

Abstract
BACKGROUND:
Vertical transmission of Zika virus leads to infection of neuroprogenitor cells and destruction of brain parenchyma. Recent evidence suggests that the timing of infection as well as host factors may affect vertical transmission. As a result, congenital Zika virus infection may only become clinically apparent in the postnatal period.
OBJECTIVE:
We sought to develop an outbred mouse model of Zika virus vertical transmission to determine if the timing of gestational Zika virus exposure yields phenotypic differences at birth and through adolescence. We hypothesized that later gestational inoculations would only become apparent in adolescence.
STUDY DESIGN:
To better recapitulate human exposures, timed pregnant Swiss-Webster dams (n = 15) were subcutaneously inoculated with 1 × 104 plaque-forming units of first passage contemporary Zika virus HN16 strain or a mock injection on embryonic day 4, 8, or 12 with bioactive antiinterferon alpha receptor antibody administered in days preceding and proceeding inoculation. The antibody was given to prevent the robust type I interferon signaling cascade that make mice inherently resistant to Zika virus infection. At birth and adolescence (6 weeks of age) offspring were assessed for growth, brain weight, and biparietal head diameters, and Zika virus viral levels by reverse transcription–polymerase chain reaction or in situ hybridization.
RESULTS:
Pups of Zika virus–infected dams infected at embryonic days 4 and 8 but not 12 were growth restricted (P < .003). Brain weights were significantly smaller at birth (P = .01) for embryonic day 8 Zika virus–exposed offspring. At 6 weeks of age, biparietal diameters were smaller for all Zika virus–exposed males and females (P < .05), with embryonic day 8–exposed males smallest by biparietal diameter and growth-restriction measurements (weight >2 SD, P = .0007). All pups and adolescent mice were assessed for Zika virus infection by reverse transcription–polymerase chain reaction. Analysis of all underweight pups reveled 1 to be positive for neuronal Zika virus infection by in situ hybridization, while a second moribund animal was diffusely positive at 8 days of age by Zika virus infectivity throughout the brain, kidneys, and intestine.
CONCLUSION:
These findings demonstrate that postnatal effects of infection occurring at single time points continue to be detrimental to offspring in the postnatal period in a subset of littermates and subject to a window of gestational susceptibility coinciding with placentation. This model recapitulates frequently encountered clinical scenarios in nonendemic regions, including the majority of the United States, where travel-related exposure occurs in short and well-defined windows of gestation. Our low rate of infection and relatively rare evidence of congenital Zika syndrome parallels human population-based data.
Keywords: congenital infection, growth restriction, ZIKV
Introduction
Zika virus (ZIKV), a mosquito-borne arbovirus of the Flaviviridae family,1 was declared a public health emergency of international concern by the director of the World Health Organization in 2016 after findings that ZIKV infection during pregnancy led to microcephaly and other congenital anomalies in affectedfetuses.2–4 Although the principal findings of congenital Zika syndrome (CZS) are microcephaly, micrencephaly, ophthalmic anomalies, and arthrogryposis,3–5 developmental delay, seizures, and visual impairment can present after the postnatal period.3,4,6 Identifying CZS is further complicated by the transient nature of both viral and serological IgM titers in mothers, and IgG cross-reactivity with other flaviviruses cocirculating in endemic areas.5,7 Indeed, a report by the Centers for Disease Control and Prevention (CDC) showed that neonates may not have microcephaly at birth, but may develop microcephaly up to 6–12 months postnatally, illustrating possible false-negative findings for CZS at the time of birth.8 It also suggests that ZIKV neuroinvasion in utero may persist undetected postnatally.9
Varying host resistance and susceptibility may also affect vertical transmission of ZIKV. Reports of exposed dichorionic twins have identified only 1 twin affected with CZS,10 while other reports in monochorionic twins suggest both may be symptomatic with CZS.11 In vitro experiments suggest primary trophoblasts exhibit modest donor-associated variability in vial permissiveness.12 Similarly, neuronal cell lines vary in permissiveness, including as a function of differentiation.12–14 Clinically, temporal associations have been made with the highest rate of ZIKV transmission observed in the first trimester at approximately 8% risk compared to 4% in the third.15 These findings suggest underlying temporal differences in the permissiveness of placental and fetal infection.
Patients from ZIKV nonendemic regions with windows of exposure (ie, women potentially exposed through travel to endemic regions) may have different appreciable risks than women residing in endemic regions. Current guidelines exclude testing of travel-exposed women who are asymptomatic. If CZS is associated with late viral exposures, it is possible that affected cases may be missed, however clinical studies are incomplete. Further, animal models have not assessed how the gestational timing of ZIKV exposure may be associated with varying postnatal outcome, including through maturation into adolescence, and further are largely limited to inbred models without host variation.16 We therefore sought to test whether gestational timing of exposure can lead to fetal consequences evident 6 weeks postnatally in an outbred mouse mode.
We hypothesized that midgestational timing of ZIKV inoculation would present more deleterious effects on offspring as compared to early infection, as detected in the postnatal period with prolonged monitoring. Using an outbred Swiss-Webster mouse model of ZIKV vertical transmission with interferon receptor signaling disruption, as described previously to be required to overcome murine resistance to ZIKV,17 we tested for differences in the presentation of ZIKV-associated defects at birth and later in adolescence with early, mid, and late gestation contemporaneous strain ZIKV HN16 inoculation.12 We established that there is a window of susceptibility as measured by growth restriction at birth and extending into adolescence.
Materials and Methods
Establishment of mouse model of CZS
Contemporary ZIKV was isolated from a symptomatic returning traveler to Honduras in 2016 in Houston, TX,18 and passaged 1 time in Vero cells. A first passage dose of the pathogenic strain was used in all experiments so as to minimize the effects of random or adaptive mutations to the propagation host cells of the highly mutagenic single stranded RNA flavivirus with extensive passaging. This first-passage strain was verified to contain the preserved virulent premembrane protein mutation associated with microcephaly and CZS in neonates the contemporary epidemic.16 The viral isolation and its similarity to pathological strains has been previously described.18 The sequenced genome is publicly available from GenBank (KY328289.1). Animal studies were conducted at Baylor College of Medicine in the Center for Comparative Medicine with review and approval by the Institutional Animal Care and Use Committee (protocol no. AN-6697). Wild-type Swiss-Webster mice were obtained from Taconic Biosciences (Rensselaer, NY). Timed pregnant dams were inoculated subcutaneously (SQ) with 1 × 104 plaque-forming units of first passage ZIKV HN16 in a 50-μL dose diluted in phosphate-buffered saline (PBS). The day preceding and proceeding ZIKV inoculation, the pregnant dam was given 2.5 mg of functional-grade antiinterferon alpha receptor 1 (MAR1–5A3) monoclonal antibody (Leinco Technologies Inc, St. Louis, MO) intraperitoneally (IP). Inoculation with ZIKV occurred at embryonic day (e) 4, e8, or e12 in the experimental arm. In the control arm, 50 μL of PBS was administered SQ as a mock, as well as 50-μL PBS injections IP the day preceding and proceeding the SQ PBS injections (Figure 1). Pregnant dams were also inoculated SQ with 50 μL of PBS on e8 with IP injections of 2.5 mg of MAR1–5A3 on days preceding and proceeding PBS inoculation as an additional control.
