

Abstract

Treatment against tuberculosis can lead to the selection of drug-resistant Mycobacterium tuberculosis strains. To tackle this serious threat, new targets from M. tuberculosis are needed to develop novel effective drugs. In this work, we aimed to provide a possible workflow to validate new targets and inhibitors by combining genetic, in silico, and enzymological approaches. CanB is one of the three M. tuberculosis β-carbonic anhydrases that catalyze the reversible reaction of CO2 hydration to form HCO3– and H+. To this end, we precisely demonstrated that CanB is essential for the survival of the pathogen in vitro by constructing conditional mutants. In addition, to search for CanB inhibitors, conditional canB mutants were also constructed using the Pip-ON system. By molecular docking and minimum inhibitory concentration assays, we selected three molecules that inhibit the growth in vitro of M. tuberculosis wild-type strain and canB conditional mutants, thus implementing a target-to-drug approach. The lead compound also showed a bactericidal activity by the time-killing assay. We further studied the interactions of these molecules with CanB using enzymatic assays and differential scanning fluorimetry thermal shift analysis. In conclusion, the compounds identified by the in silico screening proved to have a high affinity as CanB ligands endowed with antitubercular activity.
1. Introduction
The spread of strains of Mycobacterium tuberculosis (Mtb) resistant to currently available antitubercular drugs is a serious global public health problem and a major challenge for researchers, clinicians, and governments involved in the fight against this disease.1 The discovery of novel antitubercular agents requires multidisciplinary efforts to find compounds that can affect new cellular targets and consequently that could be active against Mtb drug-resistant strains. Thus, drug discovery can be performed following two different approaches: drug-to-target and target-to-drug.2 The new antitubercular drugs approved for tuberculosis (TB) therapy (bedaquiline, pretomanid, and delamanid) were discovered by following the first strategy.3
For target-based drug discovery, the knowledge of the Mtb genome sequencing has accelerated the discovery of new potential drug targets.4 In this case, compounds are screened against an Mtb cellular target enzyme. This ensures target suitability, but the activity of these compounds must be evaluated also against cell growth to test if the drugs are able to enter the mycobacterial cells and then reach their target(s).2 Unfortunately, for the antitubercular drugs, it is a challenging point due to the peculiar cell wall. For this reason, this approach has not been very successful in TB drug discovery so far, although the possibility of using inhibitors of a novel essential cellular target remains an appealing option in the fight against TB.
Mtb carbonic anhydrases (CAs) have long been considered as a possible target for antitubercular drug development. The CAs are metalloenzymes present in all living organisms that catalyze the essential reversible hydration of CO2 to form bicarbonate. Mammalian enzymes are necessary for intermediary metabolism, facilitating CO2 diffusion, pH homeostasis, and ion transport. CAs are divided into five families (α, β, γ, δ, and ε); mammals have α-CAs, while β-CAs are widespread in eubacteria, archaea, algae, and plants. Mtb presents three β-CAs (Rv1284, Rv3588c, and Rv3273) and one γ-CA (Rv3525c).5,6 These enzymes differ from the mammal enzymes.7 CanB, which is coded by Rv3588c, is the β-CA that shows greater catalytic activity for CO2 hydration than the other two mycobacterial ones.7 From previous genome-wide studies, the essentiality of CanB has not been clearly determined,8 and indeed, CanB was classified as essential by Griffin and colleagues9 but non-essential by two other recent studies.10,11 On the other hand, CAs are predicted to be required for Mtb virulence in mice.12 Therefore, the possible essentiality of CanB needs to be demonstrated experimentally.
Because mycobacterial CAs are different from those of mammals and could be essential for Mtb growth, they may have the potential to be good drug targets. However, to our knowledge, so far, while several compounds have been found to be highly active against CanB/CanA enzymatic activity, only a few inhibitors are effective in vitro against Mtb growth.8,11,13,14 Consequently, again, the drug-to-target strategy has not been very successful for TB drug discovery.
In this work, we used a multidisciplinary approach (microbiology, genetics, biochemistry, medicinal chemistry, and bioinformatics) to validate the target-based whole-cell phenotypic high-throughput screening for looking for new CanB inhibitors active against Mtb growth. This provided the possibility of using a combined approach for the validation of other possible new targets. Additionally, we found two CanB inhibitors endowed with antitubercular activity, demonstrating the validity of the approach used.
2. Results
2.1. canB Is Essential for In Vitro Mtb Growth
To prove the essentiality of Rv3588c (canB) for Mtb H37Rv growth and validate CanB as a possible cellular target against TB, two conditional knockdown (cKD) mutants were constructed in which the canB native promoter was substituted by the repressible mutagenized promoters Pptr15,16 by insertional duplication in a strain carrying the TetR-Pip-OFF repressible system, TB61 (pFRA61). The repressible promoter Pptr was mutagenized by Boldrin et al.16 to obtain differentiated promoter strength; the promoters used in this study are weaker than the wild-type Pptr. In particular, Pptr11 has 4.2% of the strength of the original Pptr, while Pptr12 has 25% of the strength.16 In the resulting strains, named TB169C and TB170A, respectively, the transcription of canB was expected to be repressed by the presence of the inducer, anhydrotetracycline (ATc).
As shown in Figure S1, Mtb canB conditional mutants TB169C and TB170A did not grow in the presence of ATc (500 ng/mL), unlike their parental strain TB61.
The growth of cKD strains in the presence of different concentrations of ATc was then evaluated in a Middlebrook 7H9 liquid medium. In the presence of ATc, an inhibition of the growth proportional to both the concentration of ATc used and the strength of the Pptr promoter of each strain was observed (Figure S2). Both TB169C and TB170A growth were inhibited at a concentration of 500 ng/mL ATc after 48 h, never reaching an exponential phase of growth (Figure S2). Thus, a concentration of 500 ng/mL was chosen for all the following experiments in the liquid medium. The growth of Mtb H37Rv cKD strains depends on the presence of the ATc inducer in a dose-dependent manner, highlighting that the canB expression affects the growth and survival of Mtb H37Rv.
To correlate canB depletion with growth arrest, the two cKD mutants and their parental strain were grown in standing liquid cultures in the presence of 500 ng/mL ATc; the number of culture rejuvenation passages required for Mtb cKD strains not to resume growth was evaluated. As a control, culture rejuvenation was also performed in an ATc-free liquid medium. Every 96 h, cultures of cKD mutants were diluted in a fresh liquid medium with/without ATc (rejuvenation). The parental strain TB61 could start the growth after each passage in the fresh medium regardless of the presence of ATc (Figure 1C). On the other hand, the growth of TB169C stopped after 96 h of exposure to ATc, while that of TB170A was arrested after the culture was refreshed twice (Figure 1A and Figure 1B, respectively), suggesting that the greater strength of the Pptr promoter influences the time required for titrating down the protein. Both strains grew well in medium without ATc.
Figure 1.

canB is essential for Mtb growth in axenic cultures. Growth curves of TB169C (A), TB170A (B), and TB61 (parental strain, C) were performed in Middlebrook 7H9 in the absence (red)/presence of ATc [500 ng/mL] (blue). The optical density (OD) was recorded at 600 nm at different time points and used to compile the growth curves. Each experiment was repeated three times, giving similar results.
Growth inhibition of canB-depleted strains shows a good correlation between growth arrest and the presence of ATc. Overall, these results confirm the essentiality of Mtb canB in vitro, under the conditions studied.
