

Key Points
-
•
Lufaxin inhibits complement in assays of aHUS and PNH as well as thrombin generation in plasma.
-
•
Lufaxin binds the C3 proconvertase of the alternative pathway and fXa at different sites, allowing simultaneous inhibition.
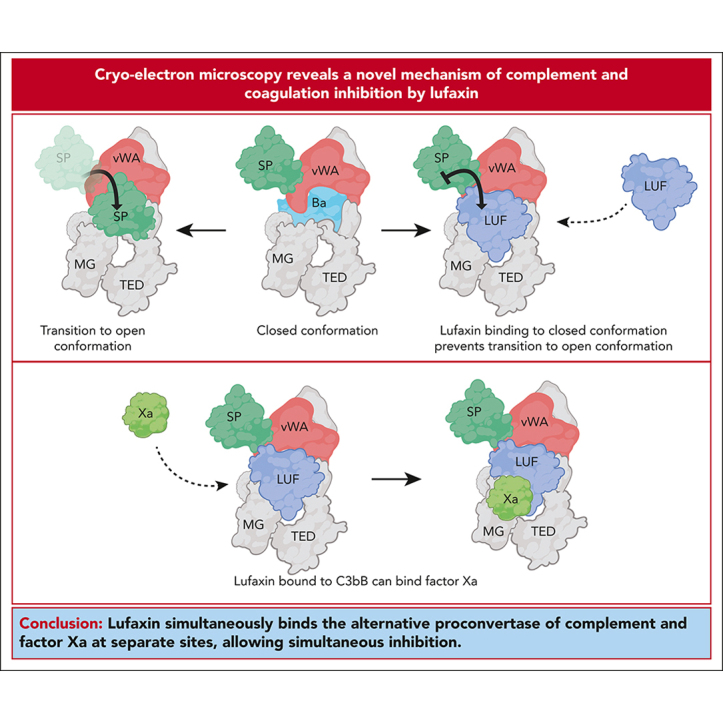
Visual Abstract
Abstract
Inhibitors of complement and coagulation are present in the saliva of a variety of blood-feeding arthropods that transmit parasitic and viral pathogens. Here, we describe the structure and mechanism of action of the sand fly salivary protein lufaxin, which inhibits the formation of the central alternative C3 convertase (C3bBb) and inhibits coagulation factor Xa (fXa). Surface plasmon resonance experiments show that lufaxin stabilizes the binding of serine protease factor B (FB) to C3b but does not detectably bind either C3b or FB alone. The crystal structure of the inhibitor reveals a novel all β-sheet fold containing 2 domains. A structure of the lufaxin-C3bB complex obtained via cryo–electron microscopy (EM) shows that lufaxin binds via its N-terminal domain at an interface containing elements of both C3b and FB. By occupying this spot, the inhibitor locks FB into a closed conformation in which proteolytic activation of FB by FD cannot occur. C3bB-bound lufaxin binds fXa at a separate site in its C-terminal domain. In the cryo-EM structure of a C3bB-lufaxin-fXa complex, the inhibitor binds to both targets simultaneously, and lufaxin inhibits fXa through substrate-like binding of a C-terminal peptide at the active site as well as other interactions in this region. Lufaxin inhibits complement activation in ex vivo models of atypical hemolytic uremic syndrome (aHUS) and paroxysmal nocturnal hemoglobinuria (PNH) as well as thrombin generation in plasma, providing a rationale for the development of a bispecific inhibitor to treat complement-related diseases in which thrombosis is a prominent manifestation.
The saliva of blood-feeding arthropods contains inhibitors of both complement and coagulation. Andersen and colleagues present the crystal structure of lufaxin, a sand fly salivary protein, and demonstrate that it binds to both C3b and factor Xa, inhibiting the activity of both. Lufaxin inhibits complement activation in models of atypical hemolytic uremic syndrome and paroxysmal nocturnal hemoglobinuria and also blocks thrombin generation, suggesting that it might be a model for an agent to treat complement-mediated diseases associated with thrombosis.
Introduction
Complement is an arm of the immune system that responds to microbial infection by opsonizing microbes and tagging them for phagocytosis as well as lysing them through the action of the membrane attack complex (MAC).1,2 Disorders including paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and antiphospholipid syndrome are linked to an abnormal activation of complement because of defects in complement regulatory systems.3,4 The classical, lectin, and alternative pathways (APs) of the complement converge at the conversion of complement factor C3 to the scaffold protein C3b. Further C3b generation is amplified by a proteolytic complex known as the alternative C3 convertase, C3bBb. The convertase is comprised of C3b and the activated serine protease factor Bb (FBb), which is formed by cleavage of the proenzyme FB into its Ba and Bb fragments by the protease factor D (FD). The complement response is amplified as more C3b is produced, leading to the accumulation of downstream pathway components and the production of the MAC. Importantly, the proinflammatory, chemoattractant anaphylatoxins C3a and C5a are produced as biproducts of proteolytic reactions involving C3bBb and the C5 convertase C3bBbC3b. The activity of convertase complexes is regulated by complement control proteins, such as decay accelerating factor and factors H and I, which disrupt the complex on the membrane surface. The plasma protein properdin is a positive regulator that binds C3bB and C3bBb and acts to stabilize them.5, 6, 7
It is now appreciated that the interplay between complement and coagulation is involved in the pathology of complement-related diseases.8,9 Proteases of the 2 pathways can activate each other, and dysregulation of 1 can result in the abnormal activation of the other. Complementopathies are often characterized by tissue damage due to MAC generation, with accompanying microvascular thrombosis.9 Like the complement, the coagulation pathway is a proteolytic cascade, with surface-assembled protein complexes playing a central role. One of these is the prothrombinase complex, consisting of coagulation factors Xa (fXa) and Va (fVa), which assembles on the anionic phospholipid surfaces of activated platelets in the presence of calcium.10, 11, 12 The complex catalyzes the cleavage of prothrombin into thrombin, leading to the formation of a fibrin clot.
Lufaxin, a naturally occurring protein found in the saliva of the blood-feeding sand fly, inhibits both the complement and coagulation cascades.13,14 The protein was first described as an inhibitor of fXa that binds the enzyme and blocks the hydrolysis of small molecule substrates and prothrombin (by the prothrombinase complex).13 Later, it was also shown to inhibit activation of the AP of the complement by binding to C3bB and preventing its conversion to C3bBb.14 Specifically, lufaxin stabilizes the binding of FB to C3b while preventing the cleavage of FB by FD. Here, we elucidate the mechanism of action of lufaxin as both an anticoagulant and complement inhibitor. We use structural analyses to show the binding mode and inhibitory mechanism of this structurally novel inhibitor. We also show that lufaxin binds to fXa at a separate site, allowing it to block coagulation and complement independently and simultaneously, thus making it a potential model for bispecific inhibition of complement-related diseases.
