

Abstract

We have developed Cp*Co(III)-catalyzed redox-neutral synthesis of 3,4-unsubstituted isoquinoline 1(2H)-ones at ambient temperature using N-chloroamides as a starting material. The reaction utilizes vinyl acetate as an inexpensive and benign acetylene surrogate. The N–Cl bond of the N-chlorobenzamides plays the role of an internal oxidant and hence precludes the need for an external oxidant. The reaction works with a wide range of substrates having various functional groups and a substrate containing a heterocyclic ring. Notably, the reaction is extended to the N-chloroacrylamides in which vinylic C–H activation occurs to furnish the 2-pyridone derivatives. Preliminary mechanistic studies were also conducted to shed light on the mechanism of this reaction.
Introduction
Isoquinolin-1(2H)-one is a privileged scaffold that often encounters many naturally occurring alkaloids, and it also constitutes a core framework in many medicinally important pharmaceutical drugs possessing anticancer, antiviral, and antidiabetic activities (Figure 1).1 Although, there are various methods available for the synthesis of isoquinolone derivatives,2 the utilization of transition-metal-catalyzed C–H annulation has gained a lot of prominence due to the step and atom-economic nature of these reactions.3 Early reports on C–H activation for the synthesis of isoquinolones rely on the oxidative-cyclization strategies, which often suffer from the drawback of using a stoichiometric amount of metal oxidants such as Cu(OAc)2 and Ag2CO3.4 These metal oxidants subsequently lead to the generation of metal wastes at the end of the reaction. In order to circumvent the issue of using an external oxidant, redox-neutral C–H annulation reactions involving oxidizing directing groups came into limelight.5
Figure 1.

Representative examples of naturally occurring and medicinally important isoquinolone derivatives.
The seminal work in the area of C–H functionalization using oxidizing groups was reported by groups of Glorius, Fagnou, Hartwig, Cui & Wu, and Yu.6 In these transformations, where an oxidizing directing group has been employed, the reactions can be conducted without any external oxidant because the directing group plays a dual role, i.e., direct the C–H metalation and oxidize the metal back to its active oxidation state. In 2014, Marsden et al. reported Cp*Rh(III)-catalyzed [4 + 2] annulation of N-pivaloyl benzamides with vinyl acetate as an acetylene equivalent for the synthesis of isoquinolones (Scheme 1a).7 In 2021, Baidya et al., during their studies on the synthesis of aminal motifs via Ru(II)-catalyzed C–H activation/annulation of N-methoxybenzamides with enamides, also reported 3 examples for isoquinolone synthesis using vinyl acetate as a coupling partner at 60 °C (Scheme 1a).8 However, the requirement of precious transition metals like Rh and Ru makes these protocols cost-intensive. Moreover, these methods are only limited to aromatic C–H functionalizations. Hence, it is highly desirable to develop a cost-efficient method for the synthesis of isoquinolone derivative as well as pyridone moiety via vinylic C–H activation.
Scheme 1. Transition-Metal-Catalyzed Synthesis of Isoquinolone Derivatives via [4 + 2] Annulation Reactions.

