

Visual Abstract

Keywords: hemolytic uremic syndrome, AKI, complement, molecular genetics, epidemiology and outcomes
Abstract
Background
The identification of complement defects as major drivers of primary atypical hemolytic uremic syndrome (HUS) has transformed the landscape of thrombotic microangiopathies (TMAs), leading to the development of targeted therapies and better patient outcomes. By contrast, little is known about the presentation, genetics, and outcomes of TMA associated with specific diseases or conditions, also referred to as secondary TMA.
Methods
In this study, we assessed the relative incidence, clinical and genetic spectra, and long-term outcomes of secondary TMA versus other TMAs in consecutive patients hospitalized with a first episode of TMA from 2009 to 2019 at two European reference centers.
Results
During the study period, 336 patients were hospitalized with a first episode of TMA. Etiologies included atypical HUS in 49 patients (15%), thrombotic thrombocytopenic purpura (TTP) in 29 (9%), shigatoxin-associated HUS in 70 (21%), and secondary TMA in 188 (56%). The main causes of secondary TMA were hematopoietic stem-cell transplantation (n=56, 30%), solid-organ transplantation (n=44, 23%), and malignant hypertension (n=25, 13%). Rare variants in complement genes were identified in 32 of 49 patients (65%) with atypical HUS and eight of 64 patients (13%) with secondary TMA; pathogenic or likely pathogenic variants were found in 24 of 49 (49%) and two of 64 (3%) of them, respectively (P < 0.001). After a median follow-up of 1157 days, death or kidney failure occurred in 14 (29%), eight (28%), five (7%), and 121 (64%) patients with atypical HUS, TTP, shigatoxin-associated HUS, and secondary TMA, respectively. Unadjusted and adjusted Cox regressions showed that patients with secondary TMA had the highest risk of death or kidney failure (unadjusted hazard ratio [HR], 3.35; 95% confidence interval [CI], 1.85 to 6.07; P < 0.001; adjusted HR, 4.11; 95% CI, 2.00 to 8.46; P < 0.001; considering atypical HUS as reference).
Conclusions
Secondary TMAs represent the main cause of TMA and are independently associated with a high risk of death and progression to kidney failure.
Introduction
Thrombotic microangiopathies (TMAs) encompass a wide spectrum of ultra-rare diseases resulting from an endothelial injury and leading to severe organ damage and dysfunction. These diseases are characterized by the triad of nonimmune microangiopathic hemolytic anemia, thrombocytopenia, and end-organ damage or by pathological evidence of microvascular injury and thrombosis.1,2 A better understanding of underlying mechanisms has led to a classification of TMA that includes primary atypical hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), shigatoxin-associated HUS, and TMA associated with specific diseases or conditions, also referred to as secondary TMA.2,3
These advances contributed to the development of targeted therapies, leading to better outcomes in patients with primary TMA. Atypical HUS is a prototype of complement-mediated disease characterized by a high prevalence (approximately 60%) of inherited or acquired defects of the alternative pathway of the complement system.1,4 In patients with atypical HUS, the use of monoclonal antibodies inhibiting complement activation drastically reduced progression to kidney failure or death and prevents relapse after kidney transplantation.5–9 Current effective treatment of acquired TTP, caused by immune-mediated deficiency of the von Willebrand factor-cleaving protease ADAMTS13, relies on the combination of daily plasma exchanges, immunosuppression, and caplacizumab.10,11
In contrast to atypical HUS and TTP, little is known about the incidence, mechanisms, and outcomes of secondary TMA. Scarce cohort studies have been reported, but their conclusions were limited by selection bias, lack of information about long-term outcomes, and/or lack of detailed genetic analysis (Supplemental Table 1).12–15 The prevalence of complement gene abnormalities and their potential contribution to the disease in patients with secondary TMA remain poorly defined, with significant variability between cohorts and the different triggers.12,16–19
To fill these gaps, we retrospectively reviewed the incidence, clinical presentation, prevalence of complement genes abnormalities, and long-term outcomes of secondary TMA versus other etiologies of TMA in consecutive patients admitted at two European reference centers over a 10-year period.
Methods
Study Design, Participants, and Adjudication
The retrospective study included consecutive patients hospitalized with a first episode of TMA between January 1, 2009, and December 31, 2019, at Cliniques universitaires Saint Luc, Brussels, and at University Hospitals Leuven, Leuven, two reference centers and members of the European Reference Network for Rare Kidney Diseases. Hospital discharge summaries were screened using the International Classification of Diseases codes of TMA, HUS, TTP, and malignant hypertension. Individual files were analyzed by four physicians (A.W., P.S., K.J.C., and J.M.) to adjudicate patients, as previously described.13 The first step consisted of validation of the diagnosis of TMA, followed by identification of the etiology of TMA, using a strict hierarchical process, including (1) identification of TTP on the basis of ADAMTS13 activity <10%, followed by (2) identification of shigatoxin-associated HUS on the basis of the presence of shigatoxin-producing enterobacteriae using polymerase chain reaction on stool and (3) identification of secondary TMA on the basis of the presence of any other disease or condition associated with TMA. Patients with evidence of TMA in the absence of TTP, shigatoxin-associated HUS, or secondary TMA were considered as atypical HUS, in line with current guidelines.2,3 In the absence of TMA, patients with hemolysis, elevated liver enzymes, low platelet count syndrome, or preeclampsia were not included.
Patients were followed until they died or progressed to kidney failure (i.e., dialysis or preemptive kidney transplantation) or until the end of the follow-up period. Information about missing data is provided in Table 1 and Supplemental Table 2.
Table 1.