Methods
For additional detailed methods, see the supplemental Data, available on the Blood website.
Proteins: sources, production, and purification
The lufaxin complementary DNA containing a 6-His tag was expressed in HEK-293 cells and purified as described previously.13 Human C3b and FB were obtained from CompTech Complement Technologies. Human fXa was obtained from Enzyme Research Laboratories. The anti-C5 monoclonal antibody (mAb) is the antibody incorporated in pharmaceutical grade eculizumab and was provided by Alexion Pharmaceuticals. For examples, see the article by Yuan et al.15
C3bB assembly in the presence and absence of lufaxin
Surface plasmon resonance measurements were performed as described previously.14 Samples were injected in 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid at pH 7.4 and 0.15 M NaCl (HEPES buffered saline [HBS]) containing either 5 mM MgCl2 or no divalent cation.
Assays of complement activation and thrombin generation
The modified Ham (mHam) assay, conventional Ham assay, thrombinoscopy, and thromboelastography were conducted as previously described.16, 17, 18, 19, 20, 21 All patients gave written informed consent. This study was approved by the institutional review board and conducted in accordance with the Declaration of Helsinki.
Crystallization and structure solution of lufaxin
Lufaxin was crystallized using the hanging drop vapor diffusion method from 100 mM Tris HCl, pH 7.0, 10% polyethylene glycol 6000. Crystals were soaked in 0.5 M potassium bromide. Diffraction data were collected at beamline 22-ID at the Advanced Photon Source. Diffraction data were processed using XDS22 Phenix Autosol and Phenix Autobuild. The structure was rebuilt and refined using Coot23 and Phenix Refine24 (Table 1).
Table 1.
Diffraction data collection and phasing and refinement statistics for lufaxin
| Crystal parameters | Lufaxin-Br | Lufaxin |
|---|---|---|
| Accession numbers | PDB:8EO2 | |
| Resolution (Å) | 36-3.48 | 42-2.31 |
| Beamline | 22-ID | 22-ID |
| Wavelength (Å) | 0.9184 | 1.0000 |
| Completeness (total/high resolution shell) | 99.5/100.0 | 100.0/100.0 |
| Average redundancy (total/high resolution shell) | 3.60/3.70 | 8.7/8.0 |
| Rmerge (total/high resolution shell, %) | 3.9/5.7 | 5.7/55.3 |
| CC1/2 | 0.99/0.99 | 0.99/0.94 |
| I/sigI (total/high resolution shell) | 19.8/4.00 | 21.5/4.12 |
| Observed reflections | 68 510 | 302 907 |
| Unique reflections | 18 896 | 34 638 |
| Space group | P212121 | P212121 |
| Unit cell dimensions (Å) | ||
| a | 64.55 | 64.48 |
| b | 71.34 | 71.31 |
| c | 167.37 | 167.32 |
| α, β, γ (°) | 90 | 90 |
| No. of Br sites | 14 | |
| FOM (phenix.autosol) | 0.29 | |
| Refinement | Lufaxin model | |
| Total non H protein atoms | 4493 | |
| Total non H solvent atoms | 143 | |
| RMS deviations | ||
| Bond lengths (Å) | 0.008 | |
| Bond angles (°) | 0.90 | |
| Mean B factors (Å2) | ||
| Protein | 60.1 | |
| Solvent | 52.6 | |
| Molprobity analysis | ||
| Ramachandran plot (favored/allowed, %) | 93.9/100 | |
| Clashscore | 4.00 | |
| Rotamer outliers (%) | 0.80 | |
| Rcryst/Rfree | 0.20/0.25 |
Br, bromide ion; CC1/2, correlation coefficient 1/2; FOM, figure of merit; RMS, root mean squared.
Cryo-EM sample preparation and data collection
The C3bB-lufaxin and C3bB-lufaxin-fXa complexes were prepared by mixing 0.57 nmol of C3b, 0.57 nmol of FB, and 1.1 nmol of lufaxin in 1.0 mL of HBS buffer containing 5 mM MgCl2, followed by concentrating the mixture. These samples were applied to freshly glow-discharged C-flat grids (Protochips, CF1.2/1.3-3Au). Grids were vitrified by plunging into liquid ethane and stored in liquid nitrogen before examination via cryo-EM. Images were recorded on a Glacios TEM at 200 kV equipped with a K3 direct electron detector in superresolution mode.
Cryo-EM image processing
Movies of the C3bB-lufaxin and C3bB-lufaxin-fXa complexes were processed with MotionCor2,25 and the contrast transfer function was estimated in Ctffind4.26 Particle picking was conducted in Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/) using references generated with EMAN2.27 The picked particles were subjected to 2D classification using RELION-3.0.8 (Table 2; supplemental Figure 1).28 A previously calculated map was used as an initial reference for 3D classification. The best class was selected for subsequent refinement (Fourier shell correlation = 0.143) in both RELION-3.0.8 and cisTEM29 for C3bB-lufaxin and RELION 3.2 for C3bB-lufaxin-fXa (Table 2; supplemental Figure 1).
Table 2.
Cryo-EM data collection and processing and refinement statistics for C3bB-lufaxin (Mg2+) and C3bB-lufaxin-fXa (Mg2+)
| Parameters | C3bB-lufaxin | C3bB-lufaxin-fXa |
|---|---|---|
| Accession numbers | PDB:8ENU, EMD: 28279 | PDB: 8EOK, EMD: 28378 |
| Data collection and processing | ||
| Magnification | 36 000× | 45 000× |
| Voltage (kV) | 200 | 200 |
| Total electron exposure/used (e/Å2) | 58.31 | 67.59 |
| Defocus range (μm) | −0.3 to −2.2 | −0.3 to −2.2 |
| Pixel size (Å2) | 0.56 | 0.46 |
| Processing program | Gautomatch, Relion 3.0.8 cisTEM, and EMAN2 | Gautomatch, Relion 3.2, and EMAN2 |
| Final particles used (number) | 80 727 | 248 281 |
| Symmetry imposed | C1 | C1 |
| Resolution (Å) (FSC threshold) | 3.22 | 3.53 |
| Refinement | ||
| Refinement program | Phenix | Phenix |
| Model composition | ||
| Nonhydrogen atoms | 20 118 | 22 275 |
| Protein residues | 2511 | 2798 |
| RMS deviations | ||
| Bond lengths (Å) | 0.003 | 0.002 |
| Bond angles (°) | 0.57 | 0.59 |
| Validation | ||
| MolProbity score | 1.74 | 1.78 |
| Clash score | 9.15 | 7.18 |
| Ramachandran plot (favored/allowed) | 96.2/100 | 94.7/100 |
| Mask CC | 0.76 | 0.81 |
CC, correlation coefficient; FSC, Fourier shell correlation; RMS, root mean squared.