FIGURE 1. Murine model of early, mid, or late gestation ZIKV exposure.
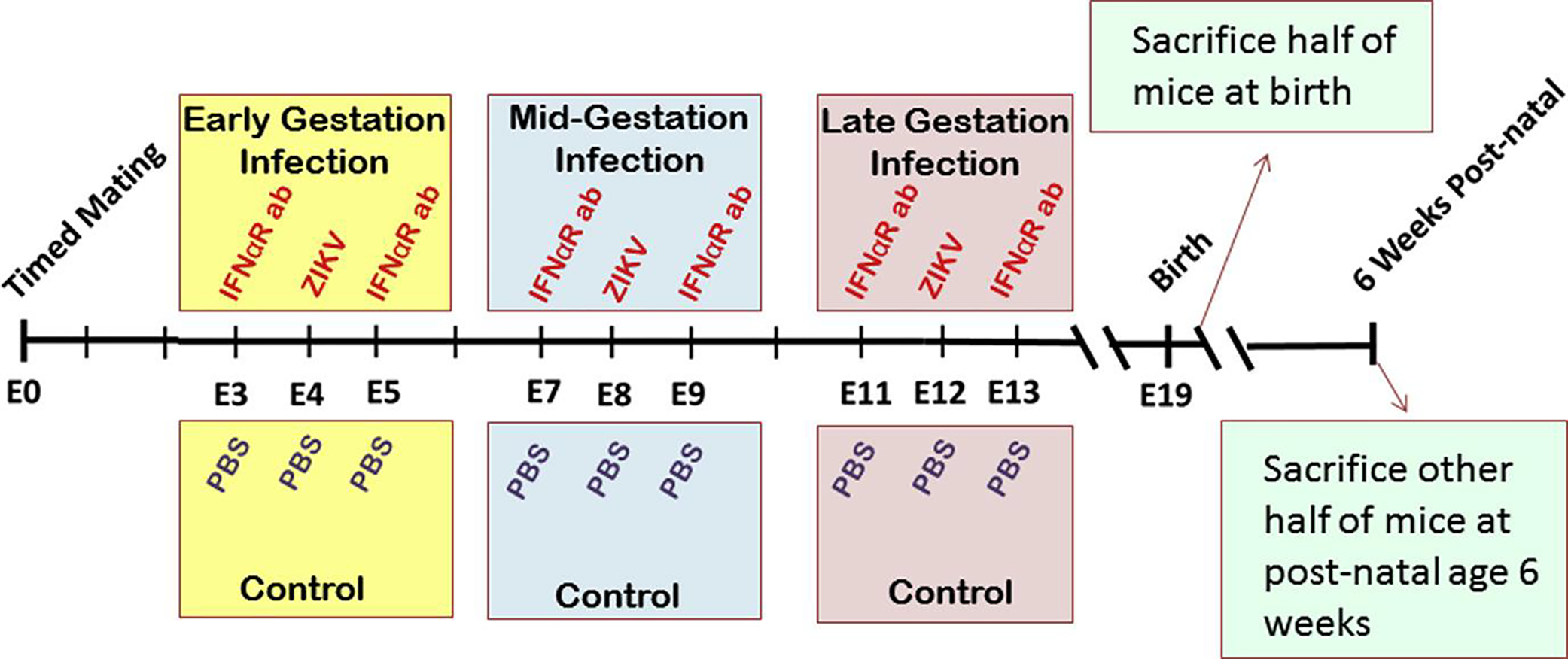
Mice were inoculated subcutaneously with ZIKV (experimental) or phosphate-buffered saline (PBS) (control) at embryonic day (E) 4, E8, or E12. Antiinterferon alpha receptor monoclonal antibody (IFNaR ab) was given intraperitoneally on days preceding and proceeding ZIKV inoculation. Half of pups were euthanized at postnatal day 1; other half were euthanized at 6 weeks of age.
One day after delivery (postnatal day 1), half of the pups were randomly assigned to be euthanized and the other assigned to continue to be housed until adolescence at 6 weeks of age. All pups were weighed on postnatal day 1 and biparietal diameter (BPD) was assessed using a traceable caliper (ThermoFisher Scientific). On postnatal day 1, half of the litter was sacrificed and brain and kidney tissues were harvested and flash frozen for later molecular analysis, or formalin fixed for histopathological ZIKV assessment. The brain weight was also measured.
The remaining pups were monitored through 6 weeks of age for signs of ZIKV infection including evaluation of paralysis or poor postnatal growth velocity. BPD was also assessed weekly.
Assessment of ZIKV viral load by reverse transcription- polymerase chain reaction
Frozen samples of brain and kidney tissues were weighed and RNA was extracted from 75 mg of tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. cDNA libraries were produced using the high-capacity cDNA reverse transcription kit (ThermoFisher Scientific) according to manufacturer’s instruction. Libraries were probed by reverse transcription (RT)- polymerase chain reaction (PCR) and GoTaq DNA polymerase (Promega, Madison, WI) in 2-μg reactions at 60°C with primer sets previously described (ZIKV 1086, 1086–1102, CCGCTGCCCAACACAAG and ZIKV 1162c, 1162–1139, CCACTAACGTTC TTTTGCAGACAT).12
In situ hybridization and high-resolution imaging
For a subset of randomly selected pups, full sagittal cross-sections of neonatal pups or organ tissue from adolescent animals was formalin-fixed and then paraffin-embedded rather than assessing by quantitative polymerase chain reaction. One pup per litter was randomly selected to be saved for histologic evaluation. All other pups were screened for ZIKV infection via quantitative polymerase chain reaction (rather than in situ hybridization [ISH]). Larger pups that were symptomatic and suspected of active ZIKV infection were dissected and organs split for both PCR and histological assessment of infection. Sections were cut and mounted onto slides and hematoxylin-eosin stained to assess for localized inflammation, while serial sections were subjected to ISH for + and − strands of genomic ZIKV ssRNA. ISH was performed using amplified RNA-scope probes (Advanced Cell Diagnostic Inc, Newark, CA) as previously described.19 Briefly, tissues were probed for both the + (stable) and − (actively replicating) RNA strands to localize viral particles and identify areas of active replication, respectively. Sections were deparaffinized and underwent antigen retrieval as per the manufacturer’s instructions (RNAscope 25 HD detection kit; Advanced Cell Diagnostics). The ZIKV sequence specific probes consisted of 87 probe pairs requiring hybridization of adjacent matching genome sites for adapter probe hybridization and amplification. 3,3’ diaminobenzidine tetrahydrochloride signal was generated after a series of amplification probes as per the manufacturer’s instructions. The slides produced were examined under bright field using a Nikon Eclipse 90i microscope (Nikon Instruments, Melville, NY). Minor adjustments for contrast and background level were made using NIS Elements Viewer 4.20 (Nikon) and Photoshop CC (Adobe, San Jose, CA).
Statistical analysis
Statistics were performed using parametric analysis of variance, 2-tailed unpaired Student t test, or Mann-Whitney as determined appropriate following an F test for normality. Fisher exact test was used to compare outcomes of exposure. All statistics were performed using software (Prism, Version 6.01; GraphPad, La Jolla, CA).
