

Key Points
-
•
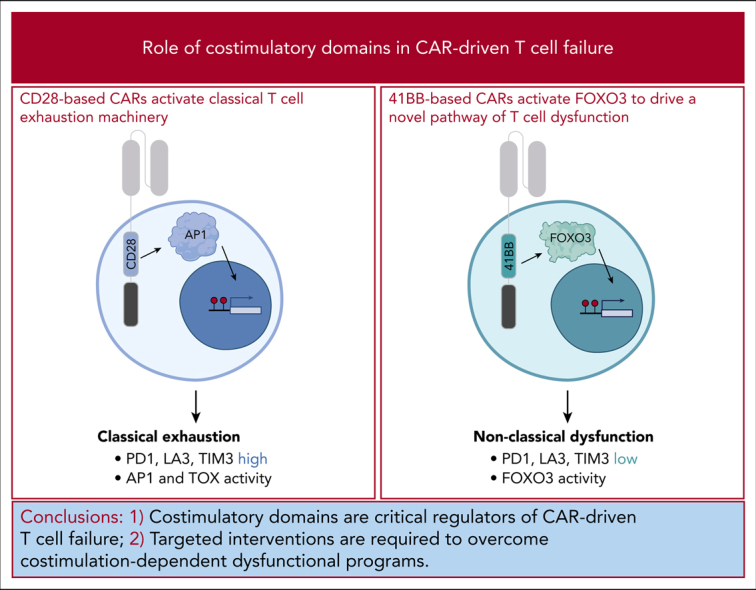
Chronic stimulation of CD28-based CARs drives T cells to become exhausted, but 41BB-based CARs direct a novel state of T-cell dysfunction.
-
•
41BB-driven T-cell dysfunction results from reactivation of the transcription factor FOXO3, and suppressing FOXO3 improves T-cell function.
Visual Abstract
Abstract
T cells engineered to express chimeric antigen receptors (CARs) targeting CD19 have demonstrated impressive activity against relapsed or refractory B-cell cancers yet fail to induce durable remissions for nearly half of all patients treated. Enhancing the efficacy of this therapy requires detailed understanding of the molecular circuitry that restrains CAR-driven antitumor T-cell function. We developed and validated an in vitro model that drives T-cell dysfunction through chronic CAR activation and interrogated how CAR costimulatory domains, central components of CAR structure and function, contribute to T-cell failure. We found that chronic activation of CD28-based CARs results in activation of classical T-cell exhaustion programs and development of dysfunctional cells that bear the hallmarks of exhaustion. In contrast, 41BB-based CARs activate a divergent molecular program and direct differentiation of T cells into a novel cell state. Interrogation using CAR T cells from a patient with progressive lymphoma confirmed the activation of this novel program in a failing clinical product. Furthermore, we demonstrate that 41BB-dependent activation of the transcription factor FOXO3 is directly responsible for impairing CAR T-cell function. These findings identify that costimulatory domains are critical regulators of CAR-driven T-cell failure and that targeted interventions are required to overcome costimulation-dependent dysfunctional programs.
Approved chimeric antigen receptor (CAR) T cells incorporate the intracellular signaling domains of either costimulatory molecules CD28 or 41BB to enhance persistence and function, but chronic continuous stimulation of T cells can cause dysfunction. Selli et al reveal that with chronic antigen stimulation, CD28-bearing CAR T cells demonstrate the cellular and biochemical hallmarks of classic T-cell exhaustion, while 41BB-bearing CAR T cells display a distinct signature with reactivation of the transcription factor, FOXO3. Suppression of FOXO3 improves CAR T-cell function, suggesting new avenues for maintaining CAR T-cell efficacy.
Introduction
Failure of chimeric antigen receptor (CAR)-engineered T cells against both hematologic and solid cancers most often results from defects in T-cell function.1 This manifests as poor CAR T-cell expansion, limited persistence, and ineffective antitumor cytotoxicity. Correlative studies have identified an association between high tumor burden and poor outcomes, linking increased receptor activation with failure.2, 3, 4 These limitations resemble the functional defects observed during exhaustion, a cellular state that develops from persistent stimulation of the native T-cell receptor (TCR) in the setting of chronic viral infection, cancer, and autoimmunity.5 Preclinical studies have confirmed that persistent CAR activation results in a dysfunctional T-cell state that shares many functional attributes of exhaustion.6, 7, 8, 9 Collectively, these findings indicate that chronic antigen receptor activation is detrimental to both TCR and CAR-driven T cells, and the prevailing presumption is that CAR T cells fail because they become exhausted.
CAR functionality is dependent on activating signals derived from the TCR and a costimulatory receptor. Of the 6 US Food and Drug Agency–approved CAR T-cell products, 2 contain the signaling domain from CD28, the paradigmatic costimulator with a central role in the endogenous T-cell response. The 4 other products contain the signaling domain from 41BB, a receptor that supports memory T-cell development. Preclinical and clinical data confirm that CAR-integrated costimulatory domains have a significant impact on T-cell function, directing distinct patterns of expansion, persistence, and toxicity.10 Consideration of CAR costimulatory domains is central to clinical decision making; however, how costimulatory domains contribute to the T-cell dysfunction has not been investigated.
