

Abstract
Motivation
With the rapid advances of RNA sequencing and microarray technologies in non-coding RNA (ncRNA) research, functional tools that perform enrichment analysis for ncRNAs are needed. On the one hand, because of the rapidly growing interest in circRNAs, snoRNAs, and piRNAs, it is essential to develop tools for enrichment analysis for these newly emerged ncRNAs. On the other hand, due to the key role of ncRNAs’ interacting target in the determination of their function, the interactions between ncRNA and its corresponding target should be fully considered in functional enrichment. Based on the ncRNA–mRNA/protein-function strategy, some tools have been developed to functionally analyze a single type of ncRNA (the majority focuses on miRNA); in addition, some tools adopt predicted target data and lead to only low-confidence results.
Results
Herein, an online tool named RNAenrich was developed to enable the comprehensive and accurate enrichment analysis of ncRNAs. It is unique in (i) realizing the enrichment analysis for various RNA types in humans and mice, such as miRNA, lncRNA, circRNA, snoRNA, piRNA, and mRNA; (ii) extending the analysis by introducing millions of experimentally validated data of RNA–target interactions as a built-in database; and (iii) providing a comprehensive interacting network among various ncRNAs and targets to facilitate the mechanistic study of ncRNA function. Importantly, RNAenrich led to a more comprehensive and accurate enrichment analysis in a COVID-19-related miRNA case, which was largely attributed to its coverage of comprehensive ncRNA–target interactions.
Availability and implementation
RNAenrich is now freely accessible at https://idrblab.org/rnaenr/.
Graphical Abstract
Graphical Abstract.
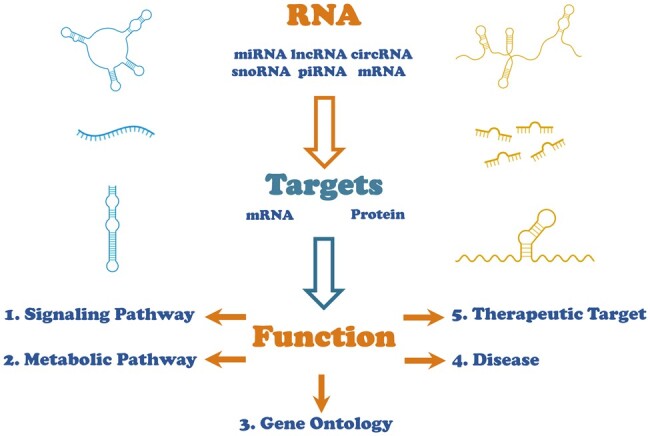
1 Introduction
Over the past quarter-century, non-coding RNAs (ncRNAs) have guided a prevalent trend in biomedical and life science research, and interest in ncRNAs and associated fields is continually expanding (Lee et al. 2020, Vidovic et al. 2020, Venkatesh et al. 2021, Yoo et al. 2021a). With the increasing amount of in depth ncRNA research, several kinds of regulatory ncRNAs, including microRNA (miRNA) (Gebert and MacRae 2019), long non-coding RNA (lncRNA) (Elling et al. 2018, Statello et al. 2021, Vollmers et al. 2021), circular RNA (circRNA) (Zeng et al. 2017, Kristensen et al. 2019, Jia et al. 2020), small nucleolar RNA (snoRNA) (Liang et al. 2019), and piwi-interacting RNA (piRNA) (Ozata et al. 2019), have received enormous attention from researchers due to their capacities to orchestrate gene expression (Chen et al. 2017, 2019b, Wang et al. 2021a). Uncovering their molecular mechanism during cellular processes is crucial for solving physiological and pathophysiological problems (Beermann et al. 2016, Goodall and Wickramasinghe 2021). Currently, the theoretical system of ncRNA regulation has been well established based on the cumulation of millions of published studies (Zeng et al. 2017, Elling et al. 2018, Gebert and MacRae 2019, Kristensen et al. 2019, Jia et al. 2020, Statello et al. 2021, Vollmers et al. 2021). For example, miRNA can bind to specific mRNAs to influence their stability or translation (Pillman et al. 2018, Gebert and MacRae 2019); lncRNA and circRNA are frequently involved in the process of sustaining mRNA and protein stability via interactions, and can also absorb miRNAs to deprive their original function (Kristensen et al. 2019, Statello et al. 2021); snoRNA modulates gene expression by controlling mRNA processing (Liang et al. 2019); and piRNA induces gene silencing in the formation of RNA–protein complexes with piwi-subfamily Argonaute proteins (Ozata et al. 2019). Therefore, a growing number of databases have been developed to accommodate the collection of ncRNA-related interactions [e.g. miRNA–mRNA (Yang et al. 2020, Huang et al. 2022) and lncRNA–miRNA (Li et al. 2014, Cruickshank et al. 2021)], as well as other associations [e.g. ncRNA–disease (Tang et al. 2018, Ning et al. 2021), and ncRNA-pathway (Kehl et al. 2020)].
Enrichment analysis is frequently applied to describe the function (involved-disease, pathway, cell process, etc.) of a gene set during the process of disease development and reveal the molecular mechanism of diverse diseases (Delorey et al. 2021). With the development of ncRNA-related association databases, some computational tools that focus on the enrichment of ncRNAs have been well constructed (Wu and Watson 2009, Hsu et al. 2011, Vlachos et al. 2015, Li et al. 2018, Kern et al. 2020, Chen et al. 2021a, Olgun et al. 2021), among them, TAM (Li et al. 2018) and miEAA (Kern et al. 2020) carry out an enrichment analysis for miRNA, and LncSEA (Chen et al. 2021a) is for lncRNA. Currently, the available enrichment strategies for ncRNA include (i) mapping a ncRNA with a function (disease or pathway) according to the literature or database (Li et al. 2018, Kern et al. 2020) and (ii) matching a ncRNA with a target mRNA/protein according to the literature or prediction data and then matching the protein with a term according to a well-established knowledge hierarchy (Kern et al. 2021). As reported, the former fails to cover the effect of diverse ncRNA interactions; therefore, this strategy may catch less valuable information (Li et al. 2018, Kern et al. 2020). Meanwhile, the predicted data of the latter may reduce the accuracy of the enrichment results (Huntley et al. 2018, Kern et al. 2021). In other words, due to the increasing interest in circRNAs, snoRNAs and piRNAs, it is essential to enable enrichment analysis of these newly identified ncRNAs. Moreover, since the function of ncRNAs depends heavily on their interacting target, the interactions between ncRNA and the corresponding target should be fully considered in functional enrichment (Kern et al. 2021). However, little tool that enables these valuable functions has yet been available for diverse ncRNAs.
