

Abstract
Background:
ERK5 is a dual kinase-transcription factor containing an N-terminal kinase domain and a C-terminal transcriptional activation domain. Many ERK5 kinase inhibitors have been developed and tested to treat cancer and inflammatory diseases. However, recent data have raised questions about the role of the catalytic activity of ERK5 in proliferation and inflammation. We aimed to investigate how ERK5 reprograms myeloid cells (MCs) to the pro-inflammatory senescent phenotype, subsequently leading to atherosclerosis.
Methods:
A ERK5 S496A (dephosphorylation mimic) knock-in (KI) mouse model was generated using CRISPR/Cas9, and atherosclerosis was characterized by hypercholesterolemia induction. The plaque phenotyping in homozygous ERK5 S496A KI and wild-type mice was studied using imaging mass cytometry. Bone marrow-derived macrophages (BMDMs) were isolated from hypercholesterolemic mice and characterized using RNA sequencing and functional in vitro approaches, including senescence, mitochondria reactive oxygen species, and inflammation assays, as well as by metabolic extracellular flux analysis.
Results:
We show that atherosclerosis was inhibited in ERK5 S496A KI mice. Furthermore, ERK5 S496 phosphorylation mediates both senescence-associated secretory phenotype (SASP) as well as senescence-associated stemness (SAS) by upregulating aryl hydrocarbon receptor (AHR) in plaque and BMDMs isolated from hypercholesterolemic mice. We also discovered that ERK5 S496 phosphorylation could induce NRF2 SUMOylation at a novel K518 site to inhibit NRF2 transcriptional activity without altering ERK5 catalytic activity and mediates oxidized LDL (oxLDL)-induced SASP. Specific ERK5 kinase inhibitors (AX15836 and XMD8-92) also inhibited ERK5 S496 phosphorylation, suggesting the involvement of ERK5 S496 phosphorylation in the anti-inflammatory effects of these ERK5 kinase inhibitors.
Conclusions:
We discovered a novel mechanism by which the macrophage ERK5-NRF2 axis develops a unique SASP/SAS phenotype by upregulating AHR to engender atherogenesis. The finding of SAS provides a molecular explanation to resolve the paradox of senescence in proliferative plaque by permitting MCs to escape the senescence-induced cell cycle arrest during atherosclerosis formation.
Keywords: Oxidized LDL, atherosclerosis, senescence-associated secretory phenotype, efferocytosis, antioxidants, p90RSK, NRF2, ERK5, imaging mass cytometry
Graphical Abstract
INTRODUCTION
Age is a significant risk factor for atherosclerosis, and cellular senescence is associated with the pro-inflammatory phenotype of macrophages, which has been described as a senescence-associated secretory phenotype (SASP) 1–3. SASP is believed to play a vital role in atherosclerosis 4–6. We have already identified the critical role of four components of SASP in the form of 1) telomere shortening-triggered DNA damage and the subsequent p53, p16, and p21 induction, 2) reactive oxygen species (ROS) induction, 3) inflammation, and 4) impairment of efferocytosis during the process of atherosclerosis 7. Senescence is characterized by cell cycle arrest, and therapy-induced senescence has long been a basis for cancer therapy to inhibit cancer cell growth 8. However, this conventional view has recently been challenged 9–11. Milanovic et al. 10 reported that senescence reprograms cancer cells to acquire a proliferative phenotype [senescence-associated stemness (SAS)] after cancer therapy, which allows them to escape senescence-induced cell cycle arrest with strongly enhanced clonogenic growth potential. SAS is different from SASP and regulated independently of senescence-induced cell cycle arrest 12,13 and death 14,15. It is now considered to be one of the critical mechanisms in developing resistance to cancer therapy 14–16. Since both senescence and proliferation of macrophages 17 play critical roles in atherosclerotic plaque formation, we hypothesized that SAS could also be involved in atherosclerosis. However, to the best of our knowledge, SAS has not been explored in cardiovascular disease.
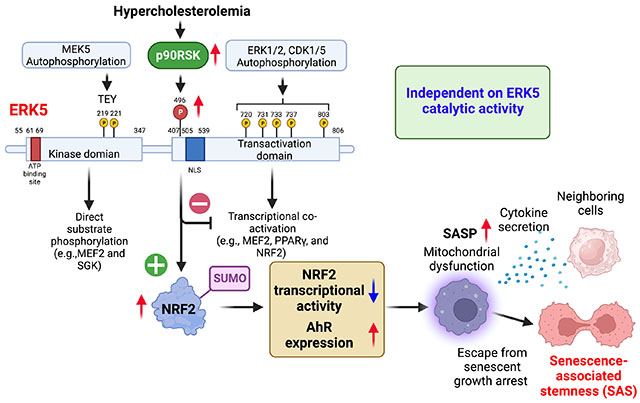
Extracellular signal-regulated kinase 5 (ERK5/BMK1/MAPK7), a member of the mitogen-activated protein kinase (MAPK) family, is unique in that it is not only a kinase but also a transcriptional co-activator with a long C-terminal transcriptional activation domain (TAD) 18,19. The ERK5 N-terminal kinase domain has a characteristic TxY (threonine-x-tyrosine, TEY) motif in the activation loop that activates both kinase and transcriptional activity once phosphorylated 18. As a negative regulator of TAD, once activated, the N-terminal kinase domain releases its inhibitory effect and activates TAD. Both the association of ERK5 with transcriptional factors and the activation of ERK5 C-terminal TAD are required for transcriptional activation of myocyte enhancer factor-2 (MEF2) 20, proliferator-activated receptor γ (PPARγ) 18, and nuclear factor-erythroid factor 2-related factor 2 (NRF2) 21. We have reported that activated p90RSK binds the ERK5 C-terminal region (amino acids 571–807), enabling p90RSK to phosphorylate ERK5 S496 7 and inhibit the transcriptional activity of ERK5 7. However, ERK5 kinase activation also can be upstream of p90RSK activation 22. Importantly, ERK5 S496 phosphorylation is not dependent on ERK5 kinase activity 1,7. p90RSK activation increased ERK5 S496 phosphorylation and induced atherosclerosis 1, but the exact role and mechanisms of ERK5 S496 phosphorylation in atherosclerosis in vivo remains unclear. Also, oxidized LDL (ox-LDL) can induce SASP in myeloid cells (MCs) 23, but how SASP is initiated by oxLDL has been incompletely characterized.
The effects of ERK5 in endothelial cells (ECs) and in the activation of monocytes on inflammation are controversial. Hellman’s group showed that ERK5 siRNA inhibits cytokine-induced inflammatory response in ECs and monocytes 24, whereas we found that depletion of ERK5 inhibits shear stress-induced anti-inflammatory effects in ECs 18. Lin et al. generated a highly selective ERK5 kinase inhibitor (AX15836) and noted that unlike ERK5 depletion, AX15836 exerts no anti-inflammatory or anti-proliferative effects 25. While, Lochhead et al. showed that AX15836 induces Kruppel-like factor 2 (KLF2) transactivation; however, no data related to the effects of this inhibitor on inflammation or proliferation was provided 26. All these differences indicate that ERK5 specific inhibitors might function differently from ERK5 genetic deletion or depletion. Lin et al. suggested that the phenotypes caused by ERK5 genetic depletion might be due to the removal of its noncatalytic function 25. However, it remains unclear what kind of noncatalytic function is removed by the deletion/depletion of ERK5 that can regulate anti-inflammatory and/or anti-proliferation effects.
In this study, we evaluate the role of ERK5 S496 phosphorylation in the induction of atherosclerosis, SASP, and SAS in vivo in ERK5 S496A (dephosphorylation mimic) knock-in (KI) mice, using a novel imaging mass cytometry (IMC) technique 27. We also investigate whether ERK5 S496 phosphorylation inhibits NRF2 transcriptional activity via a noncatalytic function of ERK5 and its regulatory mechanism.
METHODS
Details of the experimental procedures are included in the online supplemental information.
Data Availability
The RNA-seq data was deposited in the NCBI’s Gene Expression Omnibus database (accession GSE210949). The data, analytic methods, and study materials that support the findings of this study are available in the Data supplement or from the corresponding authors upon reasonable request.
RESULTS
ERK5 S496 phosphorylation promotes plaque size and vulnerable plaque formation
To determine the role of ERK5 S496 phosphorylation in atherosclerosis, we generated ERK5 S496A KI mice. We performed Western blotting and confirmed that ERK5 S496 phosphorylation is absent but ERK1/2 and p90RSK are still activated in bone marrow-derived macrophages (BMDMs) isolated from ERK5 S496A KI mice (Fig. 1A, B and Fig.S1A). Conversely, we did not observe any changes in ERK5 TEY phosphorylation, which represents ERK5 kinase activation, after oxLDL stimulation (Fig. 1A, B). We fed ERK5 S496A KI and wild type (WT) C57BL6 control mice a high-fat diet (HFD) for 16 weeks after injecting all mice with a single dose of adeno-associated-virus-8 overexpressing pro-protein convertase subtilisin/Kexin type 9 gain-of-function D377Y mutant (AAV8-PCSK9)34 as we reported previously. No differences in body weight, low-density lipoprotein (LDL), or high-density lipoprotein (HDL) cholesterol levels, and cardiac function evaluated by echocardiography were noted (Fig. S1), whereas the area of atherosclerotic lesions evaluated by Oil-Red-O staining in en face preparations of the ERK5 S496A KI aortas was smaller than that of the WT control aortas (Fig. 1C and D). The size of necrotic cores (Fig. 1E) and the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells were decreased in ERK5 S496A KI aortas compared to that of the WT control aortas (Fig. 1F), suggesting that ERK5 S496 phosphorylation increases atherosclerotic plaque size and promotes the formation of vulnerable plaques.
Figure 1: ERK5 S496 phosphorylation plays a crucial role in vulnerable plaque formation and SASP induction.
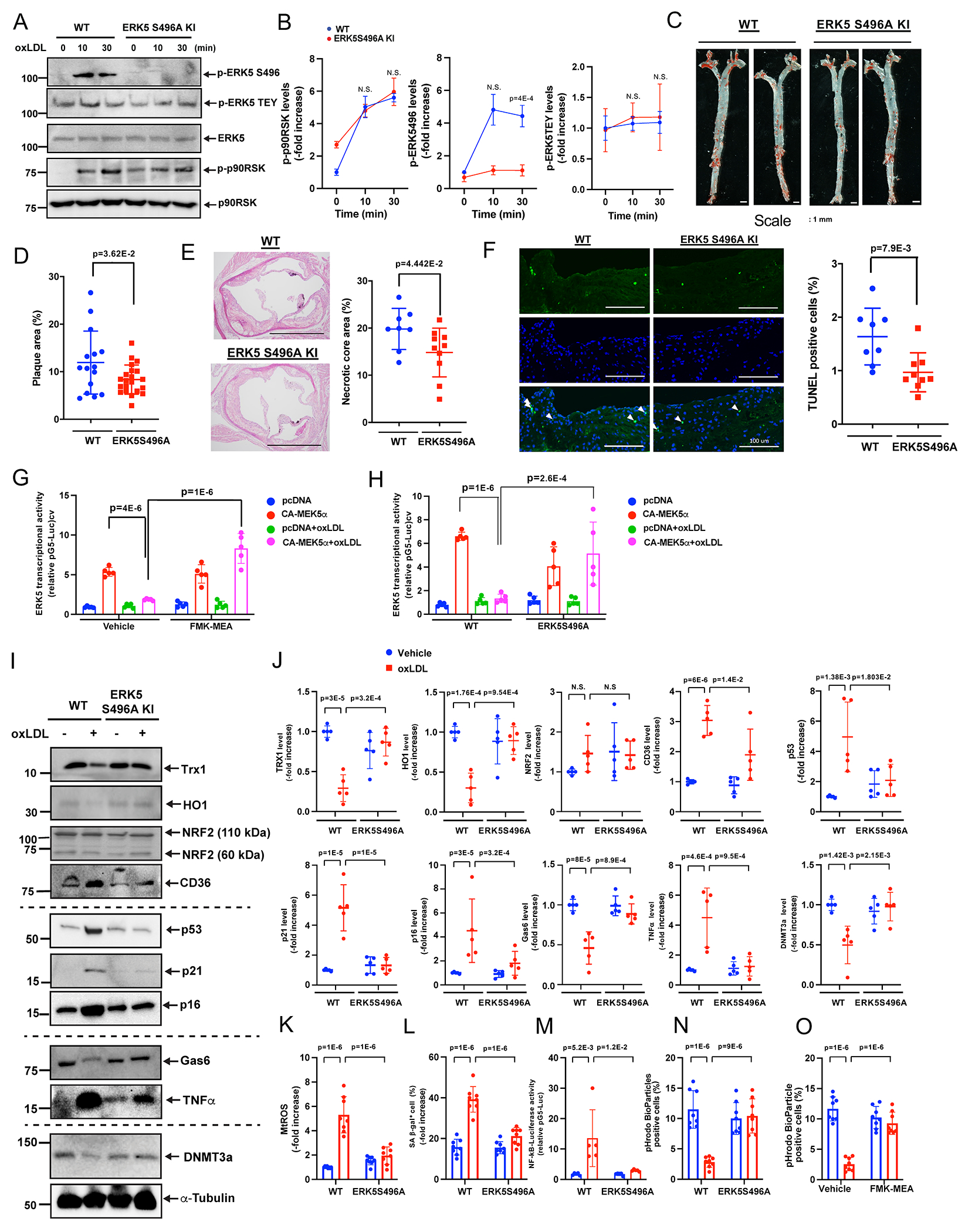
WT BMDMs and ERK5 S496A KI BMDMs were treated with oxLDL (10 μg/ml) for 0-30 minutes (A, B), and an immunoblotting analysis was performed using antibodies against the indicated proteins in vitro. (B) The graphs represent densitometry data from 3 independent gels, one of which is shown in A. (C, D) 16 weeks after AAV-PCSK9 injection and fed an HFD, ERK5 S496A KI mice exhibited fewer oil-red O-stained atherosclerotic lesions in the en face whole aorta, scale bars=1 mm. (D) Quantified oil-red O-stained lesions are shown. (n=15, 21, all male). (E) The area occupied by the necrotic core (acellular lipid core) is shown as the percentage of the total lesion area (E, right). Scale bar=1 mm. (n=8, 10). (F) Sections of proximal aortas from each group were labeled using TUNEL reagents to detect apoptotic cells and counterstained with DAPI to detect nuclei. Only double-positive cells (TUNEL and DAPI) were counted. Scale bar=100 μm. (F, right) The graph shows the percentage of TUNEL-positive cells (TUNEL+ cells/total cells counted) in the lesion area. Over 200 cells were counted for each group. (n=8, 9). (G, H) BMDMs treated with vehicle or FMK-MEA (G) or WT BMDMs and ERK5 S496A KI BMDMs (H) were incubated with oxLDL (10 μg/mL) or vehicle, and ERK5 transcriptional activity was detected. (I, J) WT BMDMs and ERK5 S496A KI BMDMs were treated with oxLDL (10 μg/ml) for 24 hours, and immunoblotting analysis was performed using antibodies against the indicated proteins in vitro. (J) The graphs represent densitometry data from 5 independent gels, one of which is shown in I. (K) WT BMDMs and ERK5 S496A KI BMDMs were incubated with oxLDL as indicated. mtROS levels were detected by MitoNeoD as described in methods section in vitro. Cells treated with oxLDL were assayed 24 hours later. (L) The percentages of cells positive for SA-β-gal staining are shown in vitro. More than 200 cells/sample were counted. (M) WT BMDMs and ERK5 S496A KI BMDMs were transfected with the NF-κB luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 hours and then incubated with oxLDL or vehicle. After 12 hours, NF-κB transcriptional activity was measured as described in Methods in vitro. (N, O) BMDMs treated with vehicle or FMK-MEA (10 μM) (N) or WT BMDMs and ERK5 S496A KI BMDMs (O) were incubated with oxLDL (10 μg/mL) or vehicle. After 24 hours, pHrodo-positive cells were quantified in vitro. The applied statistical tests, sample number, and results in all figures are summarized in Table S3. All data are expressed as mean±SD, **P<0.01, *P<0.05.