We developed an in vitro system to chronically activate CD19-directed CAR T cells with CD19+ acute lymphoblastic leukemia cells.11 We observed similar functional defects for both CD28- and 41BB-based CAR T cells but divergent transcriptional, epigenetic, and phenotypic attributes. Although CD28-based CAR T cells bore the hallmarks of T-cell exhaustion, 41BB-based CARs activated a distinct molecular program not previously associated with T-cell dysfunction. Evaluation of CAR T cells collected from a patient at the time of lymphoma progression confirmed expression of this gene signature in an actively failing clinical product. We identified the transcription factor FOXO3 as a driver of 41BB-driven CAR T-cell dysfunction, and observed that disruption of FOXO3 improved function. Together, these data identify a unique CAR-driven dysfunction program activated by 41BB.
Methods
Please see supplemental Materials, available on the Blood website, for detailed methods. Chronic stimulation cultures were established by combining CAR T cells (0.5 × 106-2 × 106) with green fluorescent protein–positive Nalm6 cells at an effector-to-target ratio (E:T) of 1:8 in standard media. Cultures were profiled every other day via flow cytometry to both count cells and evaluate changes in protein expression. The cocultures were refed with additional green fluorescent protein–positive Nalm6 cells every other day to maintain an E:T of 1:8 until the onset of dysfunction.11 CAR T cells were purified on day 0, day 6 or 7 when they were at the peak of activation, and from days 13 to 17 when they became dysfunctional for downstream analyses. Cytometry by time of flight, bulk RNA sequencing (RNAseq), assay for transposase-accessible chromatin with sequencing (ATACseq), and single-cell RNA sequencing (scRNAseq) were performed and analyzed using standard methods.12, 13, 14, 15, 16, 17, 18, 19, 20, 21 CRISPR editing was performed using the Lonza-4D system with sequencing-based confirmation of gene disruption.22 Animal studies were performed in nonobese diabetic-severe combined immunodeficiency (NOD-SCID-γc−/−) mice, and disease progression was monitored as previously described.9
Results
Persistent exposure to CD19+ tumors drives CAR T-cell dysfunction
To understand how prolonged costimulatory domain activation affects CAR T-cell function, we developed an in vitro model in which CD19-directed CAR T cells containing either CD28 (19/28) or 41BB (19/BB) domains (purified to ensure that all cells expressed the CAR) were combined with the CD19+ acute lymphoblastic leukemia cell line Nalm6. These cocultures were established at an E:T of 1:8. Because CAR T cells eliminated Nalm6 and proliferated in response to activation, we replenished cultures with additional Nalm6 to reestablish an E:T of 1:8 every ∼48 hours, ensuring chronic CAR stimulation (Figure 1A).11 As previously reported,6 CAR expression declined modestly during coculture (supplemental Figure 1A). Longitudinal measurement of T-cell proliferation, a core attribute of function, revealed robust expansion of both cell types with greater expansion of 19/28 cells than that of 19/BB cells (Figure 1B), as has been observed with clinical products.10 After ∼13 days of chronic stimulation, both 19/28 and 19/BB cell types lost proliferative capacity and contracted, reflecting loss of function. Because cumulative Nalm6 survival could not be longitudinally measured in this refeeding model, we used the E:T over time as a measure of cytotoxic function. Both 19/28 and 19/BB cells demonstrated robust cytotoxicity against persistently high tumor burdens until approximately day 13, at which time both cell types lost the ability to kill (Figure 1C). We isolated 19/28 and 19/BB cells after 7 days of chronic stimulation and restimulated them with fresh Nalm6, which elicited potent secretion of interferon γ and tumor necrosis factor α (Figure 1D-E). In contrast, cells isolated after 15 days of chronic stimulation secreted almost no cytokine in response to restimulation. We repeated these assays using T cells derived from 4 independent donors, and although each donor demonstrated distinct kinetics of cytotoxicity, we broadly found no difference in the timing of dysfunction onset (Figure 1F), defined as either expansion of Nalm6 or CAR T-cell contraction, based on CAR costimulatory domain.
Figure 1.


Chronic CAR stimulation results in T-cell dysfunction. (A) Schematic flow of in vitro chronic stimulation assay. (B) Expansion of 19/28 and 19/BB CAR T cells over the course of chronic stimulation. (C) Target Nalm6 cells per CAR T cells over the course of chronic stimulation. (D-E) Production of (D) interferon γ and (E) tumor necrosis factor α by 19/28 and 19/BB cells isolated at days 7 and 15 of chronic stimulation cultures upon restimulation. (F) Kinetics of dysfunction onset as reflected by first day of failure as measured by T-cell contraction or loss of tumor control. Data from n = 4 independent donors. (G) Change in memory phenotype of CD8+ CAR T-cell products after either acute (single combination with Nalm6 cells) or chronic stimulation. (H) Surface expression of PD1 during chronic stimulation. (I) Heatmap of median signal intensities from all cytometry by time of flight markers evaluated. (J) Violin plots of normalized signal intensity for T-cell markers on day-15 cells. Panels B-E,G-H show representative data from n = 4 independent donors; data in panel J are representative of n = 2 donors. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 using two-tailed analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons.
