

Abstract
We describe a very unusual cervical tumor in a 12-year-old patient with a clinical history indicative of DICER1 syndrome. Morphologic, immunohistochemical and molecular genetic analysis together helped to diagnose this lesion as a cervical pleuropulmonary blastoma-like tumor, associated with TP53 and DICER1 mutations.
The tumor displayed usual histologic features including mixtures of embryonal rhabdomyosarcoma, sarcomatous cartilage, compact blastema, primitive spindle cells and anaplasia, akin to Type III pleuropulmonary blastoma, and trabecular and retiform patterns. In addition to expanding the phenotypic spectrum of DICER1-associated conditions, we draw attention to genotype-phenotype correlations in DICER1-associated tumors, particularly as they relate to the discovery of a heritable tumor predisposition syndrome.
Keywords: cervix, DICER1-mutation, pleuropulmonary blastoma-like tumor
Introduction
DICER1 syndrome, also called pleuropulmonary blastoma familial tumor susceptibility syndrome, represents an autosomal dominant inherited multineoplasia disorder caused by germline DICER1 gene mutations. It is characterized by an array of topographically and phenotypically diverse benign, low-grade or malignant neoplasms1–3. Apart from macrocephaly in some individuals, DICER1 syndrome is typically not associated with specific physical features. Pleuropulmonary blastoma is the most characteristic tumor associated with germline mutations4, but the list of manifestations of the DICER1 syndrome is growing steadily and encompasses lung (cyst and pleuropulmonary blastoma), intracranial (pituitary blastoma, pineoblastoma, primitive neuroectodermal tumor-PNET, sarcomas), sinonasal (chondromyxoid hamartomas), thyroid (multinodular goiter, thyroid adenoma, differentiated thyroid cancer, poorly differentiated carcinomas, thyroblastoma), bowel (juvenile-type intestinal polyp), gonadal (sex cord stromal tumors like Sertoli-Leydig cell tumor and gynandroblastoma), extra-gonadal (pleuropulmonary-like Sertoli-Leydig cell tumor), genital (cervical and uterine corpus embryonal rhabdomyosarcoma, Müllerian adenosarcoma), renal (cystic nephroma, Wilms tumor and anaplastic sarcoma of kidney) and other neoplasms3, 5–7.
Case report
A 12-year-old patient was admitted to the Gynecology Department for acute urinary retention due to a cervical mass compressing the bladder. The clinical history revealed that at the age of 1 year, the patient had been diagnosed with an inguinal cystic tumor (cystic lymphangioma) followed by a left cystic nephroma at 4 years and a right cystic nephroma at the age of 9 years, all tumors being surgically removed. At the age of 11, a computed tomography (CT) examination revealed multiple bilateral pulmonary nodules, multinodular hyperplasia of the thyroid and a predominantly solid tumor originating from the cervix, occupying the vagina and compressing both uterine corpus and bladder. The cervical tumor doubled its volume within 3 months and was associated with vaginal discharge and urinary retention. When the patient was admitted to the Gynecology Department, computed tomography examination revealed a cervical tumor, 140 mm in diameter, as well as multiple bilateral pulmonary nodules of up to 5mm diameter (Figure 1A). Clinical examination revealed grape-like clusters filling the vaginal cavity and originating in the cervix. The cervical tumor was surgically removed in pieces. Multiple fragments of grey tissue of various sizes (5-25mm diameter) and textures (focally soft and focally firm) were received for pathologic examination (Figure 1B).
Figure 1: Morphology.