Results
At birth, e4-exposed pups were 10% smaller in overall pup weight (P =.003) even though there was no significant difference in brain weight when compared to control (Figure 2). The midgestation, e8, ZIKV-exposed pups demonstrated even greater disparity compared to mock control, with brain weights reduced by 14% and overall weight smaller by 19% (P = .01 and P < .0001, respectively). However, the late-gestation, e12, ZIKV-exposed pups did not demonstrate any weight differences compared to control. Overall there was no difference in the brain-to-weight ratio for any ZIKV-exposed group compared to mock. The mean litter size was also not different for ZIKV-exposed dams.
FIGURE 2. Neonatal pup and brain weights vary with timing of ZIKV inoculation.
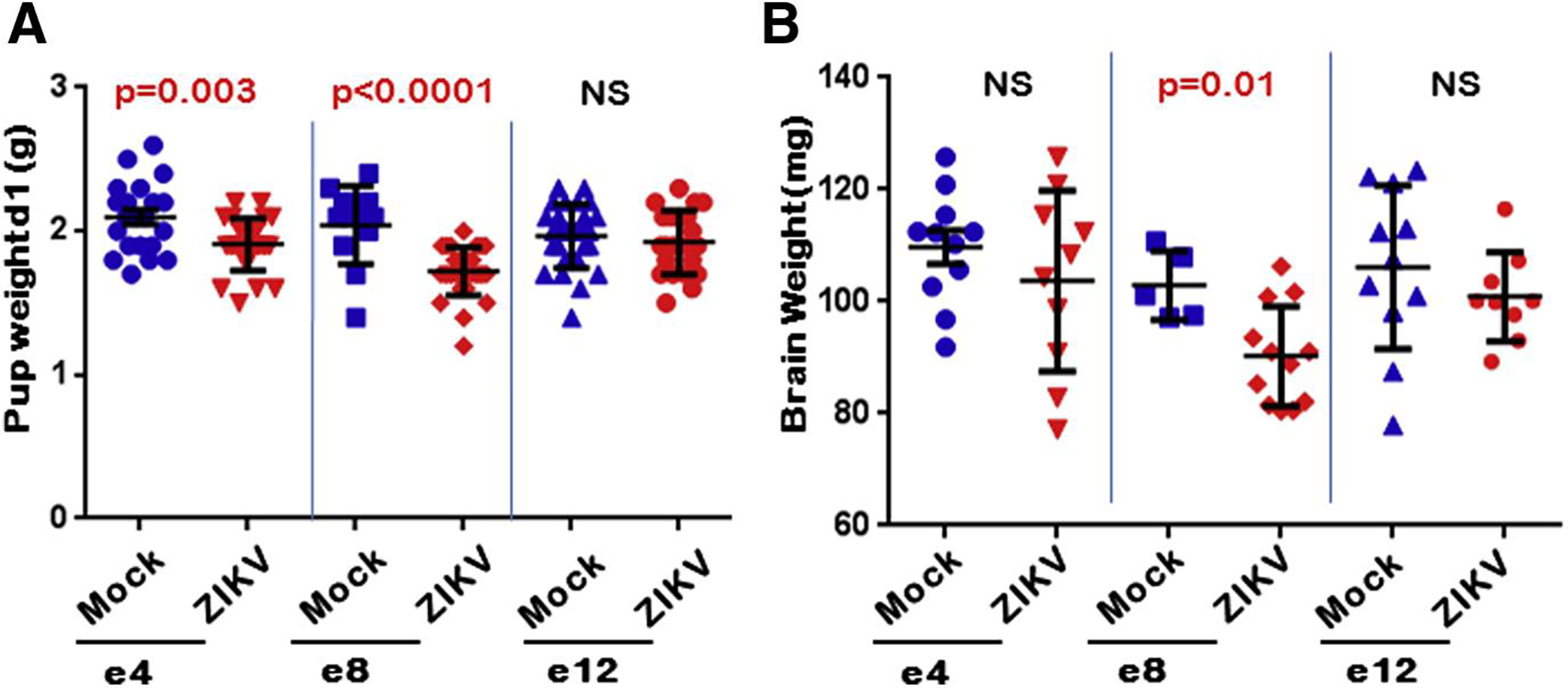
Pups were weighed on postnatal day 1. Pups of ZIKV-inoculated dams were A, significantly smaller when inoculated at embryonic day (e) 4 and e8 compared to mock injection and B, had significantly smaller brains compared to mock, which was only seen when ZIKV was inoculated at e8 time point. All other time points were found to be not statistically significant (NS).
To assess for postnatal changes as a result of potential infection, BPDs were evaluated weekly as the mice matured into adolescence (Figure 3). To account for sex and the impact gender can have on measurements (as male mice are usually larger in size and weight when compared to their female counterparts), we evaluated the mice as males or females as they matured. Male mice exposed to ZIKV in early gestation, e4, did not have any significant difference, while female pups at 1 week of age had a significantly smaller head size that reverted to normal thereafter (Figure 3, A i and B i). Male mice exposed to ZIKV in midgestation (e8), however, had significantly smaller BPDs on weeks 1, 4, and 5 (P = .02, .007, and .04, respectively), while the females trended to smaller size throughout the 6 weeks and had significantly smaller BPDs at week 4 (P = .04) (Figure 3, A and B ii). Finally, mice exposed to ZIKV at e12 showed no significant difference in BPDs between ZIKV and mock groups (Figure 3, A iii and B iii).
FIGURE 3. Midgestation ZIKV-infected litters had smaller biparietal diameters.