Next, we analyzed T-cell phenotypes over the course of chronic stimulation. CD8 memory differentiation was largely similar for 19/28 and 19/BB cells and confirmed that 41BB promotes earlier-lineage memory phenotypes23 (Figure 1G). This was most pronounced in the setting of acute stimulation, in which cocultures were established but never replenished with Nalm6. A similar trajectory was observed for CD4 T cells (supplemental Figure 1B). Next, we used Jurkat cells engineered to express distinct fluorescent proteins when the transcription factors NFAT, NF-κB, and activator protein 1 (AP1) are activated to assess changes in transcriptional regulation. Chronic stimulation of 19/28 Jurkats resulted in increased NFAT and AP1 activity whereas 19/BB Jurkats had higher NF-κB activity (supplemental Figure 1C). Persistent activation of NFAT and AP1 reportedly drives exhaustion in models of chronic viral infection24 and cancer.8,25 Next, we evaluated expression of PD1, the paradigmatic marker of exhaustion. Both 19/28 and 19/BB cells demonstrated an initial increase in PD1 expression, reflecting activation but only 19/28 cells demonstrated resurgent expression with the onset of dysfunction (Figure 1H). To interrogate T-cell phenotypes with increased granularity, we profiled 39 proteins that define T-cell lineages (supplemental Table 1). Cytometry by time of flight analysis revealed that prestimulation (day 0) and peak-stimulation (day 6) 19/28 and 19/BB cells had very similar phenotypes, however dysfunctional (day 15) samples occupied different spaces by nearest neighbor (t-distributed stochastic neighbor embedding) analysis (supplemental Figure 1D-E). Evaluation of individual markers revealed that day-15 19/28 cells expressed higher levels of PD1 and TIGIT than all other samples, whereas day 19/BB cells expressed higher CD62L (Figure 1I; supplemental Figure 1F-H). Dedicated comparison of the day-15 samples further revealed that 19/28 cells expressed higher levels of exhaustion markers CTLA4, LAG3, and TIM3, whereas 19/BB cells were higher for CD25 (Figure 1J), reflecting that dysfunctional 19/28 cells were phenotypically similar to exhausted T cells.
Dysfunctional CD28- and 41BB-based CAR T cells are programmatically distinct
To interrogate the gene expression programs associated with these distinct phenotypic states, we performed RNA sequencing of CAR T cells at days 0, 6, and 15 of chronic stimulation. Principal component analysis demonstrated minimal differences based on donor (supplemental Figure 2A) or between 19/28 and 19/BB cells at days 0 and 6 but a divergence at day 15 (Figure 2A). Although day-6 cells segregated from day 0 cells on both PC1 and PC2, day-15 cells were similar to day-0 cells on PC1, suggesting a regression to a resting-like state. Pooled analysis of samples from all donors demonstrated no differentially expressed genes (DEGs) on day 0 and only 60 DEGs at day 6 (supplemental Figure 2B-C). After the onset of dysfunction, however, we identified 365 DEGs that segregated into 6 clusters (Figure 2B). Clusters (3 and 6) with higher expression in dysfunctional 19/28 cells were highly enriched for exhaustion-associated genes, whereas clusters (2 and 4) with higher expression in dysfunctional 19/BB cells were instead enriched for memory markers and class II HLA genes.26 Visualization by volcano plot underscored the significantly greater expression of exhaustion-associated genes in 19/28 cells (Figure 2C). Tracing expression of exhaustion markers PDCD1, CTLA4, LAG3, and HAVCR2 over time demonstrated a significant rise in expression from day 6 to day 15 in 19/28 cells, as is observed in classical exhaustion, whereas expression of these genes in 19/BB cells only mildly increased over time (Figure 2D). Pathway enrichment analysis of DEGs at day 15 demonstrated some overlap but also enrichment of unique pathways, including TCR signaling, JAK-STAT signaling, and PDL1 and PD1 expression in 19/28 cells, and graft-versus-host disease and antigen processing and presentation in 19/BB cells (supplemental Figure 2D).
Figure 2.

Chronic activation of costimulatory domains directs distinct genomic activity. (A) Principal component analysis of bulk RNAseq of 19/28 and 19/BB cells on days 0, 6, and 15. (B) Heatmap of DEGs between day-15 19/28 cells and day-15 19/BB cells. (C) Volcano plot of day-15 DEGs. n = 1 donor for day-0 samples and n = 4 donors for days 6 and 15, all performed in technical triplicates. (D) Normalized transcript counts of key exhaustion markers over time. Significance determined using two-way ANOVA. (E-F) Gene set enrichment analysis of genes present in panel D >2 of 4 gene sets (E) and all 4 human TIL exhaustion gene sets (F). (G) Heatmap of day-15 sample expression of genes present in all 4 human TIL exhaustion gene sets. (H) Principal component analysis of ATAC sequencing of 19/28 and 19/BB cells at days 0, 6, and 15. n = 2 donors for all time points performed in technical triplicates. (I) Gene tracks at transcriptional start sites of PDCD1 and TOX2 reflecting chromatin accessibility. (J) Correlated transcript count and transcriptional start site accessibility for PDCD1 over time. (K) Overlap of genes with increased accessibility in exhausted T cells (as defined22) and genes with increased accessibility in dysfunctional 19/28 or 19/BB cells. ∗∗∗∗P < .0001.
To quantify the similarity of dysfunctional CAR T-cell states to exhausted human T cells we turned to 4 recent studies that profiled gene expression in exhausted tumor-infiltrating lymphocytes (TILs).27, 28, 29, 30 We found significantly greater overlap between dysfunctional 19/28 cells and all human exhaustion gene sets than for dysfunctional 19/BB cells (supplemental Figure 2E-H). We crossreferenced these TIL gene sets to identify genes shared by at least 2 gene sets (n = 89), and genes shared by all 4 gene sets (master exhaustion gene set, n = 17; supplemental Table 2). Gene set enrichment analysis demonstrated significant enrichment of both shared gene sets in dysfunctional 19/28 cells as compared with dysfunctional 19/BB cells (Figure 2E-F) and higher overall expression of nearly all 17 master exhaustion genes in dysfunctional 19/28 cells (Figure 2G).