Model building and refinement of C3bB-lufaxin and C3bB-lufaxin-fXa complexes
Models used to build the structures include the C3bB complex with nickel ion (Protein Data Bank accession code 2XWJ30), the C3bB-FD complex with magnesium ion (2XWB30), wild-type FB (2OK531), and the structure of lufaxin. fXa was added to the model using coordinates from 1HCG32 and 1FAX.33 Positioning and real space refinement were performed in Phenix24 combined with manual rebuilding using Coot.23 Model quality was evaluated using MolProbity.34 Structural figures were produced with the PyMOL Molecular Graphics System, version 2.0 Schrodinger, Inc. or UCSF Chimera X (https://www.rbvi.ucsf.edu/chimerax/).35
Results
Efficacy in ex vivo models of aHUS and PNH: inhibition of the AP
Lufaxin has previously been shown to inhibit the formation of the AP C3 convertase (C3bBb) by binding and stabilizing the proconvertase (C3bB; supplemental Figure 2) in a form that is poorly cleaved by FD.14 The protein was also shown to inhibit the lysis of rabbit erythrocytes by diluted human serum with an IC50 of ∼60 nM.14 We evaluated the efficacy of lufaxin in preventing complement activation in ex vivo models of aHUS and PNH using the mHam and conventional Ham tests and compared its efficacy with that of the mAb component of eculizumab. In the mHam test, serum from patients with aHUS was used to activate the AP on a PIGA-mutant TF-1 cell line deficient in the GPI-anchored regulators CD55 and CD59. In addition to healthy human serum, Shiga toxin (Stx) and sialidase (Sia) are also potent activators of the complement in the mHam test and serve as positive controls (Figure 1A). Responses to Stx and Sia are inhibited by anti-C5 mAb at 0.34 μm and lufaxin at 2 μM (Figure 1A). Sia-induced activation is AP-specific (because it removes the receptor for factor H) and is inhibited by both mAb and lufaxin to a greater degree than Stx-induced activation, which involves contributions from the lectin pathway (Figure 1A). In the mHam test, lufaxin also significantly reduced the percentage of cell death in response to aHUS serum in a concentration-dependent manner, from 29.5% in the absence of lufaxin to 17.3% at 2 μM of inhibitor, making it approximately as effective as mAb (15.2% cell death) at a concentration of 0.34 μm (Figure 1B). Both inhibitors reduced cell death below the 20% level accepted as the threshold for a positive test.20 In the conventional Ham test, lufaxin inhibited the lysis of erythrocytes from patients with PNH (relative to that in a healthy control) in an acidified healthy human serum (Figure 1C). Again, at 2 μM lufaxin, the degree of lysis (5.8%) was nearly identical to that observed with mAb at 0.34 μM (6.6%), and both inhibitors significantly reduced erythrocyte lysis from the level observed with serum alone (22.4%; Figure 1C).
Figure 1.

Inhibition of the AP by lufaxin in complementopathy models. (A) Inhibition of complement activation and cell killing of a PIGA-mutant TF-1 cell line by lufaxin (Luf) and anti-C5 mAb in the mHam test. Activation in healthy human serum (NHS) was potentiated by Shiga toxin (Stx; 10 μg/mL) or Sia (50 U/mL). Anti-C5 mAb (0.34 μM) and lufaxin (Luf; 2 μM) strongly inhibited the AP-specific Sia-induced activation (P < .01 [in comparison with Sia alone]) and inhibited Stx-induced activation less strongly. (B) In the mHam test, mAb and Luf significantly (P < .01 and .05, respectively) at concentrations of 2 and 0.34 μM, respectively, inhibited activation by serum from a patient with aHUS relative to aHUS serum without an inhibitor. (C) Lysis of erythrocytes from a healthy individual (red bars) and a patient with PNH (blue bars) in acidified NHS and inhibition by mAb and Luf. The sizes of the erythrocyte clones are type I, 65.1% and type III, 33.2%. At a concentration of 2 μM, Luf significantly inhibited lysis (P < .05).
Crystal structure of lufaxin
The lufaxin family of proteins is present in the saliva of new (Lutzomyia)– and old (Phlebotomus)–world sand flies but shows no significant sequence homology to any protein family outside of the sand flies.13 We crystallized recombinant lufaxin and determined its structure using single-wavelength anomalous diffraction methods after soaking crystals in a precipitant solution containing potassium bromide (Table 1). The protein consists of 2 β-sandwich domains oriented at approximate right angles to one another (Figure 2A). The N-terminal domain (114 residues) is made up of 2 4-stranded β-sheets and is stabilized by 3 disulfide bonds linking Cys 29 and 37, Cys 55 and 114, and Cys 79 and 89 (Figure 2A). The C-terminal domain (164 residues) contains 6-stranded and 3-stranded β-sheets, with 1 disulfide bond linking Cys 235 with Cys 242. A C-terminal coil region is present, which is disordered after Asp 274. The 2 domains contact one another at an interface running along the lengths of βA and βH, including connecting loops from the N-terminal domain as well as termini and connecting loops of βA∗-E∗ of the C-terminal domain (Figure 2A). Glycan modifications are present at Asn 40 of the N-terminal domain and Asn 239 of the C-terminal domain (Figure 2A). Structural searches using DALI36 did not detect any close structural homologs to lufaxin.
Figure 2.

The crystal structure of lufaxin. (A) The N-terminal domain is colored light blue and C-terminal domain, copper. In the left-hand structure, β-strands of the N-terminal domain are labeled A-G, and β-strands of the C-terminal domain are labeled A∗-H∗. Cysteine residues are shown as sticks, with sulfur atoms colored yellow. Disulfide bonds (DS1-DS4) are labeled (left). Visible glycan chains are colored red, and their positions of attachment (Asn 40 and Asn 239) are labeled (right). (B) Superposition of the lufaxin crystal structure with the structure predicted by Alphafold2 (magenta). The overall structures are very similar (root mean squared deviation = 0.69 Å for 227 Cα atoms), but they differ in the backbone conformation of loop B-C, which interacts with the C3bB complex (see "Cryo-EM studies of the C3bB-lufaxin complex").