ERK5 S496 phosphorylation induces SASP without affecting ERK5 catalytic activity
OxLDL reduced a constitutively active form of MEK5 (CA-MEK5)-induced ERK5 transcriptional activity detected by luciferase reporter assay, which could be reversed by FMK-MEA, a specific inhibitor of p90RSK 7 (Fig. 1G), and in BMDMs isolated from ERK5 S496A KI mice (Fig. 1H). However, oxLDL showed no significant effects on baseline ERK5 transcriptional activity in the MCs transfected with the control plasmid (empty pcDNA without CA-MEK5). Next, we examined the effects of ERK5 S496 phosphorylation on the following four SASP characteristics: 1) excessive ROS production with reduction of antioxidants, 2) senescence, 3) reduction of efferocytosis and 4) pro-inflammation 1. Although oxLDL showed no effect on baseline ERK5 transcriptional activity (Fig. 1G), it decreased the expression of antioxidant molecules such as thioredoxin 1 (TRX1) and heme oxygenase 1 (HO1); this decrease was completely reversed in BMDMs isolated from ERK5 S496A KI mice (Fig. 1I, J). We also found that oxLDL increased mitochondrial ROS (mtROS) production in WT BMDMs, which was inhibited in ERK5 S496A KI BMDMs as detected by two separate MitoNeoD and MitoSOX Red assays as described in the method section (Fig. 1K (MitoNeoD) and Fig.S1F, G (MitoSOX)). OxLDL increased expression of p16, p21, and p53 detected by Western blotting (Fig. 1I, J) and induced senescence-associated β-gal staining (SA-βgal) (Fig. 1L), all of which were absent in ERK5 S496A KI BMDMs. GAS6 and DNA damage response (DDR)-related molecules DNA methyltransferase 3a (DNMT3a) expression were inhibited, but TNFα was increased by oxLDL in WT BMDMs (Fig. 1I, J). In WT BMDMs, oxLDL increased NF-κB activation (Fig.1M) while reducing efferocytosis detected by using pHrodo dye27 (Fig. 1N, O); all of these effects observed in WT BMDMs were reversed in ERK5 S496A KI BMDMs or by FMK-MEA (Fig.1N, O). Of note, oxLDL had no effect on both ERK5 kinase activity and transcriptional activity (Fig. 1A, G, H). These data suggest that ERK5 S496 phosphorylation regulates SASP independent of ERK5 catalytic activity.
Divergent role of ERK5 S496 phosphorylation in SASP in MCs and ECs
IMC is a technique that uses antibodies conjugated with rare-earth-metal isotopes of defined atomic masses that allows the antibody-bound-proteins to be separated by a mass cytometer instead of fluorescent or enzymatic moieties that are used in conventional immunohistochemistry (IHC) and immunohistofluorescence (IHF). We prepared tissue sections from the plaques of the WT fed a HFD (WT HFD) and the ERK5 S496A KI mice fed a HFD (ERK5 S496 KI HFD) or a normal chow diet (NCD) as a control for 12 weeks after AAV8-PCSK9 injection, and then generated high-dimensional images of these tissue sections (Fig. 2A, B, upper). Using the VISIOPHARM program (Hoersholm, Denmark), single-cell features were computationally segmented by a watershed algorithm (Fig. 2B, lower). Single-cell markers or molecule expression data were extracted and analyzed by the Phenomap™ module, and t-SNE plots were generated by a phenotyping algorithm (Fig. 2D). Based on these analyses, we found 9 clusters in both WT and ERK5 S496A KI plaques (Fig. 2C, E). Using CD31, von Willebrand factor, and ETS-related genes as EC markers and CD11b and CD107b as MC markers, we identified 2 EC-like clusters (#5 and #6) and 5 MC-like clusters (#1, #3, and #7-9) (Fig. 2C, E). No statistically significant difference in the relative cellularity (% of total cells) of these clusters between WT and ERK5 S496A KI plaques was observed (Fig. 2F).
Figure 2. Single cells-based clustering by imaging mass cytometry and SASP markers in WT and ERK5 S496A KI plaques in vivo.
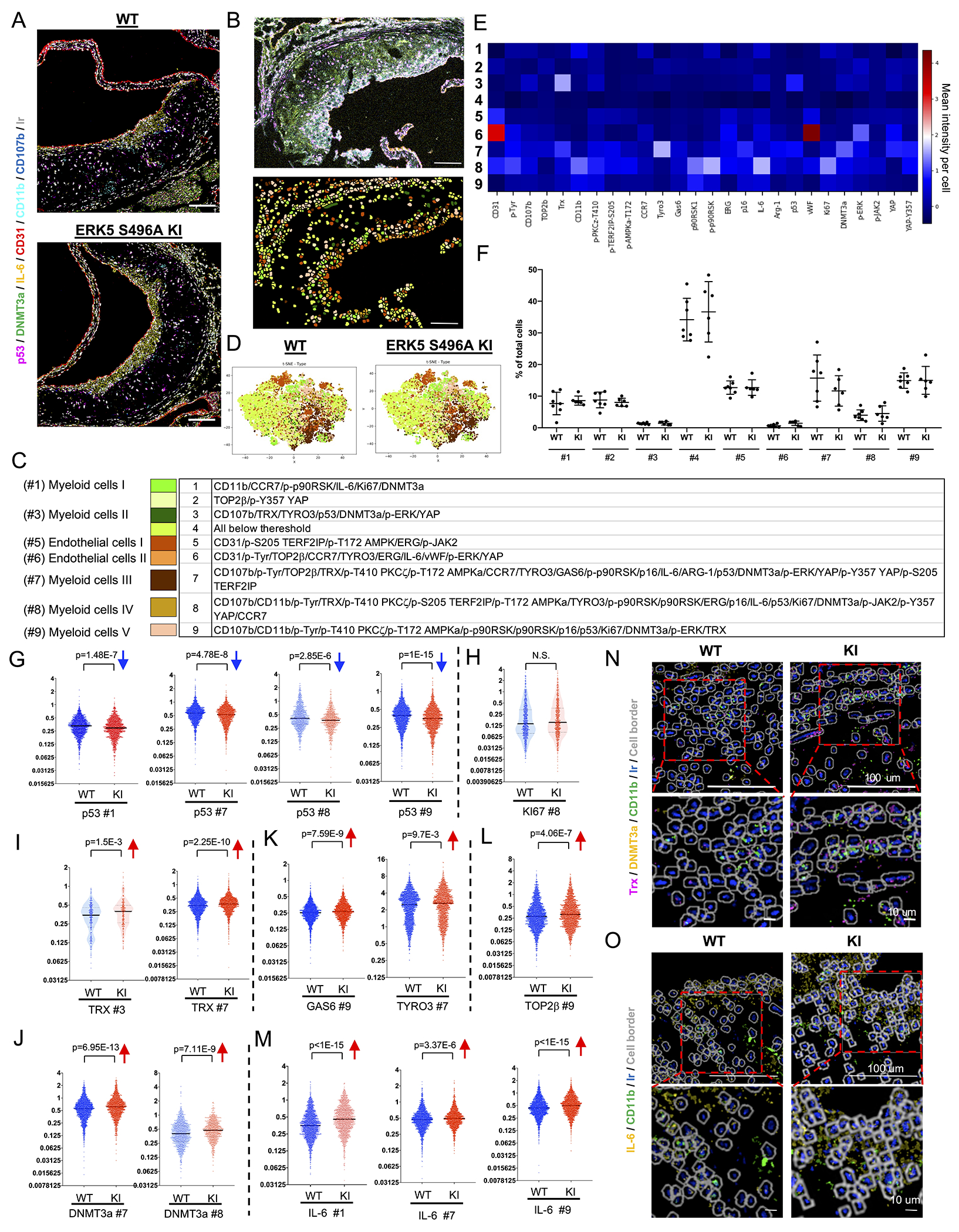
(A) Six transient and single isotope signals from the markers and the Ir DNA-Intercalator signal were plotted. (B, upper) IMC analysis of tissues sectioned from WT and ERK5 S496A KI plaques. All 26 transient and single isotope signals from the markers and the Ir DNA-Intercalator signal were plotted. (B, lower, and C) After cell segmentation and phenotyping, 9 phenotypic clusters were identified in WT and ERK5 S496A KI plaque tissues. Each color indicates a cell cluster in C. (D) t-SNE plots of WT and ERK5 S496A KI plaque tissues. (E) Heatmap with 9 phenotypic clusters showed differentially regulated 26 molecules. (F) Cell number (%) in each cluster of total cells in each region of interest (ROI). We measured 18 ROIs from 7 WT samples and 18 ROIs from 6 ERK5 S496A KI samples and averaged the values from the same sample (n=7, 6). Data are expressed as mean±SD. Statistical significance was assessed by one-way ANOVA, and showed no difference between WT and ERK5 S496A KI. (G-M) The single-cell expression level of each marker in MC-like clusters between WT and ERK5 S496A KI plaque tissues. p53 expression levels in #1, #7, #8, and #9 were decreased in ERK5 S496A KI cells compared to in WT cells (G). Ki67 expression in the most inflammatory MC-like cluster (#8) was not changed between WT and ERK5 S496A KI cells (H). TRX expression levels in #3 and #7; DNMT3a in #7 and #8; TYRO3 in #7; GAS6 and TOP2β in #9; and IL-6 in #1, #7, and #9 were increased in ERK5 S496A KI cells compared to in WT cells (I-M). The Y-axis indicates the mean intensity, and solid black lines indicate the median value in each violin plot. (N, O) Staining images of each marker acquired by IMC were exported by the MCD viewer and merged with cell border images (gray) exported from VIS software. Ir (blue) is DNA. Upper images are low magnification (scale bar=100 μm), and lower images are high magnification (scale bar=10 μm). (N) Trx (magenta) and DNMT3a (yellow) in CD11b (lime)-positive myeloid cells were induced in ERK5 S496A KI compared to WT cells. (O) IL-6 (yellow) in CD11b (lime)-positive myeloid cells was induced in ERK5 S496A KI compared to WT cells. The applied statistical tests, sample number, and results in all figures are summarized in Table S3.