2.2. Virtual Screening to Find Possible CanB Inhibitors
In aqueous solution, CanB establishes a pH-dependent equilibrium between tetrameric and dimeric forms that affect its enzymatic activity, as demonstrated experimentally by Covarrubias et al.17 In particular, at physiological pH, the CanB structure is predominantly folded into dimers (84% as the dimeric form and 16% as the tetrameric form) and the enzyme is inactive. On the contrary, at higher pH, the dimers assemble into a fully active tetrameric structure (at pH 8.4, the dimer corresponds to 31% and the tetramer to 69%). Moreover, the tetrameric form of CanB shows an open conformation of each active site (one per monomer, a total of four), where a hydroxide ion coordinates a zinc ion (the enzyme cofactor) by displacement of a Cys side chain. The presence of the hydroxide ion and the carbon dioxide is essential for triggering the enzymatic reaction.
This experimental evidence, combined with the fact that orthosteric binders of CanB (such as ethoxzolamide)18 are non-selective inhibitors of both CanB and the human carbonic anhydrase isoenzymes (i.e., hCAII),19 suggested us the rationale to be applied for searching selective inhibitors of CanB. We focused our attention on the identification of ligands for CanB shallow binding sites (away from the conserved orthosteric pocket) that are defined as superficial pockets where proteins are supposed to bind for the establishment of intermolecular interactions. Such ligands should be intended as a protein–protein interaction inhibitor that could affect CanB dimer and tetramer formation, thus reducing or abrogating CanB enzymatic activity that depends on the presence of the tetrameric form.
Based on these premises, a molecular docking-based virtual screening approach was applied to the three-dimensional structure of CanB determined by X-ray crystallography experiments (PDB entry 1YM3)20 with the aim of selecting possible CanB inhibitors. First, the SiteMap routine of the Schrödinger suite21 was used to identify putative binding sites for small molecules at the interface of CanB monomers. Next, compounds of the National Cancer Institute (NCI) database (https://dtp.cancer.gov) were iteratively docked within two of the binding sites identified in the previous step (Figure 2).
Figure 2.

Crystal structure of tetrameric CanB and its binding sites. Chains are depicted in different colors (blue, yellow, green, and cyan) with a cartoon-like representation, while the binding sites 2 (orange) and 4 (magenta) are placed in clefts defining interfaces between monomers. Black spheres represent zinc ions and are located within the catalytic sites of CanB. Atomic three-dimensional coordinates were taken from the Protein Data Bank entry 2A5V.17
As a result, 53 compounds were prioritized as putative CanB ligands and were tested for their possible activity against the Mtb H37Rv strain. Unfortunately, all the 42 compounds predicted to bind Site 4 were inactive or showed solubility issues (Table S1). On the other hand, among the 11 putative binders of Site 2, three compounds (namely, NSC118741, NSC55150, and NSC637994) were active with minimum inhibitory concentration (MIC) values comprised between 1 and 16 μg/mL (in bold in Table S1, while their structure is shown in Figure S3). Based on the better activity of NSC55150 (22, MIC = 1 μg/mL) in comparison to the other two hit compounds (MIC of 8 and 4 μg/mL, respectively) and on the availability of several derivatives of 22 within the NCI database, a small and focused library was compiled for this compound, while both NSC118741 and NSC637994 were not further considered.
Small molecules bearing amidino moieties as found for 22 are known as versatile classes of biologically interesting compounds, showing hydrogen bond ability and the possibility to undergo protonation at physiological pH. Compound 22 and its analogues belong to the class of N,N′-diphenylterephthalamide derivatives. Interestingly, some active compounds are already known for their antimicrobial activity. A representative example is constituted by pentamidine, a bisbenzamidine with a seven-atom alkoxy bridge linking the two aromatic molecular edges (Figure S3). Pentamidine is indicated in the case of Pneumocystis carinii pneumonia in immunocompromised patients and as a secondary agent in various forms of leishmaniasis and other pathogenic protozoa.22 Moreover, pentamidine showed micromolar activity toward whole-cell mycobacteria.23,24
Antimycobacterial activity values of compounds belonging to the class of N,N′-diphenylterephthalamide derivatives (analogues of 22) allowed us to derive several SAR considerations. The most simple compound was represented by the N,N′-diphenylterephthalamide itself (1; Table 1) that showed a low activity (MIC about 40–20 μg/mL).
Table 1. Structure and Antimycobacterial Activity of the 25 N,N′-Diphenylterephthalamide Derivatives Belonging to the Focused Small Library Built around the Hit Compound NSC55150 (22).

| compound | R | MIC (μg/mL) |
|---|---|---|
| 1, NSC73489 | H | 20–40 |
| 2, NSC164131 | o-F | >40 |
| 3, NSC111339 | p-F | >40 |
| 4, NSC73488 | p-Cl | >40 |
| 5, NSC204225 | m,p-di-Cl | >40 |
| 6, NSC71193 | p-Me | >40 |
| 7, NSC110298 | p-Et | >40 |
| 8, NSC110302 | m-Cl,p-Me | >40 |
| 9, NSC110317 | p-FSO2 | >40 |
| 10, NSC55146 | p-C(=NH)NH2 | >40 |
| 11, NSC53313 | p-C(=NH)NHCH3 | 0.31 |
| 12, NSC70138 | p-C(=NH)NHCH2CH3 | 0.62 |
| 13, NSC71201 | p-C(=NH)NH(CH2)2CH3 | 0.62 |
| 14, NSC80122 | p-C(=NH)NHCH(CH3)2 | 0.62 |
| 15, NSC80976 | p-C(=NH)NH(CH2)3CH3 | 1.25 |
| 16, NSC80975 | p-C(=NH)NH(CH2)5CH3 | 5 |
| 17, NSC53312 | p-C(=NCH3)NHCH3 | 0.62 |
| 18, NSC64897 | p-C(=NH)NHNH2 | >40 |
| 19, NSC77889 | 10 | |
| 20, NSC55148 | >10 | |
| 21, NSC55147 | >40 | |
| 22, NSC55150 | p-CH2C(=NH)NH2 | 1 |
| 23, NSC55151 | p-NHC(=NH)NH2 | 0.16 |
| 24, NSC72583 | 2.5 | |
| 25, NSC65376 | 0.62 |
Addition of halogens or small alkyl substituents with different substitution patterns at the terminal phenyl rings (2–8) completely abolished activity (MIC values >40 μg/mL). In a similar way, the p-F sulfonyl and the carbamimidoyl derivatives 9 and 10 (a bisbenzamidine derivative), respectively, were inactive too. Addition of linear alkyl groups to the carbamimidoyl terminal moieties led to seven additional bisbenzamidine derivatives 11–17 (Glide scores and the major interactions of these compounds are reported in Figure S4). In general, they were characterized by a significant improvement of activity, even if increasing the length and bulkiness of the substituent was detrimental. In fact, the methyl derivative 11 showed an MIC value of 0.31 μg/mL that increased to 0.62 μg/mL in the case of the ethyl, propyl, and i-propyl analogues 12–14, respectively. The butyl and the hexyl analogues 15 and 16 showed activities further reduced to 1.25 and 5 μg/mL, respectively.
Further decorations and chemical modifications of the structure of 11 allowed deduction of additional SAR considerations. As an example, double methylation at both carbamimidoyl moieties as in 17 resulted in a 2-fold reduction of activity in comparison to the parent compound 11 (0.62 vs 0.31 μg/mL). Moreover, also, the presence of a hydrazineyl(imino)methyl terminal group as in 18 or changing the amide spacer as in 19 (which gained a non-symmetrical structure) was detrimental for activity (MIC values of 40 and 10 μg/mL, respectively). On the contrary, the non-symmetrical insertion of a double bond to transform the central benzamide moiety of 11 into the cinnamamide spacer of 25 led to a 2-fold decrease in activity, from 0.31 to 0.62 μg/mL.
On the other hand, changing the substitution pattern at the terminal or at the central phenyl rings of 10 (as in 20 and 21, respectively) did not improve activity.