Several studies have demonstrated recurrent epigenetic alterations that define T-cell exhaustion.31,32 To probe how chromatin accessibility evolved over the course of chronic CAR stimulation, we performed longitudinal assay for transposase-accessible chromatin with sequencing. Consistent with our transcriptional data, we found no difference based on donor (supplemental Figure 2I) but did observe a divergence in chromatin accessibility only after the onset of dysfunction at day 15 and, again, observed that day-15 cells drifted back toward day-0 cells on PC1 (Figure 2H). Interrogation of the exhaustion-associated genes PDCD1 and TOX2 revealed significantly greater accessibility at transcriptional start sites in dysfunctional 19/28 cells as compared with all other groups (Figure 2I). We focused on PDCD1, a gene whose regulation is a defining feature of exhaustion, and simultaneously traced chromatin accessibility and transcript counts to profile gene regulatory dynamics at this site. As in exhausted T cells,31,32 19/28 cells increased PDCD1 expression over time and preserved chromatin accessibility as they progressed from activated to dysfunctional. In contrast, 19/BB cells closed this site to prestimulation levels as they progressed from activated to dysfunctional (Figure 2J), suggesting divergent regulation at this site. Finally, we quantitated the similarity of these 2 cell states to a previously defined exhaustion-associated chromatin signature33 and, again, found significantly greater overlap between open chromatin sites in classically exhausted cells and dysfunctional 19/28 cells (P = 8.82 × 10−5) and minimal overlap with dysfunctional 19/BB cells (P = .419; Figure 2K). Collectively, these data indicate that dysfunctional 19/28 cells and exhausted T cells share similar transcriptional and epigenetic identities whereas dysfunctional 19/BB cells are programmatically distinct.
Chronic stimulation of 19/BB cells promotes development of a unique dysfunctional cell state
To clarify the distinct cell states that made up the dysfunctional populations we performed longitudinal scRNAseq. We observed that cells collected at days 0, 6, and 15 segregated into 12 clusters and that clusters (Figure 3A) were largely defined by the collection time point (supplemental Figure 3A). Pathway analysis confirmed representation of predictable biological programs in each cluster (ie, cell cycle and metabolism genes in activated clusters 0, 1, 7, and 10, supplemental Figure 3B). We focused our analysis on clusters 8 and 9, which were almost entirely composed of dysfunctional cells (cluster 8, 85% day-15 cells; cluster 9, 88% day-15 cells). Notably, no other cluster comprised >5% day-15 cells. Although cluster 9 contained roughly equivalent quantities of cells from both day-15 samples (44% 19/BB cells; 42% 19/28 cells), cluster 8 was heavily skewed toward day-15 19/BB cells (83% 19/BB cells; 17% 19/28 cells; supplemental Figure 3A). Evaluation of each sample individually again demonstrated largely equivalent cluster distributions at days 0 and 6 but distinct cluster distribution at day 15 (Figure 3B). Of dysfunctional 19/28 cells, 59% occupied cluster 9, which was defined by expression of exhaustion-associated genes. In contrast, 69% of dysfunctional 19/BB cells were in cluster 8, whose identity was defined by a distinct set of genes with varied roles in lymphocyte function including cytotoxicity (GNLY, CCL5, PRF1, GZMA, GZMK, and CTSW), natural killer (NK) cell identity (KLRK1 and KLRC2), and T-cell differentiation (ID2). The expression of NK markers has previously been reported in classically exhausted T cells34,35 and dysfunctional CAR T cells.6 Consistent with the modest increase in expression of exhaustion genes (Figure 2D), we observed that a fraction of dysfunctional 19/BB cells occupied cluster 9, suggesting that dysfunctional 19/BB cells are heterogenous and contain some classically exhausted cells and that most have a novel transcriptional identity.
Figure 3.

Single-cell analysis reveals that dysfunctional 41BB CAR T cells are a unique terminal state. (A) Uniform manifold approximation and projection (UMAP) of 19/28 and 19/BB cells over time. (B) Proportion of each cluster present in each sample. (C) CD8A expression in each cluster. (D) Reclustering of CD8A-expressing 19/28 and 19/BB cells from days 0, 6, and 15. (E) Pseudotime analysis of CD8 cells. (F) Mapping of CD8 clusters onto pseudotime. (G) Proportion of each sample contained in terminal CD8 clusters 0 and 1.
Because cluster 8 cells expressed uniformly high levels of CD8A (Figure 3C), we reclustered only CD8 cells from all 6 samples. CD8 cells distributed to 9 new clusters (Figure 3D), and we found that dysfunctional 19/28 cells primarily occupied CD8-cluster 7, defined by expression of GZMB, IL2RA, ENO1, CCL3, and BATF3 (supplemental Figure 4A). Dysfunctional 19/BB cells were highly enriched for CD8-cluster 0, defined by expression of GNLY, KLRB1, CCL5, ID2, and GZMK, reflecting similarity in identity of CD8-cluster 0 to cluster 8 from the whole population analysis. To trace the transcriptional evolution of CD8+ CAR T cells, we performed pseudotime analysis.18 This demonstrated a divergence along 2 developmental trajectories (Figure 3E). Mapping of the CD8 clusters onto pseudotime revealed 2 clusters at the termination of these trajectories: CD8-cluster 0 and CD8-cluster 1 (Figure 3F). CD8-cluster 0 was predominantly composed of day-15 19/BB cells (59%, Figure 3G) and a small fraction of day-15 19/28 cells (6.8%). CD8-cluster 1 was defined by expression of activation markers MKI67, BHLHE40, and TOP2A and, consistently, was almost entirely composed of cells from day 6 (92%; Figure 3G). These findings suggest that dysfunctional 19/BB cells acquire a terminal T-cell identity.