In 5 MC-like clusters (#1, #3, and #7-9), we found less expression of p53 and more expression of TRX1 in clusters #3 and 7, DNMT3a in clusters #7 and 8, TYRO3 in cluster #7, and GAS6 and TOP2β in cluster #9 in ERK5 S496A KI plaques compared to WT plaques (Fig. 2G–O). We also verified the IMC findings using IHC and found a decreased expression of p53 in macrophages [LAMP2 (also known as Mac3)-positive cells (demarcated area)] and an increased expression of TRX1 and DNMT3A in ERK5 S496A KI plaques compared to WT plaques (Fig. S2A–C). These data suggest that the SASP-related events, including p53, efferocytosis (TYRO3), anti-oxidation (TRX1), and DDR (TOP2β) are regulated by ERK5 S496 phosphorylation in MCs localized in the atherosclerotic plaques in vivo.
In the 2 EC-like clusters, the intensity of signals associated with the SASP events was weaker than that in the 5 MC-like clusters. However, we still found an increased expression of TRX1, GAS6, TYRO3, and DNMT3A in ERK5 S496A KI plaques compared to WT plaques (Fig.S2D). Alternatively, we did not detect any differences in the expression of p53 and TOP2β in EC-like clusters between ERK5 S496A KI plaques and WT plaques (Fig. S2D). Interestingly, IL6 expression was higher in MC-like clusters (#1, 7, and 9) and EC-like cluster (#5) in ERK5 S496A KI plaques than in those of the WT plaques (Fig. 2M and Fig.S2D). IL6 athero-protective effects may contribute to less plaque formation in ERK5 S496A KI mice 28.
Since the distribution of MCs in relation to blood vessels can provide information about macrophage infiltration and migration to the plaque, we performed a morphometric analysis of the distance between MCs and ECs in WT and ERK5 S496A KI plaques. Although matching plaque sizes were selected for analysis, the mean distance from the EC-like cluster #5 to the MC-like cluster #8 was shorter in ERK5 S496A KI plaques than in WT plaques (Fig. S2E). We also found that the migration of ERK5 S5496A KI BMDMs detected by Boyden chamber assay was significantly less than wild-type (Fig. S2F). These data indicate that ERK5 S496 phosphorylation promotes MC-like cell infiltration and migration.
ERK5 S496 phosphorylation promotes SAS
Milanovic et al. have defined SAS as “an unexpected, cell-autonomous feature that exerts its detrimental, highly aggressive growth potential upon escape from the cell-cycle blockade”10. p53 plays a crucial role in controlling senescence-induced cell cycle arrest29, especially in macrophages during atherosclerosis formation30. IMC data analysis shows that p53 is expressed at relatively higher level among all senescent markers tested (Fig. S2G). To determine whether senescence, particularly SAS, is induced during the process of atherosclerosis, we investigated the relationship between p53 and Ki67 (as a proliferation marker in the atherosclerosis lesions 31) in the plaques in vivo by calculating the logarithm of the Ki67 expression to p53 expression ratio at a single-cell level [log10(Ki67: p53 ratio of selected cells] in IMC data. Because the one-dimension density distribution of this parameter was trimodal, MCs were divided into 3 groups based on different patterns of p53:Ki67 expression (Fig. 3A) with the cutoff values −1.01 and −0.024 (Fig. 3B). Anti-proliferative effects of p53 appear to explain the (Ki67: p53) ratio in MC groups 1 and 3; however, p53 and Ki67 were linearly co-expressed in group 2 (y = 0.92x + 0.81; F test p – value < 2.2e – 16), indicating that this group likely escaped from anti-proliferative effects of p53 (Fig. 3A). Quantitative data analysis showed the percentage of ERK5 S496A KI cells in group 2 was lower than that of the WT cells (Fig. 3D). In contrast, the percentage of ERK5 S496A KI cells in group 3 was higher than that of the WT cells and was not different from that of the WT cells in MC group 1 (Fig. 3C, E). Collectively, these data implicate a unique role of ERK5 S496 phosphorylation in regulating SAS in MCs during atherogenesis.
Figure 3. ERK5 S496 phosphorylation provokes SAS in vivo and ex vivo.
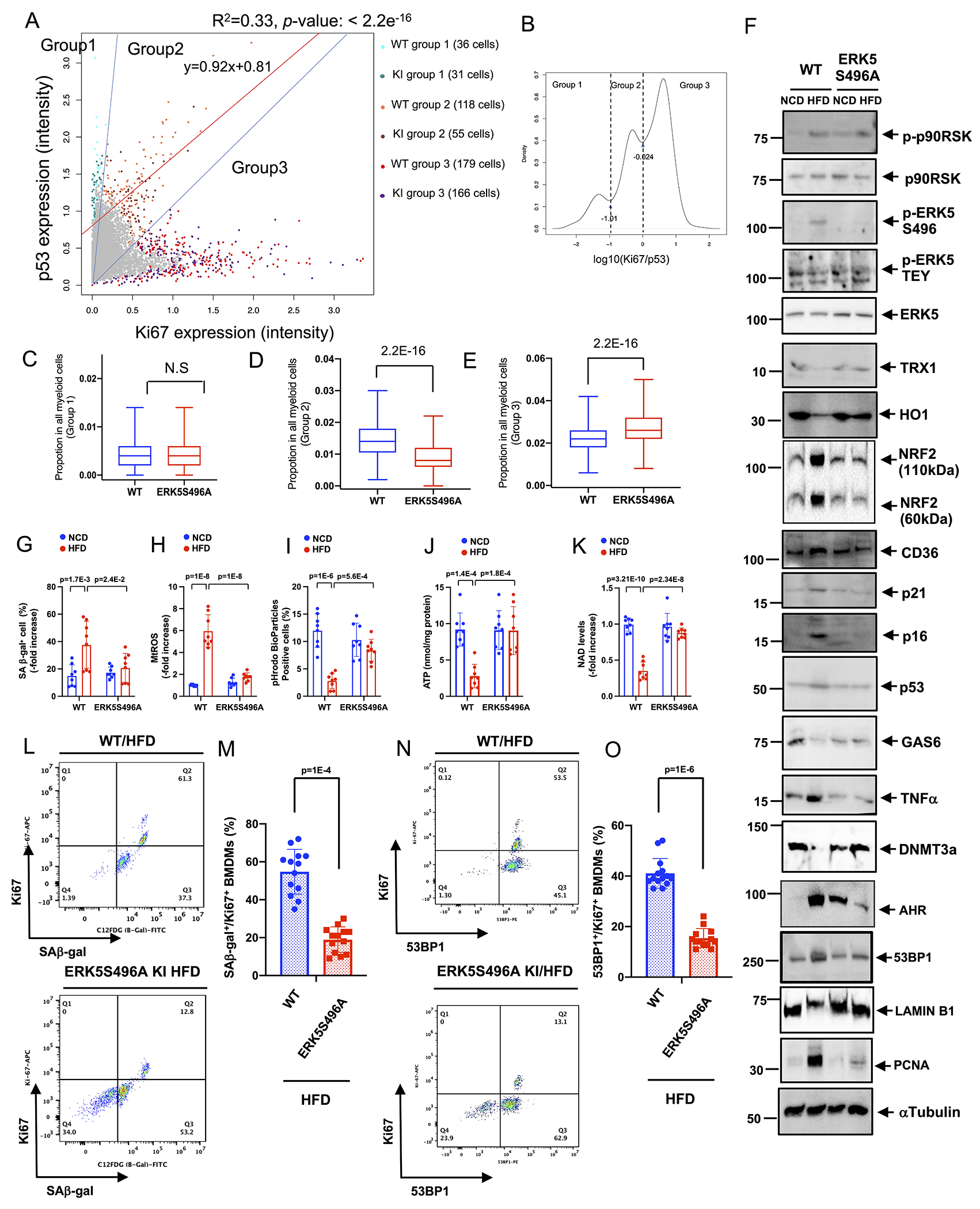
(A-E) Single-cell analysis of p53 and Ki67 expression in vivo. (B) The log scaled ratio of Ki67 and p53 of selected cells by log10(expression of Ki67/ expression of p53), and the cutoff of ratio with log10(−1.01) and log10(−0.024). (C-E) The % of ERK5 S496A KI cells in groups 1-3. KI and WT cells were resampled for 1,000 rounds to calculate the % of cells in groups 1-3. In each resampling round, 500 KI (or WT) cells were selected to calculate the % in group 1 (or group 2 or 3) by # of cells in group 1 (or group 2 or 3)/# of total cells. Wilcoxon test was performed as described in the methods. (F-O) BMDMs isolated from WT and ERK5 S496A KI mice fed a NCD or HFD for 4 months were cultured with the same medium ex vivo. (F) Immunoblotting analysis was performed using antibodies against the indicated proteins. We used the α-Tubulin expression as a loading control for each protein. (Quantification data was shown in Fig. S3.) (G) The percentages of cells positive for SA-β-gal staining are shown. (H) mtROS levels were detected by MitoNeoD as described in Methods. (I) Efferocytosis was detected by the quantification of pHrodo-positive cells (%). ATP (J) and NAD+ (K) were measured in BMDMs isolated from WT and ERK5S496A KI mice fed a NCD or HFD. (L) Co-expression of the fluorescent SA β-gal marker and Ki67 in BMDMs isolated from WT and ERK5 S496A KI mice after 4 months of HFD. (M) The graph showed % of double-positive SA-β-gal and Ki67 cells. (N) Co-expression of the fluorescent 53BP1 marker and Ki67 in BMDMs isolated from WT mice after 4 months of HFD. (O) The graph showed % of double-positive 53BP1 and Ki67 cells. n=13-14. The applied statistical tests, sample number, and results in all figures are summarized in Table S3. All data are expressed as mean±SD, **P<0.01, *P<0.05.
In contrast to MCs, we could not find the apparent SAS phenotype in ECs (Fig. S2I). We also performed IMC analysis with vascular smooth muscle cells (VSMCs) by using α-SMA antibody and found group 1 (high p53 and low Ki67) and group 3 (low p53 and high Ki67) phenotypes, but VSMCs did not show SAS phenotype of group 2 (the linear relationship between p53 and Ki67 expression) (Fig.S2J). We also evaluated the SASP phenotype in VSMCs and found an upregulation of DNMT3a and GAS6 expression in VSMCs from the plaque of ERK5 S496A KI mice. However, we could not find other SASP molecules changes between wild-type and ERK5 S496A KI mice in VSMCs (Fig.S2K). These data indicate that the SAS phenotype was present in myeloid cells, but it was less clear in ECs and VSMCs.
Hypercholesterolemia induce MC phenotypic changes to SASP/SAS via ERK5 S496 phosphorylation
Although cell cycle arrest is a hallmark of senescence, senescent cells can escape cell cycle arrest and acquire the SAS phenotype 10. Since MC proliferation is a crucial component of plaque formation 17, SAS can play a critical role in atherosclerosis. We demonstrated the potential existence of SAS in the plaque (Fig.3A–E). To investigate how SAS was regulated in MCs by hypercholesterolemia, we isolated bone marrow cells from WT and ERK5 S496A KI mice fed a HFD or NCD for 16 weeks after AAV-PCSK9 injection and differentiated them to BMDMs for evaluation of SASP and SAS induction.
We found 1) an increase of p53, p21, and p16 and SA-βgal positive cells (Fig. 3F, G, and Fig. S3), 2) a decrease of TRX1 and HO1 (Fig. 3F, and Fig. S3), an increase of mtROS (Fig. 3H (MitoNeoD), and Fig. S1G (MitoSOX)), 3) a decrease of GAS6 (Fig. 3F, and Fig. S3) and efferocytosis (Fig. 3I), 4) an increase of TNFα and various cytokine and chemokine secretion (Fig. 3F, and Fig. S3 and S4) in BMDMs isolated from hypercholesterolemic (HC) WT HFD mice compared to that of normocholesterolemic (NC) WT NCD mice, were inhibited in BMDMs isolated from HC ERK5 K496A KI HFD mice. We also found an increase in arginase-1 and Fizz1 mRNA in BMDMs isolated from HC ERK5 S496A KI HFD mice (Fig. S4B), suggesting the shift from M1 to an M2-like phenotype in MCs by the ERK5 S496A mutation. Furthermore, levels of IL-1α, IL-1β, and CXCL3 cytokines in serum obtained from ERK5 S496A KI HFD mice were lower than those from WT HFD mice (Fig. S4C), also supporting the role of ERK5 S496 phosphorylation in SASP induction.
Figure 4. ERK5 S496 phosphorylation regulates the genetic profile of HC-mediated reprogrammed MCs and the critical role of AHR on SAS ex vivo.