Characteristics of patients hospitalized with a first episode of thrombotic microangiopathy between 2009 and 2019 at Cliniques universitaires Saint Luc, Brussels, or University Hospitals Leuven, Leuven, at the time of diagnosis and according to the etiology of thrombotic microangiopathy
| Characteristics | Whole Cohort n=336 |
Atypical HUS n=49 (15%) |
TTP n=29 (9%) |
Shigatoxin-Assoc. HUS n=70 (21%) |
Secondary TMA n=188 (56%) |
|---|---|---|---|---|---|
| Age, median (IQR), yr | 40 (16–56) | 39 (21–48) | 43 (30–52) | 4 (2–9) | 51 (36–61) |
| Female, no. (%) | 197 (59) | 31 (63) | 19 (66) | 44 (63) | 103 (55) |
| Ethnicity, no. (%) | |||||
| White | 311/332 (94) | 47 (96) | 25 (86) | 69 (99) | 170/184 (92) |
| Black | 16/332 (5) | 1 (2) | 3 (10) | 1 (1) | 11/184 (6) |
| Other | 5/332 (1) | 1 (2) | 1 (4) | 0 (0) | 3/184 (2) |
| Comorbidities, no. (%) | |||||
| Diabetes | 19 (6) | 1 (2) | 1 (3) | 1 (1) | 16 (9) |
| Hypertension | 88/335 (26) | 11 (22) | 4 (14) | 0 (0) | 73/187 (39) |
| CKD | 76/242 (31) | 1/25 (4) | 2/19 (11) | 0/30 (0) | 73/168 (43) |
| Coronary artery disease | 6 (2) | 1 (2) | 0 (0) | 0 (0) | 5 (3) |
| Heart failure | 6 (2) | 0 (0) | 0 (0) | 0 (0) | 6 (3) |
| Native kidneys, no. (%) | 300 (89) | 49 (100) | 29 (100) | 70 (100) | 152 (81) |
| Organ dysfunction, no. (%) | |||||
| Kidney | 312 (93) | 49 (100) | 16 (55) | 70 (100) | 177 (94) |
| Incl. AKI requiring dialysis | 113 (34) | 33 (47) | 3 (10) | 36 (51) | 51 (27) |
| Gastrointestinal tract | 154 (46) | 28 (57) | 12 (41) | 69 (99) | 45 (24) |
| Central nervous system | 112 (33) | 11 (22) | 19 (66) | 30 (43) | 52 (28) |
| Hemoglobin, median (IQR), g/dl | 7.9 (6.7–9.3) | 7.7 (5.9–8.9) | 7.2 (6.2–9.1) | 7 (6.3–8.0) | 8.3 (7.5–9.8) |
| Platelets, median (IQR), ×103/µl | 57 (23–104) | 70 (38–115) | 15 (10–17) | 32 (20–50) | 77 (45–134) |
| LDH, median (IQR), IU/L | 848 (492–2119) | 1350 (690–2430) | 903 (578–1443) | 2523 (1928–3315) | 595 (381–938) |
| Schistocytes, median (IQR), % | 4 (2–5) | 4 (2–6) | 5 (3–6) | 5 (3–8) | 3 (2–4) |
| sCreatinine, median (IQR), mg/dl | 2.9 (1.5–4.9) | 4.6 (2.5–7.4) | 1.0 (0.8–1.8) | 3.2 (1.6–4.6) | 2.7 (1.6–4.9) |
| UPCR, median (IQR), g/g | 1.3 (0.6–3.7) | 1.7 (0.8–5.1) | 0.9 (0.3–2.4) | 3.1 (0.9–9.8) | 1.1 (0.5–2.5) |
| ADAMTS13 activity, median (IQR), % | 63 (43–80) | 80 (62–95) | 5 (0–5) | 69 (49–91) | 63 (50–77) |
| C3, median (IQR), g/L | 0.92 (0.75–1.12) | 0.84 (0.66–0.99) | 1.1 (0.81–1.19) | 0.93 (0.79–1.10) | 0.92 (0.76–1.14) |
| Treatments, no. (%) | |||||
| Patients with plasma exchange | 141 (42) | 39 (80) | 26 (90) | 11 (16) | 65 (35) |
| Patients with eculizumab | 41 (12) | 26 (53) | 0 (0) | 7 (10) | 8 (4) |
HUS, hemolytic uremic syndrome; TTP, thrombotic thrombocytopenic purpura; Shigatoxin-assoc. HUS, shigatoxin-associated HUS; TMA, thrombotic microangiopathy; IQR, interquartile range; LDH, lactate dehydrogenase; sCreatinine, serum creatinine; UPCR, urinary protein-to-creatinine ratio.
The study was conducted in accordance with the World Medical Association's Declaration of Helsinki; the Belgian law related to experiments in humans dated May 7, 2004; the General Data Protection Regulation 2016/679; and the Belgian law of July 30, 2018, regarding the protection of personal data. The Ethical Review Boards of Cliniques universitaires Saint-Luc/UCLouvain and University Hospitals Leuven approved the study and waived the need for an informed consent because of its retrospective design.
Data Collection and Definitions
Demographic, clinical, and biological characteristics were obtained from patients' electronic charts. The worst biological values within the first week after admission were recorded. Preexisting CKD was defined as a glomerular filtration rate of <60 ml/min per 1.73 m2 estimated using the creatinine-based Chronic Kidney Disease Epidemiology Collaboration equation, for 3 months or more.
Organ involvement was defined as follows: kidney involvement, as the presence of AKI, according to the creatinine-based criteria of the Kidney Disease Improving Global Outcomes guidelines20; gastrointestinal tract involvement, as the presence of diarrhea, upper or lower gastrointestinal bleeding, and/or abdominal pain; and involvement of the central nervous system, as the presence of epilepsy, altered consciousness, stroke, or confusion.
Complement Workup
Complement workup and genotyping were performed as part of routine clinical care. All patients provided informed consent for genetic testing.
Mutation screening was performed by Next-Generation Sequencing (NGS) using a SeqCap EZ Choice custom panel of genes (Roche), including CFH (NM_000186.3), CFI (NM_000204.3), CD46 (NM_002389.4), CFB (NM_001710.5), C3 (NM_000064.2), CFHR1 (NM_002113.2), and CFHR3 (NM_021023.5) genes. The enriched library was sequenced on Illumina Miseq (or NextSeq) with an average coverage of 1500 reads. Variants were called with Genome Analysis ToolKit HaplotypeCaller, UnifiedGenotyper, varscan, platypus, freebayes, and bcftools for sodium nitroprussides and small indels. Copy number variations were assessed by multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA P236 A1 ARMD mix 1 kit (MRC Holland, The Netherlands). Screening for DGKE variants has been added to the NGS panel in 2015.
Minor allele frequencies (MAFs) were obtained from the Genome Aggregation Database (https://gnomad.broadinstitute.org). Rare variants were defined by a MAF in the general population of <0.1%, as previously described,12 while pathogenic or likely pathogenic variants were defined according to the classification of the American College of Medical Genetics and Genomics (ACMG),21 last accessed on July 22, 2022.
Statistical Analyses
Results are presented as median and interquartile range (IQR) for continuous variables and as number and proportion for categorical variables. Comparisons between groups were performed using the Kruskal–Wallis test, followed by Dunn correction for multiple testing, or chi-squared test, as appropriate. Kaplan–Meier estimates and Cox proportional hazard regressions assessed the time to kidney failure or death, considering atypical HUS as the reference. Adjusted analyses included the following prespecified covariates: age and sex; creatinine, platelets, and hemoglobin at presentation; and diabetes, hypertension, previous CKD, coronary artery disease, and heart failure. Collinearity between variables was quantified using variance inflation factors. Statistical analyses were performed using Stata (version 17.0) and graphed with GraphPad Prism (version 9.0). All tests were two-tailed, and a P value of <0.05 was considered significant.