In this study, a novel online tool named RNAenrich was developed to enable comprehensive functional enrichment analysis of diverse human and mouse ncRNAs. First, the interaction data between ncRNAs (circRNA, lncRNA, miRNA, piRNA, snoRNA, etc.) and their corresponding targets were collected using a systematic literature review in PubMed and various existing databases, which resulted in ∼1.87 million experimentally validated interactions. Second, enrichment analysis was realized by supporting a variety of functional categories, such as signaling pathway, metabolic pathway, Gene Ontology, disease, and therapeutic target. To the best of our knowledge, this tool is unique in that (i) it provides enrichment analysis for the most diverse types of ncRNA (not only lncRNA & miRNA, but also circRNA, piRNA & snoRNA) compared with existing tools; (ii) it extends the analysis by introducing millions of experiment-validated ncRNA–target interactions as a built-in database; and (iii) it provides an interacting network among various RNAs and different targets to facilitate the mechanistic study of ncRNA function. RNAenrich is now freely accessible without any login requirement at https://idrblab.org/rnaenr/.
2 Materials and methods
2.1 The functional enrichment strategy adopted in RNAenrich
Currently, the following two types of enrichment strategies for ncRNA sets are commonly used: putting an ncRNA into a disease, pathway, function, tissue location, etc., or matching an ncRNA with target functional mRNA/protein and then mapping the mRNA with various given terms. To ensure the diversity and reliability of the analysis, in this study, the second strategy was adopted. Therefore, the strategy of this study is matching an ncRNA with a target mRNA/protein according to experimentally validated data and then matching the protein with function according to well-established knowledge hierarchy, in which experimentally validated data ensure the accuracy of the enrichment results. Besides, the experimentally validated ncRNA interaction data were included high-confidence (low-throughput experimental data) and low-confidence (high-throughput experimental data) interactions. Users can choose confidence level according to their preferences. The tool has been developed so that an RNA list by RNA sequencing can be enriched to analyze associated functions, such as pathways and diseases.
RNAenrich can enrich five types of ncRNAs to analyze their function, including miRNAs, lncRNAs, circRNAs, snoRNAs, and piRNAs. All these ncRNAs generally interact with specific mRNAs or proteins and control their expression or activity, ultimately affecting the protein-induced signaling pathway. The following summary was well concluded from millions of studies: (i) miRNA generally interacts with specific mRNA and affects its stability and translation; (ii) lncRNA and circRNA can interact with miRNA to deprive its original functions and can also interact with mRNA and protein to affect their expression or activity; (iii) snoRNA regulates gene expression by controlling mRNA processing; and (iv) piRNA induces gene silencing by forming RNA–protein complexes with piwi-subfamily Argonaute proteins. In short, the functions of these five types of ncRNA heavily rely on interacting mRNAs or proteins. Therefore, the first step for RNAenrich analysis was matching the target mRNA/protein of the query ncRNA; all ncRNA–mRNA/protein interaction data were from diverse databases. After obtaining the targets of query ncRNA, the second step was carrying out an enrichment analysis via traditional enrichment databases, such as KEGG and Gene Ontology.
2.2 The ncRNA–target interaction data in RNAenrich
A variety of databases were included to determine the experimentally validated interactions, such as the miRNA–mRNA interactions from miRTarBase (Huang et al. 2022), miRecords (Xiao et al. 2009), miRSponge (Wang et al. 2015), OncomiRDB (Wang et al. 2014), Gene Ontology (Huntley et al. 2016, Gene Ontology Consortium 2021), IntAct (Del Toro et al. 2022), StarBase (Li et al. 2014), and TarBase (Karagkouni et al. 2018); lncRNA–mRNA, lncRNA–protein, and lncRNA–miRNA interactions in LncTarD (Zhao et al. 2020), LncACTdb (Wang et al. 2022b), LncRNA2Target (Cheng et al. 2019), and StarBase (Li et al. 2014); circRNA–mRNA, circRNA–protein, and circRNA–miRNA interactions circRNADisease (Zhao et al. 2018), StarBase (Li et al. 2014); snoRNA–mRNA and snoRNA–miRNA in SnoDB (Bouchard-Bourelle et al. 2020), StarBase (Li et al. 2014); and piRNA–mRNA and piRNA–protein in piRBase (Wang et al. 2022a). Among these, all interactions were recorded as strong- or weak-validation based on the selection of coefficient level. Overall, a total of 1.87 million ncRNA–target interaction data were collected and included in RNAenrich. These data are rich resources that are waiting for machine-learning tools (Chen et al. 2018b, Li et al. 2020, Meyer et al. 2020, Liu et al. 2021, Wang et al. 2021b, Hu et al. 2022a, Xia et al. 2022a, b, Li et al. 2023) to analyze, especially feature selection (Chen et al. 2018c, 2020, 2021b, Hu et al. 2021, 2022b, Too et al. 2022, Zhang et al. 2023) methods have great potential in this scenario.
2.3 The diverse functions that can be enriched in RNAenrich
Moreover, five popular databases were used in RNAenrich to facilitate the functional enrichment, including the KEGG (Kanehisa et al. 2021) and Reactome (Gillespie et al. 2022), which provided protein-directed signaling pathways that the ncRNA participate in; the SMPDB (Jewison et al. 2014), which offered metabolite-based pathways or reactions that ncRNAs regulate; Gene Ontology (Gene Ontology Consortium 2021), which contributed descriptions of the functional role of ncRNAs, their contribution to biological processes and location in the cell (especially, direct ncRNA biological process, cellular component, and molecular function annotations of Gene Ontology database are also included in built-in database of RNAenrich for enrichment analysis); TTD (Zhou et al. 2022) and KEGG (Kanehisa et al. 2021), which described the RNA-mediated occurrence and development of disease indication; and TTD (Zhou et al. 2022), which illustrated the therapeutic targets that ncRNAs regulate. Such diverse functional data can significantly enhance the capacity of ncRNA enrichment.
2.4 Server implementation details and required format of input
RNAenrich is deployed on a web server running Cent OS Linux v7.4.1708, Apache HTTP web server v2.4.6, and Apache Tomcat servlet container. Its web interface was developed by R v3.4.1 and Shiny v0.13.1 running on Shiny-server v1.4.1.759. Various R packages were utilized in the background processes. RNAenrich can be readily accessed by all users with no login requirement, and by diverse and popular web browsers, including Google Chrome, Mozilla Firefox, Safari, and Internet Explorer. The input is the ncRNA/gene set, which can be selected from the ncRNA/gene list of RNA sequencing/microarray data or the ncRNA/gene list of prediction data. To enhance the tolerance of RNAenrich analysis, RNAenrich allows diverse ID types as input RNA format for each RNA type. Five types of ncRNA (miRNA, lncRNA, circRNA, snoRNA, and piRNA) and mRNA lists can be analyzed in RNAenrich. RNAenrich allows users to select different ID lists (Table 1). RNAcentral is an authoritative and comprehensive ncRNA database in which each ncRNA is endowed with a unique ID (RNAcentral Consortium 2021). Therefore, all RNAs in RNAenrich are mapped to an RNAcentral ID if it is applicable in the RNAcentral database (RNAcentral does not include circRNAs and their corresponding information). In summary, six miRNA inputs can be accepted in RNAenrich, including RNAcentral ID, miRBase ID (mature), miRBase ID (stem-loop), Official Symbol, Gene ID, and miRNA name; five lncRNA inputs are accepted, including RNAcentral ID, Official Symbol, Gene ID, Ensembl ID, and lncRNA name; circRNA ID and circRNA name can be acceptable input format; five snoRNA inputs can be accepted, including RNAcentral ID, Official Symbol, Gene ID, Ensembl ID, and snoRNA name; RNAcentral ID and piRNA name can be input formats for mouse data; mRNA analysis allows five input formats, including Gene ID, Official Symbol, UniProt ID, Ensembl ID, and RefSeq ID. In addition, RNAenrich supports one type of RNA ID to convert to another type of ID in the ID conversion module, which can be downloaded on the web page as a text file.
