients had an increased problematic and subjective fatigue as measured using the CIS. Contrarily to their parents, children did not report fatigue themselves through the PedsQL MFS. We could not fully explain the absence of reported fatigue by children with LAMA2-MD, yet the absence of cognitive fatigue and no need for sleep or rest might play an important role.
The wide range in age and disease severity, the high participation rate, and the broad range of clinical and functional outcome measures are the major strengths of our cross-sectional study, leading to a high external validity. The wide range in age and disease severity, however, results in frequent missing data because some patients were too young or were physically not able to perform some of the functional tests, limiting uniformity. Details are summarized in eTable 1 (links.lww.com/NXG/A619). All tests were adapted to the patients' ages. Some patients participated by home visits, therefore missing essential data from ancillary investigations. These patients are generally more severely affected, therefore underestimating disease severity in tests solely performed in the hospital. In patients with scoliosis surgery material and/or severe hip contractures, generally most severely affected patients, mineral bone density measurements were not possible, leading to overestimation of the bone quality. Only 1 patient refrained from spirometry.
In line with general consensus statements on clinical care, we advise routine cardiorespiratory screening and bone quality assessment in all patients with LAMA2-MD for prevention, early detection, and treatment of complications. The recommendations might be refined after the results of the longitudinal study. Based on our cross-sectional data, we propose the MFM-20/32, accelerometry, and muscle ultrasound to assess disease severity and progression in longitudinal follow-up and clinical trials. For a more definite selection of outcome measures, we need longitudinal data from our ongoing 1.5-year natural history study LAST STRONG, of which data collection will be finished in 2023. We aim to reach consensus on the selection of outcome measures in a consortium meeting involving key leader physicians, researchers, and the European patient association for LAMA2-MD (LAMA2 Europe), which will take place in March 2023.
Acknowledgment
The authors thank all patients and their relatives for participation in our study. The authors thank Marit Boxum and Daniëlle Franken for their help in contacting patients and requesting medical data from other hospitals. Several authors of this publication are members of the Radboudumc Neuromuscular Center (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD), and European Reference Network for rare neuromuscular diseases (EURO-NMD).
Glossary
- 6MWT
6-Minute Walk Test
- 10MWT
10-Meter Walk Test
- BMI
body mass index
- CIS
Checklist Individual Strength
- DEXA
dual-=energy X-ray absorptiometry
- DMD
Duchenne muscular dystrophy
- EK2
Egen Klassifikation version 2
- ECG
electrocardiogram
- FEV1%
percentage of predicted forced expiratory volume in 1 second
- FVC%
percentage of predicted forced vital capacity
- FVC
forced vital capacity
- GLS
global longitudinal strain
- GTFT
Graded and Timed Function Tests
- HFMS
Hammersmith Functional Motor Scale
- HHD
Handheld dynamometry
- INQoL
Individualized Neuromuscular Quality of Life
- IPA
Impact on Participation and Autonomy
- LAMA2-MD
LAMA2-related muscular dystrophy
- LVEF
left ventricular ejaction fraction
- MDC1A
merosin-deficient congenital muscular dystrophy type 1A
- MEP
Maximum Expiratory Pressure
- MFM-20/32
Motor Function Measure 20/32
- miniBEST
Mini Balance Evaluation Systems Test
- MIP
Maximum Inspiratory Pressure
- MRC
Medical Research Council
- PBS
Pediatric Balance Scale
- PCF
peak cough flow
- PedsQL
Pediatric Quality of Life Inventory
- PedsQL NMM
Pediatric Quality of Life Inventory Neuromuscular Module
- PedsQL MFS
Pediatric Quality of Life Inventory Multidimensional Fatigue Scale
- RAND-36
Research and Development-36
- SNIP
Sniff Nasal Inspiratory Pressure
- SVMgs