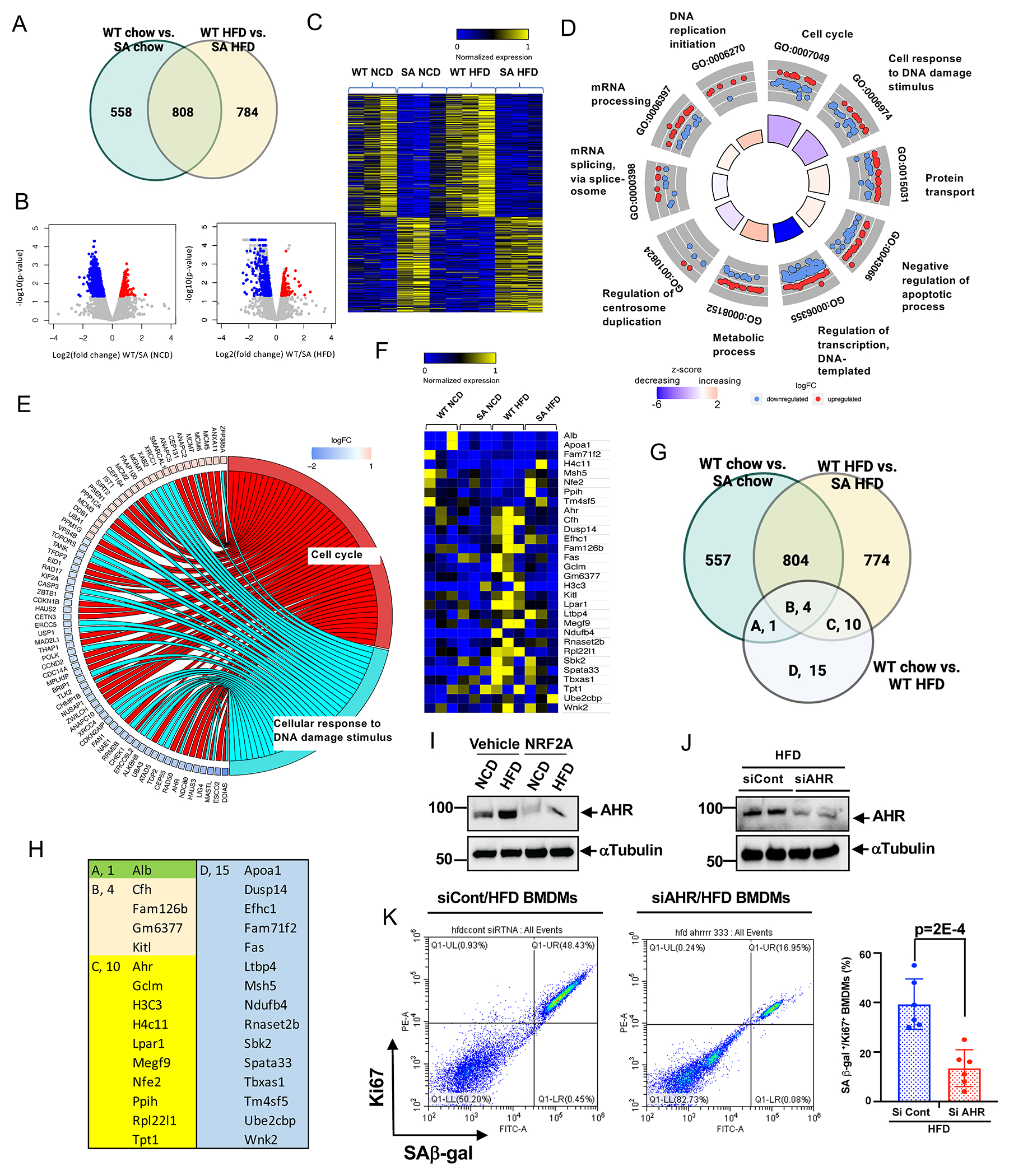
(A) Venn diagram illustrating the gene expression patterns in each experimental group. (B) Volcano plot of BMDMs from WT and ERK5 S496A KI fed a normal chow diet (NCD) or high fat diet (HFD) RNA-seq adjusted p-value < 0.01, 1039 genes upregulated and 327 downregulated in WT vs. SA NCD, and 1174 genes upregulated, and 418 downregulated in WT vs. SA HFD. 3 samples/group. (C) Heatmap differentially regulated 784 genes only between WT HFD and ERK5 S496A KI HFD (SA HFD). (D) Functional enrichment analyses. GOcircle plots display scatter plots of log fold change (logFC) for selected GO terms. Red dots represent upregulated genes, and blue dots represent downregulated genes. The inner circles display z-scores calculated as the number of up-regulated genes minus the number of down-regulated genes divided by the square root of the count for a WT HFD and ERK5 S496A KI HFD. Up-regulated means that expression is higher in the ERK5 S496A KI HFD. (E) Circle plot of select genes indicated ontologies. Gene expression relative difference (log2 fold change). (F) Heatmap differentially regulated 30 genes only between WT NCD and WT HFD (WT HFD). (G) Venn diagram including the genes listed in E, and (H) the list of the genes. (I) AHR expression in BMDMs from WT fed a NCD or HFD after 24 hours of the vehicle or NRF2-KEAP1-binding inhibitory peptide, CAS 1362661 (NRF2A; 2 μM) treatment. (J) AHR expression in BMDMs from WT fed a HFD after AHR siRNA or control siRNA transfection. (K) Co-expression of the fluorescent SA-β-gal marker and Ki67 in BMDMs isolated from WT HFD after transfection of control siRNA and AHR siRNA Graph showed % of double-positive cells of SA-β-gal and Ki67. The applied statistical tests, sample number, and results in all figures are summarized in Table S3. Mean±SD, **P<0.01.
Because mitochondrial dysfunction plays a critical role in the induction of SASP 32, we examined oxidative phosphorylation (OXPHOS) and glycolysis in cultured BMDMs isolated from WT or ERK5 S496A KI mice after NCD or HFD. Whereas OXPHOS was decreased, glycolysis was increased in BMDMs from HC WT HFD mice compared to NC WT NCD mice, an effect which was completely reversed in BMDMs from ERK5 S496A KI mice (Fig. S5). Ox-LDL reduced the ATP and NAD+ levels in WT BMDMs but not in ERK5 S496A KI BMDMs (Fig. 3J, K).
Since the data in Fig.3F–K supported the idea that BMDMs from NCD and HFD-fed mice showed significant phenotypic differences in SASP, we also evaluated two senescence markers (p53-binding protein 1 (53BP1) and LAMIN B1) and cell growth marker (proliferating cell nuclear antigen (PCNA)) as evidence for SAS in HC-mediated reprogrammed BMDMs. We observed an increase of 53BP1 and PCNA, and a decrease of LAMIN B1 expression in HC WT-HFD mice (HFD BMDMs) compared to NC WT-NCD mice (NCD BMDMs), an effect that was reversed in ERK5 S496A KI BMDMs (Fig. 3F and Fig. S3). Furthermore, we examined the amount of double SAβ-gal and Ki67 positive (SA β-gal+Ki67+) (Fig. 3L, M), and double 53BP1 and Ki67 positive (53BP1+Ki67+) macrophages (Fig. 3N, O), and found a significant decrease of both SA β-gal+Ki67+ and 53BP1+Ki67+ macrophages obtained from HC ERK5 S496A KI-HFD mice compared to those from the HC WT-HFD mice (Fig. 3L–O). These data indicate that SAS is increased in HC-mediated reprogrammed MCs in an ERK5 S496 phosphorylation-dependent manner.
Aryl hydrocarbon receptor (AHR) is required for ERK5 S496 phosphorylation-induced SAS
To investigate the molecular mechanism by which ERK5 S496 phosphorylation reprograms MCs to SAS, we performed RNA sequencing (RNA-seq) for WT and ERK5 S496A KI BMDMs under NCD or HFD. We identified 784 differentially expressed genes (DEGs) regulated only by HC-induced ERK5 S496 phosphorylation (Fig. 4A–C). The Gene Ontology (GO) analysis revealed that these DEGs are involved in critical senescence-related processes such as cell cycle, cellular response to DNA damage, protein transport, and negative regulation of apoptosis (Fig. 4D, and Fig. S6). GO Bubble analysis showed the strong involvement of ERK5 S496 phosphorylation in nuclear events, also supporting its role in regulating senescence events (Fig. S6). Gene-annotation enrichment analysis (GO Circle) showed that z-scores of both cell cycle and cellular response to DNA damage were negative in ERK5 S496A KI (Fig. 4D and E), suggesting the role of cell cycle and DNA damage response in inducing SAS. Interestingly, we found only 30 DEGs between WT NCD and WT HFD BMDMs (Fig. 4F), among which 15 DEGs were regulated by ERK5 S496 phosphorylation (Fig. 4G and H). These observations support the critical role of ERK5 S496 phosphorylation in HC-mediated MC reprogramming. We identified 10 core genes (Ahr, Gclm, H3C3, H4c11, Lpar1, Megf9, Nfe2, Ppih, Rpl22l1, and Tpt1) regulated by HC-mediated ERK5 S496 phosphorylation that might be crucial for HC-induced SAS.
Since Ahr showed the most significant changes among the 10 core genes, and its contribution to both cell proliferation 33 and senescence 34 had already been reported, we next investigated its expression in BMDMs. We found an increase in AHR protein expression in WT HFD compared to WT NCD BMDMs ex vivo. This increase was no longer apparent in ERK5 S496A KI HFD BMDMs (Fig. 3F, and Fig. S3). In addition, NRF2-KEAP1-binding inhibitory peptide (NRF2A) inhibited AHR expression (Fig. 4I) in BMDMs of WT HFD mice. The depletion of AHR inhibited the increase of SAβ-gal+Ki67+ macrophages in WT HFD BMDMs (Fig. 4J, K), supporting the pivotal role of AHR in HC-induced SAS.
ERK5 S496 phosphorylation, but not ERK5 catalytic activity, is crucial for inflammation and mtROS induction
Since we did not find an increase in ERK5 TEY motif phosphorylation by oxLDL (Fig. 1A), we used granulocyte-macrophage colony-stimulating factor (GM-CSF) to detect ERK5 TEY motif phosphorylation. We transfected BMDMs with an ERK5b splice variant cDNA mutant (aa78-806), which lacked an ATP binding site and showed no ERK5 kinase or transcriptional activity 18,35. We also studied the effect of a dual phosphorylation site mutant of ERK5 (ERK5TEYm), which inhibited stimuli-induced ERK5 kinase activation 36 after TEY motif phosphorylation (Fig. 5A). In BMDMs transfected with the ERK5b or the ERK5TEYm, GM-CSF-induced ERK5 TEY motif phosphorylation and the subsequent KLF2 induction were inhibited, whereas ERK5 S496 phosphorylation was unaffected (Fig. 5B, C and Fig. S7A, B). In contrast, ERK5 kinase inhibitors (XMD8-92 and the highly selective ERK5 kinase inhibitor, AX-1583625) both suppressed ERK5 TEY motif phosphorylation-mediated KLF2 induction and S496 phosphorylation (Fig. 5D, E and Fig. S7C, D). Lastly, the ERK5 S496A mutant inhibited ERK5 S496 phosphorylation but had no effect on KLF2 induction mediated by ERK5 TEY phosphorylation (Fig. 5F and Fig. S7E). These data support the crucial role of ERK5 TEY motif phosphorylation, but not ERK5 S496 phosphorylation, in ERK5 kinase activation-mediated KLF2 induction.
Figure 5. ERK5 TEY motif mutant and ERK5 specific kinase inhibitors inhibited ERK5 kinase activity and ERK5 S496 phosphorylation in vitro.