Table 1.
Summary of input formats that RNAenrich supports.
| Species | RNA type | ID type | |||||
|---|---|---|---|---|---|---|---|
| Human | miRNA | RNAcentral ID | miRBase ID (mature) | miRBase ID (stem-loop) | Official symbol | Gene ID | miRNA name |
| lncRNA | RNAcentral ID | Official symbol | Gene ID | Ensembl ID | lncRNA name | ||
| circRNA | circRNA ID | circRNA name | |||||
| snoRNA | RNAcentral ID | Official symbol | Gene ID | Ensembl ID | snoRNA name | ||
| mRNA | Gene ID | Gene name | Uniprot ID | Ensembl ID | RefSeq ID | ||
| Mouse | miRNA | RNAcentral ID | miRBase ID (mature) | miRBase ID (stem-loop) | Official symbol | Gene ID | miRNA name |
| lncRNA | RNAcentral ID | Official symbol | Gene ID | Ensembl ID | lncRNA name | ||
| circRNA | circRNA ID | circRNA name | |||||
| snoRNA | RNAcentral ID | Official symbol | Gene ID | Ensembl ID | snoRNA name | ||
| piRNA | RNAcentral ID | piRNA name | |||||
| mRNA | Gene ID | Official symbol | Uniprot ID | Ensembl ID | RefSeq ID | ||
3 Results
3.1 The realization of the enrichment in RNAenrich
Studies in ncRNAs are rapidly increasing. Uncovering the biological meaning of regulatory RNA molecules in living organisms is an important trend in the field of ncRNA research (Covarrubias et al. 2017, Zhang et al. 2018, Gregory 2019, Slack and Chinnaiyan 2019). Enrichment analysis is a well-used strategy to apply to explore molecular biological mechanisms (Delorey et al. 2021). A few emerging enrichment web servers focus on functional annotation and enrichment of ncRNAs (Li et al. 2018, Cardenas et al. 2020, Kern et al. 2020, Chen et al. 2021a); however, these servers have shown a series of limitations. First, the RNA types that these servers can analyze are not diverse, only covering miRNA (Li et al. 2018, Kern et al. 2020) or lncRNA (Chen et al. 2021a). Few tools can carry out enrichment analysis for other types of ncRNAs that also play crucial roles in living organisms, such as circRNA, piRNA, and snoRNA. Second, some existing tools (Li et al. 2018, Kern et al. 2020) conduct the enrichment analysis by directly matching each ncRNA with a term (pathway or disease) according to the literature, which may miss some key information, others (Cardenas et al. 2020) employ predicted data in built-in databases, which lead to low confidence in the enrichment results. Therefore, both types of mentioned tools cannot enable to conduct a comprehensive and accurate enrichment analysis for a list of ncRNAs.
In this study, many improvements have been made to overcome these limitations, including coverage of the most diverse types of ncRNA, enhanced enrichment accuracy, and introducing functional information about RNA–RNA interactions. Now, the web server can conduct a comprehensive analysis for any ncRNA set (Fig. 1). First, RNAenrich allows users to upload an RNA list, which can be a filtered RNA list from RNA sequencing or microarray, an interacting RNA list that is predicted by a computational approach, and so on. RNA types can be miRNA, lncRNA, circRNA, snoRNA, piRNA, or mRNA/gene, and the input format can be RNAcentral ID, Gene ID, Accession ID, Official Symbol, Ensembl ID, RNA name, miRbase ID (Mature miRNA), miRbase ID (Stem-loop miRNA), UniProt ID, or RefSeq ID (Table 1). Second, the input RNAs will be matched to their target mRNA or protein using the built-in database from 14 independent databases [miRTarBase (Huang et al. 2022), miRecords (Xiao et al. 2009), miRSponge (Wang et al. 2015), OncomiRDB (ling Chen et al. 2014), Gene Ontology (Huntley et al. 2016, Gene Ontology Consortium 2021), IntAct (Del Toro et al. 2022), StarBase (Fu et al. 2022), TarBase (Karagkouni et al. 2018), LncTarD (Zhao et al. 2020), LncACTdb (Wang et al. 2022b), LncRNA2Target (Cheng et al. 2019), circRNADisease (Zhao et al. 2018), SnoDB (Bouchard-Bourelle et al. 2020), and piRBase (Wang et al. 2022a)], which have experimentally validated ncRNA–target interactions of 1.87 million pairs covering all regulatory RNA types. Third, the mapped mRNAs will be enriched in five ways based on information from nine databases, namely, (i) signaling pathway, (ii) metabolic pathway, (iii) Gene Ontology, (iv) disease, and (v) therapeutic target; all of enrichment categories have attracted increasing attention in the field of RNA (Matsui and Corey 2017, Anastasiadou et al. 2018, Chen et al. 2018a, Zhang et al. 2019a, Zhu et al. 2019). Then, the enriched results will be presented as downloadable tables and visualized pictures, including bar plots, bubble plots, pathway correlation plots, and protein–protein interaction (PPI) networks. Specifically, the added PPI network will describe the regulatory molecular mechanism at the protein level in which the ncRNAs are involved. The network reveals PPIs that ncRNAs regulate via multiple signaling pathways. Finally, if users want to explore the RNA network that a query ncRNA regulates, RNA–RNA interaction module can provide an RNA–RNA regulatory network profile to show further detailed mechanisms.
Figure 1.