Recent developments in the area of C–H functionalization have shifted the focus toward the utility of inexpensive and earth-abundant first-row transition metals (e.g., Co, Cu, Fe, Ni, and Mn) in these transformations.9 In this regard, Cp*Co(III)-catalysis has seen significant development in the area of C–H functionalization.10 This is accounted for the low cost, easy preparation, and air stability of Cp*Co(III) catalysts. Various elegant approaches for the synthesis of isoquinolones were also reported under Cp*Co(III)-catalysis utilizing N-methoxyamide as a substrate, wherein the N–O bond works as an internal oxidant.11 In 2017, Zhu et al. introduced N-chlorobenzamide as a novel oxidizing group, in which the role of internal oxidant is played by the N–Cl bond. They have reported [4 + 2] annulation reactions of N-chlorobenzamides with alkyne and alkenes using Cp*Co(III)-catalysis.12,13 When alkyne has been used as a coupling partner, it resulted in the formation of isoquinolone derivatives, whereas the use of alkene leads to the generation of 3,4-dihydroisoquinolones (Scheme 1b). Following these reports, N-chlorobenzamide has been effectively used as an efficient oxidizing directing group for various transformations.14 In continuation of our interest in high-valent Co(III)-catalysis15 and mild C–H functionalizations,16 herein, we report the synthesis of isoquinolones via Cp*Co(III)-catalyzed annulation between N-chlorobenzamides and vinyl acetate at ambient temperature (Scheme 1d). During the preparation of the manuscript, Jeganmohan‘s group reported a similar synthetic protocol for the synthesis isoquinolones via Cp*Co(III)-catalyzed [4 + 2] C–H activation/annulation of N-chlorobenzamides with vinyl acetate and vinyl ketones (Scheme 1c).17 However, their scope is limited to only arene C–H functionalization, whereas our protocol can also be applied successfully for vinylic C–H bond activation along with arene C–H functionalization. In this reaction vinyl acetate acts as a synthetic equivalent for acetylene. The reaction demonstrated a good functional group and broad scope. The reaction was also applicable to the N-chloroacrylamide derivative where the reaction proceeds via vinylic C–H activation, which was otherwise elusive by previously reported methods.7,8
Results and Discussion
In order to optimize the reaction parameters, we conducted a series of reactions between N-chlorobenzamide (1a) and vinyl acetate (2) using [Cp*Co(CO)I2] (10 mol %) as a catalyst keeping the temperature constant at 30 °C (Table 1). We were glad to see the formation of isoquinolone 3aa in 56% yield in our very first attempt when the reaction was performed between 1a (0.1 mmol) and 2 (5 equiv) in the presence of 10 mol % of [Cp*Co(CO)I2] and NaOAc (1.2 equiv) using 2,2,2-trifluoroethanol (TFE) as a solvent for 12 h (Table 1, entry 1). An increase in the amount of NaOAc to (1.5 equiv) led to a slight increase in the product formation (Table 1, entry 2). Surprisingly, the introduction of Ag(I) salts, such as AgSbF6 and AgOTf, led to diminished yields (Table 1, entries 3–4). However, we were pleased with the formation of isoquinolone 3aa in an excellent yield (89%), when AgOAc (20 mol %) was used (Table 1, entry 5). Various other additives such as CsOAc, KOAc, LiOAc, Ca(OH)2, and PivOH were ineffective compared with NaOAc (Table 1, entries 6–10). The reaction was found to be sluggish in other solvents like 1,2-DCE, MeOH, and 1,4-dioxane (Table 1, entries 11–13).
Table 1. Optimization Studya.