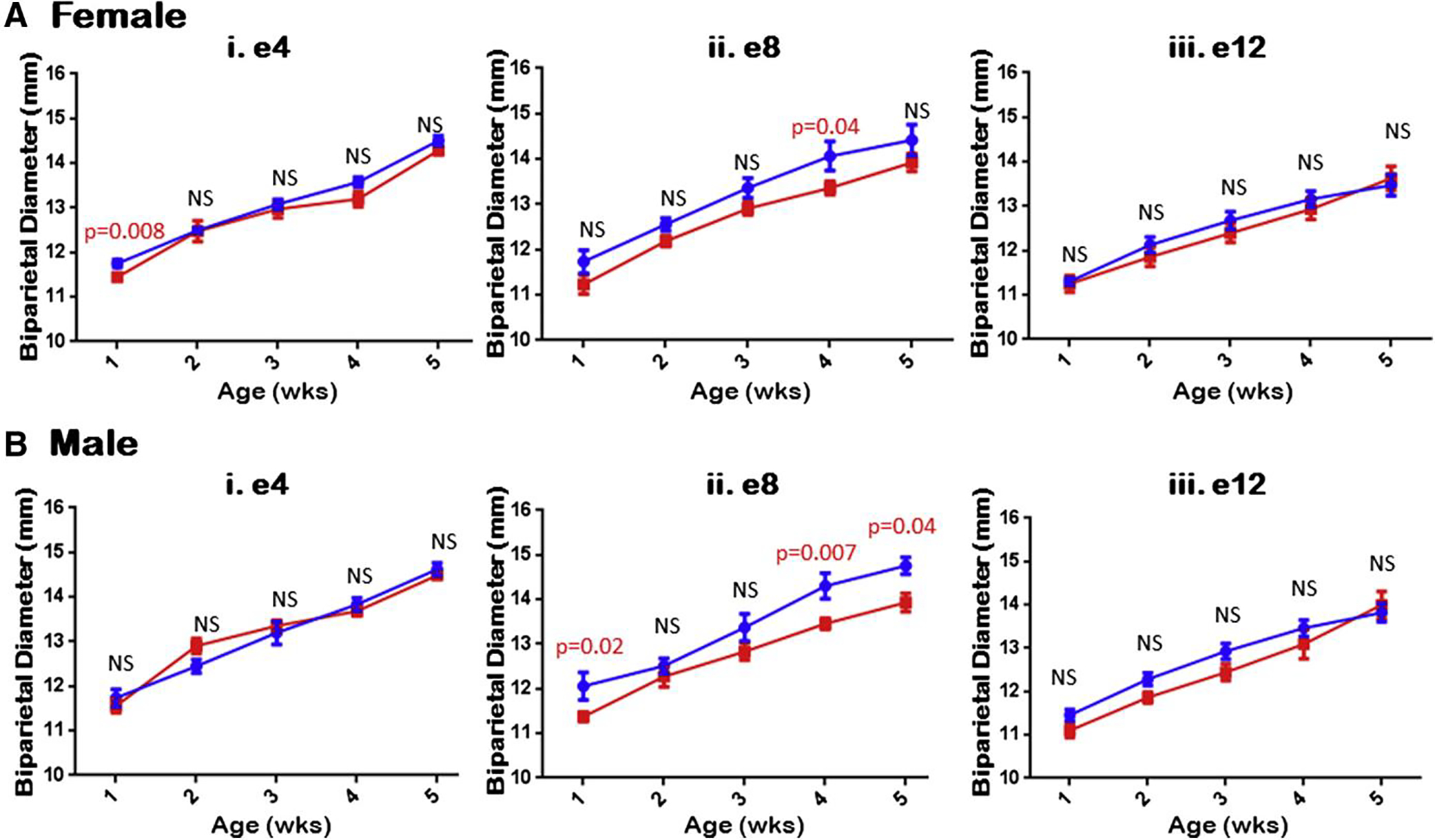
Mice were evaluated weekly for their BPD and evaluated based upon mouse sex. A, Females appeared to consistently trend smaller for overall BPD for midgestational ZIKV-infected littermates, compared to control, but not in early or late inoculation. B, Similarly, but more significantly, male mice exhibited consistently smaller BPD for midgestation ZIKV-infected littermates compared to control, but not early or late inoculation, consistent with changes for female mice. All other time points were found to be not statistically significant (NS).
When assessed at adolescence (6 weeks of life), the weights of mice exposed to ZIKV in early gestation (e4) were found to be higher overall when compared to mock for both males and females (Figure 4, A i). For the midgestation (e8) group, however, male mice exposed to ZIKV were found to be 9.3% smaller in overall weight when compared to control (P = .0007), while females trended smaller, but did not reach significance (Figure 4, A ii). When sexes were assessed together, growth restriction was still apparent with the ZIKV-infected e8 litter being 12% smaller than mock (P = .029). Finally, neither males nor females ZIKV exposed at e12 showed any differences in overall mouse weight when compared to control (Figure 4, A iii). Furthermore, brain weights were unchanged for males and females for any ZIKV exposure group (e4, e8, or e12) (Figure 4, B). When mice were assessed for viral infection of ZIKV none of the offspring from the e4 or e12 gestational exposure groups were positive for ZIKV, however 4.8% (2/42) of the e8 offspring were positive as assessed by RT-PCR and ISH.
FIGURE 4. Postnatal growth restriction persists in midgestation ZIKV-infected litters through adolescence.
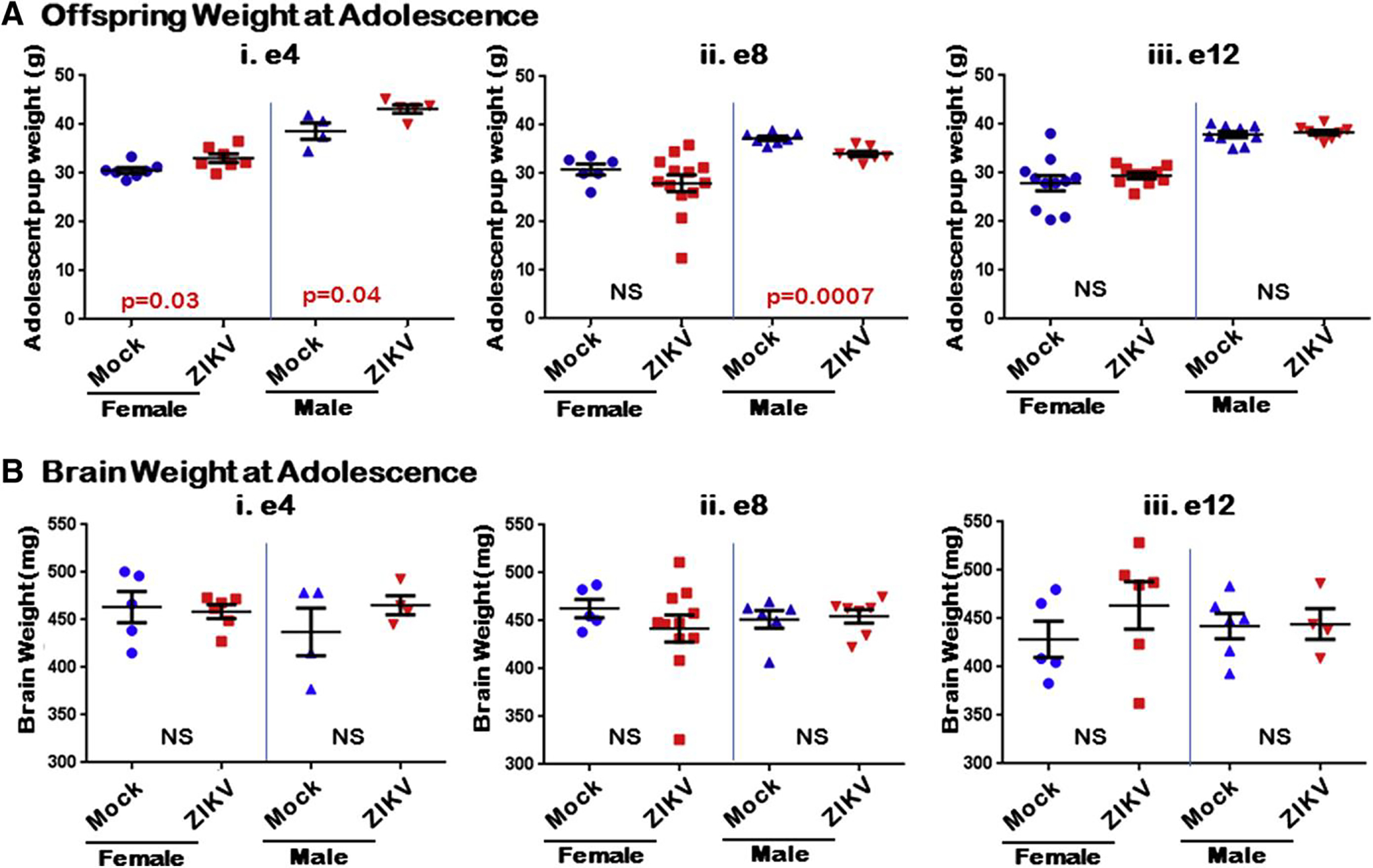
A i, Pups at 6 weeks of age in mock and early-gestation ZIKV-infected litters; ii, Midgestation ZIKV-infected litters, specifically males, were smaller overall compared to control; iii, Females appeared to trend smaller but were not statistically significant. Late-gestation ZIKV-infected litters were unaffected. B i-iii, Brain weights at 6 weeks of age were not statistically different (NS) between ZIKV and mock inoculation groups for any of time points overall.