CAR T cells that fail clinically acquire a dysfunctional 19/BB gene signature
Using our bulk RNAseq and scRNAseq data sets, we generated a gene signature of dysfunctional 19/BB cells (TBBD; supplemental Figure 5; supplemental Table 3). To determine whether this transcriptional state developed in CAR T-cell products that failed to induce remission in patients, we analyzed circulating CAR T cells collected from a patient with diffuse large B-cell lymphoma who received tisagenlecleucel, a 41BB-based CD19 CAR T-cell product. This patient had a partial response 1 month after treatment but progressive disease at 3-month evaluation, a common clinical scenario that leads to persistent CAR stimulation. We evaluated circulating T cells at the time of partial antitumor activity (day 14) and after therapeutic failure (day 100; Figure 4A). Day-14 CAR+ T cells clustered separately from day-100 cells, reflecting a transition in transcriptional identity (Figure 4B). Although the day-14 sample contained both CD4 and CD8 cells, the day-100 sample was composed almost entirely of CD8 cells (Figure 4C). We assessed the overlap between the TBBD signature and the genes that defined the identity of day-100 CAR T cells (upregulated compared with day 14; n = 232) and observed significant overlap between these 2 gene sets (P = 2.8 × 10−47; Figure 4D). Furthermore, we found that 8 of the top 10 drivers of the TBBD signature were expressed at higher levels in day-100 cells (Figure 4E). To assess whether day-100 cells also acquired features of exhaustion, we profiled expression of the exhaustion genes defined from TIL data sets.27, 28, 29, 30 Although expression of 12 of these 17 genes were higher in day-100 cells, these differences were very modest (Figure 4F) and mirror the changes in exhaustion-associated gene expression we observed in vitro (Figure 2D). We observed minimal overlap between day-100 marker genes and TIL exhaustion signatures (Figure 4G), suggesting that failing 41BB-based CAR T cells evolve a transcriptional profile that more closely resembles the TBBD gene profile than an exhausted gene profile.
Figure 4.

Failing CAR T cells express a dysfunctional 41BB CAR T-cell signature. (A) Peripheral blood cells from a patient who received tisagenlecleucel for diffuse large B-cell lymphoma were collected 14 and 100 days after infusion and purified for CAR-expressing cells. (B) UMAP of day-14 and day-100 cells. (C) Expression of CD4 and CD8A in peripheral blood CAR T cells. (D) Overlap between in vitro–defined signature of 41BB-driven dysfunction and genes defining day-100 cell identity. Significance of overlap determined using Fisher exact test. (E) Expression of top 10 driver genes in day-14 and day-100 cells. (F) Log2 fold change in expression of master exhaustion genes in day-100 cells compared with that in day-14 cells. (G) Overlap between genes defining day-100 cell identity and TIL exhaustion signatures. Significance of overlap determined using Fisher exact test.
Dysfunctional 19/BB cells reactivate FOXO3
To identify the pathways responsible for promoting the development of this transcriptionally unique state we interrogated how transcription factor binding site accessibility evolved as 19/28 and 19/BB cells progressed from resting to dysfunctional. We found that although many of the sites (32/100) with increased accessibility were shared between 19/28 and 19/BB cells, more (68/100) were specific to each. As has been demonstrated previously8 dysfunctional 19/28 cells had increased accessibility for Jun:Fos (AP1) binding (Figure 5A). In contrast, 19/BB cells opened homeobox (HOX) and forkhead box (FOX) sites (Figure 5B). Pathway analysis of the unique binding sites observed for each group confirmed that dysfunctional 19/28 cells were enriched for binding of basic leucine zipper domain–containing factors (which includes many proteins including AP1, Figure 5C). Dysfunctional 19/BB cells were enriched for HOX and FOX sites, with some specificity for FOXO sites (Figure 5D). Interrogation of our RNAseq data revealed nearly no expression of any HOX genes in day-15 cells but a resurgence in FOXO (Figure 5E) and some FOXP (supplemental Figure 6A) factors at day 15, which was more significant for 19/BB cells. Our data also confirmed higher expression of basic leucine zipper domain factors in dysfunctional 19/28 cells8 (supplemental Figure 6B).
Figure 5.

TBBD cells demonstrate reactivation of FOXO3. (A-B) Transcription factor motif analysis demonstrates increased accessibility of (A) AP1 sites in 19/28 cells and (B) HOX and FOX sites in 19/BB cells as cells progress from resting to dysfunctional. (C-D) Pathway enrichment analysis of unique sites with increased accessibility in (C) 19/28 cells and (D) 19/BB cells. (E) Expression of FOXO1, FOXO3, and FOXO4 transcripts over time. Significance determined using two-way ANOVA. (F) Expression of the FOXO3 regulon in 19/28 and 19/BB cells collected on days 0, 6, and 15. (G) FOXO3 regulon score for each scRNAseq cluster (top) and normalized FOXO3 transcript count for each cluster (bottom). (H) Expression of FOXO3 regulon in day-14 and day-100 cells collected from patient peripheral blood. Significance determined using Mann-Whitney test.