Worth noting, linking the carbamimidoyl groups and the terminal phenyl rings of 10 with a short bridge resulted in high antimycobacterial activity. In particular, insertion of a methylene spacer led to 22 with an MIC value of 1 μg/mL, while transformation of the 2-amino-2-iminoethyl moiety of 22 into the guanidino group of 23 further increased activity to 0.16 μg/mL, thus resulting in the most active compound of this series. On the contrary, structural rigidification of the terminal substituents of 22 into the tetrahydropyrimidinyl ring of 24 caused a drop in activity (MIC of 2.5 μg/mL).
In summary, activity data on this class of compounds clearly showed that halogens and small alkyl substituents added with various substitution patterns to the terminal phenyl rings of the N,N′-diphenylterephthalamide 1 resulted in inactive derivatives. Following this trend, the carbamimidoyl analogue 10 was also inactive, while its mono- and di-alkyl analogues gained a significant activity. In particular, within the mono-alkyl subseries, activity decreased with substituents longer than a propyl chain. In fact, the butyl and hexyl analogues 15 and 16 showed a 4- and 60-fold reduction of activity, respectively, in comparison to the methyl analogue 11.
Moreover, increasing the length of the terminal carbamimidoyl groups of the inactive parent compound 10 played a fundamental role for activity. In fact, the insertion of a methylene spacer led to an MIC of 1 μg/mL in 22, and the subsequent transformation into the guanidino derivative 23 resulted in the best compound of this series.
Finally, variations of the chemical features of the N,N′-diphenylterephthalamide scaffold were detrimental for activity, with the sole exception of 25 where the insertion of an unsaturated spacer maintained a significant activity.
Molecular docking simulations showed that the terminal carbamimidoyl moieties of the ligands played a fundamental role in the stabilization of the complex with the target. In fact, such substituents formed hydrogen bonds with the terminal carboxyl group of Glu173 and the side chain OH of Ser86 (or the carbonyl moiety of Ala82).
As an example of the interaction pathway between the ligands and CanB, the most active compound (23) formed a network of hydrogen bonds with amino acids of Site 2 (Figure 3).
Figure 3.

Top-scored docking pose of NSC55151 (23) within Site 2. For the sake of clarity, only residues (sticks) that interact by hydrogen bonds (green dashed lines) and by salt bridges (magenta dashed lines) with the ligand (ball-and-stick) are shown. Amino acids involved in hydrophobic contacts with the ligand are also represented.
The terminal carbamimidoyl moieties were hydrogen bond donors for the interactions with the carboxylic moiety of Glu173 and the carbonyl group of Ala82. A salt bridge between Glu173 and one of the carbamimidoyl groups further stabilized the complex. An additional hydrogen bond was formed between an amide nitrogen atom of 23 and the carboxylic moiety of Glu88, which served as the acceptor. Several hydrophobic interactions were also found between all aromatic rings of the ligand and side chains of surrounding residues (Ala170, Thr92, Val93, Tyr89, and Ala82) and the Ser86 backbone. On the other hand, the higher conformational freedom of the ethanimidamide moiety of 22 (in comparison to the quasi-planar guanidino group of 23) resulted in slightly different interactions with CanB. In fact, while the hydrogen bonds with the side chains of both Glu173 and Glu88 were maintained, the interaction of one of the guanidino moieties of 23 with Ala82 was replaced by a hydrogen bond between the terminal carbamimidoyl group of 22 and the OH of the Ser86 side chain (Figure 4).
Figure 4.
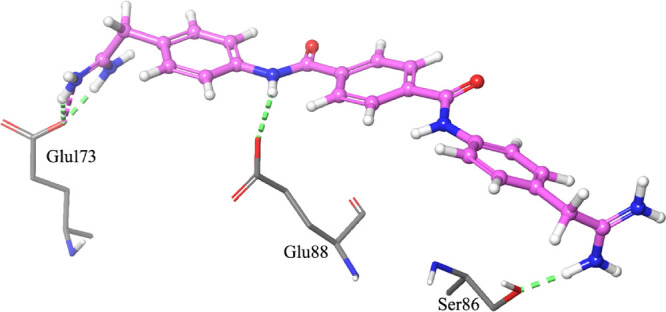
Top-scored docking pose of NSC55150 (22) within Site 2. For the sake of clarity, only residues (sticks) that interact by hydrogen bonds (green dashed lines) and by salt bridges (magenta dashed lines) with the ligand (ball-and-stick) are shown.
Moreover, hydrophobic interactions similar to those found for 23 were established, especially between all aromatic rings of the ligand and side chains of surrounding residues, as well as Ser86 and Gly85 backbones.
A comparison of the best docked pose of 22 and 23 is shown in Figure S5.
In conclusion, new putative CanB ligands belonging to the N,N′-diphenylterephthalamide class were identified and further characterized.
2.3. Construction of canB cKD Mutants Using the Pip-ON System to Validate CanB Inhibitors
To validate CanB inhibitors, we developed a whole-cell screening by means of cKD mutants. This simple tool has allowed us to identify compounds that were active in both inhibiting the mycobacterial growth and in affecting a specific target or pathway in which this enzyme is involved.16,25
To this aim, Pip-ON conditional mutants in canB were constructed.26 Unlike the TetR-Pip-OFF mutants, the construction of the recombinant vectors with the Pip-ON system allowed us to modulate the canB transcription through the addition of the pristinamycin (Pi) as an inducer; this means that if we do not add Pi to the culture medium, then canB is not transcribed. Compared to the ATc inducer, Pi has the advantage of remaining stable over time, allowing longer experiments to be performed, thus rendering Pip-ON mutants preferable for these experiments.
Mutants harboring the Pip-ON system at the attB site were obtained by a direct replacement technique that allows plasmid switching to excise the integrated TetR-Pip-OFF system from the Mtb H37Rv chromosome and to insert the Pip-ON system (derived from pFRA71, an integrative plasmid with a different marker).27 Two conditional Pip-ON canB mutants with different strengths of the Pptr promoter were obtained, namely, TB172 (TB71-canB/PPtr 25%, Pptr12) and TB173 (TB71-canB/PPtr 4%, Pptr11). The different canB expression in TB172 and TB173 mutants was also validated by real-time PCR (Figure S6). The resulting strains were Pi-dependent: the Pi-sensitive transcriptional repressor Pip, which is constitutively expressed, inhibits transcription from Pptr, while the presence of Pi in the medium allows canB transcription in a dose-dependent manner. The mutant TB173 is less viable than TB172 (Figure S7).
2.4. Two Putative CanB Ligands Are Also Active against Mtb canB cKD Mutants
NSC55150 (22, MIC = 1 μg/mL), which is the hit compound, and the two best derivatives NSC53313 (11, MIC = 0.31 μg/mL) and NSC55151 (23, MIC = 0.16 μg/mL) were chosen for further characterization (Table 1). In particular, the compounds were assayed in a target-based screening that exploits the canB Pip-ON conditional mutant TB173, characterized by the weakest promoter (PPtr 4%, Pptr11), to validate their cellular target. The rationale for this assay is that, if CanB is the target, then a low Pi concentration should lead to a low canB transcript level; therefore, the Pip-ON TB173 mutant should be hypersensitive to the selected compounds compared to the H37Rv strain, resulting in a lower MIC value. We performed the resazurin microtiter assay (REMA) to determine the activity of the lead compound 22 and its derivatives 11 and 23 against the conditional mutant TB173, at different Pi concentrations to modulate canB transcription. Both Mtb H37Rv and TB71 parental strains were included as controls. An MIC shift of the selected putative CanB inhibitors was observed; in particular, the TB173 mutant was more sensitive to 22 and 23 in comparison with Mtb H37Rv and TB71, especially in the presence of the lowest Pi concentration (Table 2). However, TB173 was slightly more resistant to 11 (Table 2). These results suggest that CanB might be the cellular target of 22 and 23, but not of 11.