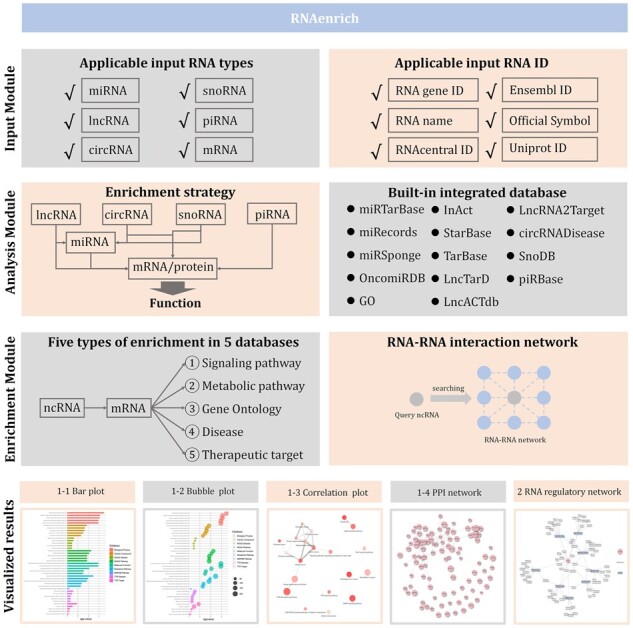
The workflow of RNAenrich. First, the users can input an RNA list; in this step, six types of RNA are optional. Then, the input RNA will be analyzed by built-in databases and a new enrichment strategy, matching a ncRNA with functional mRNA and then to a term. As a result, the tool will generate an enrichment analysis and an RNA–RNA interaction analysis, in which the former will be presented based on five different enrichments and the latter will profile a directional RNA–RNA regulatory network
3.2 The statistics of RNAenrich and current ncRNA enrichment server
Some existing tools employ a direct strategy, mapping an ncRNA to a term (a disease, a pathway, and so on) to generate an enrichment analysis (Table 2). Mechanically, as all ncRNAs execute their function depending on their associated coding RNAs or proteins, such a strategy may lead to the missing of target-associated functional information. Therefore, RNAenrich uses experiment-supported ncRNA–target interactions as built-in data to map their targets of query ncRNA list and capture the function of these targets by databases, such as KEGG and Reactome, which will generate a more comprehensive enrichment analysis relying on associated targets. Furthermore, these existing tools, such as TAM and MEAA, focus on the enrichment analysis of a single type of ncRNA, while RNAenrich includes five types of popular ncRNAs (miRNA, lncRNA, circRNA, snoRNA, and piRNA) and has sufficient coverage of ncRNA number in humans and mice (Table 3).
Table 2.
Main content and function of RNAenrich and other representative web servers.
| Server | RNA type | Evidence | Enriched strategy | Respective features | Reference |
|---|---|---|---|---|---|
| RNAenrich | Six types | Experiment-supported | ncRNA-mRNA-term | Pathway; metabolism; function; disease; therapeutic target; network | This study |
| TAM | miRNA | Experiment-supported | miRNA-term | Function; disease; transcription factor; classification; tissue and cell | (Li et al. 2018) |
| miEAA | miRNA | Experiment-supported | miRNA-term (most) | Function; disease; transcription factor; classification; tissue and cell; pathway; biomarker; localization | (Kern et al. 2020) |
| LncSEA | lncRNA | Experiment-supported | lncRNA-term | Function; disease; transcription factor; cancer; biomarker; regulation; drug | (Chen et al. 2021a) |
Table 3.
Comparison of RNAenrich and other representative web servers in coverage of RNA types and species.
| Server | miRNA |
lncRNA |
circRNA |
snoRNA |
piRNA |
mRNA |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | Mouse | Human | Mouse | Human | Mouse | Human | Mouse | Human | Mouse | Human | Mouse | |
| RNAenrich | √ | √ | √ | √ | √ | √ | √ | √ | × | √ | √ | √ |
| TAM | √ | × | × | × | × | × | × | × | × | × | × | × |
| miEAA | √ | √ | × | × | × | × | × | × | × | × | × | × |
| LncSEA | × | × | √ | × | × | × | × | × | × | × | × | × |
Taking advantage of these existing tools and adding popular trends to ncRNA research, the following five important aspects were simplified and summarized by RNAenrich to serve researchers (Table 2): (i) signaling pathway, to describe the protein-directed signaling conduction pathway that RNAs participate in, such as lncRNA AK023391 for the PI3K/Akt signaling pathway (Huang et al. 2017) and circIKBKB for the NF-κB pathway (Xu et al. 2021); (ii) metabolic pathway, to describe the metabolite-based pathway or reaction that RNAs regulate, such as lncRNA HISLA for aerobic glycolysis (Chen et al. 2019a) and miR-147b for the TCA cycle (Zhang et al. 2019b); (iii) Gene Ontology, to describe the functional role of ncRNAs and their contribution to biological processes and location in the cell, such as lncRNA SLERT for transcription (Wu et al. 2021) and lncRNA LETN for nucleolar structure (Wang et al. 2021c); (iv) disease, to describe involvement of ncRNA in the occurrence or development of disease or complications, such as miRNA-342-3p for hepatocellular carcinoma (Komoll et al. 2021) and LINC01123 for non-small cell lung cancer (Hua et al. 2019); and (v) therapeutic target, such as miRNA let-7 for immunotherapy (Gilles and Slack 2018) and lncRNA H19 for therapeutic target of pancreatic cancer (Wang et al. 2020). Each of these aspects has contributed to a bulk of publications, to reveal the physiological and pathophysiological mechanism, by exploration of ncRNA associations.
3.3 Comparing RNAenrich with existing tools based on COVID-19 data
To date, two online tools have already been developed for conducting an enrichment analysis for miRNAs, including TAM (Li et al. 2018) and miEAA (Kern et al. 2020). TAM is a very popular tool for enrichment analysis of a list of miRNAs, and has been applied to the research on molecular mechanism and signaling pathway of miRNAs in diverse diseases (Soleimani Zakeri et al. 2020). Compared with the TAM, miEAA is a more comprehensive web server for miRNA enrichment and annotation, which has made many improvements, such as providing the most coverage of miRNAs, enhancing functional diversity, and integrating multiple species (Kern et al. 2020). As reported, the significance and abundance of enrichment results are key indicators of enrichment analysis (Yang et al. 2021). In other words, the more significant and abundant results a server can enrich, the more valuable information it can provide, facilitating further discovery and analysis of biological molecular mechanisms for ncRNAs (Yang et al. 2021). Therefore, in this study, we chose miEAA to compare with RNAenrich in term of the significance and abundance of enrichment results.
We use a miRNA list from Khan’s study (Khan et al. 2020) as the test data, in which 106 human miRNAs are predicted to interact with SARS-CoV-2 genomic RNA. This study concluded that these miRNAs could regulate immune-signaling pathways and viral infection processes. In other words, the enrichment report of these miRNAs should be related to immune-signaling pathways, inflammation-related pathways, and virus-related diseases. Therefore, we analyzed the test list with RNAenrich and miEAA and compared the enrichment results. For the involved-signaling pathway, we summarized the pathways directly related to COVID-19. According to the authoritative studies by Wiersinga et al. (2020) and Blanco-Melo et al. (2020), COVID-19 presents as a viral infection and replication in the early stage, and as excessive inflammatory responses and immunological stress (typical as interleukin-induced pathways) in the late stage of infection. Therefore, we filtered virus infection-, interleukin-, and immune system-related pathways in the enrichment results from both tools.