Four mice from midgestation (e8) ZIKV inoculated were severely growth restricted (>2 SD below mean mock control weight) (Figure 5, A i). At 6 weeks of age, tissues were harvested and analyzed for ZIKV infection in one of the severely growth-restricted mice, and the other 3 were assessed via PCR. In the mouse assessed by ISH, only the brain was found to have ZIKV infection upon staining for the positive-sense RNA strand of ZIKV (Figure 5, A ii). The spleen (Figure 5, A iii) and kidneys did not show signs of ZIKV infection. The other 3 mice that were assessed via RT-PCR returned negative for ZIKV infection. Another mouse from a separate midgestation (e8) ZIKV-inoculated dam developed hind-limb paralysis and significant extrauterine growth restriction at postnatal day 8 (Figure 5, B). Tissues were harvested for active ZIKV infection. Both RT-PCR and qRT-PCR demonstrated significant ZIKV infection (Figure 5, B vi). ISH against the ZIKV RNA genome demonstrated strongly positive ZIKV within the spinal column (brown staining in Figure 5, B ii and iii). Positive signal for genomic ZIKV RNA was adjacent to the nucleus, which was consistent with an establishment of flaviviral replication factories in the endoplasmic reticulum. Despite evidence of neural infection by ZIKV, no leukocyte infiltration or inflammation was evident on hematoxylin-eosin staining. In comparison, no animals from the e4 or e12 groups became moribund and none showed evidence of ZIKV replication at 6 weeks of age by either PCR or in a subset assessed by ISH. Further, a subset of neonatal and adolescent animals of normal weight that were assessed by ISH were all negative for ZIKV by ISH and PCR.
FIGURE 5. Congenital Zika Syndrome found only in offspring from midgestation ZIKV inoculations.
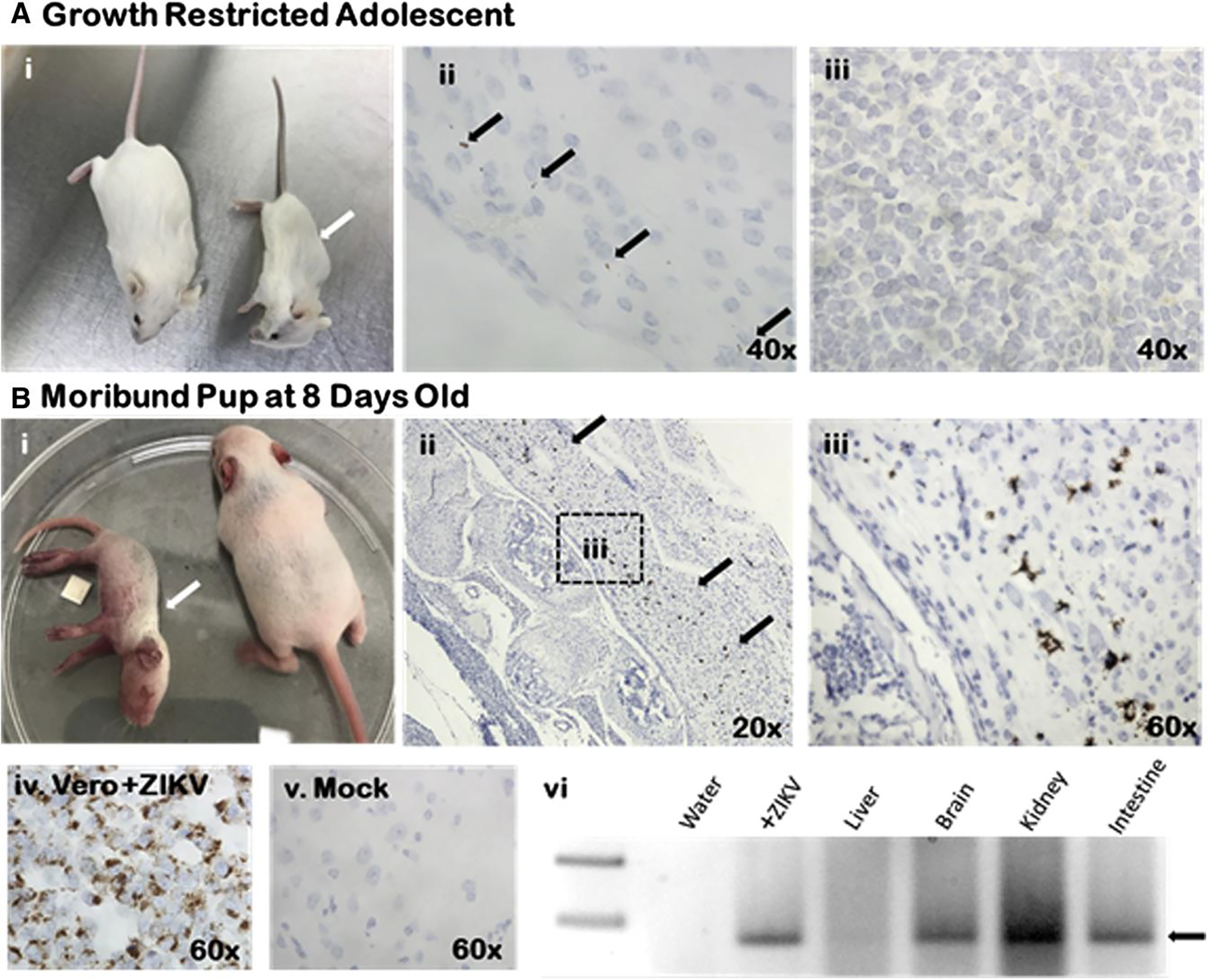
A i, Six-week-old mouse was >2 SD below mean weight (white arrow, compared with littermate) weighing only 12.5 g; ii, Brain tissue showed ZIKV positivity (black arrows) by in situ hybridization; iii, No ZIKV was apparent in spleen. B i, Eight-day-old mouse developed hind-limb paralysis and severe growth restriction (white arrow); ii, ZIKV infection present throughout spinal cord (black arrows); iii, Higher-power image showing staining throughout spinal column; iv and v, ZIKV replicating Vero cells and mock infected are positive and negative hybridization controls, respectively; vi, ZIKV polymerase chain reaction from moribund mouse showed significant positivity throughout all tissues assayed (black arrow demonstrates ZIKV band at 74 nucleotides).
Given the association of postnatal growth restriction with ZIKV infection following e8 exposure, we set out to determine the number of animals that were growth restricted at birth and those that were otherwise small compared to mock controls at adolescence (Figure 6). At 6 weeks of age, there was a significant association between postnatal growth restriction and ZIKV infection in midgestation, which was significant by Fisher exact test (P = .0007) (Figure 6). There were no animals from any other time point of gestational ZIKV exposure that were >2 SD below the mean for birthweight of mock-infected controls.