Table 2. Whole-Cell Target-Based Screena.
| MIC (μg/mL) |
||||
|---|---|---|---|---|
| Mtb strains | 22 | 23 | 11 | streptomycin |
| H37Rv | 1 | 0.3 | 0.25 | 0.25 |
| TB71 | 0.25 | 0.03–0.06 | 0.125 | 0.25 |
| TB173 Pi [25 ng/mL] | 0.125 | 0.06 | 0.125 | 0.25 |
| TB173 Pi [15 ng/mL] | 0.125 | 0.01 | 0.125 | 0.25 |
Activity of 22 and its derivatives against Mtb H37Rv, parental TB71, and conditional mutant TB173 strains (determined by REMA).
Based on these results and to confirm them, time-killing assays were performed using Mtb conditional strains TB172 and TB173 in the presence of different concentrations of hit compound 22 as well as different concentrations of Pi (Figure 5C and Figure 5D, respectively). Mtb H37Rv and parental TB71 strains were used as control (Figure 5A and Figure 5B, respectively). In time-killing assays, multiple colony-forming unit (CFU) determinations over time provide accurate information on compound activity. In this specific case, in parallel, strain growth kinetics also informed us about the vulnerability of CanB as a target.
Figure 5.

Determination of bactericidal activity of compound 22 against Mtb. Time-killing assays were performed with Mtb H37Rv (A), TB71 (B), TB172 (C), and TB173 (D) strains, testing 22 at the MIC value (black circles), 10× MIC (red squares), and 40× MIC (blue diamonds). Open symbols correspond to growth without Pi, dotted symbols indicate growth with 15 ng/mL Pi, and filled symbols indicate growth with 25 ng/mL Pi. Moxifloxacin (in green) was included as the internal control of activity for the Mtb H37Rv strain.
In the time-killing assay, the TB173 mutant had a marked decrease in CFU counts at earlier time-points (Figure 5D) in comparison with the other strains harboring different constructs (TB71 and TB172) and the wild-type strain, especially using 22 at the highest concentration. The limit of detection (2-log CFU/mL) was indeed reached at day 7 at the highest concentration of 22. On the other hand, this limit was only reached at day 21 with Mtb H37Rv and TB71 control strains (Figure 5A,B).
This result was probably affected by the weakest promoter present in the TB173 mutant. The TB172 mutant indeed exhibited a bacteriostatic behavior in the presence of 22, probably due to its higher promoter strength and consequent higher canB expression (Figure 5C). Furthermore, TB173 already experienced an impairment in the growth at 1× and 10× MIC concentrations of 22 compared to the control strains. The Pi inducer did not affect the growth of the control strains, while it was effective in the titration of CanB for the conditional mutants.
In conclusion, 22, which is the hit compound belonging to the N,N′-diphenylterephthalamide class, has a bactericidal activity at the highest concentrations used against both control strains and canB conditional mutants, confirming CanB as its target.
2.5. Hit Compound 22 and Its Derivative 23 Are CanB Ligands and Inhibit Esterase Activity
To verify whether the selected compounds (the hit compound 22 and its derivative 23) are indeed able to bind CanB, the recombinant enzyme was produced in Escherichia coli and purified (Figure S8) and its affinity for the compounds was determined by the thermal shift assay, using differential scanning fluorimetry (DSF) (Figure 6).
Figure 6.

Differential scanning fluorimetry assays of Mtb CanB. (A) Differential scanning fluorimetry (DSF) assays in the absence (black) and in the presence of 100 μM 22 (light blue), 23 (light green), 11 (yellow), and ACZ (orange). (B–D) DSF assays in the presence of different concentrations of ACZ, 22, and 23, respectively. Black, no add; pink, 0.01 μM; green, 0.1 μM; blue, 1 μM; red, 10 μM, purple, 100 μM.
As a positive control, we used the acetazolamide (ACZ), a known potent inhibitor of the hydratase activity of CAs. As a negative control, we used the compound 11, which does not target CanB. As described in Figure 6A, the presence of ACZ, 22, and 23 (100 μM) caused a significant shift of the melting temperature of the enzyme, while no significant effects were found for 11, confirming the previous result (Table 2). Consequently, to determine whether 22, 23, and ACZ are able to bind CanB even at lower concentrations, DSF assays were performed in the presence of different compound concentrations (0.01–100 μM) (Figure 6B–D). Alike for the control ACZ (Figure 6B), the thermal shift was concentration-dependent for both 22 (Figure 6C) and 23 (Figure 6D), which were able to induce a significant melting temperature shift even at sub-micromolar concentrations.
To confirm that the bindings of 22 and 23 to CanB effectively lead to an inhibition of the biological activity of this enzyme, we investigated their effects against the esterase activity of CanB, using indoxyl acetate as a substrate. Since this reaction is performed into the same active site of the hydratase reaction, it is indicative of the CanB hydratase activity.28 Also in this case, we used 11 as the negative control. We first performed a steady state kinetic analysis, which confirmed a hyperbolic response toward the substrate concentration with a Vmax value of 4.1 ± 0.2 s–1 and a Km of 4.5 ± 0.6 mM (Figure S9A). Then, to evaluate the inhibitory potential of the compounds against CanB activity, their effects were initially evaluated at 1 mM. At such a concentration, 11 did not show any effect; this result, which is in agreement with those achieved with the conditional mutants, confirms the hypothesis that this compound does not target CanB (data not shown). By contrast, 22 and 23 inhibited CanB activities at 42 and 47%, respectively. A similar value was found also for the positive control ACZ, which at 1 mM was able to inhibit the esterase activity of CanB to 31%. The IC50 values of the compounds were then determined (Figure S9B); both 22 and 23 inhibited the enzyme activity in a concentration-dependent manner, with IC50 (0.77 ± 0.05 and 0.84 ± 0.03 mM, respectively) in the same range of the value found for ACZ (0.46 ± 0.03 mM). Notably, ACZ is known to inhibit the hydratase activity in a low/sub-micromolar range of concentration, while for the esterase activity assay, the IC50 is in the mM range. Likely, this discrepancy reflects the differences in the two mechanisms of action. Nonetheless, the fact that 22 and 23 showed IC50 in the same range of ACZ strongly suggests that these compounds act similarly.
These results effectively demonstrate that 22 and 23 are high affinity ligands of Mtb CanB and are able to significantly interfere with its biological activity, in a similar way to that of the positive control ACZ. These data confirm the finding achieved by using canB Pip-ON conditional mutants by validating the approach.
3. Discussion
Mtb CanB was considered a potential pharmacological target, although its predicted essentiality has never been definitively proven, just as few CanB inhibitors have so far been active against Mtb growth.8−14 Nevertheless, CanB is the β-CA that shows greater catalytic activity for CO2 hydration than the other two mycobacterial ones.19
In this work, we aimed to demonstrate the canB in vitro essentiality and to set up a multidisciplinary target-to-drug approach to select inhibitors highly active against Mtb growth.
The possible in vitro essentiality of CanB was demonstrated by the construction of two Mtb conditional mutants using the TetR-Pip-OFF system.15 Interestingly, even if Mtb presents three β-Cas, CanB was found to be highly essential for in vitro growth underlining the potential of this enzyme as a druggable target.
Therefore, due to the availability of the CanB crystal structure,17 a molecular docking-based virtual screening was performed against the NCI database, finding 53 putative CanB ligands. Three selected compounds were active against Mtb growth; in particular, the MIC of 22, belonging to the N,N′-diphenylterephthalamide class, was 1 μg/mL. Furthermore, we tested 24 derivatives of 22, finding seven out of them with an MIC <1 μg/mL. This result confirms that some compounds belonging to N,N′-diphenylterephthalamide are possible CanB ligands (Table 1).