Results
Study Population and Etiologies of TMAs
We screened a total of 704,615 unique patients (corresponding to 984,914 hospitalizations) hospitalized at two large reference centers in Belgium over a 10-year period to identify 336 consecutive patients (0.05%) with a first episode of acute TMA (Figure 1).
Figure 1.
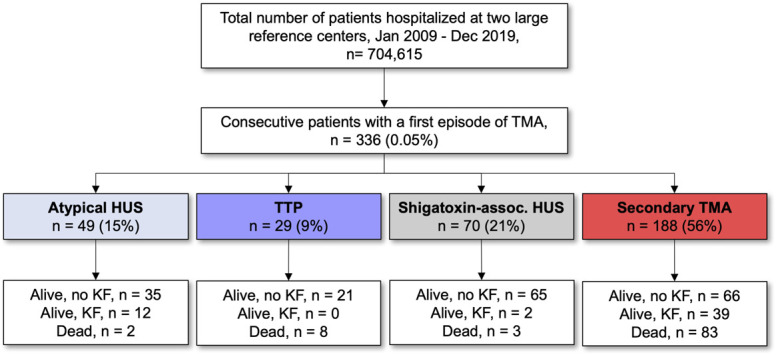
Flow chart of the study. HUS, hemolytic uremic syndrome; KF, kidney failure; shigatoxin-assoc. HUS, shigatoxin-associated HUS; TMA, thrombotic microangiopathy; TTP, thrombotic thrombocytopenic purpura.
Etiologies of TMA included atypical HUS in 49 (15%), TTP in 29 (9%), shigatoxin-associated HUS in 70 (21%), and secondary TMA in 188 (56%) patients (Figure 1, Table 1). The relative incidence and distribution of TMA etiologies were similar in both centers (Supplemental Table 3). The median age (IQR) at admission was 40 (16–56) years; 197 patients (59%) were women; most patients (94%) were White; and the majority occurred in native kidneys (300, 89%). Shigatoxin-associated HUS mainly affected children (66/70, 94%), with a median age of 4 (2–9) years, while TTP mostly occurred in adults (28/29, 97%), with a median age of 43 (30–52) years. Atypical HUS and secondary TMA were diagnosed in all age groups, with a median age at diagnosis of 39 (21–48) and 51 (36–61) years, respectively (Figure 2; Table 1). Comorbidities, including a history of diabetes, hypertension, or CKD, were more common among patients with secondary TMA (Table 1). As compared with patients with HUS, those presenting with TTP had lower serum creatinine levels and lower platelet counts. Kidney dysfunction was present in all patients with atypical HUS and shigatoxin-associated HUS and in half (55%) of those with TTP. Severe AKI was common because half of the patients with atypical HUS or shigatoxin-associated HUS and one third of those with secondary TMA required dialysis during initial hospitalization. Extrarenal organ damage was prevalent in all forms of TMA, with gastrointestinal tract and central nervous system involvement in 24%–99% and 22%–66% of the patients, respectively (Figure 2, Table 1).
Figure 2.

Baseline characteristics according to the etiology of TMA. (A) Age at diagnosis; (B) serum creatinine; (C) platelet count; (D) ADAMTS13 activity; (E) kidney involvement; (F) AKI requiring dialysis; (G) gastrointestinal tract involvement; (H) central nervous system involvement. *P < 0.05; **P < 0.01; ***P < 0.001. sCreat, serum creatinine.
Etiologies of secondary TMA were diverse and mainly included hematopoietic stem-cell transplantation (56/188, 30%), solid-organ transplantation (44/188, 23%), malignant hypertension (25/188, 13%), drugs (20/188, 11%), neoplasia (12/188, 6%), and pregnancy (7/188, 4%) (Table 2). Kidney involvement was virtually constant in HUS associated with malignant hypertension, solid-organ transplant, pregnancy, or drugs (90%–100%) (Supplemental Table 4). Serum creatinine levels on admission were higher in patients with HUS secondary to malignant hypertension compared with the three other main categories of secondary TMA. Additional information about solid-organ transplant recipients with TMA is presented in Supplemental Figure 1 and Supplemental Table 5.
Table 2.
Etiologies of secondary thrombotic microangiopathy
| Diagnosis | No. (%) |
|---|---|
| Hematopoietic stem-cell transplantation | 56 (30) |
| Solid-organ transplantationa | 44 (23) |
| Malignant hypertension | 25 (13) |
| Drugsb | 20 (11) |
| Neoplasiac | 12 (6) |
| Pregnancy | 7 (4) |
| Infectionsd | 7 (4) |
| Glomerular diseasese | 7 (4) |
| Systemic diseasesf | 6 (3) |
| Cobalamin C deficiency | 2 (1) |
Solid-organ transplantation includes kidney (35), lung (7), and liver (2) transplantation.
Drugs include gemcitabine (11), carfilzomib (2), interferon-beta (2), mitomycine C (1), capecitabine (1), cabazitaxel (1), sunitinib (1), and tacrolimus (1).
Neoplasia includes breast adenocarcinoma (3), pancreatic adenocarcinoma (2), prostatic adenocarcinoma (2), multiple myeloma (2), lymphoma (2), Ewing sarcoma (2), gastric adenocarcinoma (1), and germinal carcinoma (1).
Infections include invasive pneumococcal infection (4), HIV (1), and other causes (2).
Glomerular diseases include immunoglobulin A nephropathy (4), ANCA-associated vasculitis (1), cryoglobulin-associated glomerulonephritis (1), and anti–glomerular basement membrane disease (1).
Systemic diseases include systemic lupus erythematosus (3), scleroderma renal crisis (2), and antiphospholipid syndrome (1).
Complement Abnormalities in Patients with HUS
Rare variants in complement genes were identified in 32 of 49 patients (65%) with atypical HUS and in eight of 64 patients (13%) with secondary TMA (P < 0.001) (Table 3). As compared with those who were not tested, the subset of patients with secondary TMA who underwent genetic screening (64/188, 35%) were younger (40 versus 54 years), had higher serum creatinine levels on admission (4.8 versus 2.0 mg/dl), and a higher proportion of them required dialysis (37% versus 22%) (Supplemental Table 6). Rare variants were mainly identified in CFH (36%), CFI (21%), and CD46 (26%) (Supplemental Table 7).
Table 3.