Cryo-EM studies of the C3bB-lufaxin complex
Conformational changes of C3b-bound FB are known to regulate its cleavage by FD, a serine protease, to produce the Ba and Bb fragments.30,37,38 Two conformers of C3b-bound FB have been observed, and their ratio is affected by the type of divalent cation bound at the metal ion–dependent adhesion site (MIDAS) of the von Willebrand factor type A (VWA) domain of FB.38 The C-terminal (αʹ-chain) carboxylate of the C345C domain of C3b is also inserted into this site and chelates the metal ion.30,37 Divalent cation binding induces allosteric changes in the complex that enhance the binding of FB with C3b. In the presence of Mg2+, C3bB exhibits a high proportion of the closed conformation (35%) of bound FB, with its scissile bond being inaccessible to FD.38 The closed conformation is exemplified by the published crystal structure of the cobra venom factor-FB complex (CVFB), which is FD-activated more slowly than C3bB (Figure 3A).37 Replacement of Mg2+ by Ni2+ dramatically shifts the conformational equilibrium of FB in C3bB to an almost exclusively open conformation (98%), with the loop containing the scissile bond projecting away from the protein, exposing the P1 arginine residue and making the scissile bond accessible to FD (Figure 3B).30,38 In the open conformation of C3bB, the serine protease domain of FB is rotated by 84° from its position in CVFB, bringing it into contact with the complement C1r/C1s, Uegf, Bmp1 (CUB) and macroglobulin-2 domains of C3b (Figure 3B), whereas in the closed conformation, the SP domain extends away from the surface and does not contact C3b (Figure 3A).30 Binding of and cleavage by FD require formation of the open conformation, as was established in the crystal structure of C3bB bound with a catalytically inactive mutant of FD in the presence of Mg2+.30
Figure 3.

Structure of the C3bB-lufaxin complex. (A,B) Semitransparent surfaces covering ribbon diagrams of previously determined structures of CVFB (A) (Protein Database [PDB] accession number 3HRZ37) and C3bB in the presence of Ni2+ (B) (PDB accession number 2XWJ30). Regions of the C3bB surface and ribbon diagram are colored as follows: C3b and CVF, light gray; FB VWA domain, red; FB SP domain, green; FB Ba fragment, cyan. (C,D) Two views of the C3bB-lufaxin 3D reconstruction related by ∼180° rotation around the vertical axis and covering a ribbon diagram of the refined C3bB-lufaxin model. Coloring: C3b α-chain, purple; C3b β-chain, light gray; FB, red; lufaxin (Luf), light blue. Domain landmarks are labeled on the figures. (E) Three-dimensional reconstruction of C3bB-lufaxin colored by local resolution (as calculated using Phenix) and oriented as in panel C. Resolution color scale is shown in the bar below panel. (F) Model of C3bB-lufaxin in surface representation and oriented as CVFB in panel A and C3bB in panel B. Comparison of the 3 shows C3bB-lufaxin having the closed conformation of FB and lufaxin occupying the position of the SP domain in the C3bB-Ni2+ structure. (G) Schematic representation of conformational changes occurring in C3bB during the activation of FB and the inhibition of these changes by bound lufaxin. Panel created with Biorender.com.
We used cryo-EM to analyze a C3bB-lufaxin complex formed in the presence of Mg2+ and refined its structure to 3.2 Å resolution (Table 2; supplemental Figure 1). Overall, the C3bB part of the complex is like that of the published crystal structure of CVFB but with C3b in place of CVF (Figure 3C-E).37 The macroglobulin (MG1-6) ring, C345C, and CUB domains of C3b are well ordered. The thioester-containing domain of C3b is visible but poorly ordered because of its high mobility, as is the complement control protein-1 (CCP1) domain of FB (Figure 3C-F). Any disorder around the MIDAS in the VWA domain of FB makes it difficult to assess its Mg2+ occupancy. Weak density corresponding in position to the SP domain of FB in the CVFB crystal structure is also present (Figure 3D). Helix α7 of the VWA domain leading to the VWA-SP linker is well ordered and contains 4 full helical turns, as that in CVFB, rather than the 3 turns in the open conformation of C3bB or in the C3bB-FD complex (supplemental Figure 3A).30,37 Helix αL of the VWA domain is shortened by 2 turns relative to the open conformation of C3bB and is like that of the CVFB complex with FB in the closed conformation (supplemental Figure 3A).37 The scissile loop itself is disordered between Thr 216 and Lys 233, but its visible part is oriented similarly to that of CVFB, with the side chain of the P1 residue (Arg 234) contacting the surface of the VWA domain where it is inaccessible to FD (supplemental Figure 3B). Together, the orientation of the scissile bond loop, the position of the visible density for the SP domain, and the structures of α7 and αL clearly show that the C3bB-lufaxin complex contains FB in the activation-resistant, closed conformation (Figure 3A-B,F; supplemental Figure 3).
Lufaxin was fit into the unoccupied density projected from the region of the map where the CUB domain of C3b, the FBa region, and the VWA domain intersect. (Figure 3C-D). The lufaxin-binding site overlaps considerably with that of the SP domain in the open conformation of the proconvertase,30 suggesting that the presence of the inhibitor blocks the conformational change of FB that is necessary for its activation (compare Figure 3A-B,F with Figure 4). The face of lufaxin contacting C3bB is exclusively contained in its smaller N-terminal domain, whereas the C-terminal domain of the inhibitor extends away from the C3bB surface between the thioester-containing domain and SP domains and does not contact the complex (Figures 2 and 3C-D).
Figure 4.

The C3bB-lufaxin–binding interface. (A) Surface representation of C3bB-lufaxin model colored with C3bB in light gray and lufaxin in light blue. Binding interface residues predicted by PISA are colored (both lufaxin and C3bB) in cyan for the Ba fragment of FB (CCP3 domain), red for the VWA domain of FB, and magenta for the CUB domain of C3b. Opening of the interface by opposite rotations of lufaxin and C3bB around the vertical axis with interaction regions colored as in the left panel. (B) Space filling representation showing details of the lufaxin surface with interface residues colored (as in panel A) (right) by C3bB interaction region. Residues forming hydrogen bond, salt bridge, or cation π interactions are labeled. (C) Details of lufaxin interactions with the Ba fragment and the VWA domains of FB. Boxed area of the ribbon structure is magnified, and interface residues are shown along with the ribbon diagram. Coloring: Ba fragment, cyan; VWA domain, red; and lufaxin, light blue. Backbone and side chain atoms are shown as sticks, with oxygen colored in red and nitrogen in blue. Hydrogen bonds and salt bridges are shown as red dashed lines. (D) Details of lufaxin interactions with the CUB domain of C3b in the C3bB-lufaxin complex. The magnified region shows lufaxin (light blue) and CUB (magenta) interactions highlighted as in panel C.