We wanted to determine whether the increased binding site accessibility and transcript quantity was accompanied by increased FOXO activity. To do this we used SCENIC, a tool designed to interrogate activity of transcription factor gene regulatory networks using scRNAseq data.19 We observed high activity of the FOXO3 regulon (target network) in several clusters (2, 4, and 5-8; Figure 5F). Quantification of regulon scores, reflecting quantity of FOXO3 target gene expression, and average FOXO3 transcript counts demonstrated that both were highest in cluster 6, composed entirely of day-0 cells and consistent with FOXO3’s known role in maintaining T-cell homeostasis,36 and cluster 8, indicating high FOXO3 activity in dysfunctional 19/BB cells (Figure 5G). Gene set enrichment analysis of cluster-8 marker genes further confirmed enrichment of the FOXO3 regulon (normalized enrichment score, 2.71; false discovery rate, 0; supplemental Figure 6C). A key target of FOXO3 is FOXP3,37,38 responsible for directing development of regulatory T cells (Treg). In addition to higher expression of CD25 (a marker of Tregs) in dysfunctional 19/BB cells (Figure 1I), we also observed higher expression of FOXP3 transcripts in these cells (supplemental Figure 6D). Evaluation of our single-cell data revealed that although FOXP3 transcripts could be detected in several clusters, they were highest in cluster 8 (supplemental Figure 6E). These data validate increased FOXO3 activity using both transcript and protein expression of a known FOXO3 target in dysfunctional 19/BB cells. Next, we evaluated FOXO3 target gene expression in peripheral blood CAR+ T cells and observed a trend toward increased activity in day-100 cells (P = .0852; Figure 5H). These data demonstrate that as 19/BB cells become dysfunctional they reactivate the transcription factor FOXO3.
Disruption of FOXO3 improves function of 19/BB cells
To determine whether FOXO3 inhibited 19/BB cell function, we disrupted genomic FOXO3 in 19/28 or 19/BB cells (Figure 6A). This resulted in >90% gene disruption and had no impact on T-cell manufacturing (supplemental Figure 7A-C). We subjected these cells to our chronic in vitro stimulation assays and found that FOXO3 knockout (FOXO3KO) had minimal impact on CAR T-cell expansion (supplemental Figure 7D-E). In contrast, FOXO3KO improved antitumor function of 19/BB cells but had no change in function of 19/28 cells, with a resultant delay in onset of dysfunction (Figure 6B-C). To substantiate the role of FOXO3 in dysfunction, we engineered our CAR constructs to include a transgenic FOXO3 (Figure 6D). We confirmed that this led to overexpression (OE) of FOXO3, which resulted in a minor improvement in cell expansion during manufacturing (supplemental Figure 7F-G). In chronic stimulation cultures, FOXO3OE did not affect 19/28 cell expansion but dramatically inhibited 19/BB cell expansion (peak expansion from baseline of 22.8-fold compared with 3.2-fold; P < .0001; supplemental Figure 7H-I). 19/BB FOXO3OE did not undergo more cell death (supplemental Figure 7J), indicating that this lower cell count was because of suppressed expansion. This was accompanied by a rapid loss of tumor control for 19/BB cells (Figure 6E) and much earlier onset of dysfunction (Figure 6F). We observed that 19/BB FOXO3OE cells were significantly smaller in size throughout chronic stimulation (supplemental Figure 7K), suggestive of a less-activated cell state.
Figure 6.

Manipulation of FOXO3 affects 41BB-based CAR T-cell function. (A) Representation of FOXO3KO cells being evaluated. (B) Target Nalm6 cells per CAR T cells over the course of chronic stimulation. Representative data from n = 3 donors. (C) Day of dysfunction onset as measured based on the first day of T-cell contraction or loss of tumor control. Data from n = 3 independent donors. (D) Representation of FOXO3OE cells being evaluated. (E) Target Nalm6 cells per CAR T cells over the course of chronic stimulation. Representative data from n = 3 donors. (F) Day of dysfunction onset as measured by first day of T-cell contraction or loss of tumor control. Data from n = 3 independent donors. (G) Survival of mice after treatment with 0.125 × 106 CAR T cells. Significance determined using log-rank test. (H) Survival of mice after treatment with 0.5 × 106 CAR T cells. Significance determined using log-rank test. For both experiments n = 5 mice per group. ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001. ns, not significant.
We proceeded to evaluate the impact of FOXO3KO on 19/BB cells in vivo. To mimic conditions of chronic stimulation, we used a validated stress model39 in which NOD-SCID/γ−/− mice were engrafted with 106 Nalm6 cells and then given subtherapeutic doses of CAR T cells 7 days later. In the first model we delivered 1.25 × 105 CAR T cells and found that FOXO3KO had a modest impact in improving antileukemic activity (supplemental Figure 8A) and animal survival (Figure 6G). In a second model we delivered 5 × 105 CAR T cells and observed significantly enhanced antitumor function of FOXO3KO cells (supplemental Figure 9B). At this higher dose, FOXO3KO 19/BB cells enabled a more impactful prolongation of animal survival (25 days compared with 36 days; P = .0162; Figure 6H). Collectively, these data indicate that FOXO3 specifically suppresses function of 41BB-based CAR T cells.
Discussion
We demonstrate that 41BB-based CARs direct a unique state of T-cell dysfunction that is, in part, reliant on FOXO3. A notable attribute of dysfunctional 19/BB cells is expression of genes encoding NK receptors, a finding that has been observed in various models of T-cell exhaustion.34,35 Interestingly, 2 recent reports identified a novel preterminal exhausted state (termed TKLR).40,41 These cells also expressed Gzma Gzmk, Ccl5, and Id2 and lacked expression of terminal exhaustion markers Pdcd1, Lag3, and Tox, reflecting high similarity to the TBBD transcriptional signature defined here. Whether TKLR and dysfunctional 19/BB cells are a similar intermediate-exhausted lineage is not clear; dysfunctional 19/BB cells lack expression of CX3CR1, a defining marker of TKLR cells. Whether TKLR development is dependent on 41BB or instead relies on a shared independent pathway is the focus of ongoing studies. Intriguingly, both studies found that TKLR cells were a fraction of the bulk exhausted T-cell population, whereas we observe that the majority of cells present at day 15 of chronic 19/BB cell stimulation bear the TBBD gene signature. If these cell states are indeed dependent on 41BB, this higher frequency may reflect the increased strength and duration of 41BB activation from CARs as opposed to natural costimulatory signals resulting from chronic infection.