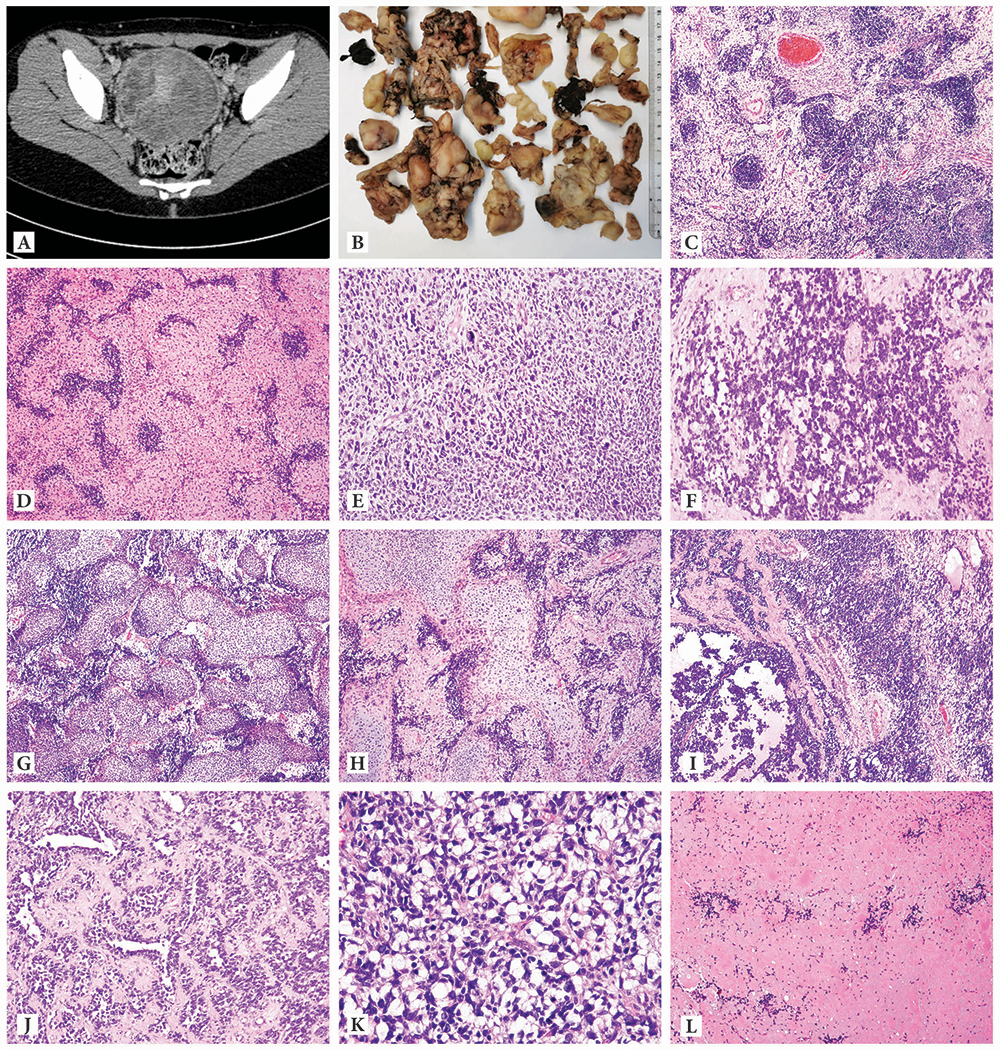
Computed tomography (CT) examination revealed the presence of a 140mm diameter solid tumor originating into the cervix, occupying the vagina and compressing both uterine corpus and bladder (A): The cervical tumor was surgically removed in pieces and multiple fragments of various sizes (5-25mm diameter) and grey color were received for pathologic examination (B): microscopic examination: tumor predominantly presented with loose, edematous and hypocellular areas surrounding hypercellular foci (balls) composed of tumor cells with primitive blastemal appearance, small in size, hyperchromatic and atypical nuclei, brisk mitotic activity with limited cytoplasm suggestive of rhabdomyoblastic differentiation (C, D); areas in which tumor cells presented bizarre nuclei and marked nuclear pleomorphism having a nondescript looking sarcoma appearance (E); areas suggesting alveolar rhabdomyosarcoma (F); massive heterologous differentiation (G) represented by islands of sarcomatous rather than fetal cartilage (H); areas with mixed embryonal and alveolar rhabdomyosarcomatous appearance (I); areas with retiform and trabecular architecture suggestive of Sertoli-Leydig differentiation (J); signet ring- like tumor cells (K); areas with a colloid-like eosinophilic or hyaline-like material and a granular pink neuropil-like background (L).