The N-terminal domain of lufaxin interacts with the C3bB surface at 3 points contained in C3b and FB identified using the program PISA,39 suggesting a bridging mechanism for binding that explains its stabilization of the proconvertase complex observed in surface plasmon resonance (SPR) and chromatographic experiments. The inhibitor is positioned where the main chains of the CCP3 domain of the Ba fragment, the VWA domain of FB, and the CUB domain of C3b lie within 9 Å of one another. At the first interaction point, loop B-C in the N-terminal domain of lufaxin (Gly 23-Ile 31) binds the CCP3 domain of FB (Figure 4A-C; supplemental Figures 4 and 5). This interface includes the loops of CCP3 containing Gln 181-Ser 185 and Gly 136-Ser 141, which are traversed by Lys 25-Ile 31 of the lufaxin loop B-C (Figure 4C). This region contains hydrogen bonds between Arg 26 of lufaxin and the carbonyl oxygen atoms of Gly 138 and Tyr 139 of FB as well as between the amide nitrogen of Lys 25 of lufaxin and the carbonyl oxygen of Gly 183 of FB (Figure 4C; supplemental Figure 5). At the second contact point, loop G-H at one end of the N-terminal domain of lufaxin contacts the VWA domain at its end opposite the MIDAS (Figure 4A-C; supplemental Figures 4 and 6). Hydrogen bonds are formed between the Arg 98 side chain of lufaxin and the hydroxyl group of Tyr 397 of FB (Figure 4C) and between the side chain of Asp 27 in loop BC of lufaxin and Asn 423 of FB. Sequence comparisons of FB from different vertebrate species show that these interface sequences are highly conserved, suggesting that lufaxin would interact with C3b-bound FB in mammals and other vertebrate classes (supplemental Figure 6). At the third interaction point, β-strands D and E of the CUB domain of C3b run parallel to β-strands B and H of the N-terminal domain of lufaxin (Figure 4A-B,D). Phe 19 of lufaxin is positioned to form a possible cation-π interaction (4.2 Å) with Arg 1288 from the CUB domain, and Glu 105 of lufaxin also forms a salt bridge with Arg 1288 of the CUB domain (Figure 4D). A second hydrogen bond is formed between the hydroxyl group of Tyr 64 from lufaxin and the amino group of Lys 1284 in the CUB domain. Sequence comparisons of C3 from mammals, reptiles, and amphibians show incomplete conservation of CUB domain–binding interface residues (supplemental Figure 7). Nevertheless, Arg 1288 is conserved across the group, including in CVF from Naja naja kaouthia (supplemental Figure 7). We performed SPR and size exclusion chromatography experiments, showing that lufaxin stabilizes binding of FB with CVF in the presence or absence of divalent cations as it does with C3b, further suggesting that interactions involving Arg 1288 (or equivalents) may play a key role in the binding of lufaxin (supplemental Figure 8).
Interaction of lufaxin with fXa
In addition to inhibiting complement, recombinant lufaxin is a potent inhibitor of coagulation in human plasma. Using the thrombinoscope, thrombin generation was found to be reduced 15-fold at a concentration of 2.5 nM, and little or no thrombin was generated at a lufaxin concentration of 5 nM (Figure 5A). Likewise, the thromboelastogram exhibits large concentration-dependent increases in the R and K values, which become difficult to measure at lufaxin concentrations above 1.5 nM (Figure 5B). In addition, size exclusion chromatography of C3bB-lufaxin-fXa mixtures shows fXa binding to lufaxin in the presence of C3bB. All components are included in a single complex, as indicated by the coelution of the C3bB components, lufaxin, and the chains of fXa in the complex peak (Figure 5C-D, bottom gel). In the absence of lufaxin, fXa does not interact detectably with C3bB (Figure 5C,D, top gel). SPR results show that fXa causes only a small reduction in the rate of complex formation and essentially no change in the dissociation rate constant, suggesting that the complex assembles freely without significant steric hindrance (Figure 5E). In accordance with this, lufaxin (30 nM) strongly inhibits hydrolysis of the substrate S-2222 by fXa in the presence of Mg2+ and Ca2+ and an excess of C3bB (Figure 5F).
Figure 5.

Binding and inhibition of fXa by lufaxin. (A) Concentration dependence of lufaxin inhibition of thrombin generation in whole plasma using the thrombinoscope. Legend shows lufaxin concentrations. (B) Inhibition of clot formation by lufaxin as measured via thromboelastography. Legend shows lufaxin concentrations. (C) Size exclusion chromatography (A280) of C3bB-fXa mixtures in the presence (blue trace) and absence (red trace) of lufaxin (luf). The elution buffer was 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic, pH 7.4, and 0.15 M NaCl (HBS) containing 5 mM MgCl2; the identified peaks are labeled. (D) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of fractions from the chromatograms in panel C with silver staining. (Top) A sample separated in the absence of lufaxin and (bottom) a sample separated in the presence of lufaxin. The retention volume scale above the gels corresponds to the x-axis of the chromatograms. The bands are labeled on the left side of the gel, and the standard sizes are shown on the right side. (E) SPR analysis of complex formation on a C3b surface in the presence and absence of fXa. Injections of 57 or 114 nM FB along with 500 nM lufaxin were made in the presence or absence of 500 nM fXa. The rate constants kon and koff refer to the observed first-order rate constants for association and dissociation of the complex at 114 nM FB, 500 nM lufaxin in the absence of fXa (purple trace) and 114 nM FB, and 500 nM lufaxin in the presence of fXa (blue trace). The buffer for all experiments was HBS containing 5 mM Mg2+. (F) Progress curves for the hydrolysis of S2222 (250 μM) using 4.3 nM fXa in the presence of 176 nM each of C3b and FB either with (red trace) or without (purple trace) 30 nM lufaxin. The experiment was performed in HBS containing 5 mM MgCl2 at 30°C.
The structure of fXa bound to C3bB-lufaxin
The structure of lufaxin bears no resemblance to that of other fXa inhibitors, and its binding mechanism is unknown. With the goal of understanding this mechanism, we determined the binding mode of lufaxin with fXa by producing a C3bB-lufaxin-fXa complex in the presence of equimolar lufaxin and fXa and determined its structure using cryo-EM (Table 2). In the 3D reconstruction, C3bB-lufaxin is present in the arrangement described earlier but with additional density contiguous with the C-terminal domain of lufaxin (Figure 6A-B). This extra density was assigned as the serine protease domain of fXa, which is well defined, whereas the epidermal growth factor and Gla domains of fXa are less well ordered and project into the solution away from the C3bB complex (Figure 6A-B). In this orientation, fXa does not interfere with lufaxin binding to C3bB because the inhibitor interacts with C3bB via its N-terminal domain and with fXa via its C-terminal domain.