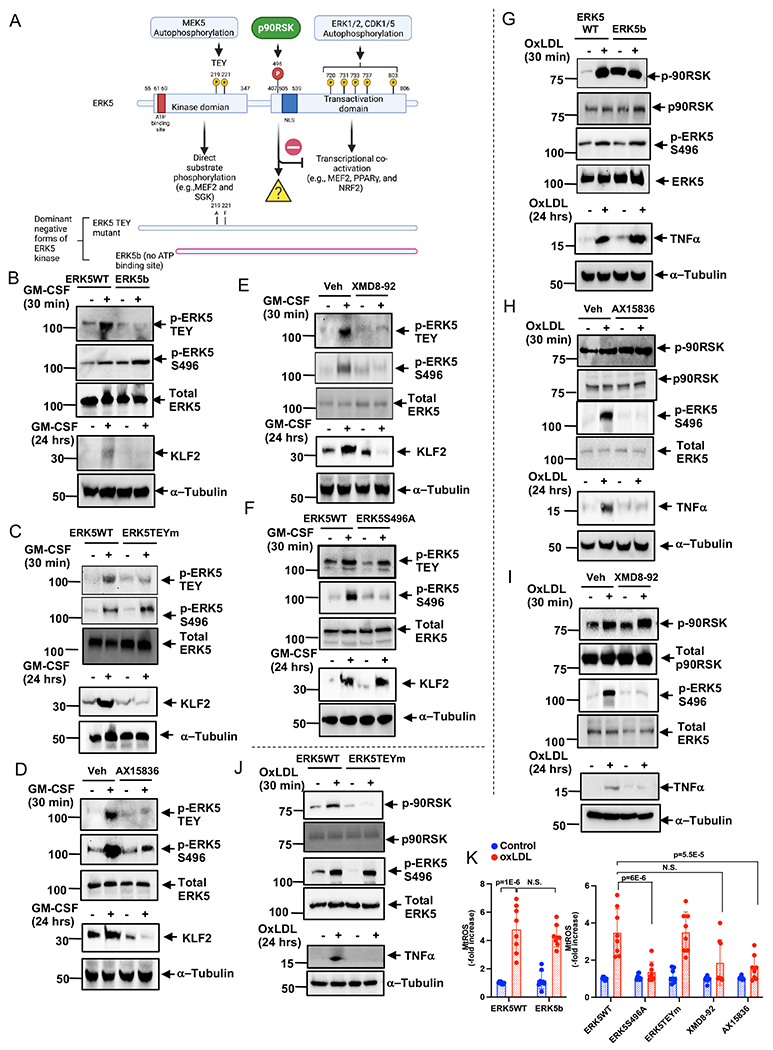
(A) The scheme of ERK5 kinase, transactivation domain, and phosphorylation sites. BMDMs were transfected with ERK5 WT or ERK5b (B, G), ERK5 TEY motif mutant (ERK5TEYm) (C, J), ERK5 S496A mutant (F), or pre-treated with AX15836 (D, H, 5 μM), XMD8-92 (E, I, 10 μM), or vehicle for 1 hour, then BMDMs were stimulated by GM-CSF (20 ng/ml, B-F), ox-LDL (10 μg/ml, G-K), or vehicle. Cell lysates were collected after 24 hours of stimulation, and western blotting was performed by the indicated antibodies. Representative images from 5 independent experiments are shown. (Quantification data was shown in Fig. S7.) (K) BMDMs were treated with indicated mutants and inhibitors as described above, and mtROS production was detected by MitoNeoD as described in the Methods. The applied statistical tests, sample number, and results in all figures are summarized in Table S3. Mean±SD, **P<0.01.
ERK5 kinase inhibitors prevented oxLDL-mediated ERK5 S496 phosphorylation and TNFα induction but showed no effect on p90RSK phosphorylation (Fig. 5H, I and Fig. S7H, I), which supports the specificity of ERK5 kinase inhibitors. In contrast, ERK5b did not inhibit oxLDL-mediated p90RSK phosphorylation, ERK5 S496 phosphorylation, and TNFα induction (Fig. 5G and Fig. S7G). These data indicate that the inhibition of ERK5 catalytic activity is dispensable for the anti-inflammatory effects of ERK5 kinase inhibitors. Interestingly, ERK5TEYm inhibited both oxLDL-induced p90RSK phosphorylation and TNFα induction (Fig. 5J and Fig. S7F), indicating a unique role for ERK5 TEY motif phosphorylation on interplay between ERK5 and p90RSK, which could subsequently regulate p90RSK activation in an ERK5 kinase activity-independent manner 37.
Lastly, we found that both ERK5b deletion mutant and ERK5TEYm could not inhibit oxLDL-induced mtROS production, but ERK5 S496A and both ERK5 kinase inhibitors inhibited mtROS production (Fig.5K and Fig.S1H). Taken together, this finding indicates a crucial role for ERK5 S496 phosphorylation in mtROS production, independent of ERK5 catalytic activity.
ERK5 S496 phosphorylation induces NRF2 SUMOylation to provoke SASP and mitochondrial dysfunction
OxLDL-induced reduction of NRF2 transcriptional activity detected by luciferase reporter assay was abolished by ERK5 S496A mutant and AX-15836 but not by the ERK5b deletion mutant (Fig. 6A). The data collectively (Fig. 5K and S1I) suggests that ERK5 kinase inhibitors might inhibit oxLDL-mediated reduction of NRF2 transcriptional activity and subsequent mtROS induction by inhibiting ERK5 S496 phosphorylation but not ERK5 catalytic activity. The role of SUMOylation in the regulation of transcriptional factor activity has already been reported 38. We investigated whether NRF2 SUMOylation can be regulated by ERK5 S496 phosphorylation. OxLDL-increased NRF2 SUMOylation in WT BMDMs was inhibited in ERK5 S496A KI BMDMs (Fig. 6C). Importantly, we also found an increase of NRF2 SUMOylation in WT HFD compared to WT NCD BMDMs but not in ERK5 S496A KI BMDMs (Fig. S8A, B). NRF2 SUMOylations at K525(6)/595(6)39 and K11040 (NXQ16236-1) have been reported (Fig. 6B). However, based on iPTMnet (#Q16236), the publicly accessible post-translational modification database, only K518 is an NRF2 SUMOylation site detected by mass spectrometry 41, and to the best of our knowledge, the functional role of NRF2 K518 SUMOylation remains unclear. Therefore, we generated the NRF2 K518R mutant and found that the NRF2 K518 to R mutation blocked oxLDL-induced NRF2 SUMOylation, indicating that NRF2 K518 is a primary SUMOylation site in MCs in response to oxLDL stimulation (Fig. 6D). Next, we found that the inhibition of NRF2 transactivation after oxLDL treatment was reversed by NRF2 K518R mutant, indicating that NRF2 K518 SUMOylation inhibited NRF2 transcriptional activity (Fig. 6E). All four components of SASP were abolished by transfection with the NRF2 K518R mutant (Fig. 6F–J and Fig. S8C). Furthermore, we also found that the NRF2 K518R mutant negated the oxLDL-induced AHR expression (Fig. 6J and Fig.S8C).
Figure 6. NRF2 K518 SUMOylation mediated by ERK5 S496 phosphorylation inhibits NRF2 transcriptional activity and subsequent SASP induction in vitro.
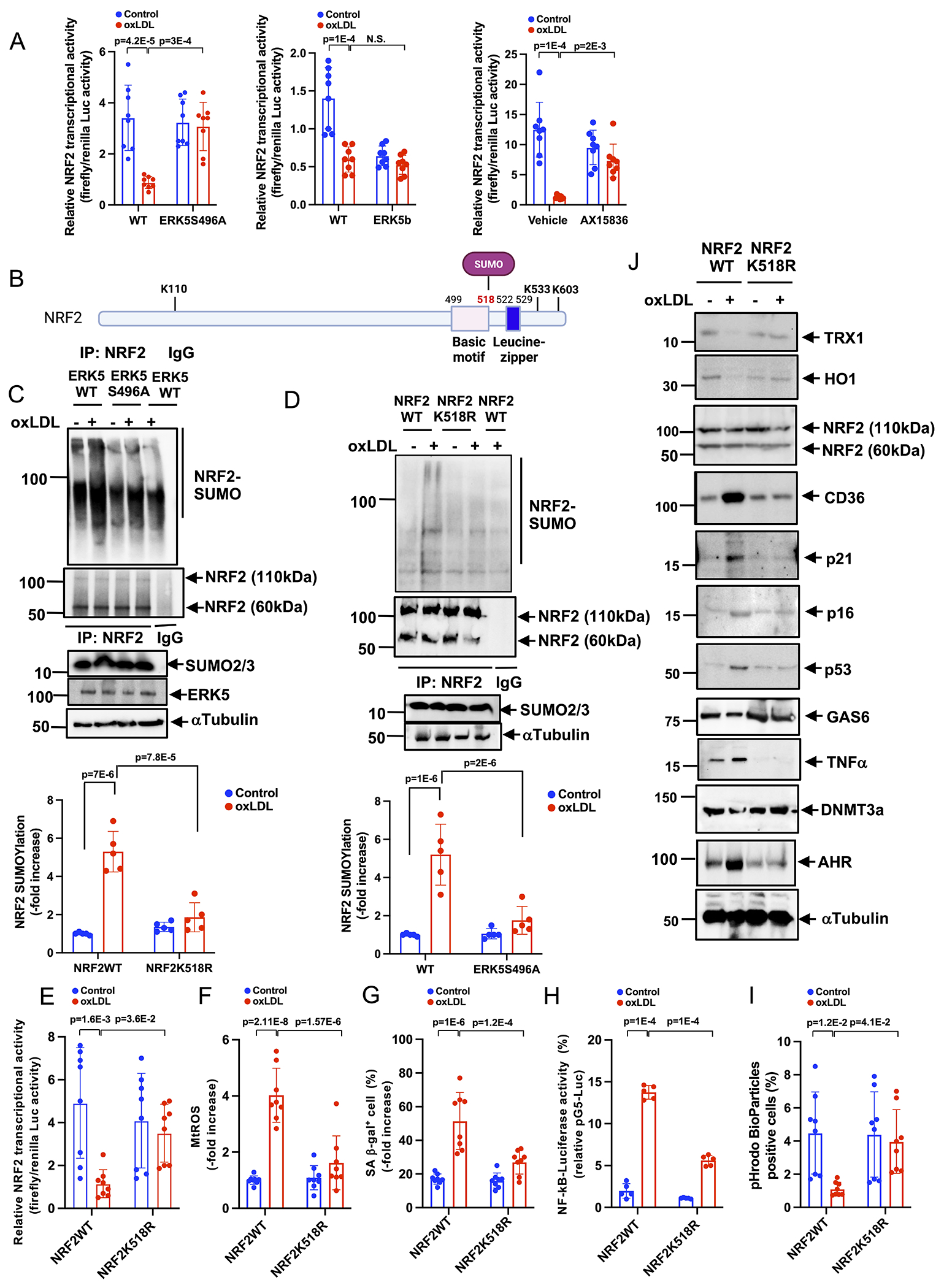
(A) BMDMs were transfected with the ERK5 WT, ERK5 S496A, or ERK5b plasmid, and ARE luciferase reporter and the constitutively expressing Renilla luciferase vector. After 16 hours of transfection, cells were pretreated with AX15836 for 1 hour and then treated with ox-LDL (10 μg/ml) or vehicle. After 12 hours, ARE transcriptional activity was measured as described in the Methods. (B) The scheme of NRF2 SUMOylation sites and DNA binding sites. (C) WT BMDMs and ERK5 S496A KI BMDMs were incubated with oxLDL or vehicle. After 0 or 30 min of oxLDL incubation, cell lysates were immunoprecipitated with anti-NRF2 or IgG control and immunoblotted with SUMO2/3 antibody. (D) BMDMs were transfected with GFP-tagged NRF2 K518R mutant or GFP tag plasmid. After 0 or 30 min of oxLDL incubation, cell lysates were immunoprecipitated with anti-NRF2 or IgG control and immunoblotted with SUMO2/3 antibody. The lower graphs represent densitometry data from 5 independent gels, one of which is shown in C and D. (E) WT BMDMs were transfected with NRF K518R mutant or NRF2 WT, the ARE luciferase reporter, and the constitutively expressing Renilla luciferase vector for 16 hours. Cells were treated with oxLDL (10 μg/mL) or vehicle, and 6 hours later, NRF2 transcriptional activity was measured as described in Methods. (F) WT BMDMs were transfected with NRF2 or control siRNA, and after 48 hours of transfection, mtROS levels were detected by MitoNeoD as described in Methods. Cells treated with oxLDL or vehicle were assayed 12 hours later. (G) The percentages of cells positive for SA β-gal staining are shown. More than 200 cells/sample were counted. (H) NF-κB transcriptional activity was measured as described in Fig. 1M in BMDMs transfected with NRF2 K518R mutant or wild type after 24 hrs of oxLDL or vehicle treatment. (I) BMDMs were incubated with oxLDL or vehicle after NRF2 K518R mutant or wild-type transfection. After 24 hours of oxLDL incubation, pHrodo-positive cells were quantified. (J) BMDMs transfected with NRF2 K518R mutant or wild type were treated with oxLDL (10 μg/mL) or vehicle. After 0-24 hours, Western blotting was performed with the indicated antibodies. Representative images from 5 independent experiments are shown. (Quantification was shown in Fig. S8C). The applied statistical tests, sample number, and results in all figures are summarized in Table S3. All data are expressed as mean±SD, **P<0.01, *P<0.05.
Mitochondrial dysfunction plays a crucial role in triggering SASP 32. Whereas levels of OXPHOS, ATP, and NAD+ were decreased, glycolysis was increased in WT BMDMs after oxLDL incubation; however, these effects were attenuated in ERK5 S496A KI BMDMs (Fig. 7A–F). Although oxLDL effects on glycolysis after plasmid transfection were less clear, we observed a decreased OXPHOS, ATP, and NAD+ after adding oxLDL to the cells overexpressing NRF2 WT; however, this decrease was no longer apparent in cells with the NRF2 K518R mutant (Fig. 7G–L). Taken together, these observations indicate a critical role of NRF2 K518 SUMOylation in SASP/SAS and AHR induction.
Figure 7. ERK5 S496 phosphorylation and NRF2 K518 SUMOylation mediate mitochondrial dysfunction in vitro.