It is worth noting that pentamidine, belonging to the N,N′-diphenylterephthalamide class, is active against several microorganisms, including P. carinii, mycobacteria, and some pathogenic protozoa.29−31 Furthermore, several of the compounds identified by virtual screening showed the bisbenzamidine feature and were already reported for their biological profile. As an example, 10, which is the simplest bisbenzamidine derivative considered in this study, showed a single-digit micromolar activity toward the D6 Sierra Leone strain of Plasmodium falciparum growth.32 In addition, similarly to pentamidine, some bisbenzamidines were active against P. carinii;3321 showed submicromolar activity (IC50 = 0.6 μM) against P. carinii pneumonia in mice.34 Further, bisbenzamidine derivatives were able to potentiate antibiotic activity against Gram-negative bacteria22 and to inhibit the virulence of Porphyromonas gingivalis.(29,30) On the other hand, it was shown that compounds bearing guanidino moieties, such as 23, had antimicrobial, antimycobacterial, antifungal, and antimalarial properties.31,35,36 As an example, streptomycin is a di-guanidino compound. So, the antimicrobial activity of this class of compounds was already shown, confirming our achieved results.
Subsequently, we would like to validate CanB as the target of this class of compounds, which is already known including some antimicrobial compounds, following different approaches. First, starting from the TetR-Pip-OFF conditional canB mutants, two new ones were constructed using the Pip-ON system; thus, canB transcription is allowed only in the presence of long-lasting inducer Pi; this strategy allows us to develop a whole-cell target-based screening.25
The Pip-ON canB conditional mutants are more sensitive to 22 and 23 in the presence of different Pi concentrations, revealing a lower MIC value with respect to wild-type and parental strains; consequently, CanB might be their target. The time-killing assays performed using conditional Pip-ON mutants grown at different Pi concentrations specifically showed a decrease in CFU counts for the TB173 mutant. These results suggest that the weakest Pptr promoter, which replaces the native canB promoter, causes a reduction in canB transcription at low Pi concentrations, thus sensitizing cells to the 22 inhibitor that likely acts on CanB.
Then, by using a biophysical assay such as DSF, we demonstrated that both 22 and 23 bind CanB, conceivably with high affinity, as suggested by the significant thermal shift that they induce in a sub-micromolar range of concentration. A similar behavior was also found for the well-known CA inhibitor ACZ, used as a positive control. Moreover, the compounds inhibited the CanB esterase activity, with IC50 values in the same range of ACZ. The esterase activity is indicative of the hydratase main activity since it is performed in the same active site.28 It is worth noting that ACZ, which inhibits the hydratase activity at a very low concentration, showed anyway an IC50 for the esterase activity in the same range of 22 and 23. This strongly suggests that 22 and 23 could behave as ACZ also for the main activity.
So, these data definitively confirm that these compounds strongly bind CanB, supporting the previous results achieved by using Pip-ON canB conditional mutants. Moreover, the fact that the conditional mutants did not show a higher sensitivity to 11, which was found to neither bind nor inhibit CanB, further confirms the selectivity of our approach. On the other hand, since 11 has antitubercular activity, it cannot be excluded that this class of compounds has multiple targets.
Thus, following this target-to-drug modified approach, we were able to discover a new class of CanB inhibitors with bactericidal activity against Mtb growth.
So far, a few CanB inhibitors were found to be active against Mtb growth: the 9-sulfonylated/sulfenylated-6-mercaptopurines (MIC = 0.39–3.39 mg/mL)37 and one C-cinnamoyl glycoside derivative [(E)-1-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-4-(3-hydroxyphenyl)but-3-en-2-one; MIC = 3.125–6.25 mg/mL)].13 Indeed, most CanB inhibitors, although very effective against the enzymatic activity, failed to kill Mtb cells.
4. Conclusions
In conclusion, the discovery of compounds belonging to the N,N′-diphenylterephthalamide class as active CanB inhibitors successfully validated the multidisciplinary target-to-drug strategy used in this work. This approach could be applied both for the validation of drug targets and for the identification of potential inhibitors. Further preclinical studies are needed to understand if these promising compounds can be new drug candidates to fight tuberculosis disease.
5. Experimental Section
5.1. Bacterial Strains, Media, and Growth Conditions
Mtb H37Rv and its derivative strains (TB61, TB169C, TB170A, TB71, TB172, and TB173) were cultured at 37 °C in either Middlebrook 7H9 (liquid medium, Difco) or 7H10 (solid medium, Difco), supplemented with 0.05% v/v Tween-80 (Sigma-Aldrich), 0.2% v/v glycerol (Sigma-Aldrich), and 10% Middlebrook OADC (Difco). For cloning procedures, E. coli DH5α was grown in Luria–Bertani (LB) broth and LB-agar.
When required, antibiotics were added at the following concentrations: kanamycin (Sigma-Aldrich), 50 μg/mL (E. coli); hygromycin (Sigma-Aldrich), 200 μg/mL (E. coli) or 50 μg/mL (Mtb); and streptomycin (Sigma-Aldrich), 50 μg/mL (E. coli) or 20 μg/mL (Mtb). Anhydrotetracycline (ATc, Sigma-Aldrich) was added at a final concentration of 500 ng/mL, if required. Pristinamycin IA (Pi, Molcan Corporation) was added, when required, at the concentrations indicated in the text. NCI compounds were all dissolved in DMSO and used for the experiments at concentrations indicated in the text.
Experiments performed with Mtb strains were conducted in the BLS3 safety laboratory by authorized researchers.
5.2. Construction of Mtb H37Rv canB Conditional Mutants
The canB conditional mutants were constructed using the TetR/Pip OFF repressible promoter system.15 Briefly, the 5′ portion (402 bp) of the canB gene was amplified, using the primers in Table S2, and directionally cloned in integrative plasmids (pFRA169-170), conferring resistance to hygromycin, downstream two different Pptr-derived promoters (Pptr11–12)(at the level of restriction enzyme sites NsiI and XhoI, present in the plasmid polylinker). The promoters Pptr11–12 were constructed by Boldrin et al.,16 starting from Pptr, a Pip-dependent promoter from Streptomyces pristinaespiralis. Pptr11 has 4.2% of the strength of the original Pptr, while Pptr12 has 25% of the strength.
Two micrograms of each recombinant plasmid was used to transform TB61 (recipient harboring the TetR/Pip-OFF system at the attB site of its genome, carrying a streptomycin-resistant determinant). Several colonies were isolated on 7H10 plates containing streptomycin and hygromycin, screened by colony PCR to check for the insertion of suicide vectors, using an upper primer in the TetR-Pptr region (GD18upperSEQf; Table S2) and a lower primer external to the homology region used for recombination (GD3rev2NEW; Table S2) (data not shown). Then, the selected colonies were tested for their responsiveness to ATc. Two strains were saved for further studies and named TB169C (TB61-canB/Pptr11, 4%) and TB170A (TB61-canB/ Pptr12, 25%), in which the canB native promoter was substituted by Pptr by insertional duplication.
To obtain the Pip-ON conditional strains, the pFRA71 plasmid was introduced into the cKD canB mutants, TB170A and TB169C, by plasmid switching18 generating TB172 (Pptr12) and TB173 (Pptr11) mutants, respectively.