Figure 6.

Structure of the C3bB-lufaxin-fXa complex. (A) Cryo-EM reconstruction of the complex with C3b, FB, and lufaxin colored as in Figure 1C, with the SP domain of fXa in light green and the light chain of fXa in magenta. Domain features of the complex are labeled. (B) The map of panel A colored by local resolution (calculated using Phenix) per the color key shown below the map. (C) Ribbon diagram of the fXa-SP domain (light green) and lufaxin (light blue) portions of the complex highlighting key features of the interface, including the C-terminal coil of lufaxin, the autolysis loop of fXa and the 220 loop of fXa. (D) Surfaces of fXa-SP domain (light green) and lufaxin (light blue) rotated around the vertical axis to show the binding interface determined by PISA. Interface residues on lufaxin are colored light green, and those on fXa-SP are colored light blue. Arg 277 of lufaxin, which interacts with the S1 subsite of fXa, is colored magenta, with nitrogen atoms in blue. The catalytic serine residue (Ser 195) of fXa is colored red. (E) Details of interactions between the C-terminal domain of lufaxin (light blue) with the SP domain of fXa (light green), with the fXa portion also shown as a semitransparent surface. Individual side chains are shown as sticks, with oxygen colored red and nitrogen colored blue. Lufaxin side chains are labeled in light blue, and fXa side chains interacting with the lufaxin surface are labeled in light green. Residues of the fXa catalytic triad (His 57, Asp 102, and Ser 195) are colored in cyan and labeled in black. The C-terminal coil structure of lufaxin-containing Arg 277 is labeled. (F) Ribbon diagram of the lufaxin-fXa-SP magnified to show electrostatic interactions of Arg 277 of lufaxin (light blue, with oxygen colored red and nitrogen blue) with residues of the S1 subsite at the active site of fXa (light green). The catalytic serine (Ser 195) of fXa is colored cyan. Hydrogen bonds and salt bridges are shown as red dashed lines. (G) Lufaxin interactions with the autolysis loop and 220 loop of fXa-SP detailed as in panel F.
Substrate entry to the active site of fXa is prevented by lufaxin, which occludes the prothrombin-binding face of the protease (Figure 6A,C-G).12 Residues forming this interface were identified using PISA and are shown in the amino acid alignments of supplemental Figure 9. The C-terminal coil of lufaxin runs toward the active site of the protease, and the side chain of Arg 277 fills the S1 subsite at the active site of fXa, participating in several hydrogen bond and salt bridge interactions with Asp 189, Ala 190, and Gly 216 of fXa (Figure 6C; Figure E-F; supplemental Figure 5). The carbonyl group (or C-terminal carboxyl group) of Arg 277 is situated adjacent to the catalytic serine residue (Ser 195; Figure 6D-F) of fXa, and Gly 278 and the 6-histidine tag immediately after it is removed by cleavage, as verified by the absence of the His-tag epitope in western blots after incubation of lufaxin with excess fXa (supplemental Figure 10). Loop A∗-B∗ (Arg 155-Glu 159), residues along β-strand G∗ and residues in the H∗-I∗ loop of lufaxin bind the autolysis loop of fXa. Tyr 228 and His 230 in β-strand G∗ pack against the apex of the loop (Figure 6C; supplemental Figure 5),40 whereas loop A∗-B∗ of the inhibitor lies along 1 face of the autolysis loop with the side chain of Gln 156 of lufaxin contacting the side chain of Phe 156 from fXa. Arg 150 in the autolysis loop forms salt bridges with Asp 247 and Asp 249 in the H∗-I∗ loop of lufaxin (Figure 6C; supplemental Figure 5). Arg 222 of fXa, which lies in a loop (220-loop) forming 1 wall of the S1 subsite that has been identified as a sodium ion–binding site,41 forms a salt bridge with Glu 234 in the G∗-H∗ loop of the inhibitor. Structure prediction of the fXa-lufaxin complex was performed after the experimental solution was determined using Alphafold 2.3.1,42 which gives improved predictions of multimeric complexes. The structure is consistent with that determined using cryo-EM (supplemental Figure 11).
The target of lufaxin in vivo is fXa contained in the prothrombinase complex rather than free fXa. To examine the possibility of C3bB-bound lufaxin interacting with this target, we produced a model of C3bB-lufaxin in complex with prothrombinase by superposition of the fXa protease domains of C3bB-lufaxin-fXa and the recently determined structure (Protein Data Bank accession 7TPP) of prothrombin-prothrombinase,12 with the prothrombin coordinates removed (fVa-fXa; Figure 7). The model suggests that lufaxin can join the 2 complexes into a larger C3bB-lufaxin-prothrombinase complex without introducing significant molecular clashes, making it possible that the inhibitor binds C3bB and prothrombinase simultaneously (Figure 7). Membrane-binding sites in this modeled complex are not perfectly coplanar, making it uncertain whether C3bB and prothrombinase can interact on cell surfaces.
Figure 7.

Simultaneous binding of C3bB and prothrombinase by lufaxin. Model of a hypothetical complex containing C3bB, lufaxin, and the prothrombinase complex (fVa and fXa) created by superimposing the SP domains of fXa in C3bB-lufaxin-fXa and prothrombinase (PDB ID 7TPP12 with prothrombin coordinates removed). The left and right images are related by rotation around the vertical axis. C3bB is colored light gray and lufaxin light blue. fVa is colored yellow and fXa light green. Membrane contact points determined by Ruben et al12 in fVa and fXa are colored red and the thioester-forming residues (Cys 988, Gln 991) in the TED domain of C3b are colored green in the right hand image. TED, thioester-containing domain.
Discussion
Complementopathies such as aHUS and PNH are associated with the activation of the AP and the coagulation cascade.9,43, 44, 45, 46 C3bB is an attractive target for therapeutic inhibition because it is the precursor of C3bBb, the central convertase of the AP, and a point of amplification for the overall complement response. Blockade at this step prevents the accumulation of C3b and anaphylatoxin C3a as well as the production of downstream terminal pathway intermediates, including anaphylatoxin C5a and components of the MAC. Small molecule FD and FB inhibitors are now available that specifically block the conversion of C3bB to C3bBb and have shown promise in treating complement-related diseases.21,47,48 Here, we describe the structure and mechanism of lufaxin, a macromolecular inhibitor of C3bB activation that has the added feature of being a potent inhibitor of coagulation through the binding of fXa.