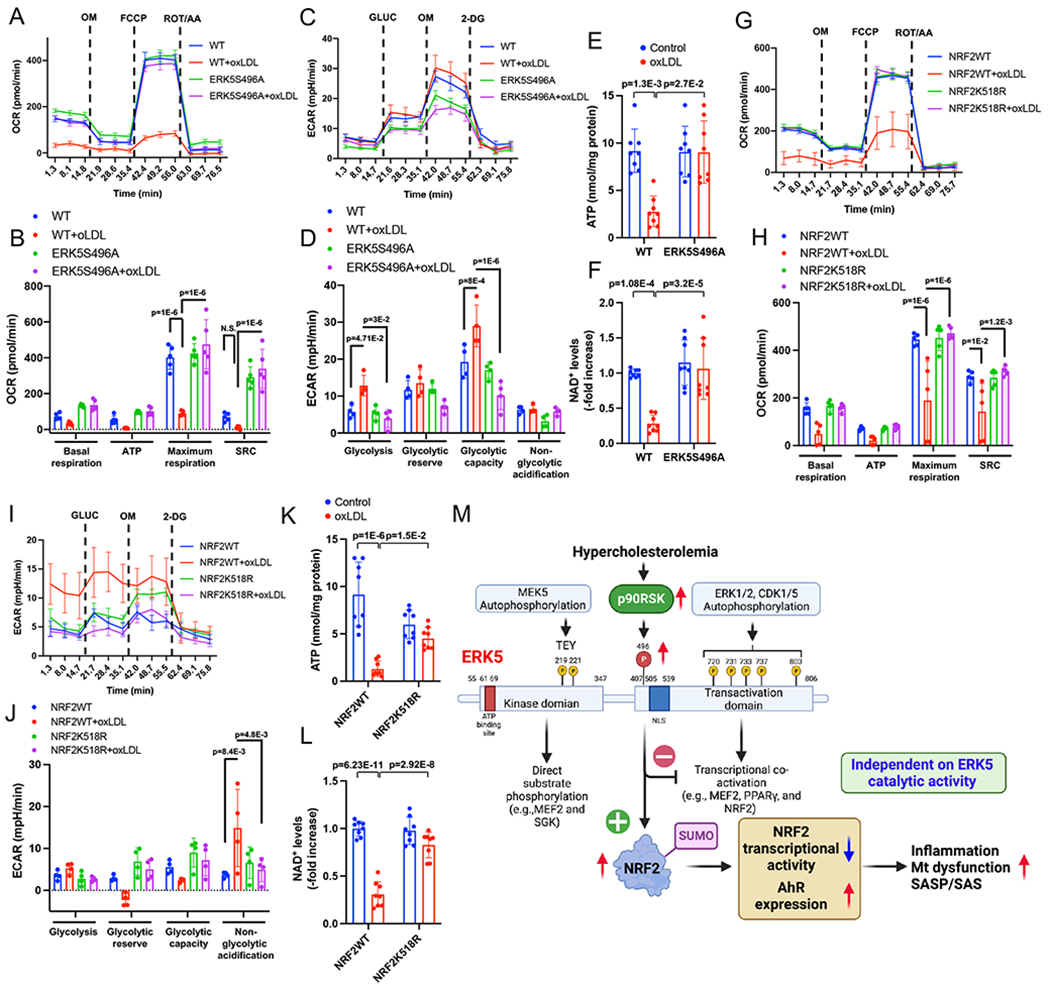
(A-F) WT BMDMs and ERK5 S496A KI BMDMs and (G-L) BMDMs transfected with NRF2 K518R and control plasmid, incubated with oxLDL (10 μg/mL) or vehicle. These cells were then seeded on Seahorse plates. After 24 hours, OXPHOS and glycolysis parameters were measured. During extracellular flux analysis, cells were sequentially treated with (A, G) oligomycin (OM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and rotenone plus antimycin A (ROT/AA) and used to assess OXPHOS parameters based on oxygen consumption rates. (B, H) The basal respiration, mt ATP production, maximal respiration, and spare respiratory capacity were calculated and plotted as oxygen consumption rates in pmoles/minutes. (C, I) Glucose (GLUC), OM, and 2-deoxyglucose (2-DG) were used to determine glycolysis parameters from extracellular acidification rates. (D, J) Glycolysis, glycolytic reserve, glycolytic capacity, and non-glycolytic acidification were calculated and plotted as the extracellular acidification rate in mpH/minutes. ATP (E, K) and NAD+ (F, L) were measured after 24 hrs of oxLDL (10 μg/mL) or vehicle treatment in WT BMDMs and ERK5 S496A KI BMDMs. (M) Proposed model of HC-mediated MC reprogramming to SASP/SAS by ERK5 S496 phosphorylation. The applied statistical tests, sample number, and results in all figures are summarized in Table S3. All data except A, C, G, and I are expressed as mean±SD, and others are expressed as mean±SEM, **P<0.01, *P<0.05.
DISCUSSION
In this study, we have used ERK5 S496A KI mice to show a decrease in the size of atherosclerosis and vulnerable plaque formation with the reduction of necrotic core formation and apoptotic cell accumulation in response to HC. To determine the role of ERK5 S496 phosphorylation in SASP, we utilized the IMC technique that performs a qualitative single-cell assessment of SASP in vivo. We found that SASP events were inhibited in MC-like clusters obtained from ERK5 S496A KI plaques, which confirmed the crucial role of ERK5 S496 phosphorylation in SASP induction. More importantly, we found the unique type of MCs, so-called SAS, that escaped from the growth-suppression effects of p53 in the plaque (Group 2 in Fig. 3A). The induction of SAS was reduced in the plaques from ERK5 S496A KI compared to WT. In addition, the increase of SAβ-gal+Ki67+ and 53BP1+Ki67+ macrophages in HC WT HFD BMDMs compared to those in NC WT NCD BMDMs was attenuated in ERK5 S496A KI mice (Fig. 3). These data are in agreement with a critical role of ERK5 S496 phosphorylation in SAS induction. Although senescence in atherosclerotic plaque has been reported 42, it is challenging to link senescence to plaque formation because of the increase of cell proliferation in the plaque. This paradox is resolved by our demonstration of SAS, which permits macrophages to escape the senescence-induced cell cycle arrest during atherosclerosis formation.
AHR is a ligand-activated transcription factor, which promotes cellular adaptation to environmental changes by sensing compounds from the environment, diet, microbiome, and cellular metabolism 33. AHR can regulate many biological processes, including angiogenesis, cell motility, and immune modulation. A role for AHR in senescence and atherosclerosis, by increasing ROS has been reported 34,43–47. Since AHR was the most significantly upregulated gene, by HC-mediated ERK5 S496 phosphorylation in MCs, we examined its role in SAS induction. We found that AHR depletion inhibited the SAS phenotype in BMDMs from HC WT HFD mice (Fig. 4K). We also found that ERK5 S496A and NRF2 K518R mutant inhibited AHR expression and induction of SAS phenotype. Taken together, these data indicate a crucial role of ERK5 S496 phosphorylation and subsequent NRF2 K518 SUMOylation on AHR induction in the initiation of SAS (Fig. 7M). Further investigation will be necessary to determine how AHR can regulate SAS phenotype.
Of significance, two ERK5 kinase inhibitors (XMD8-92 and a highly selective inhibitor, AX15836) inhibited ERK5 S496 phosphorylation (Fig. 5D, E, H, I and Fig. S7C, D, H, I). There are still conflicting results regarding the functional role of ERK5 catalytic activity. ERK5 contains not only kinase and transcriptional activity, but also possesses noncatalytic functions. Therefore, the function of ERK5 may be different depending on which component (kinase, transcriptional activity, and ERK5 S496 phosphorylation-NRF2 K518 SUMOylation) is dominant in the condition under study. For example, Wilhelmsen et al. reported that the pro-inflammatory role of ERK5 is induced by pro-inflammatory cytokines such as IL-1β, TNFα, and Toll-like receptor 2 agonists 24,48. However, we 18 and others 49 found that ERK5 has anti-inflammatory effects in the context of shear stress. We found that shear stress increased only ERK5 TEY motif phosphorylation and had no effect on p90RSK and ERK5 S496 phosphorylation 7. In contrast, cytokines, and oxLDL increased p90RSK activation 50 and ERK5 S496 phosphorylation, but the effects on ERK5 TEY motif phosphorylation were minimum (Fig.1A). Therefore, the difference in ERK5 post-translational modification in response to each stimulus could be the key to explaining these controversies. These data also indicate that not only ERK5 catalytic activity but also ERK5 S496 phosphorylation-NRF2 K518 SUMOylation should be considered when assessing the role of ERK5 in various situations (Fig. 7M).
The depletion of NRF2 in bone marrow-derived cells accelerated atherosclerosis formation in LDLR−/− mice by inducing pro-inflammatory gene expression 51. There are several potential mechanisms that induce crosstalk between NRF2 and NF-κB signaling. For example, the increase of ROS caused by NRF2 depletion can activate IKK and increase pro-inflammatory gene expression 52. Furthermore, NRF2 could intervene in the senescence process by disrupting proteostasis, altering genomic stability, and causing telomere dysfunction and apoptosis 53. We and others 54,55 reported a crucial role of NRF2 in upregulating efferocytosis. Previously, we also showed that the NRF2 activator abolished SASP events in BMDMs 1. In the current study, we found a role for ERK5 S496 phosphorylation in reducing NRF2 transcriptional activity and AHR-SAS induction via upregulation of NRF2 K518 SUMOylation. The role of NRF2 in up-regulating CD36 expression has been implicated in the depletion of NRF2-mediated reduction of atherosclerotic plaque formation 56. We detected that NRF2 K518R inhibited oxLDL-induced CD36 expression (Fig. 6J), which points to different roles for NRF2 depletion and NRF2 K518 SUMOylation in the regulation of CD36 expression. Taken together, these data indicate a critical role of ERK5 S496 phosphorylation-mediated NRF2 SUMOylation in SASP/SAS and consequently accelerated atherosclerosis (Fig. 7M).
We found that the mutation of ERK5 S496A inhibited unstable plaque formation and SASP events in MCs in the plaque. However, ERK5 S496 phosphorylation instigated not only SASP but also SAS via upregulating AHR expression. The involvement of senescence in atherosclerosis formation has already been suggested57, but the exact molecular mechanisms of senescence-induced atherosclerosis remain unclear. SASP is usually claimed to favor atherosclerosis formation by producing numerous inflammatory cytokines 58 and growth factors59 that lead to neighboring cells’ proliferation 60,61. To the best of our knowledge, our study provides the first evidence in favor of the hypothesis that the senescent myeloid cells themselves can proliferate by escaping p53-mediated cell cycle arrest and contribute to plaque formation. This is important for understanding the persistent effects of stress-induced senescence (SIS), which can be caused by internal and external stimuli such as stress, radiation, and cancer therapy 62. It is already known that many stressors or stressful events that are transient can have a persistent impact on atherosclerosis formation 63. If SAS does not take place and senescent cells cannot proliferate, these senescent cells should disappear as they would be removed from the plaque shortly after termination of the transient stress 64. However, we found evidence for the appearance of SAS in the plaque. Therefore, a transient stress can have long-lasting effects during plaque formation because SAS myeloid cells induced by transient stress can proliferate, thereby maintaining their SASP phenotype. These findings point to the importance of phenotypic changes and reprogramming of myeloid cells in atherosclerosis formation that occurs years after the termination of transient stress such as cancer treatments 62 and sepsis63. Further investigation is necessary to clarify this putative mechanism.
We are aware of the fact that our study has several limitations. First, we could not use MC-specific ERK5 S496A KI mice. Therefore, we cannot conclude that the SASP/SAS phenotype observed in this mouse model is uniquely due to MC ERK5 S496 phosphorylation. However, since we could not observe apparent SASP and SAS phenotype in vascular smooth muscle cells and endothelial cells (Fig. S2D, and G–K), we can still propose that myeloid cells ERK5 S496 phosphorylation plays a critical role in the induction of SASP/SAS phenotype in atherosclerotic plaque. Another limitation is the possible difference in the MC phenotype observed in the plaque and the one occurring in BMDMs 65. However, our data show that the phenotype of bone marrow cells in HFD mice is quite different from the one observed in NCD mice. Therefore, MC SASP/SAS phenotypic changes already take place in the bone marrow, before entering the vessel wall. To determine the MC phenotype independently of the environmental effects taking place in the plaque, it will be necessary to isolate macrophages from the plaque and check and compare gene expression levels in BMDMs and plaque-derived macrophages. The third limitation is that we do not yet show the real impact on plaque formation of the relatively small number of SAS cells we observed in our conditions (1-1.5% in the plaque). In the current study, we were able to demonstrate the critical role played by ERK5 S496A phosphorylation and AHR expression in the induction of SAS. However, these events are not specific to SAS induction. Therefore, we still need to develop an appropriate tool to specifically inhibit SAS induction, before being able to ensure that SAS plays an essential role in atherosclerosis. This step is still necessary to investigate the molecular mechanism by which SAS is regulated and to define the pathophysiological role of SAS in plaque formation.
Supplementary Material
Novelty and Significance.
What is Known?