the gravity subtracted sum of vector magnitudes
- TUG
Timed Up and Go Test
- TDI
tissue Doppler imaging
- TTE
transthoracic echocardiography
- VC
vital capacity
Appendix. Authors

Study Funding
This study is funded by Stichting Spieren voor Spieren (SVS19001) and Stichting Voor Sara.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures
References
- 1.Sarkozy A, Foley AR, Zambon AA, Bönnemann CG, Muntoni F. LAMA2-related dystrophies: clinical phenotypes, disease biomarkers, and clinical trial readiness. Front Mol Neurosci. 2020;13:123. doi: 10.3389/fnmol.2020.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geranmayeh F, Clement E, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010;20(4):241-250. doi: 10.1016/j.nmd.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 3.Barraza-Flores P, Bates CR, Oliveira-Santos A, Burkin DJ. Laminin and integrin in LAMA2-related congenital muscular dystrophy: from disease to therapeutics. Front Mol Neurosci. 2020;13:1. doi: 10.3389/fnmol.2020.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kemaladewi DU, Bassi PS, Erwood S, et al. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature. 2019;572(7767):125-130. doi: 10.1038/s41586-019-1430-x [DOI] [PubMed] [Google Scholar]
- 5.Jain MS, Meilleur K, Kim E, et al. Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology. 2019;93(21):e1932-e1943. doi: 10.1212/wnl.0000000000008517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zambon AA, Ridout D, Main M, et al. LAMA2-related muscular dystrophy: natural history of a large pediatric cohort. Ann Clin Transl Neurol. 2020;7(10):1870-1882. doi: 10.1002/acn3.51172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bérard C, Payan C, Hodgkinson I, Fermanian J. A motor function measure for neuromuscular diseases. Construction and validation study. Neuromuscul Disord. 2005;15(7):463-470. doi: 10.1016/j.nmd.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 8.Bouman K, Groothuis JT, Doorduin J, et al. Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: protocol of the LAST STRONG study. BMC Neurol. 2021;21(1):313. doi: 10.1186/s12883-021-02336-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beenakker EA, van der Hoeven JH, Fock JM, Maurits NM. Reference values of maximum isometric muscle force obtained in 270 children aged 4-16 years by hand-held dynamometry. Neuromuscul Disord. 2001;11(5):441-446. doi: 10.1016/s0960-8966(01)00193-6 [DOI] [PubMed] [Google Scholar]
- 10.van der Ploeg RJ, Fidler V, Oosterhuis HJ. Hand-held myometry: reference values. J Neurol Neurosurg Psychiatry. 1991;54(3):244-247. doi: 10.1136/jnnp.54.3.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roscoe CMP, James RS, Duncan MJ. Calibration of GENEActiv accelerometer wrist cut-points for the assessment of physical activity intensity of preschool aged children. Eur J Pediatr. 2017;176(8):1093-1098. doi: 10.1007/s00431-017-2948-2 [DOI] [PubMed] [Google Scholar]
- 12.Phillips LRS, Parfitt G, Rowlands AV. Calibration of the GENEA accelerometer for assessment of physical activity intensity in children. J Sci Med Sport. 2013;16(2):124-128. doi: 10.1016/j.jsams.2012.05.013 [DOI] [PubMed] [Google Scholar]
- 13.Esliger DW, Rowlands AV, Hurst TL, Catt M, Murray P, Eston RG. Validation of the GENEA accelerometer. Med Sci Sports Exerc. 2011;43(6):1085-1093. doi: 10.1249/MSS.0b013e31820513be [DOI] [PubMed] [Google Scholar]
- 14.van Doorn JLM, Wijntjes J, Saris CGJ, Ottenheijm CAC, van Alfen N, Doorduin J. Association of diaphragm thickness and echogenicity with age, sex, and body mass index in healthy subjects. Muscle Nerve. 2022;66(2):197-202. doi: 10.1002/mus.27639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietrasik G, Zaręba W. QRS fragmentation: diagnostic and prognostic significance. Cardiol J. 2012;19(2):114-121. doi: 10.5603/cj.2012.0022 [DOI] [PubMed] [Google Scholar]
- 16.Kosaraju A, Goyal A, Grigorova Y, Makaryus AN. Left ventricular ejection fraction. StatPearls. StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.; 2022. [PubMed] [Google Scholar]