Rare and pathogenic or likely pathogenic variants in complement genes among patients with atypical hemolytic uremic syndrome or secondary thrombotic microangiopathy
| Gene Variants | Atypical HUS n=49 |
Secondary TMA n=64 |
P Value |
|---|---|---|---|
| Rare variants, no. (%) | 32 (65) | 8 (13) | <0.001 |
| Pathogenic or likely pathogenic variants, no. (%) | 24 (49) | 2 (3)a | <0.001 |
HUS, hemolytic uremic syndrome; TMA, thrombotic microangiopathy.
The two patients with secondary hemolytic uremic syndrome and pathogenic/likely pathogenic variants had postpartum hemolytic uremic syndrome and de novo hemolytic uremic syndrome after kidney transplantation. Rare variants were defined by a minor allele frequency in the general population of <0.1%, while pathogenic or likely pathogenic variants were defined according to the classification of the American College of Medical Genetics and Genomics.
Likely pathogenic or pathogenic variants were identified in 24 of 49 (49%) and two of 64 (3%) patients with atypical HUS and secondary TMA, respectively (P < 0.001) (Table 3). In secondary TMA, the two patients with pathogenic/likely pathogenic variants had pregnancy-associated HUS and de novo HUS after kidney transplantation, two conditions known to be complement-mediated, like atypical HUS (Supplemental Table 8).2 Detailed information about the variants identified in this cohort, including MAF and ACMG classification, is provided in Supplemental Table 9. Anti-CFH antibodies were detected in four patients (8%) with atypical HUS. There was no patient with DGKE-TMA in the cohort.
Treatments and Outcomes
Therapeutic plasma exchanges were used in 39 of 49 (80%) and 26 of 29 (90%) patients with atypical HUS and TTP, respectively (Table 1). Of note, three patients with TTP died in the first few hours after admission, before any treatment could be initiated. Plasma exchanges were also used in 11 patients (16%) with shigatoxin-associated HUS and in 65 (35%) with secondary TMA.
Eculizumab was initiated in 26 (53%) of the 49 patients with atypical HUS (Table 1). The remaining patients with atypical HUS did not receive eculizumab because they presented before eculizumab was available (n=11, 22%), achieved remission spontaneously or were under plasma exchanges (n=7, 14%), presented with kidney failure and extensive chronic damage on kidney biopsy (n=3, 6%), were initially misdiagnosed and later reclassified (n=1, 2%), or had life-threatening Pneumocystis jirovecii infection (n=1, 2%). Of the 26 patients with atypical HUS treated with eculizumab, 22 (85%) discontinued treatment after a median (IQR) of 12 (6–28) months. After a median follow-up (IQR) of 43 (35–60) months after eculizumab discontinuation, two patients (2/22, 9%) had a relapse of atypical HUS. Eculizumab was also used in a minority of patients with severe, life-threatening shigatoxin-associated HUS (7/70, 10%) or secondary TMA (8/188, 4%) (Table 1). Among these eight patients with secondary TMA, two received eculizumab for HUS associated with pregnancy (n=2, 25%) and two for de novo HUS after kidney transplantation (n=2, 25%). The remaining had TMA associated with scleroderma renal crisis, cancer, or malignant hypertension.
During a median (IQR) follow-up of 1157 (360–2126) days, 148 patients (44%) died or progressed to kidney failure (Table 4; Supplemental Table 10). Patients with secondary TMA showed the worst outcomes, as death or kidney failure occurred in 65% (122/188) of them, while those with shigatoxin-associated HUS had the lowest rates of death or kidney failure (5/70, 7%) (Figure 3; Table 4). Patients with atypical HUS and TTP showed an intermediate composite outcome (death or kidney failure in 14/49, 29%, and 8/29, 28%, respectively), mainly driven by progression to kidney failure in patients with atypical HUS and by death in those with TTP (Figure 3; Table 4). Of the 13 patients with atypical HUS who progressed to kidney failure, five presented before the era of eculizumab and eight presented with severe kidney dysfunction requiring dialysis. Among those, five were treated by eculizumab but failed to recover from kidney failure and three did not receive eculizumab because of late referral, extensive chronic lesions on kidney biopsy, and the absence of extrarenal complications.
Table 4.
Cox regression analyses for kidney failure or death according to the etiology of thrombotic microangiopathy
| Outcome | Unadjusted | Adjusted | |||||
|---|---|---|---|---|---|---|---|
| n (%) with Outcome | HR | 95% CI | P Value | Adj. HR | 95% CI | P Value | |
| Kidney failure or death | |||||||
| Atypical HUS | 14 (29) | 1.00 (ref.) | — | — | 1.00 (ref.) | — | — |
| TTP | 8 (28) | 1.16 | 0.47 to 2.85 | 0.74 | 1.60 | 0.57 to 4.44 | 0.37 |
| Shigatoxin-assoc. HUS | 5 (7) | 0.21 | 0.07 to 0.65 | 0.007 | 0.39 | 0.12 to 1.34 | 0.14 |
| Secondary TMA | 122 (65) | 3.35 | 1.85 to 6.07 | <0.001 | 4.11 | 2.00 to 8.46 | <0.001 |
| Death | |||||||
| Atypical HUS | 2 (4) | 1.00 (ref.) | — | — | 1.00 (ref.) | — | — |
| TTP | 8 (28) | 12.55 | 2.25 to 69.93 | 0.004 | 6.00 | 0.71 to 50.60 | 0.10 |
| Shigatoxin-assoc. HUS | 3 (4) | 1.60 | 0.25 to 10.20 | 0.62 | 3.49 | 0.35 to 34.55 | 0.29 |
| Secondary TMA | 83 (44) | 21.91 | 4.48 to 107.09 | <0.001 | 15.21 | 2.07 to 111.71 | 0.007 |
| Kidney failure | |||||||
| Atypical HUS | 13 (26) | 1.00 (ref.) | — | — | 1.00 (ref.) | — | — |
| TTP | 2 (7) | 0.32 | 0.07 to 1.44 | 0.14 | 0.52 | 0.06 to 4.29 | 0.55 |
| Shigatoxin-assoc. HUS | 5 (7) | 0.23 | 0.07 to 0.72 | 0.01 | 0.41 | 0.12 to 1.48 | 0.17 |
| Secondary TMA | 61 (33) | 1.65 | 0.87 to 3.15 | 0.13 | 2.51 | 1.15 to 5.50 | 0.02 |
The multivariable model is adjusted for age, sex, biological values of severity at presentation (creatinine, platelets, and hemoglobin), and comorbidities (diabetes, hypertension, previous CKD, coronary artery disease, and heart failure). HR, hazard ratio; 95% CI, 95% confidence interval; adj., adjusted; HUS, hemolytic uremic syndrome; ref., reference; TTP, thrombotic thrombocytopenic purpura; shigatoxin-assoc. HUS, shigatoxin-associated HUS; TMA, thrombotic microangiopathy.