A recent study similarly interrogated the trajectory of chronically stimulated 41BB-based CAR T cells.6 Although the authors also observed expression of NK receptors, they did not identify the same transcriptional features or regulatory pathways as we did in our studies. Whether this is a result of a context dependence to this dysfunctional circuitry (their CARs targeted low-abundance mesothelin on pancreatic cancer, whereas our model targets high-abundance CD19 on leukemia) remains unknown. The authors also observed increased expression of classical exhaustion markers in dysfunctional as compared with resting CAR T cells, as have others.42,43 We, too, observe an increase in exhaustion marker expression as these cells progress from resting to dysfunctional, but identified that this expression is modest compared with CD28-based CAR T cells and is restricted to a distinct, smaller subset of dysfunctional cells. These data highlight that low-level expression of exhaustion markers may not be the same as commitment to the classical exhaustion program; indeed, we found dysfunctional 19/BB cells to have closed chromatin at the PDCD1 locus, which would preclude defining these cells as classically exhausted. Notably, we did not observe expression of NK receptor genes in CD28-based CAR T cells, suggesting that this is not a broad feature of CAR-driven dysfunction but is dependent on costimulatory structure.
FOX factors have broad roles in an array of cellular functions across tissue types. By inducing FOXP3 expression, FOXO3 promotes differentiation of induced Tregs.37,38 Our data demonstrate an enrichment of CD25+ and FOXP3+ cells in dysfunctional 19/BB cells. Two recent reports identified that enrichment of Tregs is associated with clinical failure of CAR T cells, directly implicating a role for this pathway in limiting antitumor efficacy. These studies evaluated preinfusion and early postinfusion CD28-based CAR T cells; thus, the contribution of induced Tregs to the impaired functionality of 19/BB cells remains to be determined. FOXO3 also reportedly suppresses antiviral T-cell function by inhibiting the formation of durable memory in patients with HIV44 and in murine models of lymphocytic choriomeningitis.45 Of particular relevance to our findings, a distinct study found that Foxo3 deletion significantly improved T-cell control of chronic lymphocytic choriomeningitis virus infection,46 bolstering our findings by corroborating a role for FOXO3 in a distinct model of chronic antigen stimulation. How FOXO3 exerts this suppressive function remains unclear, and of specific interest to the development of next-generation CAR therapies is the link between 41BB and FOXO3. We were surprised to observe that manipulation of FOXO3 did not impact T-cell manufacturing or the function of CD28-based CARs, both of which are driven by CD3/CD28 signaling. These findings support the hypothesis that FOXO3’s suppressive effect is dependent on 41BB and underscore that elucidating the relationship between 41BB, FOXO3, and activation of the TBBD dysfunction program is of critical importance.
Although beneficial, disruption of FOXO3 resulted in modest delay in the onset of 41BB-based CAR T-cell failure. This is consistent with the improvements noted in other studies that disrupt T-cell transcription factors in an effort to improve CAR efficacy6 and suggests that an individual factor is unlikely to completely rescue CAR-driven T-cell dysfunction. Multiplexed manipulations that exploit vulnerabilities in distinct pathways of CAR-driven circuitry are likely necessary to propel improvements in function. Another limitation of this study is the inclusion of clinical samples from only 1 patient. Given the nature of our question, we prioritized sequencing of cells undergoing chronic stimulation for a prolonged period. Most studies evaluate CAR T cells collected between days 7 and 30 after infusion,47,48 or cells collected at later time points from patients with durable responses, a clinical setting in which CAR T cells are not chronically stimulated and do not persist longer.43,49 To our knowledge, this is the first study to report data on CAR T cells isolated months after infusion from a patient with progressive lymphoma, a scenario in which CAR T-cell persistence is highly limited beyond 30 days. Although we demonstrate feasibility of evaluating these late–time point cells, this analysis required a significant volume of peripheral blood, which is not available for many patients enrolled on current banking protocols. Next, we aim to improve collection procedures to enhance our ability to evaluate these valuable tissues.
Conflict-of-interest disclosure: M.E.S. is an employee of Wugen, Inc. M.M.B.-E. and T.A.F. report equity in, consulting for, and research grants from, Wugen, Inc. M.M.B.-E. and T.A.F. are coinventors on patents licensed by Washington University to Wugen, Inc. T.A.F. also discloses equity in Orca Bio and Indapata Therapeutics, and research funding from HCW Biologics and Affimed (all unrelated to this report). N.S. holds several patents related to CAR therapy and is listed on a patent application related to some of the technology presented here. The remaining authors declare no competing financial interests.
The current affiliation for M.E.S. is Wugen, Inc, St. Louis, MO.
Acknowledgments
The authors thank Abby Green, Julie Ritchey, Matthew Cooper, Alun Carter, John DiPersio, Hamza Celik, Grant Challen, Nicole Helton, Tim Ley, Carl DeSelm, Josephine Giles, and John Wherry for helpful discussions and technical assistance.
This work was supported by National Cancer Institute, National Institutes of Health grants P50CA171963 and R01CA205239 (T.A.F.), K08CA237740, a Be The Match Amy Strelzer Manasevit Research Award, a Gabrielle’s Angel Foundation Cancer Research Award, and a Damon Runyon Clinical Investigator Award (N.S.).