Interestingly, a series of COVID-19 related pathways were significantly enriched by RNAenrich (shown in Fig. 2 and Table 4), e.g. human interleukin-4 and interleukin-13 signaling (P = 5.74E-14) (P refers to P.adjust), interleukin-6 signaling (P = 8.64E-04), and so on (shown in Table 4 and Supplementary Tables S1 and S2). However, miEAA enrichment identified some unrelated pathways (shown in Fig. 2 and Supplementary Tables S3 and S4). For disease enrichment, RNAenrich captured some SARS-CoV-2-related or similar diseases (shown in Table 4), including COVID-19 (P = 3.49E-02), and lupus erythematosus (a type of immune system disease) (P = 8.28E-03); however, miEAA failed to significant diseases (shown in Supplementary Table S5), with some non-significant related terms. As shown in Table 4, a series of COVID-19-related diseases and signaling pathways were significantly enriched in RNAenrich but not in miEAA (shown in Supplementary Table S6). Overall, RNAenrich has illustrated its enhanced performance in enriching the COVID-19-related miRNA list compared with miEAA.
Figure 2.
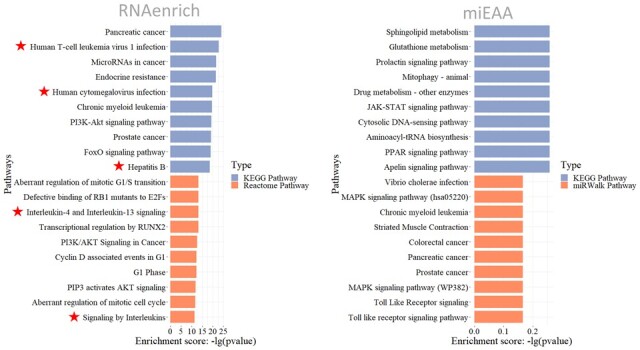
Pathway enrichment results (top 10) of RNAenrich (left) and miEAA (right) for a COVID-19-related miRNA list as input. Pathway enrichment patterns included KEGG and Reactome by RNAenrich and KEGG and miRWalk by miEAA
Table 4.
SARS-CoV-2-related pathways and diseases based on enrichment results from RNAenrich with a significant difference (P.adjust < 0.05).
| Description | Rate | P-value | P.adjust |
|---|---|---|---|
| Human T-cell leukemia virus 1 infection | 41/222 | 3.07E−24 | 2.53E−22 |
| Human cytomegalovirus infection | 39/225 | 5.20E−22 | 2.14E−20 |
| Hepatitis B | 32/162 | 6.23E−20 | 1.40E−18 |
| Human papillomavirus infection | 42/331 | 2.33E−18 | 3.39E−17 |
| Epstein–Barr virus infection | 32/202 | 6.02E−17 | 6.76E−16 |
| Kaposi sarcoma-associated herpesvirus infection | 30/194 | 1.23E−15 | 1.21E−14 |
| Hepatitis C | 27/157 | 2.40E−15 | 2.20E−14 |
| Interleukin-4 and interleukin-13 signaling | 23/108 | 1.91E−16 | 5.74E−14 |
| Signaling by interleukins | 41/462 | 3.83E−14 | 3.68E−12 |
| Human immunodeficiency virus 1 infection | 20/212 | 5.11E−07 | 1.58E−06 |
| Interleukin-6 signaling | 4/11 | 7.61E−05 | 8.64E−04 |
| Interleukin-10 signaling | 7/47 | 8.39E−05 | 9.41E−04 |
| Interleukin-3, interleukin-5, and GM-CSF signaling | 7/48 | 9.63E−05 | 1.04E−03 |
| Interleukin-6 family signaling | 5/24 | 1.75E−04 | 1.63E−03 |
| Interleukin-15 signaling | 4/14 | 2.19E−04 | 1.95E−03 |
| Coronavirus disease—COVID-19 | 15/232 | 9.80E−04 | 2.03E−03 |
| Interleukin receptor SHC signaling | 5/27 | 3.14E−04 | 2.60E−03 |
| Interleukin-2 family signaling | 6/44 | 4.47E−04 | 3.53E−03 |
| Interleukin-7 signaling | 5/36 | 1.24E−03 | 8.16E−03 |
| Lupus erythematosus | 9/45 | 8.57E−04 | 8.28E−03 |
| Cutaneous lupus erythematosus | 4/11 | 2.66E−03 | 1.93E−02 |
| COVID-19 | 10/70 | 6.26E−03 | 3.49E−02 |
3.4 Visualization of enrichment results in RNAenrich
Currently, two functions can be carried out by RNAenrich for an ncRNA list, RNA functional enrichment and RNA–RNA interaction network analysis. According to the RNAenrich procedure, users first input the RNA list and choose some related options and then obtain the result page after 5–60 s (the waiting time depends on the number of selected databases). The result page first shows the following information: (i) an enrichment analysis report with term name, database, link ID, P-value, P.adjust, and so on, which is presented as a table and can be downloaded as CSV file; (ii) visualized plots of the enrichment results (bar plot, bubble plot, and correlation plot for selectable one and more databases) shown in Fig. 3A–C; and (iii) a PPI network plot (shown in Fig. 3D) to describe which PPI pairs are regulated by these ncRNAs, and these nodes with more neighbors may be key proteins that ncRNA can regulate.
Figure 3.
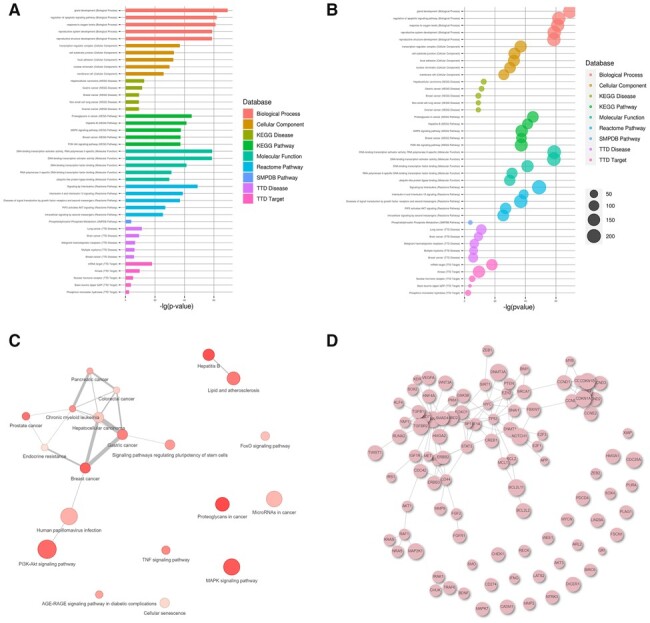
Visualization of the enrichment results by RNAenrich. Bar (A) and bubble (B) plots of enrichment results in five databases. (C) The correlation plot of enrichment results in a single database. (D) The PPI network of proteins that ncRNAs regulate is shown
As the regulation among different RNAs is of great concern, e.g. miRNA–mRNA (Yoo et al. 2021b), lncRNA–miRNA–mRNA (Wasson et al. 2021), and circRNA–miRNA–mRNA (Neumann et al. 2018), our server also provides RNA–RNA interaction information in a downloadable table and picture to describe the regulatory network that a ncRNA is involved in. As shown in Fig. 4A, the miRNA regulatory network is shown in this way because its regulatory mode is simple. For lncRNAs, the mechanism of action is more diverse, such as binding to DNA promoters, miRNAs, mRNAs, and proteins, therefore, the regulatory network of lncRNA is complicated and crosslinked (shown in Fig. 4B), which may provide more regulatory information to uncover molecular mechanisms in diseases for users.