Figure 3.

Survival free from kidney failure and overall survival according to the etiology of TMA. Kaplan–Meier curves of survival free from kidney failure (A) and overall survival (B).
Survival analysis using Cox proportional regressions confirmed poor outcomes associated with secondary TMA as compared with patients with atypical HUS, both in unadjusted (hazard ratio [HR], 3.35; 95% confidence interval [CI], 1.85 to 6.07; P < 0.001) and adjusted (adjusted HR, 4.11; 95% CI, 2.00 to 8.46; P < 0.001) analyses (Table 4). Analysis of the rates of death or kidney failure considered separately confirmed the higher risk of both outcomes in this population (adjusted HR for death: 15.21; 95% CI, 2.07 to 111.71; P = 0.007; and adjusted HR for kidney failure: 2.51, 95% CI, 1.15 to 5.50, P = 0.02) (Table 4). Sensitivity analyses performed after exclusion of patients with TMA associated with hematopoietic stem-cell transplantation or cancer showed that remaining patients with other causes of secondary TMA also had very poor outcomes, with high rates of death or progression to kidney failure (Supplemental Figure 2; Supplemental Table 11). Analyses restricted to the adult population confirmed the poor outcomes associated with secondary TMA compared with other TMAs (Supplemental Figure 3).
At the last follow-up, most of the patients (61/66, 92%) with secondary TMA who did not reach kidney failure had an estimated glomerular filtration rate below 60 ml/min per 1.73 m2.
Altogether, these observations highlight the poor outcomes in patients with secondary TMA, with a significant risk of death or progression to kidney failure that is independent of disease severity and comorbidities. Among those who survived and did not reach kidney failure, an overwhelming majority had significant kidney dysfunction at the last follow-up.
Discussion
In this study, we screened more than 700,000 unique patients at two European reference centers over a 10-year period to identify 336 consecutive patients hospitalized with a first episode of TMA. We observed that secondary TMA accounted for more than half (approximately 55%) of the etiologies of TMA and were associated with a high risk of death or progression to kidney failure. Pathogenic or likely pathogenic variants in complement genes were identified in a minority of patients with secondary TMA, contrasting with their high prevalence in those with atypical HUS.
The high prevalence of secondary TMA in our cohort is in line with recent studies, in which secondary TMA represented 49%–88% of all TMAs.12–15 Etiologies of secondary TMA varied widely across studies, reflecting differences in inclusion criteria, definitions, and expertise of the different centers (Supplemental Table 1). In our cohort, hematopoietic stem-cell transplantation, solid-organ transplantation, and malignant hypertension were identified as the main causes of secondary TMA, representing two thirds of all secondary TMAs.
Secondary TMA was associated with the highest risk of a composite of death or kidney failure, which occurred in 64% of the patients during follow-up. In addition, 94% of those who survived without kidney failure had CKD at the last follow-up. Poor outcomes in this group were independent of age, sex, disease severity, and comorbidities, suggesting they might be partly explained by the lack of specific treatment.
Patients with secondary TMA showed a much lower prevalence of genetic defects in complement genes than those with atypical HUS. Pathogenic or likely pathogenic variants were found in 49% of patients with atypical HUS but only in 3% of patients with secondary TMA. Importantly, if we exclude patients with pregnancy-associated HUS and de novo HUS after kidney transplantation, none of the remaining patients with secondary TMA had pathogenic or likely pathogenic variants in complement genes. These data are similar to those from the French registry where 2% of the patients with secondary TMA showed pathogenic or likely pathogenic variants in complement genes12; of note, pregnancy-associated HUS and de novo HUS after kidney transplantation were excluded from this report from the French registry because these conditions are associated with a higher prevalence of rare variants in complement genes (56% and 29%, respectively) and may actually be considered as a first presentation of complement-mediated TMA.22–24
Even in the absence of constitutive genetic defects, secondary TMA was commonly (60%–100%) linked with complement activation, in two independent cohorts that used ex vivo complement testing on endothelial cells.25,26 These data provide a rationale to consider the use of complement inhibitors in selected patients with secondary TMA, that is, those with ongoing active disease despite removal of the trigger and/or associated with life- or kidney-threatening complications. Although case reports and case series suggested that some of these conditions may respond to eculizumab,27,28 robust evidence supporting the benefit of complement blockade in this population is still lacking. Unfortunately, a phase 3 trial investigating the efficacy and safety of ravulizumab (Alexion Pharmaceuticals, Boston, MA), a long-acting complement C5 inhibitor, in adult participants with secondary TMA has been terminated prematurely because of continued enrollment challenges (NCT04743804).
Data from clinical trials showed that the incidence of the composite of death or kidney failure among patients with atypical HUS dropped from approximately 50% to approximately 15% after the introduction of eculizumab.6 In our cohort, a similar proportion of patients died or progressed to kidney failure during the eculizumab period, validating these findings in real life. This emphasizes the importance of timely referral, diagnosis, and treatment of atypical HUS to avoid the development of chronic and irreversible lesions causing kidney failure.
In our cohort, the subgroup of patients with TTP was characterized by a very high risk of death, early during hospitalization. Of note, caplacizumab, an anti–von Willebrand factor humanized single-variable domain immunoglobulin inhibiting the interaction between von Willebrand factor multimers and platelets, was not available during the study period. Used on top of plasma exchanges, steroids, and rituximab, caplacizumab was recently found to shorten the time to normalization of the platelet count and to improve outcomes in patients with immune-mediated TTP,11 supporting its use in reducing early complications of TTP. In our cohort, a significant proportion of patients with TTP (55%) also had AKI, in line with previous reports (approximately 60%).29
Our study has several strengths. We included consecutive patients without selection bias, and we did not enroll patients with conditions known to mimic TMA, such as preeclampsia. We provided detailed information about phenotypes, long-term outcomes, and results of genetic screening for a subset of patients with secondary TMA. Importantly, our two centers, which have the largest recruitment of patients with TMA/HUS in Belgium, apply a unified approach for the classification and management of TMA, on the basis of currently available international and national guidelines,2,3 resulting in similar incidences and distribution of the different etiologies of TMA. We also acknowledge limitations, including the retrospective design; the absence of systematic complement workup in patients with secondary TMA, reflecting real-life conditions; and our inability to formerly assess the potential benefit of eculizumab in patients with secondary TMA, a condition for which complement inhibitors are currently not reimbursed in Belgium. Additional risk haplotypes in CFH (CFH-H3) and CD46 (MCPaaggt) may modulate complement activity,30 but were not systematically performed throughout the study period and could, therefore, not be included in the analysis.