The complement inhibitory mechanism of lufaxin is novel. The inhibitor binds at a site that includes elements of C3b and FB and prevents activation of FB by blocking the conformational changes necessary for interaction with FD. The complex is stabilized even in the absence of normally required divalent cations through bridging interactions between lufaxin, C3b, and FB. Lufaxin itself shows little interaction with the individual components of complex C3b and FB. The binding of lufaxin14 in the absence of Mg2+ suggests the formation of a low-affinity encounter complex between C3b and FB in its closed conformation that allows less hindered access by lufaxin than in the presence of Mg2+. When bound to C3bB, lufaxin directs its C-terminal domain, using its fXa-binding site, away from the surface of the complex, where it is free to interact with fXa, allowing the inhibitor to bind to both targets simultaneously. The structure of lufaxin is not like that of other naturally occurring proteinaceous fXa inhibitors, including various serpins and the tick anticoagulant peptide. The tick anticoagulant peptide is a Kunitz-type inhibitor that interacts with fXa in a noncanonical manner via an N-terminal tyrosine residue whose side chain inserts into the S1 subsite of the protease,49 whereas the mechanism of lufaxin is centered on a C-terminal arginine residue that inserts at the active site of fXa.
The co-occurrence of complement and fibrin deposition in many complement-related disorders suggests that concomitant inhibition of complement and coagulation may be an effective avenue for treatment.43 The 2 pathways are activated reciprocally in that tissue damage caused by the MAC results in the generation of procoagulant surfaces and subsequently activated coagulation proteases are capable of activating complement in a noncanonical manner.50 In addition, MAC formation on platelet surfaces or microparticles and the activation of platelets by anaphylatoxin C3a lead to the exposure of anionic phospholipids and binding of coagulation complexes.51,52 Because of the direct interplay between the 2 pathways, complement deposition and thrombosis are colocalized. Our ex vivo assays clearly show that lufaxin limits complement activation in sera from patients with aHUS and red blood cells from patients with PNH to a similar degree as the anti-C5 mAb. Assays of thrombin generation and fXa activity in human plasma and reconstituted enzyme systems demonstrate that lufaxin also potently inhibits coagulation. With separate binding sites for C3bB and fXa allowing simultaneous binding of the 2 targets, lufaxin may serve as a model for the development of bispecific therapeutics. Successes with bispecific antibodies developed for tumor immunotherapy and the treatment of A-type hemophilia suggest that other systems involving multiple extracellular targets can be effectively treated in this manner.53,54
Conflict-of-interest disclosure: R.A.B. has served on an advisory board for Alexion Pharmaceutical Inc. G.F.G serves on a medical advisory board and has received an honorarium from Apellis Pharmaceuticals. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the staff of the Southeast Regional Collaborative Access Team at the Advanced Photon Source for assistance with diffraction data collection. Elizabeth Fischer and her team in the National Institute of Allergy and Infectious Diseases Research Technologies Branch assisted with cryo-EM data collection. This work required the use of the computational resources of the NIH High-Performance Computing Biowulf cluster (http://hpc.nih.gov).
This work was supported by the intramural research program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health; National Institutes of Health, National Heart, Lung, and Blood Institute, grant R56HL133113 (R.A.B.); Johns Hopkins Hospital grant 80053630 (I.M.B.F.); and an American Society of Hematology Research Training Award for Fellows (G.F.G).
Authorship
Contribution: J.F.A., I.M.B.F., and R.A.B. designed the study; J.F.A., H.L., E.C.S., I.M.B.F., T.K., G.F.G., X-Z.P., O.A.A., and P.H.A. performed experiments and analyzed data; V.P. provided technical assistance; J.F.A. drafted the manuscript; and J.G.V. and J.M.C.R. contributed to subsequent drafts.
Footnotes
Structural data reported in this article have been deposited in the Protein Data Bank (accession numbers 8EO2 [lufaxin crystal structure], 8ENU [C3bB-lufaxin cryo-EM structure], and 8EOK [C3bB-lufaxin-fXa cryo-EM structure]). Cryo-EM maps reported in this article have been deposited in the Electron Microscopy Database (accession numbers 28279 [C3bB-lufaxin] and 28378 [C3bB-lufaxin-fXa]).
Data are available on request from the corresponding author, John F. Andersen (john.andersen@nih.gov).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. 2017;18(12):1288–1298. doi: 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - the "Swiss army knife" of innate immunity and host defense. Immunol Rev. 2016;274(1):33–58. doi: 10.1111/imr.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niederreiter J, Eck C, Ries T, et al. Complement activation via the lectin and alternative pathway in patients with severe COVID-19. Front Immunol. 2022;13:835156. doi: 10.3389/fimmu.2022.835156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong EKS, Kavanagh D. Anticomplement C5 therapy with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Transl Res. 2015;165(2):306–320. doi: 10.1016/j.trsl.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol. 2010;28:131–155. doi: 10.1146/annurev-immunol-030409-101250. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen DV, Gadeberg TAF, Thomas C, et al. Structural basis for properdin oligomerization and convertase stimulation in the human complement system. Front Immunol. 2019;10:2007. doi: 10.3389/fimmu.2019.02007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van den Bos RM, Pearce NM, Granneman J, Brondijk THC, Gros P. Insights into enhanced complement activation by structures of properdin and its complex with the C-terminal domain of C3b. Front Immunol. 2019;10:2097. doi: 10.3389/fimmu.2019.02097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185(9):5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baines AC, Brodsky RA. Complementopathies. Blood Rev. 2017;31(4):213–223. doi: 10.1016/j.blre.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnaswamy S, Jones KC, Mann KG. Prothrombinase complex assembly. Kinetic mechanism of enzyme assembly on phospholipid vesicles. J Biol Chem. 1988;263(8):3823–3834. [PubMed] [Google Scholar]
- 11.Mann KG. Prothrombinase: the paradigm for membrane bound enzyme complexes; a memoir. J Thromb Thrombolysis. 2021;52(2):379–382. doi: 10.1007/s11239-021-02402-w. [DOI] [PubMed] [Google Scholar]