Microscopic examination revealed that the tumor had a complex morphology, with most tumor fragments partially lined by squamous epithelium and consisting of loose, edematous hypocellular areas surrounding hypercellular foci (blue balls), some of them with entrapped endocervical glands. The hypercellular balls, typical of rhabdomyoblasts in embryonal rhabdomyosarcoma, were composed of tumor cells with a primitive appearance, small in size, round to spindle in shape, with scant cytoplasm, hyperchromatic and atypical nuclei, brisk mitotic activity and apoptosis. There were also areas in which the tumor cells were bizarre and pleomorphic, such as that seen in some TP53-mutated sarcomas, including embryonal rhabdomyosarcoma with anaplasia7. Areas with a nondescript sarcomatous appearance were also identified. Another subgroup of primitive cells was arranged in solid, dyshesive nests, reminiscent of alveolar rhabdomyosarcoma. Interestingly, areas with a trabecular and retiform pattern, suggesting a moderately-poorly differentiated Sertoli-Leydig tumor were also found. Nests of malignant cartilage were present in variable amounts throughout the tumor, but in some sections, chondrosarcoma was the predominant finding. Fetal-type cartilage was not present. Extensive tumor necrosis was present in multiple sections, as well as myxoid stromal areas, signet ring-like cells, sheets of large polygonal cells with eosinophilic cytoplasm, areas with an eosinophilic colloid-like, hyaline-like material and a granular pink neuropil-like background (Figure 1C-L).
Immunohistohemically, the areas suggesting rhabdomyoblastic differentiation expressed desmin, myogenin and Myo-D1, while areas suggesting Sertoliform differentiation were focally positive for CD56 and FOXL2. CD56 was diffusely positive and p53 was overexpressed in all areas. Diffuse non-membranous CD99 and cytoplasmic WT1 expression was also present. However, tumor cells were negative for SMA, CD10, cyclin D1, TTF1, S100 protein, GFAP, HMB45, synaptophysin, inhibin, calretinin, thyroglobulin, MUC4, ALK1, pan CK and CK 7. The trichrome stain highlighted the pink-staining loose myxo-edematous zones (Figure 2A-J).
Figure 2: Ancillary markers assessed by immunohistochemistry :
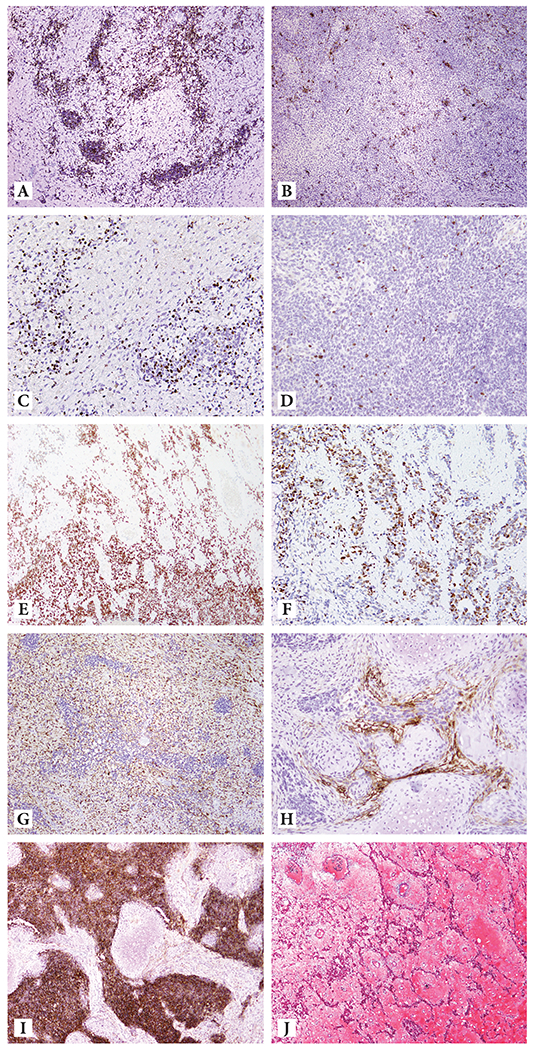
Hypercellular foci positive for Desmin (A); Desmin was also positive in undifferentiated mesenchyme (B); Myogenin-positive in desmin-positive areas (C); Scant myogenin expression in desmin-negative areas (D); p53 overexpression (E); SF-1 positivity in retiform and trabecular architecture suggestive for sex cord differentiation (F); CD10 positivity in undifferentiated mesenchyme (G); CD10 positive cells condensing around lobules of cartilage (H); diffuse CD56 positivity (I); Trichrome stain highlighting the pink-staining loose myxo-edematous zones (J).