Finally, our observations also highlight the need for an update in the nomenclature of TMA/HUS because the current terminology mixes various and often complicated terms to describe TMA caused by diverse mechanisms. A revised TMA nomenclature would have the potential to facilitate the understanding of the disease; to increase awareness; and, potentially, to improve the diagnosis, management, and outcomes in patients with TMA. A novel, pathophysiology-based terminology of TMA might, for instance, include “low ADAMTS13-TMA,” “shigatoxin-TMA,” “secondary TMA,” and “primary TMA” (Supplemental Figure 4). The term “primary TMA” would correspond to primary atypical HUS and would remind that it is a diagnosis of exclusion, where ADAMTS13 activity is normal, there is no shigatoxin-associated infection, and no disease or condition known to cause TMA. Importantly, a subset of primary and secondary TMAs (i.e., those associated with pregnancy, de novo TMA after kidney transplantation, or malignant hypertension) shows underlying defects in the complement system, such as pathogenic or likely pathogenic gene variants or anti-CFH antibodies, and such information should be added to the diagnosis (e.g., primary TMA with a pathogenic variant in the CFH gene; pregnancy-associated TMA with a likely pathogenic variant in CFI). We feel such proposal is timely and may serve as a basis for future discussions with the whole TMA/HUS community, including physicians, scientists, patients, and caregivers, to reach a consensus.
In conclusion, using a large cohort of consecutive patients with TMA, this study showed that secondary TMA represent the main cause of TMA and is associated with a high risk of death and progression to kidney failure. Although complement activation is common in these diseases, genetic defects in complement genes are very unusual, except in some conditions such as pregnancy and de novo HUS after kidney transplantation. Future studies are needed to better understand the mechanisms and to improve outcomes of patients with secondary TMA.
Supplementary Material
Acknowledgments
The funders had no role in study design, data collection, analysis, reporting, or the decision to submit for publication.
We thank all participating patients and families. We also thank the collaborators from the UCLouvain and UZ Leuven Kidney Disease Networks for the referral and care of the patients included in this study.
Footnotes
Collaborators from the UCLouvain TMA/HUS Network: Selda Aydin (Pathology, Cliniques universitaires Saint-Luc, Brussels), Charles Cuvelier (Nephrology, CHU UCLouvain-Namur, Namur), Pierre-Yves Decleire (Nephrology, Vivalia, Hôpital d'Arlon, Arlon), Nathalie Demoulin (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Arnaud Devresse (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Ludovic Gérard (Intensive Care Medicine, Cliniques universitaires Saint-Luc, Brussels), Gaëlle Gillerot (Nephrology, Clinique Saint-Pierre Ottignies, Ottignies), Valentine Gillion (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Eric Goffin (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Philippe Hantson (Intensive Care Medicine, Cliniques universitaires Saint-Luc, Brussels), Michel Jadoul (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Jean Jamez (Nephrology, Clinique Saint-Pierre Ottignies, Ottignies), Nada Kanaan (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Laura Labriola (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Jean-Philippe Lengelé (Nephrology, Grand Hôpital de Charleroi, Gilly), Lionel Mazzoleni (Nephrology, CHU Ambroise Paré, Mons), Yves Pirson (Nephrology, Cliniques universitaires Saint-Luc, Brussels), Jean-Michel Pochet (Nephrology, CHU UCLouvain-Namur, Namur), Nadejda Ranguelov (Pediatrics, Cliniques universitaires Saint-Luc, Brussels), Marie-Astrid van Dievoet (Hematology, Department of Laboratory Medicine, Cliniques universitaires Saint-Luc, Brussels), Elliott van Regemorter (Nephrology, Cliniques universitaires Saint-Luc, Brussels), and Xavier Wittebole (Intensive Care Medicine, Cliniques universitaires Saint-Luc, Brussels). Collaborators from the KU Leuven TMA/HUS Network : Bert Bammens (Nephrology, UZ Leuven, Leuven), Greet De Vlieger (Intensive Care Unit, UZ Leuven, Leuven), Koenraad Devriendt (Genetics, UZ Leuven, Leuven), Katrien De Vusser (Nephrology, UZ Leuven, Leuven), Pieter Evenepoel (Nephrology, UZ Leuven, Leuven) Laurent Godinas (Pneumology, UZ Leuven, Leuven), Priyanka Koshy (Pathology, UZ Leuven, Leuven), Dirk Kuypers (Nephrology, UZ Leuven, Leuven), Evelyne Lerut (Pathology, UZ Leuven, Leuven), Björn Meijers (Nephrology, UZ Leuven, Leuven), Maartens Naesens (Nephrology, UZ Leuven, Leuven), Patrick Schöffski, (Medical oncology, UZ Leuven, Leuven), Ben Sprangers (Nephrology, ZOL, Genk), Dirk Timmerman (Gynecology and Obstetrics, UZ Leuven, Leuven), Amaryllis Van Craenenbroeck (Nephrology, UZ Leuven, Leuven), Alexander Wilmer (Intensive care unit, UZ Leuven, Leuven), and all the collaborators/nephrologists from the Leuvense Samenwerkende Groep voor Nefrologen (LSGN).
K.J.C. and J.M. cosupervised this work.
A.W. and P.S. contributed equally to this work.
See related editorial, “Hemolytic Uremic Syndrome: A Question of Terminology,” on pages 831–833.