Figure 4.
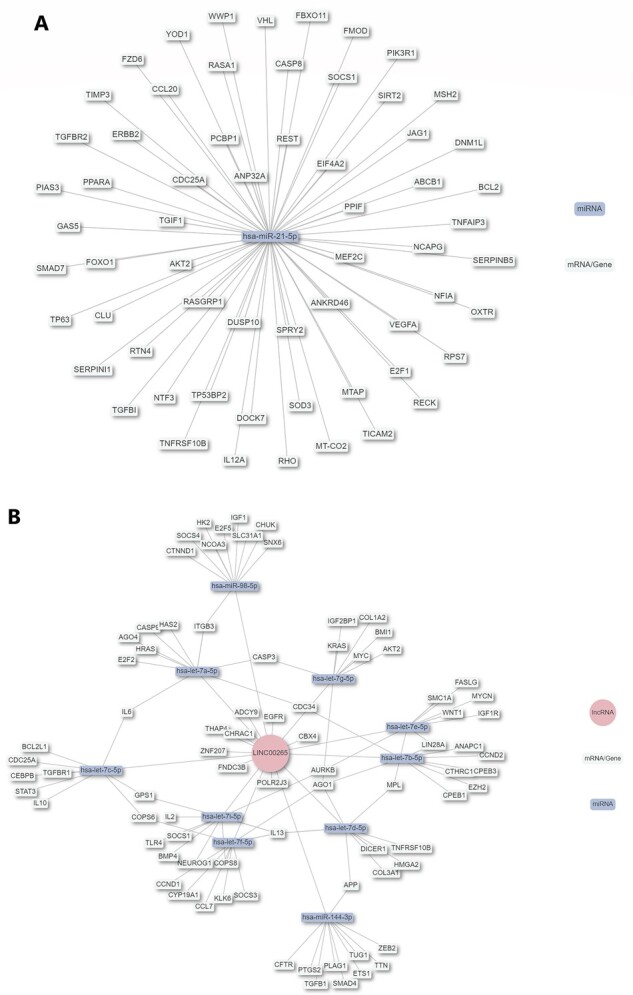
Visualization of the RNA–RNA interaction network. The regulatory networks that miRNAs (A) and lncRNAs (B) direct. (A) The miRNA-central network consists of a series of miRNA–mRNA regulations, and (B) the lncRNA-central network consists of diverse regulations, such as lncRNA–miRNA, miRNA–mRNA, lncRNA–mRNA, and lncRNA–protein.
4 Conclusions
In this study, an online tool, RNAenrich, was developed to enable comprehensive enrichment analysis of ncRNAs. This tool is unique in that (i) it can be used for enrichment analysis for various RNA types, such as circRNAs, snoRNAs, piRNAs, miRNAs, lncRNAs, and mRNAs; (ii) the analysis can be extended by introducing millions of experimentally validated RNA–target interactions as a built-in database; and (iii) the results provide a comprehensive interacting network among various ncRNAs and targets to facilitate the mechanistic study of ncRNA function. Interest in the exploration of ncRNAs is gradually extending and transforming from interest in miRNAs and lncRNAs into interest in newly identified ncRNAs, such as circRNAs, piRNAs, and snoRNAs. RNAenrich now enables enrichment analysis of all RNA types, which will be beneficial for researchers from different ncRNA fields. Studies on ncRNAs are ongoing and expanding, and RNAenrich will also be improving to better serve the field of ncRNA research.
Supplementary Material
Acknowledgements
The authors would like to thank RNAenrich users for providing feature requests and identifying bugs.
Contributor Information
Song Zhang, Center for Clinical Pharmacy, Cancer Center, Department of Pharmacy, Zhejiang Provincial People’s Hospital, Affiliated People's Hospital, Hangzhou Medical College, Hangzhou, Zhejiang 310014, China; College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Kuerbannisha Amahong, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Yintao Zhang, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Xiaoping Hu, Center for Clinical Pharmacy, Cancer Center, Department of Pharmacy, Zhejiang Provincial People’s Hospital, Affiliated People's Hospital, Hangzhou Medical College, Hangzhou, Zhejiang 310014, China.
Shijie Huang, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Mingkun Lu, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Zhenyu Zeng, Innovation Institute for Artificial Intelligence in Medicine of Zhejiang University, Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare, Hangzhou, Zhejiang 330110, China.
Zhaorong Li, Innovation Institute for Artificial Intelligence in Medicine of Zhejiang University, Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare, Hangzhou, Zhejiang 330110, China.
Bing Zhang, Innovation Institute for Artificial Intelligence in Medicine of Zhejiang University, Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare, Hangzhou, Zhejiang 330110, China.
Yunqing Qiu, State Key Laboratory for Diagnosis and Treatment of Infectious Disease, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Zhejiang Provincial Key Laboratory for Drug Clinical Research and Evaluation, The First Affiliated Hospital, Zhejiang University, Hangzhou, Zhejiang 310000, China.
Haibin Dai, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Jianqing Gao, College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China; Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang 310030, China.
Feng Zhu, Center for Clinical Pharmacy, Cancer Center, Department of Pharmacy, Zhejiang Provincial People’s Hospital, Affiliated People's Hospital, Hangzhou Medical College, Hangzhou, Zhejiang 310014, China; College of Pharmaceutical Sciences, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058, China.
Supplementary data
Supplementary data are available at Bioinformatics online.
Conflict of interest
None declared.