- 12.Ruben EA, Summers B, Rau MJ, Fitzpatrick JAJ, Di Cera E. Cryo-EM structure of the prothrombin-prothrombinase complex. Blood. 2022;139(24):3463–3473. doi: 10.1182/blood.2022015807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collin N, Assumpção TCF, Mizurini DM, et al. Lufaxin, a novel factor Xa inhibitor from the salivary gland of the sand fly Lutzomyia longipalpis blocks protease-activated receptor 2 activation and inhibits inflammation and thrombosis in vivo. Arterioscler Thromb Vasc Biol. 2012;32(9):2185–2198. doi: 10.1161/ATVBAHA.112.253906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendes-Sousa AF, do Vale VF, Silva NCS, et al. The sand fly salivary protein lufaxin inhibits the early steps of the alternative pathway of complement by direct binding to the proconvertase C3b-B. Front Immunol. 2017;8:1065. doi: 10.3389/fimmu.2017.01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan X, Yu J, Gerber G, et al. Ex vivo assays to detect complement activation in complementopathies. Clin Immunol. 2020;221:108616. doi: 10.1016/j.clim.2020.108616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francischetti IMB, Toomer K, Zhang Y, et al. Upregulation of pulmonary tissue factor, loss of thrombomodulin and immunothrombosis in SARS-CoV-2 infection. EClinicalMedicine. 2021;39:101069. doi: 10.1016/j.eclinm.2021.101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavriilaki E, Yuan X, Ye Z, et al. Modified Ham test for atypical hemolytic uremic syndrome. Blood. 2015;125(23):3637–3646. doi: 10.1182/blood-2015-02-629683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerber GF, Yuan X, Yu J, et al. COVID-19 vaccines induce severe hemolysis in paroxysmal nocturnal hemoglobinuria. Blood. 2021;137(26):3670–3673. doi: 10.1182/blood.2021011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haddaway K, Bloch EM, Tobian AAR, et al. Hemostatic properties of cold-stored whole blood leukoreduced using a platelet-sparing versus a non-platelet-sparing filter. Transfusion. 2019;59(5):1809–1817. doi: 10.1111/trf.15159. [DOI] [PubMed] [Google Scholar]
- 20.Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136(18):2080–2089. doi: 10.1182/blood.2020008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan X, Gavriilaki E, Thanassi JA, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017;102(3):466–475. doi: 10.3324/haematol.2016.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Mooney P, Zheng S, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10(6):584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192(2):216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang G, Peng L, Baldwin PR, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180(3):519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grigorieff N. Frealign: an exploratory tool for single-particle cryo-EM. Methods Enzymol. 2016;579:191–226. doi: 10.1016/bs.mie.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forneris F, Ricklin D, Wu J, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science. 2010;330(6012):1816–1820. doi: 10.1126/science.1195821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milder FJ, Gomes L, Schouten A, et al. Factor B structure provides insights into activation of the central protease of the complement system. Nat Struct Mol Biol. 2007;14(3):224–228. doi: 10.1038/nsmb1210. [DOI] [PubMed] [Google Scholar]
- 32.Padmanabhan K, Padmanabhan KP, Tulinsky A, et al. Structure of human des(1-45) factor Xa at 2.2 A resolution. J Mol Biol. 1993;232(3):947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 33.Brandstetter H, Kuhne A, Bode W, et al. X-ray structure of active site-inhibited clotting factor Xa. Implications for drug design and substrate recognition. J Biol Chem. 1996;271(47):29988–29992. doi: 10.1074/jbc.271.47.29988. [DOI] [PubMed] [Google Scholar]
- 34.Williams CJ, Headd JJ, Moriarty NW, et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27(1):293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm L. Using dali for protein structure comparison. Methods Mol Biol. 2020;2112:29–42. doi: 10.1007/978-1-0716-0270-6_3. [DOI] [PubMed] [Google Scholar]
- 37.Janssen BJC, Gomes L, Koning RI, et al. Insights into complement convertase formation based on the structure of the factor B-cobra venom factor complex. EMBO J. 2009;28(16):2469–2478. doi: 10.1038/emboj.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torreira E, Tortajada A, Montes T, Rodriguez de Cordoba S, Llorca O. Coexistence of closed and open conformations of complement factor B in the alternative pathway C3bB(Mg2+) proconvertase. J Immunol. 2009;183(11):7347–7351. doi: 10.4049/jimmunol.0902310. [DOI] [PubMed] [Google Scholar]
- 39.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Manithody C, Yang L, Rezaie AR. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry. 2002;41(21):6780–6788. doi: 10.1021/bi0255367. [DOI] [PubMed] [Google Scholar]
- 41.Yang L, Manithody C, Qureshi SH, Rezaie AR. Factor Va alters the conformation of the Na+-binding loop of factor Xa in the prothrombinase complex. Biochemistry. 2008;47(22):5976–5985. doi: 10.1021/bi800319r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2022;139(25):3571–3582. doi: 10.1182/blood.2021012860. [DOI] [PubMed] [Google Scholar]
- 44.Merrill SA, Brodsky RA. Complement-driven anemia: more than just paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2018;2018(1):371–376. doi: 10.1182/asheducation-2018.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Risitano AM, Frieri C, Urciuoli E, Marano L. The complement alternative pathway in paroxysmal nocturnal hemoglobinuria: from a pathogenic mechanism to a therapeutic target. Immunol Rev. 2023;313(1):262–278. doi: 10.1111/imr.13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida Y, Kato H, Ikeda Y, Nangaku M. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb. 2019;26(2):99–110. doi: 10.5551/jat.RV17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schubart A, Anderson K, Mainolfi N, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci U S A. 2019;116(16):7926–7931. doi: 10.1073/pnas.1820892116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schubart A, Flohr S, Junt T, Eder J. Low-molecular weight inhibitors of the alternative complement pathway. Immunol Rev. 2023;313(1):339–357. doi: 10.1111/imr.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei A, Alexander RS, Duke J, Ross H, Rosenfeld SA, Chang CH. Unexpected binding mode of tick anticoagulant peptide complexed to bovine factor Xa. J Mol Biol. 1998;283(1):147–154. doi: 10.1006/jmbi.1998.2069. [DOI] [PubMed] [Google Scholar]
- 50.Eriksson O, Mohlin C, Nilsson B, Ekdahl KN. The human platelet as an innate immune cell: interactions between activated platelets and the complement system. Front Immunol. 2019;10:1590. doi: 10.3389/fimmu.2019.01590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28(4):184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. 2010;47(13):2170–2175. doi: 10.1016/j.molimm.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen RP, Shinoda K, Rampuria P, et al. Bispecific antibodies for immune cell retargeting against cancer. Expert Opin Biol Ther. 2022;22(8):965–982. doi: 10.1080/14712598.2022.2072209. [DOI] [PubMed] [Google Scholar]
- 54.Mazurkiewicz Ł, Czernikiewicz K, Rupa-Matysek J, Gil L. Emicizumab: a novel drug in hemophilia A prophylaxis - a narrative review. Expert Rev Hematol. 2022;15(10):933–942. doi: 10.1080/17474086.2022.2131526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.