Contributor Information
Collaborators: Selda Aydin, Charles Cuvelier, Pierre-Yves Decleire, Nathalie Demoulin, Arnaud Devresse, Ludovic Gérard, Gaëlle Gillerot, Valentine Gillion, Eric Goffin, Philippe Hantson, Michel Jadoul, Jean Jamez, Nada Kanaan, Laura Labriola, Jean-Philippe Lengelé, Lionel Mazzoleni, Yves Pirson, Jean-Michel Pochet, Nadejda Ranguelov, Marie-Astrid van Dievoet, Elliott van Regemorter, Xavier Wittebole, all the collaborators of UCLouvain Kidney Disease Network, Bert Bammens, Greet de Vlieger, Koenraad Devriendt, Katrien de Vusser, Pieter Evenepoel, Laurent Godinas, Priyanka Koshy, Dirk Kuypers, Evelyne Lerut, Björn Meijers, Maartens Naesens, Patrick Schöffski, Ben Sprangers, Dirk Timmerman, Amaryllis van Craenenbroeck, Alexander Wilmer, and all the collaborators/nephrologists from the Leuvense Samenwerkende Groep voor Nefrologen (LSGN)
Disclosures
C. Beguin reports employment with Cliniques Universitaires Saint Luc—Université catholique de Louvain. J. Bernards reports employment with ZNA Middelheim, Antwerpen, Belgium. J.-F. Cambier reports consultancy for Vifor and honoraria from AstraZeneca and Boehringer Ingelheim. K.J. Claes reports consultancy for Alexion Pharmaceuticals, Astellas, Fresenius Kabi, GSK, and Sanofi; advisory or leadership roles for Alexion, Astellas, and Fresenius Kabi; and speaker's fee for AstraZeneca and Vifor Pharma. K. Dahan reports employment with Institut de Pathologie et de Génétique de Gosselies and Universite Catholique de Louvain; consultancy for AstraZeneca and CHIESI; and advisory or leadership roles as President and a member of advisory committees for three nonprofit patient organizations: AIRG, FAPA, and Retina, and an advisory or leadership role for the Board of Directors of IPG. D. Dierickx reports employment with University Hospitals Leuven, consultancy for Sanofi Genzyme, and honoraria from Sanofi Genzyme. C. Lambert reports employment with Cliniques universitaires Saint-Luc. E. Levtchenko reports consultancy for Chiesi, KKI, and Recordati and advisory or leadership roles for Chesi, Advicenne, KKI, and Recordati. T. Meyskens reports employment with AZ Klina/AZ Voorkempen. J. Morelle reports employment with Cliniques universitaires Saint-Luc, Brussels, Belgium, and UCLouvain, Brussels, Belgium; consultancy for Alexion Pharmaceuticals, AstraZeneca, Bayer, GlaxoSmithKline, and Sanofi-Genzyme; research funding from Alexion Pharmaceuticals, AstraZeneca, and Baxter Healthcare; advisory or leadership roles for Alexion Pharmaceuticals, AstraZeneca, Bayer, Sanofi-Genzyme, and Versantis; speaker honoraria for AstraZeneca, Baxter Healthcare, and Fresenius Medical Care; funding from the National Fund for Scientific Research (FRS-FNRS, Brussels, Belgium), the Association pour l’Information et la Recherche sur les Maladies Rénales Génétiques (AIRG, Brussels, Belgium), and the Saint-Luc Foundation (Brussels, Belgium); and travel grants from Sanofi-Genzyme and Vifor Pharma. X. Poiré reports employment with Cliniques Universitaires St-Luc and consultancy for MSD and Novartis. P. Storms's spouse reports employment (currently and during the past 24 months) with Puilaetco, a Quintet Private Bank (Europe) SA Branch. Y. Zizi reports employment with Cliniques universitaires Saint-Luc. All remaining authors have nothing to disclose.
Funding
The study was supported in part by grants from the Association pour l’Information et la Recherche sur les Maladies Rénales Génétiques (AIRG, Brussels, Belgium), AstraZeneca Rare Disease/Alexion, Fonds de la Recherche Scientifique (FRS-FNRS, 40000157 and 4003771), Fondation Saint-Luc, Fondation Louvain, and Fonds de Recherche Clinique des Cliniques universitaires Saint-Luc. The Divisions of Nephrology at Cliniques universitaires Saint-Luc and UZ Hospitals Leuven are members of the European Rare Kidney Disease Reference Network (project 739532).
Author Contributions
Conceptualization: Kathleen J. Claes, Catherine Lambert, Johann Morelle.
Data curation: Kathleen J. Claes, Johann Morell, Pauline Storms, Alexis Werion.
Formal analysis: Kathleen J. Claes, Karin Dahan, Daan Dierickx, Nathalie Godefroid, Pascale Hilbert, Catherine Lambert, Elena Levtchenko, Thomas Meyskens, Johann Morelle, Xavier Poiré, Pauline Storms, Lambert van den Heuvel, Alexis Werion.
Funding acquisition: Johann Morelle.
Investigation: Claire Beguin, Jelle Bernards, François Cambier, Kathleen J. Claes, Karin Dahan, Daan Dierickx, Nathalie Godefroid, Pascale Hilbert, Catherine Lambert, Elena Levtchenko, Thomas Meyskens, Johann Morelle, Xavier Poiré, Pauline Storms, Lambert van den Heuvel, Alexis Werion, Ysaline Zizi.
Methodology: Kathleen J. Claes, Johann Morelle.
Project administration: Kathleen J. Claes, Johann Morelle.
Supervision: Kathleen J. Claes, Johann Morelle.
Validation: Kathleen J. Claes, Johann Morelle.
Visualization: Kathleen J. Claes, Johann Morelle, Pauline Storms, Alexis Werion.
Writing – original draft: Kathleen J. Claes, Johann Morelle, Pauline Storms, Alexis Werion.
Writing – review & editing: Claire Beguin, Jelle Bernards, Jean-François Cambier, Karin Dahan, Daan Dierickx, Nathalie Godefroid, Pascale Hilbert, Catherine Lambert, Elena Levtchenko, Thomas Meyskens, Xavier Poiré, Lambert van den Heuvel, Ysaline Zizi.
Data Sharing Statement
All data are included in the manuscript and/or supporting materials.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B763.
Supplemental Table 1. Comparison of cohort studies of secondary TMA.
Supplemental Table 2. Information about available and missing data (continuous variables).
Supplemental Table 3. Incidence and distribution of TMA etiologies at each study center.
Supplemental Table 4. Baseline characteristics of patients with the main etiologies of secondary TMA.
Supplemental Table 5. Additional information about solid-organ transplant recipients with TMA.
Supplemental Table 6. Comparison of patients with secondary TMA who underwent genetic testing versus those who did not.
Supplemental Table 7. Distribution of rare variants in complement genes and CFHR rearrangements in the cohort.
Supplemental Table 8. Characteristics of patients with secondary TMA associated with pathogenic or likely pathogenic variants in complement genes.
Supplemental Table 9. Rare complement gene variants identified in the cohort.
Supplemental Table 10. Detailed outcomes at 1 month and 1 year.
Supplemental Table 11. Cox regression analyses for kidney failure or death after exclusion of patients with HUS associated with hematopoietic stem-cell transplantation or cancer.
Supplemental Figure 1. Etiologies of endothelial damage in patients with HUS associated with solid-organ transplantation or hematopoietic stem-cell transplantation.
Supplemental Figure 2. Kaplan–Meier curves of kidney failure–free survival after exclusion of patients with HUS associated with hematopoietic stem-cell transplantation or cancer.