Funding
This work was supported by Natural Science Foundation of Zhejiang Province [LR21H300001]; National Natural Science Foundation of China [22220102001, U1909208]; Leading Talent of the “Ten Thousand Plan”—National High-Level Talents Special Support Plan of China; Fundamental Research Fund for Central Universities [2018QNA7023]; “Double Top-Class” University Project [181201*194232101]; and Key R&D Program of Zhejiang Province [2020C03010]. This work was also supported by Westlake Laboratory (Westlake Laboratory of Life Sciences and Biomedicine); Alibaba-Zhejiang University Joint Research Center of Future Digital Healthcare; Alibaba Cloud; and Information Technology Center of Zhejiang University.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
References
- Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer 2018;18:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beermann J, Piccoli M-T, Viereck J et al. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev 2016;96:1297–325. [DOI] [PubMed] [Google Scholar]
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020;181:1036–45.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard-Bourelle P, Desjardins-Henri C, Mathurin-St-Pierre D et al. snoDB: an interactive database of human snoRNA sequences, abundance and interactions. Nucleic Acids Res 2020;48:D220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas J, Balaji U, Gu J. Cerina: systematic circRNA functional annotation based on integrative analysis of ceRNA interactions. Sci Rep 2020;10:22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Chen J, Yang L et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol 2019a;21:498–510. [DOI] [PubMed] [Google Scholar]
- Chen HL, Yang B, Wang SJ et al. Towards an optimal support vector machine classifier using a parallel particle swarm optimization strategy. Appl Math Comput 2014;239:180–97. [Google Scholar]
- Chen J, Zhang J, Gao Y et al. LncSEA: a platform for long non-coding RNA related sets and enrichment analysis. Nucleic Acids Res 2021a;49:D969–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Cai J, Wang Q et al. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/beta-catenin pathway by scaffolding EZH2. Clin Cancer Res 2018a;24:684–95. [DOI] [PubMed] [Google Scholar]
- Chen X, Yan CC, Zhang X et al. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform 2017;18:558–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Guan NN, Sun YZ et al. MicroRNA-small molecule association identification: from experimental results to computational models. Brief Bioinform 2018b;21:47–61. [DOI] [PubMed] [Google Scholar]
- Chen X, Xie D, Zhao Q et al. MicroRNAs and complex diseases: from experimental results to computational models. Brief Bioinform 2019b;20:515–39. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhao P, Li C et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res 2021b;49:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhao P, Li F et al. iFeature: a python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018c;34:2499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhao P, Li F et al. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 2020;21:1047–57. [DOI] [PubMed] [Google Scholar]
- Cheng L, Wang P, Tian R et al. LncRNA2Target v2.0: a comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res 2019;47:D140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias S, Robinson EK, Shapleigh B et al. CRISPR/cas-based screening of long non-coding RNAs (lncRNAs) in macrophages with an NF-kappaB reporter. J Biol Chem 2017;292:20911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank BM, Wasson M-CD, Brown JM et al. LncRNA PART1 promotes proliferation and migration, is associated with cancer stem cells, and alters the miRNA landscape in triple-negative breast cancer. Cancers 2021;13:2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Toro N, Shrivastava A, Ragueneau E et al. The IntAct database: efficient access to fine-grained molecular interaction data. Nucleic Acids Res 2022;50:D648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorey TM, Ziegler CGK, Heimberg G et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021;595:107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling R, Robinson EK, Shapleigh B et al. Genetic models reveal cis and trans immune-regulatory activities for lincRNA-Cox2. Cell Rep 2018;25:1511–24.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Zhang Y, Wang Y et al. Optimization of metabolomic data processing using NOREVA. Nat Protoc 2022;17:129–51. [DOI] [PubMed] [Google Scholar]
- Gebert LF, R, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 2019;20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 2021;49:D325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles ME, Slack FJ. Let-7 microRNA as a potential therapeutic target with implications for immunotherapy. Expert Opin Ther Targets 2018;22:929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie M, Jassal B, Stephan R et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res 2022;50:D687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall GJ, Wickramasinghe VO. RNA in cancer. Nat Rev Cancer 2021;21:22–36. [DOI] [PubMed] [Google Scholar]
- Gregory PA. The miR-200-Quaking axis functions in tumour angiogenesis. Oncogene 2019;38:6767–9. [DOI] [PubMed] [Google Scholar]
- Hsu JB-K, Chiu C-M, Hsu S-D et al. miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics 2011;12:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Chen H, Heidari AA et al. Orthogonal learning covariance matrix for defects of grey wolf optimizer: insights, balance, diversity, and feature selection. Knowl Based Syst 2021;213:106684. [Google Scholar]
- Hu J, Gui W, Heidari AA et al. Dispersed foraging slime mould algorithm: continuous and binary variants for global optimization and wrapper-based feature selection. Knowl Based Syst 2022a;237:107761. [Google Scholar]
- Hu J, Han Z, Heidari AA et al. Detection of COVID-19 severity using blood gas analysis parameters and Harris hawks optimized extreme learning machine. Comput Biol Med 2022b;142:105166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Q, Jin M, Mi B et al. LINC01123, a c-Myc-activated long non-coding RNA, promotes proliferation and aerobic glycolysis of non-small cell lung cancer through miR-199a-5p/c-Myc axis. J Hematol Oncol 2019;12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-Y, Lin Y-C-D, Cui S et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 2022;50:D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang J, Hou L et al. LncRNA AK023391 promotes tumorigenesis and invasion of gastric cancer through activation of the PI3K/akt signaling pathway. J Exp Clin Cancer Res 2017;36:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley RP, Kramarz B, Sawford T et al. Expanding the horizons of microRNA bioinformatics. RNA 2018;24:1005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley RP, Sitnikov D, Orlic-Milacic M et al. Guidelines for the functional annotation of microRNAs using the gene ontology. RNA 2016;22:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewison T, Su Y, Disfany FM et al. SMPDB 2.0: big improvements to the small molecule pathway database. Nucleic Acids Res 2014;42:D478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Bi Y, Chen J et al. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics 2020;36:4276–82. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Sato Y et al. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res 2021;49:D545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagkouni D, Paraskevopoulou MD, Chatzopoulos S et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res 2018;46:D239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl T, Kern F, Backes C et al. miRPathDB 2.0: a novel release of the miRNA pathway dictionary database. Nucleic Acids Res 2020;48:D142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern F, Aparicio-Puerta E, Li Y et al. miRTargetLink 2.0-interactive miRNA target gene and target pathway networks. Nucleic Acids Res 2021;49:W409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern F, Fehlmann T, Solomon J et al. miEAA 2.0: integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res 2020;48:W521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA-A-K, Sany MRU, Islam MS et al. Epigenetic regulator miRNA pattern differences among SARS-CoV, SARS-CoV-2, and SARS-CoV-2 world-wide isolates delineated the mystery behind the epic pathogenicity and distinct clinical characteristics of pandemic COVID-19. Front Genet 2020;11:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoll R-M, Hu Q, Olarewaju O et al. MicroRNA-342-3p is a potent tumour suppressor in hepatocellular carcinoma. J Hepatol 2021;74:122–34. [DOI] [PubMed] [Google Scholar]
- Kristensen LS, Andersen MS, Stagsted LVW et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 2019;20:675–91. [DOI] [PubMed] [Google Scholar]