FIGURE 6. Growth restriction most evident in midgestation ZIKV inoculation offspring at adolescence.
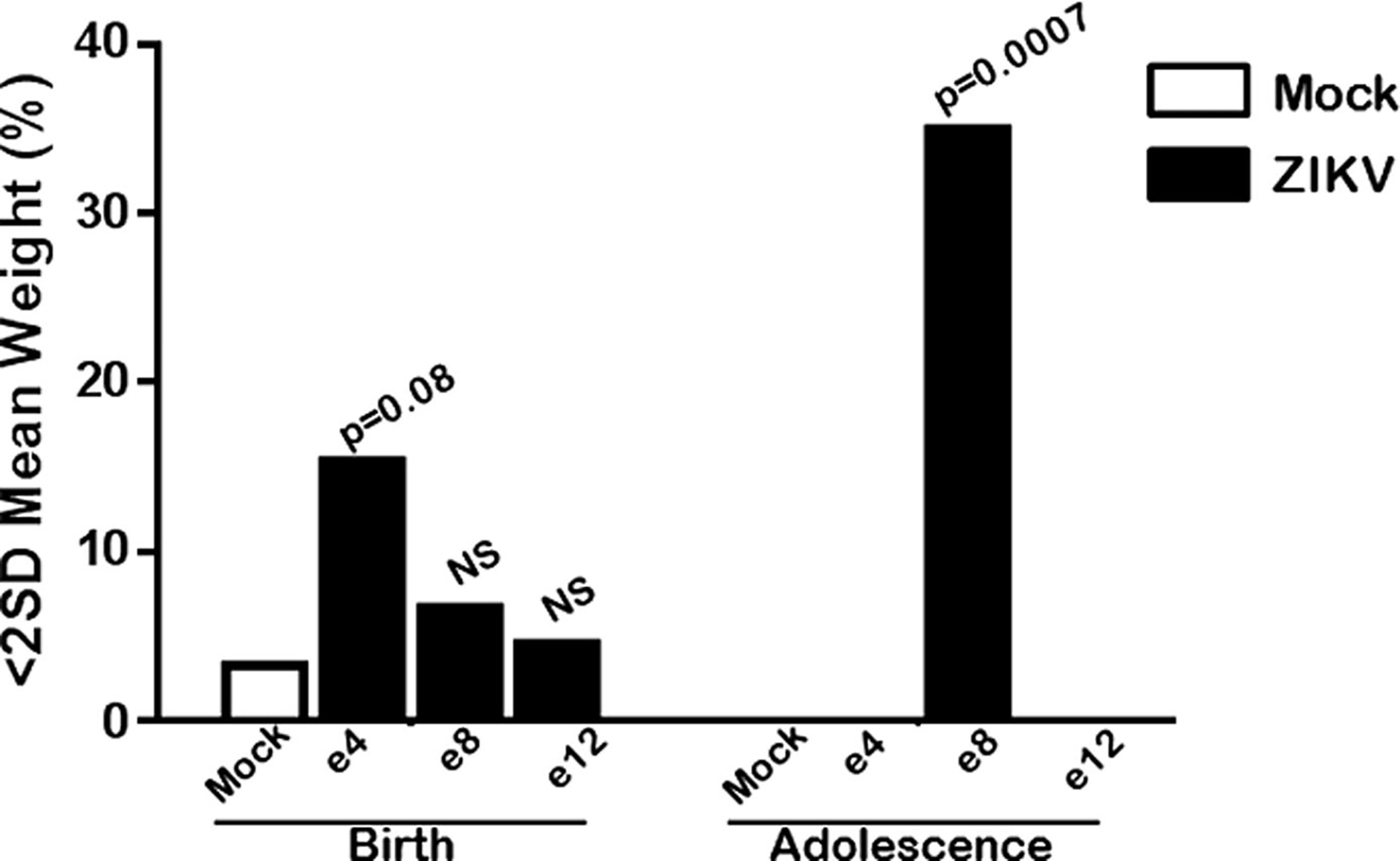
While all ZIKV-exposed offspring had some growth restriction at birth, it was not significantly increased over baseline mock. However, by adolescence, only offspring of mothers infected midgestation were severely underweight compared to mock. In total, 7 of 20 were >2 standard deviation below mean of mock embryonic day (e) 8, which was significantly associated with exposure by Fisher exact test.
Comment
Principal findings
Key findings of this study include an association between midgestation (e8) ZIKV infection and postnatal growth restriction, alongside evidence of CZS. Specifically, 35% were apparently affected by e8 congenital exposure when assessed at 6 weeks. Notably, only the midgestation, ZIKV-infected group had pups that became moribund with congenitally acquired ZIKV infection. Our findings demonstrate that the gestational timing of ZIKV inoculation of pregnant Swiss-Webster mice leads to significant differences in offspring outcomes, and these findings persist at 7 weeks after exposure (6 weeks postnatal age). Further, differences at 6 weeks of age were attributable to a very small subset that were severely growth restricted, recapitulating suspected differences in vertical transmission rates including low rates of vertical transmission and CZS associated with acute and time-limited exposure in a human population. We speculate that the differences are associated with single periods of inoculation, the stages of placentation, and fetal genetic variably of the outbred model here used.
Results in the context of what is known
This observed window of susceptibility may be due to the timing of inoculation in relation to the concurring embryogenic events occurring in the murine embryo. The early gestation ZIKV inoculation (e4) corresponds to the murine embryonic stage of implantation with limited blood supply.20,21 The late gestational ZIKV inoculation (e12) occurs at the timing where the mouse has an active circulatory system and placental maturity.22 The maturing fetus and placenta may likely have a greater capacity for viral defense and is therefore unaffected compared to e4. Conversely, the e8 window may correspond with relative permissiveness combined with hematogenous access.20,21 This time point corresponds to the human first trimester, with organogenesis, and early neural plate formation along with formation of the anterior and posterior neuropores.23
Furthermore, recently published animal studies on rhesus macaques have evaluated the role of the placenta in gestational ZIKV infection. Hirsch and colleagues24 found that even with normal fetal growth, there could be persistent placental infection that leads to uterine and placental vasculitis leading to a decreased oxygen permeability of the placental villi. Additionally, another study by Nguyen and colleagues25 found that gestational ZIKV infection led to neutrophilic infiltration in the placenta and membranes as well as in the fetal eye when ZIKV infection occurred in the first trimester. Finally, Martinot and colleagues26 described early infection in rhesus macaques led to maternal viremia, fetal neuropathology, and placental vascular changes leading to vascular insufficiency. These insights further the impact that the placental infection plays a key role in fetal infection. The heterogeneity of the offspring outcomes in our study potentially are reflective upon differences in placental infection. In contrast, in other nonhuman primate models, including our common marmoset model, infection of the placenta did not lead to significant inflammation although significant infection and even fetal loss still occurred.27 The lack of placental inflammation other than macrophage hyperplasia has also been described in human beings as well.28 Therefore, although it is established that the placenta acts as a reservoir and conduit for fetal infection, the implication of placental dysfunction with ZIKV infection in the development of CZS in human beings is less clear.