5.3. Determination of Minimal Inhibitory Concentration (MIC)
The drug susceptibility of Mtb strains was determined by the resazurin microtiter assay (REMA), as previously described.38 Positive and negative growth controls were included in every plate. Serial 2-fold dilutions of compounds were performed in a 96-well black plate (Fluoronuc, Thermo Fisher Scientific), and then, bacterial cultures in the log phase were diluted and added to the wells. After 7 days of incubation at 37 °C, 10 μL of resazurin (0.025% w/v) was added to the wells and fluorescence was measured after a further overnight incubation by a Fluoroskan Microplate Fluorometer (Thermo Fisher Scientific; excitation = 544 nm; emission = 590 nm). Bacterial viability was calculated as a percentage of resazurin turnover in the absence of compound (internal negative control). Experiments were performed in duplicate at least three times. MIC90 values were obtained.
In the case of whole-cell target-based screening, the TB172 and TB173 strains were grown in the presence of Pi (15–25 ng/mL).
5.4. Time Killing Assay (NSC55150, 22)
Bacterial cultures were inoculated at a final cell density of about 106 cells/mL and a final volume of 2.5 mL of the 7H9 medium without Tween-80 in 10 mL tubes. Compound 22 was added at the following concentrations based on MIC values of each strain: MIC, 10× MIC, and 40× MIC. Pi was added to TB172 and TB173 at 15 and 25 ng/mL to allow the growth of the strains, while it was added as the control to the Mtb H37Rv and TB71 strain at 25 ng/mL to assure that it does not interact with 22. Cultures were then incubated at 37 °C for 21 days. At every time point (0, 7, 14, and 21 days), bacterial suspensions were thoroughly mixed and serially diluted 10-fold in PBS and 10 μL aliquots plated in triplicates on 7H10 Pi (50 ng/mL) agar quad plates. CFUs were enumerated after 14 and 21 days of incubation at 37 °C. Moxifloxacin (0.6 μg/mL, corresponding to 10× MIC) was included for the Mtb H37Rv strain as a control. Experiments were performed in duplicate.
5.5. Computational Details
The crystal structures of CanB, obtained by X-ray diffraction both in monomeric (PDB: 1YM3, 1.7 Å resolution)20 and homotetrameric (a dimer of dimers, PDB: 2A5V, 2.2 Å resolution)17 forms, were retrieved from the protein data bank39 and prepared according to the Protein Preparation Wizard routine of the Schrödinger suite, version 2019-2.40 Bond orders were assigned, while hydrogens were removed and then readded to fix potential problems in the original crystallographic coordinates. Missing side chains were modeled, and loops were refined with Prime software. After an optimization procedure to avoid overlapping of atoms, both systems were minimized with the most recent OPLS3e force field until heavy atoms converged to a root mean square deviation of 0.30 Å. All water molecules were retained up to the restrained minimization step, to avoid alterations of volume of the binding sites, and were then removed prior to the receptor grid generation protocol.
With the aim of identifying ligands able to limit or prevent the formation of CanB dimers of dimers that represent the active form of the enzyme, shallow binding sites on the surface of CanB were identified. Being shallow pockets, the portions of the CanB monomer surface required for monomer–monomer interactions and subsequent formation of CanB active tetramers, ligands able to affect CanB protein–protein interactions are expected to reduce or abrogate CanB enzymatic activity.
In this context, we decided to identify the top five shallow binding pockets on the CanB monomer surface by using the SiteMap routine implemented within the Glide software of the Schrödinger suite. A standard spacing grid was used for the spatial search (0.7 Å distance between the lines), and more restrictive definition of hydrophobicity was selected. All other options were set to default. Only two out of the five potential binding sites were retained for the following virtual screening, namely, Site 2 and Site 4 (Figure 2). Their name was relative to their order in the SiteMap ranking list, based on the software scoring function of druggability (SiteScore was 0.67 for Site 2 and 0.60 for Site 4). These binding sites were selected because they were located at the interface between single chains of the CanB tetrameric structure. As a consequence, compounds able to bind these pockets are expected to interfere with the CanB polymerization process and to block CanB enzymatic activity that is a prerogative of the tetrameric form. Indeed, our purpose was to find small molecules able to bind within these interfaces and therefore to potentially block the multimerization process.
Site 2 has a total surface area and a volume of approximately 747 and 23,667 Å3, while the surface and volume of Site 4 are approximately 930 and 28,812 Å3, respectively. Intriguingly, both selected binding sites included a few interesting residues (Arg73, His104, and Leu164 for Site 2; Cys107 and Arg73 for Site 4): Arg73 is a highly conserved residue among mycobacterial anhydrases, and His104 and Cys107 lie at the center of the monomer where the catalytic activity of the enzyme resides, while Leu164 is part of an extremely conserved hydrophobic region20 (Suarez Covarrubias et al.).
The Receptor Grid Generation tool was used to build grids suitable for molecular docking, using the corresponding SiteMap entry as reference. The resulting boxes were 14 Å long each side. All rotatable OH and SH side chains were allowed to vary during the simulation.
The NCI All Open database (updated to June 2016, as available from the DTP section of the National Cancer Institute, comprising 284,176 compounds)41 was downloaded in sdf format. The database was fully recompiled into a multi-conformational Phase database, generating up to 100 minimized conformers for each entry of the database. Ionization states were computed with Epik software at pH 7.00 ± 1.00. All tautomers and up to 16 stereoisomers were generated, retaining only the already specified chiral centers. Up to four conformations were generated for five- and six-membered rings, while larger rings were sampled with Macromodel software. All high-energy conformers were discarded. For each compound, physico-chemical and ADME-Tox properties were predicted with QikProp software.
Glide software was used to perform the virtual screening. In the first step, a high throughput virtual screening protocol was applied to speed up computational time for calculations. Afterward, the poses of the top 462 compounds were refined with a standard precision protocol. Flexible molecular docking was performed, disabling the canonicalization option. Conformation of amide bonds was allowed to vary. Ionization penalties and strain correction terms were applied to the final list of compounds generated by the scoring functions. All calculations were performed on a 16-CPU x86_64 workstation running a Debian OS release 4.9.303-1.
The final list of the top-ranked compounds was based on the score value (a cutoff of −6.00 kcal/mol for the Glide Score was arbitrarily set) and on the availability for free at the NCI. A general evaluation of physico-chemical and ADME-Tox properties of the selected compounds was also carried out. Top ranking compounds with known antimycobacterial activity were discarded. Finally, a Tanimoto similarity index of 0.8 was applied to guarantee to cover the chemical space as much as possible, thus leading to the final list of 53 compounds.
NSC compounds were provided as solid by the NCI. Since, in most cases, such compounds have not been analyzed by DTP for accuracy of the structure or for the purity of the sample, 1H NMR spectra were recorded for 22 and 23 and are reported in Figure S10 as illustrative examples of the entire compound library.
SMILES notations of 1–25 are reported in Table S3.
5.6. CanB Cloning Expression and Purification
To obtain the recombinant Mtb β-carbonic anhydrase (CanB), the Rv3588c gene was inserted into the pET-28a + expression vector, using an In-fusion HD Cloning Kit (Takara). For this purpose, the gene was amplified by PCR, using the primers CanB-For and CanB-Rev (Table S2), designed according to the In-fusion HD Cloning Kit protocol. The forward primer carried the sequence encoding the TEV protease cleavage site for the removal of the 6× His-tag. The purified PCR fragment was recombined into the BamHI-HindIII digested pET-28a+ following the manufacturer’s instructions, affording the pET28a-canB vector.
E. coli BL21(DE3) cells were then transformed with the pET28a-canB vector and grown at 37 °C up to OD600nm = 0.6, and then, CanB synthesis was induced by the addition of 0.5 mM IPTG followed by overnight induction at 25 °C. Cells were harvested by centrifugation at 4000g for 15 min, resuspended in 50 mM potassium phosphate buffer pH 8.0, 500 mM KCl (buffer A), supplemented with 1 mM phenylmethylsulfonyl fluoride, and lysed by sonication. The cell-free extract, obtained by centrifuging the lysate at 50,000g for 20 min at 4 °C, was loaded on a His-Trap column (1 mL, GE Healthcare) equilibrated in buffer A, the column was washed with 20 mM imidazole in buffer A, and CanB eluted with 500 mM imidazole in buffer A.