Senescence-associated secretory phenotype (SASP) is vital in instigating atherosclerosis and tumorigenesis.
Senescence reprograms cancer cells to acquire a proliferative phenotype [senescence-associated stemness (SAS)] after cancer therapy, which allows them to escape senescence-induced cell cycle arrest.
ERK5 is a dual kinase-transcription factor containing an N-terminal kinase domain and a C-terminal transcriptional activation domain, and ERK5 S496 phosphorylation inhibits ERK5 transcriptional activity and subsequently induces SASP.
What New Information Does This Article Contribute?
We have identified the SAS phenotype of myeloid cells (MCs) that escaped the growth-suppression effects of p53 in the plaque.
ERK5 S496 phosphorylation triggered not only SASP but also SAS by upregulating aryl hydrocarbon receptor (AHR) in MCs and promoting atherosclerosis formation.
ERK5 S496 phosphorylation induces a novel site (K518) of NRF2 SUMOylation and inhibits NRF2 transcriptional activity without affecting ERK5 catalytic activity.
The involvement of senescence in atherosclerosis formation is now well established; however, cell cycle arrest is the hallmark of senescence, and myeloid cell (MC) proliferation plays a crucial role in developing atherosclerosis. Therefore, it is challenging to link MC senescence to plaque formation. Using an imaging mass cytometry approach, we discovered the unique MC phenotype of SAS in the plaque, which permits MCs to escape the senescence-induced cell cycle arrest during atherosclerosis formation. This can resolve the paradox of MC senescence observed in proliferative plaque formation. We also demonstrated the crucial role of ERK5 S496 phosphorylation in upregulating MC SAS phenotype in the plaque. RNA sequencing from bone-marrow-derived macrophages isolated from normal and hypercholesterolemia (HC) mice indicated the possible role of AHR. The depletion of AHR inhibited the increase of senescence-associated β-gal+Ki67+ macrophages, supporting the pivotal role of AHR in HC-induced SAS.
ERK5 S496 phosphorylation upregulates NRF2 SUMOylation (K518) and inhibits NRF2 transcriptional activity, which induces AHR in an ERK5 catalytic activity-independent manner. These data indicate a critical role of ERK5 S496 phosphorylation-mediated NRF2 SUMOylation in both SASP and SAS induction and, consequently, accelerated atherosclerosis. These data provide a novel framework for therapeutic intervention to inhibit atherosclerosis by controlling senescence.
Acknowledgements
We thank Scientific Publications, Research Medical Library at The University of Texas MD Anderson Cancer Center for editing and Carolyn J. Giancursio for her technical assistance and Darrell R. Schroeder for helping with statistical analysis.
Funding Sources
This work was partially supported by grants from the National Institutes of Health (NIH) to Drs. Abe (HL-149303 and AI-156921), and Cooke (HL-149303), Le (HL-134740 and HL-149303), and from Cancer Prevention and Research Institute of Texas (CPRIT) to Drs. Abe and Schadler (RP190256). This work is also partially supported by the University of Texas MD Anderson Cancer Center Institutional Research Grant (IRG) Program to Dr. Kotla. This research was performed in the Flow Cytometry & Cellular Imaging Core Facility, which is supported in part by the National Institutes of Health through M. D. Anderson’s Cancer Center Support Grant CA016672, the NCI’s Research Specialist 1 R50 CA243707-01A1, and a Shared Instrumentation Award from the Cancer Prevention Research Institution of Texas (CPRIT), RP121010.
Non-standard Abbreviations and Acronyms:
- 53BP1
p53-binding protein 1
- AAV8-PCSK9
adeno-associated-virus-8 overexpressing pro-protein convertase subtilisin/Kexin type 9 gain-of-function D377Y mutant
- AHR
aryl hydrocarbon receptor
- AX-15836
ERK5 kinase inhibitor
- BM
bone marrow
- BMDMs
bone marrow-derived macrophages
- CA-MEK5
constitutively active form of MEK5
- DEGs
differentially expressed genes
- DNMT3A
DNA methyltransferase 3a
- ECs
endothelial cells
- ERK5/BMK1/MAPK7
extracellular signal-regulated kinase 5
- ERK5b
ERK5 splice variant cDNA mutant (aa78-806)
- ERK5TEYm
ERK5 dual phosphorylation site mutant
- FMK-MEA
a potent and selective p90RSK inhibitor
- GAS6
growth arrest-specific protein 6
- GO
gene ontology
- GCLM
glutamate-cysteine ligase modifier subunit
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- H3C3
H3 clustered histone 3
- H4C11
H4 clustered histone 11
- HC
hypercholesterolemic
- HO1
heme oxygenase 1
- HFD
high-fat diet
- IMC
imaging mass cytometry
- IHC
immunohistochemistry
- IHF
immunohistofluorescence
- Ki67
marker of proliferation Ki-67
- KI
knock-in
- KLF2
kruppel-like factor 2
- LAMP2
(Mac3) lysosomal associated membrane protein 2
- LPAR1
lysophosphatidic acid receptor 1
- MAPK
mitogen-activated protein kinase
- MCs
myeloid cells
- MEF2
myocyte enhancer factor 2
- MEGF9
multiple EGF like domain 9
- mtROS
mitochondrial reactive oxygen species
- NAD+
nicotinamide adenine dinucleotide
- NC
normocholesterolemic
- NCD
normal chow diet
- NFE2
nuclear factor erythroid 2
- NRF2
NFE2-related factor 2
- NRF2A
NRF2-KEAP1-binding inhibitory peptide
- NF-κB
nuclear factor kappaB
- OXPHOS
oxidative phosphorylation
- oxLDL
oxidized LDL
- p90RSK
p90 ribosomal S6 kinase
- p53
tumor protein p53
- p16
cyclin-dependent kinase inhibitor 2A
- p21
cyclin-dependent kinase inhibitor 1
- PPARγ
proliferator-activated receptor γ
- PPIH
peptidyl-prolyl cis-trans isomerase H
- ROS
reactive oxygen species
- RNA-seq
RNA sequencing
- SA-βgal
senescence-associated β-gal
- SAS
senescence-associated stemness
- SASP
senescence-associated secretory phenotype
- TAD
C-terminal transcriptional activation domain
- TEY
threonine-x-tyrosine
- TRX1
thioredoxin 1
- TNFα
tumor necrosis factor α
- TOP2β
DNA topoisomerase IIβ
- TPT1
tumor protein translationally-controlled 1
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- TYRO3
tyrosine-protein kinase receptor TYRO3
- WT
wild type
- XMD8-92
ERK5 kinase inhibitor
Footnotes
Disclosures
S.H.L is an Advisory Board member of AstraZeneca, Beyond Spring Pharmaceuticals, STCube Pharmaceuticals. The other authors report no conflicts.
References
- 1.Singh MV, Kotla S, Le NT, Ae Ko K, Heo KS, Wang Y, Fujii Y, Thi Vu H, McBeath E, Thomas TN, et al. Senescent Phenotype Induced by p90RSK-NRF2 Signaling Sensitizes Monocytes and Macrophages to Oxidative Stress in HIV-Positive Individuals. Circulation. 2019;139:1199–1216. doi: 10.1161/CIRCULATIONAHA.118.036232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitada M, Ogura Y, Koya D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging (Albany NY). 2016;8:2290–2307. doi: 10.18632/aging.101068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du W, Wong C, Song Y, Shen H, Mori D, Rotllan N, Price N, Dobrian AD, Meng H, Kleinstein SH, et al. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell. 2016;15:766–777. doi: 10.1111/acel.12488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circulation research. 2012;111:245–259. doi: 10.1161/CIRCRESAHA.111.261388 [DOI] [PubMed] [Google Scholar]
- 5.Li W Phagocyte dysfunction, tissue aging and degeneration. Ageing research reviews. 2013;12:1005–1012. doi: 10.1016/j.arr.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22:R741–752. doi: 10.1016/j.cub.2012.07.024 [DOI] [PubMed] [Google Scholar]
- 7.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;127:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019. doi: 10.1038/s41568-019-0156-2 [DOI] [PubMed] [Google Scholar]
- 9.Milanovic M, Yu Y, Schmitt CA. The Senescence-Stemness Alliance - A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018;28:1049–1061. doi: 10.1016/j.tcb.2018.09.001 [DOI] [PubMed] [Google Scholar]
- 10.Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, Dorr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553:96–100. doi: 10.1038/nature25167 [DOI] [PubMed] [Google Scholar]
- 11.Dou Z, Berger SL. Senescence Elicits Stemness: A Surprising Mechanism for Cancer Relapse. Cell Metab. 2018;27:710–711. doi: 10.1016/j.cmet.2018.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrand M, Kirsh O, Griveau A, Vindrieux D, Martin N, Defossez PA, Bernard D. Screening of a kinase library reveals novel pro-senescence kinases and their common NF-kappaB-dependent transcriptional program. Aging (Albany NY). 2015;7:986–1003. doi: 10.18632/aging.100845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saleh T, Tyutynuk-Massey L, Cudjoe EK Jr., Idowu MO, Landry JW, Gewirtz DA. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front Oncol. 2018;8:164. doi: 10.3389/fonc.2018.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saleh T, Tyutyunyk-Massey L, Murray GF, Alotaibi MR, Kawale AS, Elsayed Z, Henderson SC, Yakovlev V, Elmore LW, Toor A, et al. Tumor cell escape from therapy-induced senescence. Biochem Pharmacol. 2019;162:202–212. doi: 10.1016/j.bcp.2018.12.013 [DOI] [PubMed] [Google Scholar]
- 16.Munoz DP, Yannone SM, Daemen A, Sun Y, Vakar-Lopez F, Kawahara M, Freund AM, Rodier F, Wu JD, Desprez PY, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. 2019;5. doi: 10.1172/jci.insight.124716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, Braza MS, Baxter S, Fay F, Sanchez-Gaytan BL, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1. doi: 10.1126/sciadv.1400223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004 24/19/8691 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000;20:8382–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M, Kim S, Lim JH, Lee C, Choi HC, Woo CH. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem. 2012;287:40722–40731. doi: 10.1074/jbc.M112.381509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearson G, English JM, White MA, Cobb MH. ERK5 and ERK2 cooperate to regulate NF-kappaB and cell transformation. J Biol Chem. 2001;276:7927–7931. doi: 10.1074/jbc.M009764200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maciejczyk M, Mikoluc B, Pietrucha B, Heropolitanska-Pliszka E, Pac M, Motkowski R, Car H. Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. 2017;11:375–383. doi: 10.1016/j.redox.2016.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilhelmsen K, Xu F, Farrar K, Tran A, Khakpour S, Sundar S, Prakash A, Wang J, Gray NS, Hellman J. Extracellular signal-regulated kinase 5 promotes acute cellular and systemic inflammation. Sci Signal. 2015;8:ra86. doi: 10.1126/scisignal.aaa3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin EC, Amantea CM, Nomanbhoy TK, Weissig H, Ishiyama J, Hu Y, Sidique S, Li B, Kozarich JW, Rosenblum JS. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc Natl Acad Sci U S A. 2016;113:11865–11870. doi: 10.1073/pnas.1609019113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lochhead PA, Tucker JA, Tatum NJ, Wang J, Oxley D, Kidger AM, Johnson VP, Cassidy MA, Gray NS, Noble MEM, et al. Paradoxical activation of the protein kinase-transcription factor ERK5 by ERK5 kinase inhibitors. Nature communications. 2020;11:1383. doi: 10.1038/s41467-020-15031-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotla S, Zhang A, Imanishi M, Ko KA, Lin SH, Gi YJ, Moczygemba M, Isgandarova S, Schadler KL, Chung C, et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox Biol. 2021;47:102132. doi: 10.1016/j.redox.2021.102132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schieffer B, Selle T, Hilfiker A, Hilfiker-Kleiner D, Grote K, Tietge UJ, Trautwein C, Luchtefeld M, Schmittkamp C, Heeneman S, et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation. 2004;110:3493–3500. doi: 10.1161/01.CIR.0000148135.08582.97 [DOI] [PubMed] [Google Scholar]
- 29.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128:1238–1246. doi: 10.1172/JCI95148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merched AJ, Williams E, Chan L. Macrophage-specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler Thromb Vasc Biol. 2003;23:1608–1614. doi: 10.1161/01.ATV.0000084825.88022.53 [DOI] [PubMed] [Google Scholar]
- 31.Sinha SK, Miikeda A, Fouladian Z, Mehrabian M, Edillor C, Shih D, Zhou Z, Paul MK, Charugundla S, Davis RC, et al. Local M-CSF (Macrophage Colony-Stimulating Factor) Expression Regulates Macrophage Proliferation and Apoptosis in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2021;41:220–233. doi: 10.1161/ATVBAHA.120.315255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, Perez-Garcia A, Kiourtis C, Dasgupta N, Lei X, et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34:428–445. doi: 10.1101/gad.331272.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta. 2013;1836:197–210. doi: 10.1016/j.bbcan.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 34.Kaiser H, Parker E, Hamrick MW. Kynurenine signaling through the aryl hydrocarbon receptor: Implications for aging and healthspan. Exp Gerontol. 2020;130:110797. doi: 10.1016/j.exger.2019.110797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan C, Luo H, Lee JD, Abe J, Berk BC. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001;276:10870–10878. [DOI] [PubMed] [Google Scholar]