- 17.Tissot C, Singh Y, Sekarski N. Echocardiographic evaluation of ventricular function-for the neonatologist and pediatric intensivist. Front Pediatr. 2018;6:79. doi: 10.3389/fped.2018.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truong VT, Phan HT, Pham KNP, et al. Normal ranges of left ventricular strain by three-dimensional speckle-tracking echocardiography in adults: a systematic review and meta-analysis. J Am Soc Echocardiogr. 2019;32(12):1586-1597.e5. doi: 10.1016/j.echo.2019.07.012 [DOI] [PubMed] [Google Scholar]
- 19.Levy PT, Machefsky A, Sanchez AA, et al. Reference ranges of left ventricular strain measures by two-dimensional speckle-tracking echocardiography in children: a systematic review and meta-analysis. J Am Soc Echocardiogr. 2016;29(3):209-225.e6. doi: 10.1016/j.echo.2015.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siris ES, Adler R, Bilezikian J, et al. The clinical diagnosis of osteoporosis: a position statement from the National Bone Health Alliance Working Group. Osteoporos Int. 2014;25(5):1439-1443. doi: 10.1007/s00198-014-2655-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachrach LK, Gordon CM. Bone densitometry in children and adolescents. Pediatrics 2016;138(4):e20162398. doi: 10.1542/peds.2016-2398 [DOI] [PubMed] [Google Scholar]
- 22.Coral-Vazquez RM, Rosas-Vargas H, Meza-Espinosa P, et al. Severe congenital muscular dystrophy in a Mexican family with a new nonsense mutation (R2578X) in the laminin alpha-2 gene. J Hum Genet. 2003;48(2):91-95. doi: 10.1007/s100380300013 [DOI] [PubMed] [Google Scholar]
- 23.Reddy HM, Cho KA, Lek M, et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. 2017;62(2):243-252. doi: 10.1038/jhg.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beytía MdlA, Dekomien G, Hoffjan S, Haug V, Anastasopoulos C, Kirschner J. High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol Cell Probes. 2014;28(4):118-122. doi. 10.1016/j.mcp.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 25.Khodaenia N, Farjami Z, Ashnaei AH, et al. Novel homozygous pathogenic mutations of LAMA 2 gene in patients with congen ital muscular dystrophy. Iran J Child Neurol. 2021;15(1):101-106. doi: 10.22037/ijcn.v15i1.21649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westra D, Schouten MI, Stunnenberg BC, et al. Panel-based exome sequencing for neuromuscular disorders as a diagnostic service. J Neuromuscul Dis. 2019;6(2):241-258. doi: 10.3233/jnd-180376 [DOI] [PubMed] [Google Scholar]
- 27.Løkken N, Born AP, Duno M, Vissing J. LAMA2-related myopathy: frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve. 2015;52(4):547-553. doi: 10.1002/mus.24588 [DOI] [PubMed] [Google Scholar]
- 28.Magri F, Brusa R, Bello L, et al. Limb girdle muscular dystrophy due to LAMA2 gene mutations: new mutations expand the clinical spectrum of a still challenging diagnosis. Acta Myol. 2020;39(2):67-82. doi: 10.36185/2532-1900-009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira J, Gruber A, Cardoso M, et al. LAMA2 gene mutation update: toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. 2018;39(10):1314-1337. doi: 10.1002/humu.23599 [DOI] [PubMed] [Google Scholar]
- 30.Oliveira J, Santos R, Soares-Silva I, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. 2008;74(6):502-512. doi: 10.1111/j.1399-0004.2008.01068.x [DOI] [PubMed] [Google Scholar]
- 31.Enright PL, Sherrill DL. Reference equations for the six-minute walk in healthy adults. Am J Respir Crit Care Med. 1998;158(5 Pt 1):1384-1387. doi: 10.1164/ajrccm.158.5.9710086 [DOI] [PubMed] [Google Scholar]
- 32.Geiger R, Strasak A, Treml B, et al. Six-minute walk test in children and adolescents. J Pediatr. 2007;150(4):395-399.e1-2. doi: 10.1016/j.jpeds.2006.12.052 [DOI] [PubMed] [Google Scholar]
- 33.Iannaccone ST, Hynan LS, Morton A, Buchanan R, Limbers CA, Varni JW. The PedsQL in pediatric patients with spinal muscular atrophy: feasibility, reliability, and validity of the pediatric quality of life inventory generic core scales and neuromuscular module. Neuromuscul Disord. 2009;19(12):805-812. doi: 10.1016/j.nmd.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang CH, Bonnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010;25(12):1559-1581. doi: 10.1177/0883073810381924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol. 2012;27(3):363-382. doi: 10.1177/0883073812436605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bianchi C, Baiardi P, Khirani S, Cantarella G. Cough peak flow as a predictor of pulmonary morbidity in patients with dysphagia. Am J Phys Med Rehabil. 2012;91(9):783-788. doi: 10.1097/PHM.0b013e3182556701 [DOI] [PubMed] [Google Scholar]