Authorship
Contribution: M.E.S., J.H.L., and N.S. designed and planned experiments; M.E.S., J.H.L., J.L., A.H., Y.-S.H., T.-C.C., J.C., J.W., H.H., and N.S. performed experiments; N.K., G.H., M.M.B.-E., M.F., S.K.-S., D.D., and T.A.F. contributed to cytometry by time of flight studies; M.S. coordinated primary sample acquisition and analysis; M.E.S. and A.G. performed bulk RNA and ATAC sequencing analysis; M.T. and M.A. performed scRNAseq analysis; M.E.S. and N.S. wrote the manuscript; and all authors reviewed the manuscript.
Footnotes
All bulk RNA, bulk ATAC, and scRNA sequencing data from preclinical studies will be available through Gene Expression Omnibus (accession number GSE231312).
Requests for clinical sample sequencing data will be reviewed by Washington University to determine whether they are subject to confidentiality obligations. All other data generated from this study will be made available upon request from the corresponding author, Nathan Singh (nathan.singh@wustl.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19(2):73–74. doi: 10.1038/s41577-018-0119-y. [DOI] [PubMed] [Google Scholar]
- 2.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 3.Locke FL, Rossi JM, Neelapu SS, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020;4(19):4898–4911. doi: 10.1182/bloodadvances.2020002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119–3128. doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Good CR, Aznar MA, Kuramitsu S, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. 2021;184(25):6081–6100.e26. doi: 10.1016/j.cell.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynn RC, Weber EW, Sotillo E, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh N, Lee YG, Shestova O, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. 2020;10(4):552–567. doi: 10.1158/2159-8290.CD-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–727. doi: 10.1038/s41571-021-00530-z. [DOI] [PubMed] [Google Scholar]
- 11.Selli ME, Landmann JH, Arveseth C, Singh N. Inducing T cell dysfunction by chronic stimulation of CAR-engineered T cells targeting cancer cells in suspension cultures. STAR Protoc. 2023;4(1):101954. doi: 10.1016/j.xpro.2022.101954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berrien-Elliott MM, Foltz JA, Russler-Germain DA, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med. 2022;14(633):eabm1375. doi: 10.1126/scitranslmed.abm1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finak G, McDavid A, Yajima M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–982. doi: 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aibar S, Gonzalez-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van de Sande B, Flerin C, Davie K, et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc. 2020;15(7):2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- 21.Moerman T, Aibar Santos S, Bravo Gonzalez-Blas C, et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019;35(12):2159–2161. doi: 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 22.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawalekar OU, O'Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Martinez GJ, Pereira RM, Aijo T, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–278. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Lopez-Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567(7749):530–534. doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boroughs AC, Larson RC, Marjanovic ND, et al. A distinct transcriptional program in human CAR T cells bearing the 4-1BB signaling domain revealed by scRNA-seq. Mol Ther. 2020;28(12):2577–2592. doi: 10.1016/j.ymthe.2020.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 28.Li H, van der Leun AM, Yofe I, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176(4):775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Yu X, Zheng L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 30.Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 31.Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sen DR, Kaminski J, Barnitz RA, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bengsch B, Ohtani T, Khan O, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity. 2018;48(5):1029–1045.e5. doi: 10.1016/j.immuni.2018.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathewson ND, Ashenberg O, Tirosh I, et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell. 2021;184(5):1281–1298.e26. doi: 10.1016/j.cell.2021.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng L, Qin S, Si W, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374(6574):abe6474. doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 36.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12(9):649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harada Y, Harada Y, Elly C, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. 2010;207(7):1381–1391. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerdiles YM, Stone EL, Beisner DR, et al. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33(6):890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feucht J, Sun J, Eyquem J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daniel B, Yost KE, Hsiung S, et al. Divergent clonal differentiation trajectories of T cell exhaustion. Nat Immunol. 2022;23(11):1614–1627. doi: 10.1038/s41590-022-01337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giles JR, Ngiow SF, Manne S, et al. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nat Immunol. 2022;23(11):1600–1613. doi: 10.1038/s41590-022-01338-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson Z, Hong C, Schauner R, et al. Sequential single-cell transcriptional and protein marker profiling reveals TIGIT as a marker of CD19 CAR-T cell dysfunction in patients with non-Hodgkin lymphoma. Cancer Discov. 2022;12(8):1886–1903. doi: 10.1158/2159-8290.CD-21-1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson TL, Kim H, Chou CH, et al. Common trajectories of highly effective CD19-specific CAR T cells identified by endogenous T-cell receptor lineages. Cancer Discov. 2022;12(9):2098–2119. doi: 10.1158/2159-8290.CD-21-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Grevenynghe J, Procopio FA, He Z, et al. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med. 2008;14(3):266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]
- 45.Sullivan JA, Kim EH, Plisch EH, Peng SL, Suresh M. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS Pathog. 2012;8(2):e1002533. doi: 10.1371/journal.ppat.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan JA, Kim EH, Plisch EH, Suresh M. FOXO3 regulates the CD8 T cell response to a chronic viral infection. J Virol. 2012;86(17):9025–9034. doi: 10.1128/JVI.00942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haradhvala NJ, Leick MB, Maurer K, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat Med. 2022;28(9):1848–1859. doi: 10.1038/s41591-022-01959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Good Z, Spiegel JY, Sahaf B, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med. 2022;28(9):1860–1871. doi: 10.1038/s41591-022-01960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheih A, Voillet V, Hanafi LA, et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat Commun. 2020;11(1):219. doi: 10.1038/s41467-019-13880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.