- 36.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. [DOI] [PubMed] [Google Scholar]
- 37.Le NT, Takei Y, Shishido T, Woo CH, Chang E, Heo KS, Lee H, Lu Y, Morrell C, Oikawa M, et al. p90RSK targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ Res. 2012;110:536–550. doi: 10.1161/CIRCRESAHA.111.254730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abe JI, Sandhu UG, Hoang NM, Thangam M, Quintana-Quezada RA, Fujiwara K, Le NT. Coordination of Cellular Localization-Dependent Effects of Sumoylation in Regulating Cardiovascular and Neurological Diseases. Adv Exp Med Biol. 2017;963:337–358. doi: 10.1007/978-3-319-50044-7_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Lai Q, Chen C, Li N, Sun F, Huang W, Zhang S, Yu Q, Yang P, Xiong F, et al. Both conditional ablation and overexpression of E2 SUMO-conjugating enzyme (UBC9) in mouse pancreatic beta cells result in impaired beta cell function. Diabetologia. 2018;61:881–895. doi: 10.1007/s00125-017-4523-9 [DOI] [PubMed] [Google Scholar]
- 40.Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K, Huang X, Fan Q, Ma J, Cheng J, et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019;466:39–48. doi: 10.1016/j.canlet.2019.09.010 [DOI] [PubMed] [Google Scholar]
- 41.Hendriks IA, D’Souza RC, Yang B, Verlaan-de Vries M, Mann M, Vertegaal AC. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol. 2014;21:927–936. doi: 10.1038/nsmb.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominic A, Banerjee P, Hamilton DJ, Le NT, Abe JI. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020;37:101614. doi: 10.1016/j.redox.2020.101614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu P, Herrmann R, Bednar A, Saloupis P, Dwyer MA, Yang P, Qi X, Thomas RS, Jaffe GJ, Boulton ME, et al. Aryl hydrocarbon receptor deficiency causes dysregulated cellular matrix metabolism and age-related macular degeneration-like pathology. Proc Natl Acad Sci U S A. 2013;110:E4069–4078. doi: 10.1073/pnas.1307574110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eckers A, Jakob S, Heiss C, Haarmann-Stemmann T, Goy C, Brinkmann V, Cortese-Krott MM, Sansone R, Esser C, Ale-Agha N, et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Scientific reports. 2016;6:19618. doi: 10.1038/srep19618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bravo-Ferrer I, Cuartero MI, Medina V, Ahedo-Quero D, Pena-Martinez C, Perez-Ruiz A, Fernandez-Valle ME, Hernandez-Sanchez C, Fernandez-Salguero PM, Lizasoain I, et al. Lack of the aryl hydrocarbon receptor accelerates aging in mice. FASEB J. 2019;33:12644–12654. doi: 10.1096/fj.201901333R [DOI] [PubMed] [Google Scholar]
- 46.Brinkmann V, Ale-Agha N, Haendeler J, Ventura N. The Aryl Hydrocarbon Receptor (AhR) in the Aging Process: Another Puzzling Role for This Highly Conserved Transcription Factor. Frontiers in physiology. 2019;10:1561. doi: 10.3389/fphys.2019.01561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerley-Hamilton JS, Trask HW, Ridley CJ, Dufour E, Lesseur C, Ringelberg CS, Moodie KL, Shipman SL, Korc M, Gui J, et al. Inherent and benzo[a]pyrene-induced differential aryl hydrocarbon receptor signaling greatly affects life span, atherosclerosis, cardiac gene expression, and body and heart growth in mice. Toxicological sciences : an official journal of the Society of Toxicology. 2012;126:391–404. doi: 10.1093/toxsci/kfs002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilhelmsen K, Mesa KR, Lucero J, Xu F, Hellman J. ERK5 protein promotes, whereas MEK1 protein differentially regulates, the Toll-like receptor 2 protein-dependent activation of human endothelial cells and monocytes. J Biol Chem. 2012;287:26478–26494. doi: 10.1074/jbc.M112.359489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr., et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng C, Cho YY, Zhu F, Xu YM, Wen W, Ma WY, Bode AM, Dong Z. RSK2 mediates NF-{kappa}B activity through the phosphorylation of IkappaBalpha in the TNF-R1 pathway. FASEB J. 2010;24:3490–3499. doi: 10.1096/fj.09-151290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Makinen PI, Heinonen SE, Laitinen HM, Heikkila J, Vatanen T, et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res. 2013;98:107–115. doi: 10.1093/cvr/cvt008 [DOI] [PubMed] [Google Scholar]
- 52.Nakajima S, Kitamura M. Bidirectional regulation of NF-kappaB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162–174. doi: 10.1016/j.freeradbiomed.2013.06.020 [DOI] [PubMed] [Google Scholar]
- 53.Tsakiri EN, Sykiotis GP, Papassideri IS, Terpos E, Dimopoulos MA, Gorgoulis VG, Bohmann D, Trougakos IP. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell. 2013;12:802–813. doi: 10.1111/acel.12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim W, Kim HU, Lee HN, Kim SH, Kim C, Cha YN, Joe Y, Chung HT, Jang J, Kim K, et al. Taurine Chloramine Stimulates Efferocytosis Through Upregulation of Nrf2-Mediated Heme Oxygenase-1 Expression in Murine Macrophages: Possible Involvement of Carbon Monoxide. Antioxid Redox Signal. 2015;23:163–177. doi: 10.1089/ars.2013.5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helou DG, Noel B, Gaudin F, Groux H, El Ali Z, Pallardy M, Chollet-Martin S, Kerdine-Romer S. Cutting Edge: Nrf2 Regulates Neutrophil Recruitment and Accumulation in Skin during Contact Hypersensitivity. J Immunol. 2019;202:2189–2194. doi: 10.4049/jimmunol.1801065 [DOI] [PubMed] [Google Scholar]
- 56.Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL, Polotsky VY, Biswal S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 2008;3:e3791. doi: 10.1371/journal.pone.0003791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dominic A, Banerjee P, Hamilton DJ, Le NT, Abe JI. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020:101614. doi: 10.1016/j.redox.2020.101614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coppe JP, Kauser K, Campisi J, Beausejour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200 [DOI] [PubMed] [Google Scholar]
- 60.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011;25:1245–1261. doi: 10.1101/gad.625811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banerjee P, Kotla S, Reddy Velatooru L, Abe RJ, Davis EA, Cooke JP, Schadler K, Deswal A, Herrmann J, Lin SH, et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front Cardiovasc Med. 2021;8:763930. doi: 10.3389/fcvm.2021.763930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merdji H, Schini-Kerth V, Meziani F, Toti F. Long-term cardiovascular complications following sepsis: is senescence the missing link? Annals of intensive care. 2021;11:166. doi: 10.1186/s13613-021-00937-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sadhu S, Decker C, Sansbury BE, Marinello M, Seyfried A, Howard J, Mori M, Hosseini Z, Arunachalam T, Finn AV, et al. Radiation-Induced Macrophage Senescence Impairs Resolution Programs and Drives Cardiovascular Inflammation. J Immunol. 2021;207:1812–1823. doi: 10.4049/jimmunol.2100284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iwata H, Manabe I, Nagai R. Lineage of bone marrow-derived cells in atherosclerosis. Circ Res. 2013;112:1634–1647. doi: 10.1161/CIRCRESAHA.113.301384 [DOI] [PubMed] [Google Scholar]
- 66.McBeath E, Parker-Thornburg J, Fujii Y, Aryal N, Smith C, Hofmann MC, Abe JI, Fujiwara K. Rapid Evaluation of CRISPR Guides and Donors for Engineering Mice. Genes (Basel). 2020;11. doi: 10.3390/genes11060628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ko KA, Wang Y, Kotla S, Fujii Y, Vu HT, Venkatesulu BP, Thomas TN, Medina JL, Gi YJ, Hada M, et al. Developing a Reliable Mouse Model for Cancer Therapy-Induced Cardiovascular Toxicity in Cancer Patients and Survivors. Front Cardiovasc Med. 2018;5:26. doi: 10.3389/fcvm.2018.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116–E7125. doi: 10.1073/pnas.1607728113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in Mice. Curr Protoc Mouse Biol. 2011;1:71–83. doi: 10.1002/9780470942390.mo100130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937 [DOI] [PubMed] [Google Scholar]
- 71.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ren JL, Chen Y, Zhang LS, Zhang YR, Liu SM, Yu YR, Jia MZ, Tang CS, Qi YF, Lu WW. Intermedin(1–53) attenuates atherosclerotic plaque vulnerability by inhibiting CHOP-mediated apoptosis and inflammasome in macrophages. Cell death & disease. 2021;12:436. doi: 10.1038/s41419-021-03712-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Venegas-Pino DE, Banko N, Khan MI, Shi Y, Werstuck GH. Quantitative analysis and characterization of atherosclerotic lesions in the murine aortic sinus. J Vis Exp. 2013;82:50933. doi: 10.3791/50933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Englen MD, Valdez YE, Lehnert NM, Lehnert BE. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J Immunol Methods. 1995;184:281–283. [DOI] [PubMed] [Google Scholar]
- 75.Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112:911–923. doi: 10.1161/CIRCRESAHA.111.300179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39:75–85. doi: 10.2144/05391RV01 [DOI] [PubMed] [Google Scholar]
- 77.Shchepinova MM, Cairns AG, Prime TA, Logan A, James AM, Hall AR, Vidoni S, Arndt S, Caldwell ST, Prag HA, et al. MitoNeoD: A Mitochondria-Targeted Superoxide Probe. Cell Chem Biol. 2017;24:1285–1298 e1212. doi: 10.1016/j.chembiol.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bornfeldt KE, Raines EW, Nakano T, Graves LM, Krebs EG, Ross R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J Clin Invest. 1994;93:1266–1274. doi: 10.1172/JCI117081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heo KS, Cushman HJ, Akaike M, Woo CH, Wang X, Qiu X, Fujiwara K, Abe J. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation. 2014;130:180–191. doi: 10.1161/CIRCULATIONAHA.113.005991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191 [DOI] [PubMed] [Google Scholar]
- 81.Sousa LG, McGrail DJ, Li K, Marques-Piubelli ML, Gonzalez C, Dai H, Ferri-Borgogno S, Godoy M, Burks J, Lin SY, et al. Spontaneous tumor regression following COVID-19 vaccination. J Immunother Cancer. 2022;10. doi: 10.1136/jitc-2021-004371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vidgen B, Yasseri T. P-Values: Misubnderstood and Misused. Front Phys. 2016;4:1–5. [Google Scholar]
- 83.Fay MP, Proschan MA. Wilcoxon-Mann-Whitney or t-test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Stat Surv. 2010;4:1–39. doi: 10.1214/09-SS051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN. Evaluating strategies to normalise biological replicates of Western blot data. PLoS One. 2014;9:e87293. doi: 10.1371/journal.pone.0087293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ortega N, Fanelli A, Serrano A, Martinez-Carrasco C, Escribano F, Tizzani P, Candela MG. Salmonella seroprevalence in wild boar from Southeast Spain depends on host population density. Res Vet Sci. 2020;132:400–403. doi: 10.1016/j.rvsc.2020.07.026 [DOI] [PubMed] [Google Scholar]
- 86.Krzywinski M, Altman N, Blainey P. Points of significance: nested designs. For studies with hierarchical noise sources, use a nested analysis of variance approach. Nat Methods. 2014;11:977–978. doi: 10.1038/nmeth.3137 [DOI] [PubMed] [Google Scholar]
- 87.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960 [DOI] [PubMed] [Google Scholar]
- 88.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. doi: 10.1084/jem.20060245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cudejko C, Wouters K, Fuentes L, Hannou SA, Paquet C, Bantubungi K, Bouchaert E, Vanhoutte J, Fleury S, Remy P, et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood. 2011;118:2556–2566. doi: 10.1182/blood-2010-10-313106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, Sharpless NE. Cells exhibiting strong p16(INK4a) promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A. 2019;116:2603–2611. doi: 10.1073/pnas.1818313116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aguilar G, Cordova F, Koning T, Sarmiento J, Boric MP, Birukov K, Cancino J, Varas-Godoy M, Soza A, Alves NG, et al. TNF-alpha-activated eNOS signaling increases leukocyte adhesion through the S-nitrosylation pathway. Am J Physiol Heart Circ Physiol. 2021;321:H1083–H1095. doi: 10.1152/ajpheart.00065.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Behmoaras J, Gil J. Similarities and interplay between senescent cells and macrophages. J Cell Biol. 2021;220. doi: 10.1083/jcb.202010162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi: 10.1093/bioinformatics/btv300 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data was deposited in the NCBI’s Gene Expression Omnibus database (accession GSE210949). The data, analytic methods, and study materials that support the findings of this study are available in the Data supplement or from the corresponding authors upon reasonable request.