The enzyme solution was dialyzed in 50 mM potassium phosphate pH 8.0, 150 mM KCl (buffer B), in the presence of 50 μg of TEV protease, to remove the 6× His-Tag. The digested protein was further purified by a second His-Trap chromatography. Sample purity was checked by SDS-PAGE and protein concentration evaluated by absorbance at 280 nm (ε: 10095 M–1 cm–1).
5.7. Thermal Shift Assay
Differential scanning fluorimetry (DSF) is a cost-effective and parallelizable technique widely used to track protein thermal stability. Since complex formation with even weakly binding ligands affects protein thermal stability, DSF has become a widespread high-throughput screening tool, known for being very powerful and rapid in early drug discovery efforts, including the studies of CA inhibitors.42
To investigate the interactions between CanB and its possible inhibitors, we performed a label-free thermal shift assay using a Tycho NT.6 instrument (NanoTemper Technologies) and intrinsic fluorescence of tryptophan and tyrosine.
Protein samples (1.0 mg/mL in buffer B) were loaded into Tycho NT.6 capillaries (TY-C001, NanoTemper Technologies) and subjected to unfolding using a thermal ramp rate of 30 °C min–1 monitoring the ratio of the intrinsic fluorescence at 350 and 330 nm (emission at 350 nm/emission at 330 nm, upon excitation at 280 nm) that was recorded as the protein unfolding probe.
5.8. CanB Enzymatic Assay
The CanB esterase activity was determined spectrophotometrically using indoxyl acetate as the substrate and measuring the production of indoxyl at 375 nm (ε: 2.54 mM–1 cm–1), as previously described.28 Briefly, assays were performed at 37 °C in a final volume of 100 μL of 17.5 mM Hepes and 17.5 mM imidazole pH 7.2, containing 20 μg of purified CanB, and the reaction was started by adding indoxyl acetate (stock solution of 100 mM in 97% ethanol). Steady-state kinetic analysis was performed by assaying the activity at different indoxyl acetate concentrations and the kinetic constants Vmax and Km were determined by fitting the data to the Michaelis–Menten equation, using the GraphPad Prism software (GraphPad software. Inc).
Inhibition assays were initially performed in the presence of the compounds at a final concentration of 500 μM (stock solution of 10 mM in DMSO) and at a final concentration of the indoxyl acetate substrate of 4 mM.
For active compounds, IC50 values were evaluated measuring the enzyme activity in the presence of different compound concentrations and fitting the values with the following equation (eq 1):
| 1 |
where A[I] is the enzyme activity in the presence of a given concentration of inhibitor [I] and A[0] is the enzyme activity in the absence of inhibitors.
The known carbonic anhydrase inhibitor acetazolamide (ACZ) was used as a positive control. All experiments were performed in triplicate.
Acknowledgments
This research was partially supported by EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (project no. PE00000007, INF-ACT) (MRP) and One Health Basic and Translational Research Actions addressing Unmet Needs on Emerging Infectious Diseases (CUP B63C22001400007) (FM). G.D. was the recipient of a fellowship earned from the University of Pavia (FRG – Fondo Ricerca e Giovani: “Assegno di ricerca di tipo A”). We thank Prof. R. Manganelli (University of Padova, Italy) for providing us with the pFRA61, pFRA71, pFRA169, and pFRA170 plasmids. We also thank Dr. F. Boldrin (University of Padova, Italy) for fruitful discussion about the construction of conditional mutants. National Cancer Institute (NCI), Division of Cancer Treatment and Diagnosis (DCTD), Developmental Therapeutics Program (DTP) (http://dtp.cancer.gov) is acknowledged as the source of the NSC compounds.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02311.
Essentiality of canB for Mtb growth on a solid medium; growth curves of conditional mutants in the presence of different ATc concentrations; molecular structure of the three hit compounds identified by virtual screening on the NCI database and pentamidine; Glide scores of 11–17 and their interactions with Site 2 of CanB; comparison of the binding mode of 22 and 23; changes in canB mRNA levels of TB71, TB172, and TB173 strains; pristinamycin induction of canB expression in cKD Mtb mutant strains; SDS-PAGE of CanB purification; enzymatic studies of CanB esterase activity; 1H NMR spectra for 22 and 23; MIC values of the hit compounds from the NCI library utilized in this study; list of oligonucleotides used; SMILES notations of the 25 N,N′-diphenylterephthalamide derivatives 1–25 (PDF)
Author Contributions
Conceptualization: M.R.P., G.D., F.M., and V.M.; methodology: G.D., B.G., D.R., G.S., G.I.T., and L.R.C.; formal analysis, data curation, and writing: M.R.P., F.M., G.D., and L.R.C.; project administration: M.R.P. and F.M. All authors reviewed and approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dartois V. A.; Rubin E. J. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat. Rev. Microbiol. 2022, 20, 685–701. 10.1038/s41579-022-00731-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams K. A.; Besra G. S. Mycobacterial drug discovery. RSC Med. Chem. 2020, 11, 1354–1365. 10.1039/d0md00261e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayev F.; Viney K.; Linh N. N.; Gonzalez-Angulo L.; Gegia M.; Jaramillo E.; Zignol M.; Kasaeva T. World Health Organization recommendations on the treatment of drug-resistant tuberculosis, 2020 update. Eur. Respir. J. 2021, 57, 2003300. 10.1183/13993003.03300-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014, 6, 158–168. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. S.; Jakubzick C.; Whittam T. S.; Ferry J. G. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 15184–15189. 10.1073/pnas.96.26.15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionetto M.; Caricato R.; Giordano M.; Schettino T. The complex relationship between metals and carbonic anhydrase: new insights and perspectives. Int. J. Mol. Sci. 2016, 17, 127. 10.3390/ijms17010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocentini A.; Capasso C.; Supuran C. T. Carbonic Anhydrase Inhibitors as Novel Antibacterials in the Era of Antibiotic Resistance: Where Are We Now?. Antibiotics 2023, 12, 142. 10.3390/antibiotics12010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspatwar A.; Kairys V.; Rala S.; Parikka M.; Bozdag M.; Carta F.; Supuran C. T.; Parkkila S. Mycobacterium tuberculosis β-carbonic anhydrases: novel targets for developing antituberculosis drugs. Int. J. Mol. Sci. 2019, 20, 5153. 10.3390/ijms20205153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus M. A.; Gerrick E. R.; Xu W.; Park S. W.; Long J. E.; Boutte C. C.; Rubin E. J.; Schnappinger D.; Ehrt S.; Fortune S. M.; Sassetti C. M.; Ioerger T. R. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio 2017, 8, e02133–e02116. 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch B.; DeJesus M. A.; Poulton N. C.; Zhang W.; Engelhart C. A.; Zaveri A.; Lavalette S.; Ruecker N.; Trujillo C.; Wallach J. B.; Li S.; Ehrt S.; Chait B. T.; Schnappinger D.; Rock J. M. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 2021, 184, 4579–4592.e24. 10.1016/j.cell.2021.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Rubin E. J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12989–12994. 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchieri M. V.; Riafrecha L. E.; Rodríguez O. M.; Vullo D.; Morbidoni H. R.; Supuran C. T.; Colinas P. A. Inhibition of the β-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: identification of the first inhibitor with anti-mycobacterial activity. Bioorg. Med. Chem. Lett. 2013, 23, 740–743. 10.1016/j.bmcl.2012.11.085. [DOI] [PubMed] [Google Scholar]