- 37.Bouman K, Gubbels M, van den Heuvel FMA, et al. Cardiac involvement in two rare neuromuscular diseases: LAMA2-related muscular dystrophy and SELENON-related myopathy. Neuromuscul Disord. 2022;32(8):635-642. doi: 10.1016/j.nmd.2022.06.004 [DOI] [PubMed] [Google Scholar]
- 38.Cho MJ, Lee JW, Lee J, Shin YB, Lee HD. Relationship between fragmented QRS complexes and cardiac status in duchenne muscular dystrophy: multimodal validation using echocardiography, magnetic resonance imaging, and holter monitoring. Pediatr Cardiol. 2017;38(5):1042-1048. doi: 10.1007/s00246-017-1616-7 [DOI] [PubMed] [Google Scholar]
- 39.Shehta M, Rayan MM, Fahmy NA, Onsy A, Bastawy I. Global longitudinal strain detects subtle left ventricular systolic dysfunction in Duchenne muscular dystrophy patients and carriers. Egypt Heart J. 2021;73(1):91. doi: 10.1186/s43044-021-00214-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alfano LLL, Berry K, Flanigan K, Cripe L, Mendell J. Role of motivation on performance of the 6-minute walk test in boys with Duchenne muscular dystrophy. Dev Med Child Neurol. 2015;57:57-58. [Google Scholar]
- 41.Heresi GA, Dweik RA. Strengths and limitations of the six-minute-walk test: a model biomarker study in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(9):1122-1124. doi: 10.1164/rccm.201012-2079ED [DOI] [PubMed] [Google Scholar]
- 42.Mazzone ES, Coratti G, Sormani MP, et al. Timed rise from floor as a predictor of disease progression in duchenne muscular dystrophy: an observational study. PLoS One. 2016;11(3):e0151445. doi: 10.1371/journal.pone.0151445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bortolani S, Brusa C, Rolle E, et al. Technology outcome measures in neuromuscular disorders: a systematic review. Eur J Neurol. 2022;29(4):1266-1278. doi: 10.1111/ene.15235 [DOI] [PubMed] [Google Scholar]
- 44.Goode AP, Hall KS, Batch BC, et al. The Impact of interventions that integrate accelerometers on physical activity and weight loss: a systematic review. Ann Behav Med. 2017;51(1):79-93. doi: 10.1007/s12160-016-9829-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakr HM, Fahmy N, Elsayed NS, et al. Whole-body muscle MRI characteristics of LAMA2-related congenital muscular dystrophy children: an emerging pattern. Neuromuscul Disord. 2021;31(9):814-823. doi. 10.1016/j.nmd.2021.06.012 [DOI] [PubMed] [Google Scholar]
- 46.Quijano-Roy S, Haberlova J, Castiglioni C, et al. Diagnostic interest of whole-body MRI in early- and late-onset LAMA2 muscular dystrophies: a large international cohort. J Neurol. 2022;269(5):2414-2429. doi: 10.1007/s00415-021-10806-0 [DOI] [PubMed] [Google Scholar]
- 47.Tordjman M, Dabaj I, Laforet P, et al. Muscular MRI-based algorithm to differentiate inherited myopathies presenting with spinal rigidity. Eur Radiol. 2018;28(12):5293-5303. doi: 10.1007/s00330-018-5472-5 [DOI] [PubMed] [Google Scholar]
- 48.Tan D, Ge L, Fan Y, et al. Muscle magnetic resonance imaging in patients with LAMA2-related muscular dystrophy. Neuromuscul Disord. 2021;31(11):1144-1153. doi: 10.1016/j.nmd.2021.09.006 [DOI] [PubMed] [Google Scholar]
- 49.Mul K, Horlings CGC, Vincenten SCC, Voermans NC, van Engelen BGM, van Alfen N. Quantitative muscle MRI and ultrasound for facioscapulohumeral muscular dystrophy: complementary imaging biomarkers. J Neurol. 2018;265(11):2646-2655. doi: 10.1007/s00415-018-9037-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pangalila RF, van den Bos GA, Bartels B, et al. Quality of life of adult men with Duchenne muscular dystrophy in The Netherlands: implications for care. J Rehabil Med. 2015;47(2):161-166. doi: 10.2340/16501977-1898 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.