- Lee TJ, Yuan X, Kerr K et al. Strategies to modulate MicroRNA functions for the treatment of cancer or organ injury. Pharmacol Rev 2020;72:639–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Yin J, Lu M et al. DrugMAP: molecular atlas and pharma-information of all drugs. Nucleic Acids Res 2023;51:D1288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han X, Wan Y et al. TAM 2.0: tool for MicroRNA set analysis. Nucleic Acids Res 2018;46:W180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-H, Liu S, Zhou H et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 2014;42:D92–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Li XX, Hong JJ et al. Clinical trials, progression-speed differentiating features, and swiftness rule of the innovative targets of first-in-class drugs. Brief Bioinform 2020;21:649–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Wen J, Huang Z et al. Small nucleolar RNAs: insight into their function in cancer. Front Oncol 2019;9:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen J, Wang Y et al. DeepTorrent: a deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief Bioinform 2021;22:bbaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov 2017;16:167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SM, Williams CC, Akahori Y et al. Small molecule recognition of disease-relevant RNA structures. Chem Soc Rev 2020;49:7167–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann DP, Goodall GJ, Gregory PA. Regulation of splicing and circularisation of RNA in epithelial mesenchymal plasticity. Semin Cell Dev Biol 2018;75:50–60. [DOI] [PubMed] [Google Scholar]
- Ning L, Cui T, Zheng B et al. MNDR v3.0: mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res 2021;49:D160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgun G, Nabi A, Tastan O. NoRCE: non-coding RNA sets cis enrichment tool. BMC Bioinformatics 2021;22:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A et al. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 2019;20:89–108. [DOI] [PubMed] [Google Scholar]
- Pillman KA, Phillips CA, Roslan S et al. miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein quaking. EMBO J 2018;37:e99016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RNAcentral Consortium. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res 2021;49:D212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell 2019;179:1033–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani Zakeri NS, Pashazadeh S, MotieGhader H. Gene biomarker discovery at different stages of Alzheimer using gene co-expression network approach. Sci Rep 2020;10:12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statello L, Guo C-J, Chen L-L et al. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 2021;22:96–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Wan S, Yang Z et al. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018;34:398–406. [DOI] [PubMed] [Google Scholar]
- Too J, Liang G, Chen H. Memory-based Harris hawk optimization with learning agents: a feature selection approach. Eng Comput 2022;38:4457–78. [Google Scholar]
- Venkatesh J, Wasson M-CD, Brown JM et al. LncRNA-miRNA axes in breast cancer: novel points of interaction for strategic attack. Cancer Lett 2021;509:81–8. [DOI] [PubMed] [Google Scholar]
- Vidovic D, Huynh TT, Konda P et al. ALDH1A3-regulated long non-coding RNA NRAD1 is a potential novel target for triple-negative breast tumors and cancer stem cells. Cell Death Differ 2020;27:363–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos IS, Zagganas K, Paraskevopoulou MD et al. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res 2015;43:W460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers AC, Covarrubias S, Kuang D et al. A conserved long noncoding RNA, GAPLINC, modulates the immune response during endotoxic shock. Proc Natl Acad Sci USA 2021;118:e2016648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Han CD, Zhao Q et al. Circular RNAs and complex diseases: from experimental results to computational models. Brief Bioinform 2021a;22:bbab286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Zhao Y, Chen X. Drug-pathway association prediction: from experimental results to computational models. Brief Bioinform 2021b;22:bbaa061. [DOI] [PubMed] [Google Scholar]
- Wang D, Gu J, Wang T et al. OncomiRDB: a database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics 2014;30:2237–8. [DOI] [PubMed] [Google Scholar]
- Wang J, Shi Y, Zhou H et al. piRBase: integrating piRNA annotation in all aspects. Nucleic Acids Res 2022a;50:D265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Guo Q, Qi Y et al. LncACTdb 3.0: an updated database of experimentally supported ceRNA interactions and personalized networks contributing to precision medicine. Nucleic Acids Res 2022b;50:D183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhi H, Zhang Y et al. miRSponge: a manually curated database for experimentally supported miRNA sponges and ceRNAs. Database (Oxford) 2015;2015:bav098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hu X, Song W et al. Mutual dependency between lncRNA LETN and protein NPM1 in controlling the nucleolar structure and functions sustaining cell proliferation. Cell Res 2021c;31:664–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang S, Li F et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res 2020;48:D1031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasson M-CD, Brown JM, Venkatesh J et al. Datasets exploring putative lncRNA-miRNA-mRNA axes in breast cancer cell lines. Data Brief 2021;37:107241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersinga WJ, Rhodes A, Cheng AC et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 2020;324:782–93. [DOI] [PubMed] [Google Scholar]
- Wu M, Xu G, Han C et al. lncRNA SLERT controls phase separation of FC/DFCs to facilitate Pol I transcription. Science 2021;373:547–55. [DOI] [PubMed] [Google Scholar]
- Wu X, Watson M. CORNA: testing gene lists for regulation by microRNAs. Bioinformatics 2009;25:832–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wang Z, Yang D et al. Performance optimization of support vector machine with oppositional grasshopper optimization for acute appendicitis diagnosis. Comput Biol Med 2022a;143:105206. [DOI] [PubMed] [Google Scholar]
- Xia J, Yang D, Zhou H et al. Evolving kernel extreme learning machine for medical diagnosis via a disperse foraging sine cosine algorithm. Comput Biol Med 2022b;141:105137. [DOI] [PubMed] [Google Scholar]
- Xiao F, Zuo Z, Cai G et al. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009;37:D105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhang S, Liao X et al. Circular RNA circIKBKB promotes breast cancer bone metastasis through sustaining NF-kappaB/bone remodeling factors signaling. Mol Cancer 2021;20:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Li B, Chen S et al. MMEASE: online meta-analysis of metabolomic data by enhanced metabolite annotation, marker selection and enrichment analysis. J Proteomics 2021;232:104023. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li B, Tang J et al. Consistent gene signature of schizophrenia identified by a novel feature selection strategy from comprehensive sets of transcriptomic data. Brief Bioinform 2020;21:1058–68. [DOI] [PubMed] [Google Scholar]
- Yoo JY, Yeh M, Kaur B et al. Targeted delivery of small noncoding RNA for glioblastoma. Cancer Lett 2021a;500:274–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J-Y, Yeh M, Wang Y-Y et al. MicroRNA-138 increases chemo-sensitivity of glioblastoma through downregulation of Survivin. Biomedicines 2021b;9:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Lin W, Guo M et al. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 2017;13:e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zou S, Deng L. Gene Ontology-based function prediction of long non-coding RNAs using bi-random walk. BMC Med Genomics 2018;11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Wells JM, Chow K-H et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat Metab 2019a;1:460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun H, Lian X et al. ANPELA: significantly enhanced quantification tool for cytometry-based single-cell proteomics. Adv Sci (Weinh) 2023;10:e2207061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shi J, Rassoulzadegan M et al. Sperm RNA code programmes the metabolic health of offspring. Nat Rev Endocrinol 2019b;15:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Shi J, Zhang Y et al. LncTarD: a manually-curated database of experimentally-supported functional lncRNA-target regulations in human diseases. Nucleic Acids Res 2020;48:D118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wang K, Wu F et al. circRNA disease: a manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis 2018;9:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhang Y, Lian X et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res 2022;50:D1398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K-P, Zhang C-L, Ma X-L et al. Analyzing the interactions of mRNAs and ncRNAs to predict competing endogenous RNA networks in osteosarcoma chemo-resistance. Mol Ther 2019;27:518–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.