Based on morphology and immunohistochemical results suggestive of a cervical pleuropulmonary blastoma-like tumor with rhabdomyoblastic and sertoliform differentiation, the tumor was subjected to next generation sequencing targeting 505 cancer-related genes (MSK-IMPACT at Memorial Sloan Kettering Cancer Center’s Integrated Genomics Operation)8,9. Mutations and copy number alterations were identified using validated bioinformatics methods9,10. This analysis revealed that the tumor harbored high levels of gene copy number alterations (fraction of genome altered 19%), and, consistent with the clinicopathologic findings, a clonal, pathogenic TP53 mutation associated with loss of heterozygosity (LOH) and two clonal DICER1 mutations, including a hotspot p.E1813G and a frameshift p.P1161Lfs*31 mutation (Figure 3). Furthermore, the tumor harbored a clonal AR missense mutation coupled with LOH, two subclonal missense mutations affecting INHA and SETD8 as well as a subclonal KMT2D in-frame small insertion and deletion (indel) (Figure 3A-B).
Figure 3: Mutations and copy number alterations identified in the cervical tumor using targeted next generation sequencing.
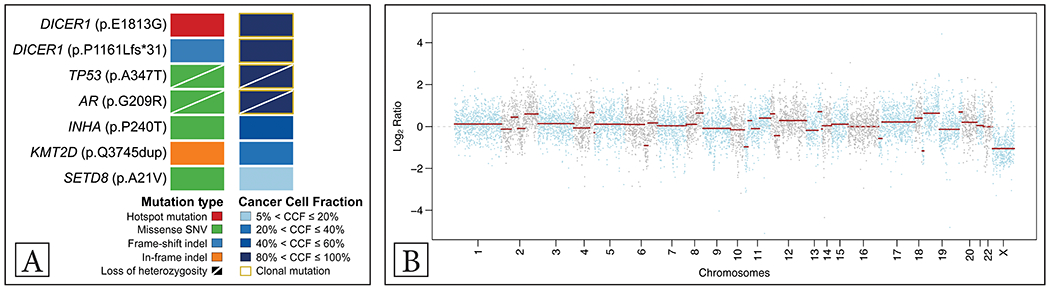
Mutations and cancer cell fractions of the mutations identified. Clonal mutations are depicted by a yellow box, loss of heterozygosity of the wild-type allele is displayed by a diagonal bar. The mutation types and cancer cell fractions are color-coded according to the legend (A); Chromosome plot with the copy number Log2-ratios plotted on the y-axis according to their genomic coordinates on the x-axis (B)
Chemotherapy including bleomycin, etoposide and carboplatin was initiated. After 5 cycles, radiologic examination revealed that the tumor reduced in size from 140mm to 10mm diameter. The multidisciplinary team subsequently decided for the patient to undergo surgical treatment to remove the remaining tumor. A CT examination of the chest thereafter showed multiple bilateral pulmonary nodules unchanged in number, size and location, which were interpreted as part of DICER1-syndrome rather than pulmonary metastases.
Discussion
Patients, especially females, with germline or mosaic DICER1 gene mutations are at an increased risk of developing a range of childhood, adolescent, and young adult-onset tumors. The present case represents a cervical pleuropulmonary blastoma-like tumor with very unusual microscopic findings including rhabdomyoblastic, chondrosarcomatous and sertoliform differentiation, and overlapping immunohistochemical features in a 12-year-old patient with clinical history indicating a DICER1- syndrome. Targeted sequencing in combination with morphologic, immunohistochemical findings and clinical correlation diagnosed this lesion as a TP53- and DICER1-mutant tumor. We are therefore expanding the phenotypic spectrum of DICER1-associated sarcomas in the female genital tract to include a unique tumor seen here in the cervix, having composite features of both pleuropulmonary blastoma and a Sertoli-Leydig cell tumor.