Clinical implications
The CDC reports that the highest risk for having a fetus or neonate with microcephaly occurs in women with confirmed ZIKV infection during their first trimester.15 We, as well as others, have demonstrated variable permissiveness of trophoblasts to ZIKV infection as a function of host factors including microRNA and interferon signaling.12,17 Moreover, a recent publication by Platt and colleagues29 demonstrates that ZIKV may not be the only neurotropic flavivirus that causes fetal symptoms and potential demise. They demonstrated that 2 other flaviviruses can cause fetal demise in wild-type mice: West Nile virus and Powassan virus.29 Given these findings, we speculate our findings may generalize to other potential congenital neurotropic viral infections.
Research implications
Human and mouse studies have shown the variation of placental viral resistance as a function of gestational age. For instance, the placental balance of Th2-type to Th1-type cytokine predilection is known to vary in pregnancy, and confer increased susceptibility to intracellular pathogens including viruses.23–29 Furthermore, Yockey and colleagues30 have shown that in interferon alpha receptor knockout mice, exposure to ZIKV while in utero leads to higher levels of ZIKV but the offspring continued to develop. However, in mice in which interferon signaling was still present via the IFNAR, these fetuses were resorbed after ZIKV infection during early pregnancy.30 These findings suggest a molecular basis for the temporal and individual variation in susceptibility to ZIKV vertical transmission. Further efforts to categorize entry receptors, placental histologic changes, and up- and down-regulation of interferons at each of these time points during placental and embryonic development is necessary to help ascertain the potential reasons for this observed window of fetal susceptibility to congenital ZIKV infection.
In addition, recently published literature from CZS in human beings elucidate a femur-sparing growth restriction can occur in ZIKV-infected neonates.31 While we did not directly measure femur lengths such as in this publication in our neonatal or adolescent pups, we did find that overall growth restriction was evident in the mice that were ZIKV infected. Furthermore, while there were no differences in overall growth restriction as pups between any of the groups, only offspring from the midgestational ZIKV-infected group had significantly higher rates of growth restriction at adolescence—reinforcing the findings that symptoms may not be present at birth but can develop weeks later.
Moreover, our model evaluated for the potential involvement of gender in the impact of the severity of phenotype in offspring outcomes. While males appeared to have more growth restriction when compared to their mock counterparts in the midgestation (e8) ZIKV-infected litters, females appeared to trend this direction. However, 1 of the 2 highly affected mice that were found to have CZS with severe growth restriction at 6 weeks of age was female. Since males are inherently larger, and therefore grow more quickly, CZS may simply be less readily apparent in adolescent females, and it seems likely that a larger study may have found significant weight differences across sexes.
Strengths and limitations
A strength of our study is its potential to more closely recapitulate human disease in nonendemic cohorts. Our experimental design used a single low-dose SQ acute exposure, and our rates of infection in midgestation were 4.8%. Care was taken to ensure that the ZIKV virus was first passage and contained the prM virion surface protein coding mutation that has been associated with vertical transmission in the recent epidemic. PrM modulates host cell interactions in flaviviruses, and the virulent mutation is absent in highly passaged African isolates that have been used in other recent experiments (notably MR766).16 This rate of infection in offspring via vertical transmission correlates to what has been found in human beings (estimated to be between 1–15%).15,32,33 A further novel aspect to our model is the evaluation of exposed pups well into the postnatal period (6 weeks of age, adolescence). In contrast, the majority of other murine studies have measured congenital disease at birth or immediately after. Our findings are pertinent based upon the CDC recent publications demonstrating that neonates may be asymptomatic with normal head circumferences at birth, but they can develop signs and symptoms of CZS months after birth.8
Our study is not without limitations. First, murine microcephaly is rare and difficult to define, and thus our primary outcome was growth restriction.17,34 Second, rodents are not natural hosts to ZIKV, presumptively due to inherently high levels of type I interferons as well as differences in the signal transducer and activator of transcription protein requiring suppression for susceptibility.34–37 However, our inoculations of interferon suppression were only maternal, and occurred at a single time interval surrounding infection (1 day pre and post). Antiinterferon receptor antibody was necessary to overcome the natural resistance of mice to ZIKV infection as has been described previously.17 Most mouse models today have in some way disrupted the interferon responses to observe an effect either using exogenous antibody or interferon receptor knockout mice. A recent publication made use of a mutated ZIKV strain,38 while the original experiments used extraordinarily high doses of virus.35 We believed it best to use an unmodified or low-passage original strain at a moderate dose, with the compromise of using exogenous antibody in this model.
Conclusion
Despite such limitations, our study reveals findings of significance. First, it recapitulates nonendemic human population exposures by virtue of study design. Accordingly, we observe a very similar rate of CZS and evidence of infectivity comparable to human epidemiologic estimates. Second, we define a midgestation window (e8) that poses unique susceptibility to gestationally acquired low-dose ZIKV infection that can persist in nonimmunocompromised offspring postnatally up to at least 6 weeks of age. Further efforts are necessary to evaluate and determine the mechanisms by which this gestational window of susceptibility to ZIKV infection exists. This preferred gestational window of virulence, which echoes epidemiological evidence of increased risk associated with trimester of exposure, coincides with critical events in placentation. This obviates the need for further correlative research to associate the timing of gestational ZIKV exposure and offspring outcomes at birth and postnatally to elucidate possible associations in ZIKV-exposed populations.
AJOG at a Glance.
Why was this study conducted?
To determine if the timing of gestational infection with Zika virus (ZIKV) leads to differences in offspring outcomes both at birth and adolescence.
Key findings
Viral exposure in the midgestational period (embryonic day 8) led to greater measures of congenital ZIKV infection and growth restriction that was apparent through maturation into adolescence in a mouse model.
What does this add to what is known?
The results reveal that while late gestation infection does not lead to postnatal growth restriction, there is a midgestational temporal window of susceptibility to ZIKV.
Acknowledgment
We would like to acknowledge the laboratory personnel in the Baylor College of Medicine germ-free facility for helping maintain the mice colonies throughout this series of experiments.
This study was supported in part by the Evangelina Whitlock grant provided to G.C.V. The funders had no role in developing the study design, data collection, and/or analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors report no conflict of interest.
Presented as poster no. 277 at the Society for Maternal-Fetal Medicine Pregnancy Meeting, Dallas, TX, Feb. 1, 2018.
Contributor Information
Gregory C. Valentine, Department of Pediatrics, Section of Neonatology, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Maxim D. Seferovic, Department of Obstetrics and Gynecology, Division of Maternal-Fetal Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Stephanie W. Fowler, Center for Comparative Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Angela M. Major, Department of Pathology, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX.
Rodion Gorchakov, Department of Pediatrics, Section of Pediatric Tropical Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Rebecca Berry, Department of Pediatrics, Section of Pediatric Tropical Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Alton G. Swennes, Center for Comparative Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Kristy O. Murray, Department of Pediatrics, Section of Pediatric Tropical Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Melissa A. Suter, Department of Obstetrics and Gynecology, Division of Maternal-Fetal Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
Kjersti M. Aagaard, Department of Obstetrics and Gynecology, Division of Maternal-Fetal Medicine, at Baylor College of Medicine and Texas Children’s Hospital, Houston, TX..