Supplemental Figure 3. Baseline characteristics and kidney failure–free survival according to the etiology of TMA in adult patients.
Supplemental Figure 4. Proposal of a novel pathophysiology-based classification of TMA.
References
- 1.Fakhouri F, Frémeaux-Bacchi V. Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics. Nat Rev Nephrol. 2021;17(8):543–553. doi: 10.1038/s41581-021-00424-4 [DOI] [PubMed] [Google Scholar]
- 2.Goodship TH Cook HT Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2017;91(3):539–551. doi: 10.1016/j.kint.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 3.Claes KJ Massart A Collard L, et al. Belgian consensus statement on the diagnosis and management of patients with atypical hemolytic uremic syndrome. Acta Clin Belg. 2018;73(1):80–89. doi: 10.1080/17843286.2017.1345185 [DOI] [PubMed] [Google Scholar]
- 4.Palomo M Blasco M Molina P, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14(12):1719–1732. doi: 10.2215/CJN.05830519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Legendre CM Licht C Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–2181. doi: 10.1056/nejmoa1208981 [DOI] [PubMed] [Google Scholar]
- 6.Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681–696. doi: 10.1016/s0140-6736(17)30062-4 [DOI] [PubMed] [Google Scholar]
- 7.Zuber J Frimat M Caillard S, et al. Use of highly individualized complement blockade has revolutionized clinical outcomes after kidney transplantation and renal epidemiology of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2019;30(12):2449–2463. doi: 10.1681/ASN.2019040331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rondeau E Scully M Ariceta G, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287–1296. doi: 10.1016/j.kint.2020.01.035 [DOI] [PubMed] [Google Scholar]
- 9.Barbour T Scully M Ariceta G, et al. Long-term efficacy and safety of the long-acting complement C5 inhibitor ravulizumab for the treatment of atypical hemolytic uremic syndrome in adults. Kidney Int Rep. 2021;6(6):1603–1613. doi: 10.1016/j.ekir.2021.03.884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graça NAG Joly BS Voorberg J, et al. TTP: from empiricism for an enigmatic disease to targeted molecular therapies. Br J Haematol. 2022;197(2):156–170. doi: 10.1111/bjh.18040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scully M Cataland SR Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;380(4):335–346. doi: 10.1056/nejmoa1806311 [DOI] [PubMed] [Google Scholar]
- 12.Le Clech A Simon-Tillaux N Provôt F, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019;95(6):1443–1452. doi: 10.1016/j.kint.2019.01.023 [DOI] [PubMed] [Google Scholar]
- 13.Bayer G von Tokarski F Thoreau B, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14(4):557–566. doi: 10.2215/CJN.11470918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schönermarck U Ries W Schröppel B, et al. Relative incidence of thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome in clinically suspected cases of thrombotic microangiopathy. Clin Kidney J. 2019;13(2):208–216. doi: 10.1093/ckj/sfz066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henry N Mellaza C Fage N, et al. Retrospective and systematic analysis of causes and outcomes of thrombotic microangiopathies in routine clinical practice: an 11-year study. Front Med (Lausanne). 2021;8:566678. doi: 10.3389/fmed.2021.566678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timmermans SAMEG Abdul-Hamid MA Potjewijd J, et al.; Limburg Renal Registry. C5b9 formation on endothelial cells reflects complement defects among patients with renal thrombotic microangiopathy and severe hypertension. J Am Soc Nephrol. 2018;29(8):2234–2243. doi: 10.1681/ASN.2018020184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timmermans SAMEG, Werion A, Morelle J, van Paassen P. Defects in complement and “secondary” hemolytic uremic syndrome. Kidney Int. 2019;96(2):517. doi: 10.1016/j.kint.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 18.Timmermans SAMEG, Wérion A, Damoiseaux JGMC, Morelle J, Reutelingsperger CP, van Paassen P. Diagnostic and risk factors for complement defects in hypertensive emergency and thrombotic microangiopathy. Hypertension. 2020;75(2):422–430. doi: 10.1161/hypertensionaha.119.13714 [DOI] [PubMed] [Google Scholar]
- 19.Timmermans SAMEG Werion A Spaanderman MEA, et al. The natural course of pregnancies in women with primary atypical haemolytic uraemic syndrome and asymptomatic relatives. Br J Haematol. 2020;190(3):442–449. doi: 10.1111/bjh.16626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81(9):819–825. doi: 10.1038/ki.2011.339 [DOI] [PubMed] [Google Scholar]
- 21.Richards S Aziz N Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Quintrec M Lionet A Kamar N, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. 2008;8(8):1694–1701. doi: 10.1111/j.1600-6143.2008.02297.x [DOI] [PubMed] [Google Scholar]
- 23.Bruel A Kavanagh D Noris M, et al. Hemolytic uremic syndrome in pregnancy and postpartum. Clin J Am Soc Nephrol. 2017;12(8):1237–1247. doi: 10.2215/CJN.00280117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fakhouri F Scully M Provôt F, et al. Management of thrombotic microangiopathy in pregnancy and postpartum: report from an international working group. Blood. 2020;136(19):2103–2117. doi: 10.1182/blood.2020005221 [DOI] [PubMed] [Google Scholar]
- 25.Galbusera M Noris M Gastoldi S, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis. 2019;74(1):56–72. doi: 10.1053/j.ajkd.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 26.Timmermans SAMEG, Damoiseaux JGMC, Werion A, Reutelingsperger CP, Morelle J, van Paassen P. Functional and genetic landscape of complement dysregulation along the spectrum of thrombotic microangiopathy and its potential implications on clinical outcomes. Kidney Int Rep. 2021;6(4):1099–1109. doi: 10.1016/j.ekir.2021.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devresse A Aydin S Le Quintrec M, et al. Complement activation and effect of eculizumab in scleroderma renal crisis. Medicine (Baltimore). 2016;95(30):e4459. doi: 10.1097/md.0000000000004459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavero T Rabasco C López A, et al. Eculizumab in secondary atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. 2017;32(3):466–474. doi: 10.1093/ndt/gfw453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zafrani L Mariotte E Darmon M, et al. Acute renal failure is prevalent in patients with thrombotic thrombocytopenic purpura associated with low plasma ADAMTS13 activity. J Thromb Haemost. 2015;13(3):380–389. doi: 10.1111/jth.12826 [DOI] [PubMed] [Google Scholar]
- 30.Rodríguez de Córdoba S. Genetic variability shapes the alternative pathway complement activity and predisposition to complement-related diseases. Immunol Rev. 2023;313(1):71–90. doi: 10.1111/imr.13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the manuscript and/or supporting materials.