The estimated prevalence of pathogenic DICER1 variants in the general population is ~1:10,600, overall and ~1:4600 in the adult cancer population11. The DICER1 gene, located on chromosome 14q32.13, plays an important role in the control of protein translation. The DICER1 protein is necessary for the production of microRNAs (miRNA) that regulate expression of genes by interacting with mRNA12. Functional DICER1 is essential for embryogenesis and its absence is incompatible with development. Syndromic patients usually carry one germline DICER1 mutation (usually loss-of-function) and acquire a secondary missense DICER1 mutation affecting one of the hotspots in the RNase IIIb domain2,11,13. Syndromic patients lacking germline hotspot mutations might harbor mosaicism for missense variants, which may be associated with more severe disease14. Molecular analysis applied in this case could not discriminate between somatic and germline DICER1 mutations because normal tissue was not available for study. However, the clinical picture strongly suggests the presence of a germline mutation.
Pleuropulmonary blastomas, a dysembryonic lung neoplasm is the best-known manifestation of the DICER1 syndrome, occurs as type I (cystic), type II (solid and cystic) and type III (solid). Type III tumors contain different components such as embryonal rhabdomyosarcoma, fetal or sarcomatous cartilage, compact blastema, primitive spindle cells and anaplastic tumor cells, all of which were identified in the present tumor. It is a possibility that this patient also has a type I pleuropulmonary blastoma in the lung, given the finding of pulmonary nodules. DICER1-associated neoplastic lesions frequently demonstrate teratoma-like mixtures of various tissue types, with benign-appearing eutopic epithelial components such as seen in Müllerian adenosarcoma, cervical embryonal rhabdomyosarcoma, and intracranial sarcomas1,4. Foci of cartilage are another feature of DICER1-associated lesions and are therefore a histologic indication of the possibility of this disorder. Many DICER1-associated neoplasms also resemble different developmental stages in organogenesis, which may impart a teratoid or blastematous appearance. Given these features, there has been a proposal to unify the variety of terms that have been used to describe such tumors as “DICER 1 sarcomas” 15.
Clinically and macroscopically (grape-like clusters), the case was initially considered to represent embryonal rhabdomyosarcoma, a disease known to occur in patients with DICER1 syndrome. This suspicion was heightened at microscopic examination where undifferentiated small blue round cells with rhabdomyoblastic differentiation were identified, with a cambium layer beneath the squamous epithelium, admixed with hypercellular tumor balls set in a myxoid hypocellular stroma. Immunohistochemical analysis of desmin, myogenin and MyoD1 confirmed rhabdomyoblastic differentiation. Areas resembling alveolar rhabdomyosarcoma were also present and were positive for the same markers. Translocations associated with this tumor type have not been documented in uterine rhabdomyosarcomas or mixed epithelial-stromal tumors containing rhabdomyosarcoma, despite histologic appearances. As mentioned, parts of the tumor were represented mostly by chondrosarcoma, which is only very rarely encountered in embryonal rhabdomyosarcoma (wherein fetal type cartilage is significantly more common). Interestingly, portions of the tumor resembled poorly differentiated Sertoli-Leydig cell tumor with heterologous elements (rhabdomyosarcoma and chondrosarcoma) and the differentiated areas expressed CD56 and FOXL2. No Leydig cells were seen, however. Sertoli-Leydig cell tumor is a well-known constituent of the DICER1 syndrome as has the exceedingly uncommon pleuropulmonary-like Sertoli-Leydig cell tumor and Sertoli-Leydig cell tumors that arise in extraovarian sites3. Whether the AR and INHA mutations are associated with this morphology is uncertain. Immunohistochemical p53 analysis was initiated given the anaplastic appearance of many elements, most notably the cartilage, blastema and seemingly undifferentiated spindle cells. P53 mutations are documented in pleuropulmonary blastoma and is associated with anaplasia, older age, positive margins and poorer prognosis. Somatic TP53 mutations have also been described in embryonal rhabdomyosarcomas with aggressive clinical features and histologic anaplasia7. DICER1 syndrome-associated cervical tumors that harbor such TP53 mutations are similarly very rare7.