References
- 1.Marcondes CB, Ximenes Mde F. Zika virus in Brazil and the danger of infestation by Aedes (Stegomyia) mosquitoes. Rev Soc Bras Med Trop 2016;49:4–10. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. WHO statement on the first meeting of the International Health Regulations (2005) Emergency Committee on Zika virus and observed increase in neurological disorders and neonatal malformations. Available at: http://www.who.int/en/news-room/detail/01-02-2016-who-statement-on-the-first-meeting-of-the-international-health-regulations-(2005)-(ihr-2005)-emergency-committee-on-zika-virus-and-observed-increase-in-neurological-disorders-and-neonatal-malformations. Accessed February 5, 2018.
- 3.Valentine G, Marquez L, Pammi M. Zika virus-associated microcephaly and eye lesions in the newborn. J Pediatric Infect Dis Soc 2016;5:323–8. [DOI] [PubMed] [Google Scholar]
- 4.Valentine G, Marquez L, Pammi M. Zika virus epidemic: an update. Expert Rev Anti Infect Ther 2016;14:1127–38. [DOI] [PubMed] [Google Scholar]
- 5.Adebanjo T, Godfred-Cato S, Viens L, et al. Update: Interim guidance for the diagnosis, evaluation, and management of infants with possible congenital Zika virus infectione–United States, October 2017. MMWR Morb Mortal Wkly Rep 2017;66:1089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura CV, Maia M, Travassos SB, et al. Risk factors associated with the ophthalmoscopic findings identified in infants with presumed Zika virus congenital infection. JAMA Ophthalmol 2016;134:912–8. [DOI] [PubMed] [Google Scholar]
- 7.Eppes C, Rac M, Dunn J, et al. Testing for Zika virus infection in pregnancy: key concepts to deal with an emerging epidemic. Am J Obstet Gynecol 2017;216:209–25. [DOI] [PubMed] [Google Scholar]
- 8.van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015-January 2016 with congenital Zika virus infection without microcephaly at birth–Brazil. MMWR Morb Mortal Wkly Rep 2016;65:1343–8. [DOI] [PubMed] [Google Scholar]
- 9.Franca GV, Schuler-Faccini L, Oliveira WK, et al. Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 2016;388:891–7. [DOI] [PubMed] [Google Scholar]
- 10.Linden VV, Linden HVJ, Leal MC, et al. Discordant clinical outcomes of congenital Zika virus infection in twin pregnancies. Arq Neuropsiquiatr 2017;75:381–6. [DOI] [PubMed] [Google Scholar]
- 11.Santos VS, Oliveira SJG, Gurgel RQ, Lima DRR, Dos Santos CA, Martins-Filho PRS. Case report: microcephaly in twins due to the Zika virus. Am J Trop Med Hyg 2017;97:151–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aagaard KM, Lahon A, Suter MA, et al. Primary human placental trophoblasts are permissive for Zika virus (ZIKV) replication. Sci Rep 2017;7:41389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheridan MA, Yunusov D, Balaraman V, et al. Vulnerability of primitive human placental trophoblast to Zika virus. Proc Natl Acad Sci U S A 2017;114:E1587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Costa H, Gouilly J, Mansuy JM, et al. ZIKA virus reveals broad tissue and cell tropism during the first trimester of pregnancy. Sci Rep 2016;6:35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro-Mendoza CK, Rice ME, Galang RR, et al. Pregnancy outcomes after maternal Zika virus infection during pregnancy–US territories, January 1, 2016-April 25, 2017. MMWR Morb Mortal Wkly Rep 2017;66:615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan L, Huang XY, Liu ZY, et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017;358:933–6. [DOI] [PubMed] [Google Scholar]
- 17.Miner JJ, Cao B, Govero J, et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 2016;165:1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray KO, Gorchakov R, Carlson AR, et al. Prolonged detection of Zika virus in vaginal secretions and whole blood. Emerg Infect Dis 2017;23:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhatnagar J, Rabeneck DB, Martines RB, et al. Zika virus RNA replication and persistence in brain and placental tissue. Emerg Infect Dis 2017;23:405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet 2001;2:538–48. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Dey SK. Roadmap to embryo implantation: clues from mouse models. Nat Rev Genet 2006;7:185–99. [DOI] [PubMed] [Google Scholar]
- 22.Chen F, De Diego C, Chang MG, et al. Atrioventricular conduction and arrhythmias at the initiation of beating in embryonic mouse hearts. Dev Dyn 2010;239:1941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland AE. Tissue morphodynamics shaping the early mouse embryo. Semin Cell Dev Biol 2016;55:89–98. [DOI] [PubMed] [Google Scholar]
- 24.Hirsch AJ, Roberts VHJ, Grigsby PL, et al. Zika virus infection in pregnant rhesus macaques causes placental dysfunction and immunopathology. Nat Commun 2018;9:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen SM, Antony KM, Dudley DM, et al. Highly efficient maternal-fetal Zika virus transmission in pregnant rhesus macaques. PLoS Pathog 2017;13:e1006378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinot AJ, Abbink P, Afacan O, et al. Fetal neuropathology in Zika virus-infected pregnant female rhesus monkeys. Cell 2018;173:1111–22.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seferovic M, Martin CS, Tardif SD, et al. Experimental Zika virus infection in the pregnant common marmoset induces spontaneous fetal loss and neurodevelopmental abnormalities. Sci Rep 2018;8:6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg AZ, Yu W, Hill DA, Reyes CA, Schwartz DA. Placental pathology of Zika virus: viral infection of the placenta induces villous stromal macrophage (Hofbauer cell) proliferation and hyperplasia. Arch Pathol Lab Med 2017;141:43–8. [DOI] [PubMed] [Google Scholar]
- 29.Platt DJ, Smith AM, Arora N, Diamond MS, Coyne CB, Miner JJ. Zika virus-related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci Transl Med 2018;10(426). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yockey LJ, Jurado KA, Arora N, et al. Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol 2018;3(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker CL, Merriam AA, Ohuma EO, et al. Femur-sparing pattern of abnormal fetal growth in pregnant women from New York city after maternal Zika virus infection. Am J Obstet Gynecol 2018. May 5. pii: S0002–9378(18) 30378–8. 10.1016/j.ajog.2018.04.047. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaenisch T, Rosenberger KD, Brito C, Brady O, Brasil P, Marques ET. Risk of microcephaly after Zika virus infection in Brazil, 2015 to 2016. Bull World Health Organ 2017;95:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johansson MA, Mier-y-Teran-Romero L, Reefhuis J, Gilboa SM, Hills SL. Zika and the risk of microcephaly. N Engl J Med 2016;375:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miner JJ, Sene A, Richner JM, et al. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 2016;16:3208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cugola FR, Fernandes IR, Russo FB, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016;534:267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazear HM, Govero J, Smith AM, et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 2016;19:720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Saucedo-Cuevas L, Shresta S, Gleeson JG. The neurobiology of Zika virus. Neuron 2016;92:949–58. [DOI] [PubMed] [Google Scholar]
- 38.Gorman MJ, Caine EA, Zaitsev K, et al. An immunocompetent mouse model of Zika virus infection. Cell Host Microbe 2018;23:672–85.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]