- Zaro M. J.; Bortolotti A.; Riafrecha L. E.; Concellón A.; Morbidoni H. R.; Colinas P. A. Anti-tubercular and antioxidant activities of C-glycosyl carbonic anhydrase inhibitors: towards the development of novel chemotherapeutic agents against Mycobacterium tuberculosis. J. Enzyme Inhib. Med. Chem. 2016, 31, 1726–1730. 10.3109/14756366.2016.1172577. [DOI] [PubMed] [Google Scholar]
- Boldrin F.; Casonato S.; Dainese E.; Sala C.; Dhar N.; Palù G.; Riccardi G.; Cole S. T.; Manganelli R. Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res. 2010, 38, e134 10.1093/nar/gkq235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrin F.; Degiacomi G.; Serafini A.; Kolly G. S.; Ventura M.; Sala C.; Provvedi R.; Palù G.; Cole S. T.; Manganelli R. Promoter mutagenesis for fine-tuning expression of essential genes in Mycobacterium tuberculosis. Microb. Biotechnol. 2018, 11, 238–247. 10.1111/1751-7915.12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias A. S.; Bergfors T.; Jones T. A.; Högbom M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 4993–4999. 10.1074/jbc.M510756200. [DOI] [PubMed] [Google Scholar]
- Di Fiore A.; Pedone C.; Antel J.; Waldeck H.; Witte A.; Wurl M.; Scozzafava A.; Supuran C. T.; De Simone G. Carbonic anhydrase inhibitors: the X-ray crystal structure of ethoxzolamide complexed to human isoform II reveals the importance of thr200 and gln92 for obtaining tight-binding inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2669–2674. 10.1016/j.bmcl.2008.03.023. [DOI] [PubMed] [Google Scholar]
- Carta F.; Maresca A.; Covarrubias A. S.; Mowbray S. L.; Jones T. A.; Supuran C. T. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg. Med. Chem. Lett. 2009, 19, 6649–6654. 10.1016/j.bmcl.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Suarez Covarrubias A.; Larsson A. M.; Högbom M.; Lindberg J.; Bergfors T.; Björkelid C.; Mowbray S. L.; Unge T.; Jones T. A. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 18782–18789. 10.1074/jbc.M414348200. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Jin Y.; Zhang L.; Wang H.; Wang X. Ninety Years of Pentamidine: The development and applications of pentamidine and its analogs. Curr. Med. Chem. 2022, 29, 4602–4609. 10.2174/0929867329666220314121446. [DOI] [PubMed] [Google Scholar]
- Kanvatirth P.; Jeeves R. E.; Bacon J.; Besra G. S.; Alderwick L. J. Utilisation of the Prestwick Chemical Library to identify drugs that inhibit the growth of mycobacteria. PLoS One 2019, 14, e0213713 10.1371/journal.pone.0213713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongaerts N.; Edoo Z.; Abukar A. A.; Song X.; Sosa-Carrillo S.; Haggenmueller S.; Savigny J.; Gontier S.; Lindner A. B.; Wintermute E. H. Low-cost anti-mycobacterial drug discovery using engineered E. coli. Nat. Commun. 2022, 13, 3905. 10.1038/s41467-022-31570-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams G. L.; Kumar A.; Savvi S.; Hung A. W.; Wen S.; Abell C.; Barry C. E. III; Sherman D. R.; Boshoff H. I.; Mizrahi V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 2012, 19, 844–854. 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti F.; Crosta A.; Ghisotti D. Pristinamycin-inducible gene regulation in mycobacteria. J. Biotechnol. 2009, 140, 270–277. 10.1016/j.jbiotec.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Pashley C. A.; Parish T. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2003, 229, 211–215. 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- Baliukynas M.; Veteikytė A.; Kairys V.; Matijošytė I. The hydrolysis of indoxyl acetate: A versatile reaction to assay carbonic anhydrase activity by high-throughput screening. Enzyme Microb. Technol. 2020, 139, 109584 10.1016/j.enzmictec.2020.109584. [DOI] [PubMed] [Google Scholar]
- Fröhlich E.; Kantyka T.; Plaza K.; Schmidt K. H.; Pfister W.; Potempa J.; Eick S. Benzamidine derivatives inhibit the virulence of Porphyromonas gingivalis. Mol. Oral. Microbiol. 2013, 28, 192–203. 10.1111/omi.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen I.; Potempa J. Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J. Oral. Microbiol. 2014, 6, 24800. 10.3402/jom.v6.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degardin M.; Wein S.; Duckert J. F.; Maynadier M.; Guy A.; Durand T.; Escale R.; Vial H.; Vo-Hoang Y. Development of the first oral bioprecursors of bis-alkylguanidine antimalarial drugs. ChemMedChem 2014, 9, 300–304. 10.1002/cmdc.201300419. [DOI] [PubMed] [Google Scholar]
- Cruz-Monteagudo M.; Borges F.; Perez González M.; Cordeiro M. N. D. S. Computational modeling tools for the design of potent antimalarial bisbenzamidines: overcoming the antimalarial potential of pentamidine. Bioorg. Med. Chem. 2007, 15, 5322–5339. 10.1016/j.bmc.2007.05.034. [DOI] [PubMed] [Google Scholar]
- Pugh B. A.; Rao A. B.; Angeles-Solano M.; Grosser M. R.; Brock J. W.; Murphy K. E.; Wolfe A. L. Design and evaluation of poly-nitrogenous adjuvants capable of potentiating antibiotics in Gram-negative bacteria. RSC Med. Chem. 2022, 13, 1058–1063. 10.1039/d2md00041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent J.; Stanicki D.; Huang T. L.; Dei-Cas E.; Pottier M.; Aliouat el M.; Vanden Eynde J. J. Bisbenzamidines as antifungal agents. are both amidine functions required to observe an anti-Pneumocystis carinii activity?. Molecules 2010, 15, 4283–4293. 10.3390/molecules15064283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti F.; Castagnolo D.; Raffi F.; Zizzari A. T.; Rajamäki S.; D’Arezzo S.; Visca P.; Cona A.; Fracasso M. E.; Doria D.; Posteraro B.; Sanguinetti M.; Fadda G.; Botta M. Synthesis of new linear guanidines and macrocyclic amidinourea derivatives endowed with high antifungal activity against Candida spp. and Aspergillus spp. J. Med. Chem. 2009, 52, 7376–7379. 10.1021/jm900760k. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Semenya D.; Castagnolo D. Antimicrobial drugs bearing guanidine moieties: A review. Eur. J. Med. Chem. 2021, 216, 113293 10.1016/j.ejmech.2021.113293. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Mastrolorenzo A.; Supuran C. T. Antimycobacterial activity of 9-sulfonylated/sulfenylated-6-mercaptopurine derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 1675–1678. 10.1016/s0960-894x(01)00266-9. [DOI] [PubMed] [Google Scholar]
- Palomino J.-C.; Martin A.; Camacho M.; Guerra H.; Swings J.; Portaels F. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protein Data Bank; RCSB; https://www.rcsb.org.
- Schrödinger Release 2019–2; Schrödinger, LLC: New York, NY, 2019. [Google Scholar]
- NCI Development Therapeutics Program; NCI; https://dtp.cancer.gov/
- Zakšauskas A.; Čapkauskaitė E.; Paketurytė-Latvė V.; Smirnov A.; Leitans J.; Kazaks A.; Dvinskis E.; Stančaitis L.; Mickevičiu̅tė A.; Jachno J.; Jezepčikas L.; Linkuvienė V.; Sakalauskas A.; Manakova E.; Gražulis S.; Matulienė J.; Tars K.; Matulis D. Methyl 2-halo-4-substituted-5-sulfamoyl-benzoates as high affinity and selective inhibitors of carbonic anhydrase IX. Int. J. Mol. Sci. 2022, 23, 130. 10.3390/ijms23010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.