Our report shows that the recognition of certain pathologic findings in a site-specific context like female genital tract can raise suspicion for a DICER1-associated tumor even in a resource-limited setting, such as developing countries, with the aid of reference centers that can confirm the diagnostic impression. Although not the subject of this report, it should be noted that somatic DICER1 mutations occur in large proportions of sporadic Sertoli-Leydig cell tumors and embryonal rhabdomyosarcomas, and less commonly in other tumors such as Mullerian adenosarcoma3. Therefore, somatic and germline testing to assess for the presence of DICER1 mutations may be performed when morphology, clinical history and age are considered. The workup should include genetic counseling, followed by germline detection, cascade testing, and surveillance, given that affected individuals remain at risk of developing additional tumors. Schultz et al recommend considering surveillance imaging until at least 40 years of age by which time 95% of Sertoli-Leydig cell tumors/gynandroblastomas will have been diagnosed4. The prognosis depends on the type of treatment with chemosensitivity noted in some cases, but further systematic studies in larger cohorts are warranted5.
Funding:
This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. BW is funded in part by Breast Cancer Research Foundation, Cycle for Survival and NIH/NCI P50 CA247749 01 grants.
Footnotes
Conflicts of Interest/Competing interests: B. Weigelt reports ad hoc membership of the scientific advisory board of Repare Therapeutics, outside the scope of the submitted work.
Ethics approval: This study was approved by the institutional review boards of each participating center. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate: Informed consent was obtained from all individual participants included in the study.
Consent for publication: All authors agreed to publish the results of the present study
Availability of data and material:
All data can be provided by the corresponding author (Simona Stolnicu).
References
- 1.Foulkes WD, Bahubeshi A, Hamel N et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat 2011; 32:1381–1384. [DOI] [PubMed] [Google Scholar]
- 2.de Kock L, Wu MK, Foulkes WD. Ten years of DICER1 mutations: provenance, distribution, and associated phenotypes. Hum Mutat 2019; 40:1939–1953. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez IA, Stewart DR, Schultz KA et al. DICER 1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Mod Pathol 2022; 35: 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz KAP, Williams GM, Kamihara J et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 2018; 24(10): 2251–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roy P, Das A, Singh A et al. Phenotypic similarities within the morphologic spectrum of DICER1-associated sarcomas and pleuropulmonary blastoma: histopathologic features guide diagnosis in the LMIC setting. Pediatric Blood Cancer 2022; 69(3); e29466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCluggage WG, Apellaniz-Ruiz M, Chong AL et al. Embryonal rhabodmyosarcona of the ovary and fallopian tube: rare neoplasms association with germline and somatic DICER1 mutations. Am J Surg Pathol 2020; 44(6): 738–747. [DOI] [PubMed] [Google Scholar]
- 7.Bennett JA, Ordulu Z, Young RH et al. Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod Pathol 2021; 34 (9): 1750–1762. [DOI] [PubMed] [Google Scholar]
- 8.Zehir A, Benayed R, Shah RH et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10000 patients. Nat Med 2017; 23(6): 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Da Cruz Paula A, da Silva EM, Segura SE et al. Genomic profiling of primary and recurrent adult granulosa cell tumor of the ovary. Mod Pathol 2020; 33(8): 1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SH, Basili T, Dopeso H et al. Recurrent WWTR1 S89W mutations and Hippo pathway deregulation in clear cell carcinomas of the cervux. J Pathol 2022; Apr 12. doi: 10.1002/path.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Schultz KAP, Hill DA et al. The prevalence of germline DICER1 pathogenic variation in cancer populations. Mol Genet Genom Med 2019; 7, e555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slade I, Bacchelli C, Davies H et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 2011; 48, 273–278. [DOI] [PubMed] [Google Scholar]
- 13.Robertson JC, Jorcyk CL, Oxford JT. DICER1 syndrome: DICER1 mutations in rare cancers. Cancers 2018; 10, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenneman M, Field SA, Yang J et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma/DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res 2015; 4, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCluggage WG, Foulkes WD: DICER1- associated sarcomas: towards a unified nomenclature. Mod Pathol 2021; 34(6): 1226–1228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data can be provided by the corresponding author (Simona Stolnicu).