| entry | Ag(I) salt | additive (equiv) | solvent | time (h) | yield (%)b |
|---|---|---|---|---|---|
| 1 | NaOAc (1.2) | TFE | 12 | 56 | |
| 2 | NaOAc (1.5) | TFE | 12 | 68 | |
| 3 | AgSbF6 | NaOAc (1.5) | TFE | 12 | 19 |
| 4 | AgOTf | NaOAc (1.5) | TFE | 12 | 21 |
| 5 | AgOAc | NaOAc (1.5) | TFE | 12 | 89 |
| 6 | AgOAc | CsOAc (1.5) | TFE | 12 | 61 |
| 7 | AgOAc | KOAc (1.5) | TFE | 12 | 66 |
| 8 | AgOAc | LiOAc (1.5) | TFE | 12 | 22 |
| 9 | AgOAc | Ca(OH)2 (1.5) | TFE | 12 | n.d. |
| 10 | AgOAc | PivOH (1.5) | TFE | 12 | trace |
| 11 | AgOAc | NaOAc (1.5) | 1,2-DCE | 12 | 44 |
| 12 | AgOAc | NaOAc (1.5) | MeOH | 12 | 22 |
| 13 | AgOAc | NaOAc (1.5) | 1,4-dioxane | 12 | trace |
| 14 | AgOAc | NaOAc (1.5) | TFE | 12 | 83c |
| 15 | AgOAc | NaOAc (1.5) | TFE | 12 | 72d |
| 16 | AgOAc | NaOAc (1.5) | TFE | 8 | 66 |
| 17 | AgOAc | NaOAc (1.5) | TFE | 4 | 54 |
| 18 | AgOAc | NaOAc (1.5) | TFE | 12 | 76e |
| 19 | AgOAc | NaOAc (1.5) | TFE | 12 | n.d.f |
| 20 | AgOAc | TFE | 12 | <5% |
Reaction conditions: 1a (0.10 mmol), 2a (0.50 mmol, 5.0 equiv), [Cp*Co(CO)I2] (10.0 mol %), Ag(I) salt (20.0 mol %), and additives in solvent (0.6 mL) for given time at 30 °C.
Yields are based on crude 1H NMR (internal standard: 1,1,2,2 tetrachloroethane).
3.0 equiv of 2 was used.
2.0 equiv of 2 was used.
[Cp*Co(CO)I2] (5.0 mol %) and AgOAc (10 mol %) were used.
Without [Cp*Co(CO)I2]. n.d. = not detected. TFE = 2,2,2-trifluoroethanol.
Reducing the equivalents of vinyl acetate resulted in a decrease in the yield of the product (Table 1, entries 14–15). A similar decline in the yield was observed when the reaction was run for a shorter duration (Table 1, entries 16–17). When the catalyst loading was reduced to 5 mol %, it furnished the annulated product in 76% yield (Table 1, entries 18). We did not observe any product formation in the absence of a cobalt catalyst and only a trace amount without NaOAc (Table 1, entries 19–20).
After optimizing the reaction parameters, we have applied this method for the synthesis of various isoquinolone derivatives using differently functionalized N-chlorobenzamides (Scheme 2). The reaction worked with N-chlorobenazmides having electron-donating substituents such as t-Bu, Me, and Ph at the para-position (3b–3d). Moreover, X-ray crystallographic analysis further confirmed the structure of the isoquinolone 3d. Gratifyingly, the reaction tolerated all four halogens at the para-position (3e–3h).18 This gives the scope for further synthetic manipulations of the isoquinolone derivatives by performing different cross-coupling reactions. Furthermore, the structure of the 4-iodo isoquinolone derivative (3h) was confirmed by X-ray analysis. The reaction also worked well with electron-withdrawing functional groups such as NO2, CF3, and CO2Me (3i–3k). The reaction was highly regioselective in the case of meta-substituted N-chlorobenzamides and was applicable to both electron-donating and electron-withdrawing substituents furnishing the required annulated products in moderate to good yields (3l–3p).
Scheme 2. Scope of N-Chlorobenzamides.

Reaction conditions: 1 (0.6 mmol), 2 (5.0 equiv), [Cp*Co(CO)I2] (10 mol %), AgOAc (20 mol %), and NaOAc (1.5 equiv) in TFE (2.5 mL) at 30 °C for 12 h. Isolated yields are given. TFE = 2,2,2-trifluoroethanol.
However, when the reaction was performed with N-chloro-3,4-dimethylbenzamide (1q), it resulted in the formation of a inseparable mixture of regioisomeric products in a 5:1 ratio. The major product (3q) is formed by the C–H functionalization at a less hindered position. Next, we tested various ortho-substituted N-chlorobenzamides. The reaction proceeded well with a 41% yield in the case of F substituent (3r). However, the reaction failed to produce any desired product in the case of ortho-methyl and ortho-phenyl substituents (3s–3t). The reaction was successfully applied to N-chloro-2-naphthamide also furnishing the isoquinolone derivative (3u) in moderate yield. We were pleased to observe that the current protocol is compatible with a substrate bearing a thiophene ring (3v). Further, this protocol was extended to the vinylic C–H activation, wherein N-chloromethacrylamide derivatives were converted into the corresponding pyridone derivatives (3w–3x) in good yields. However, the reaction failed to produce any product with (E)-N-chloro-2-methylbut-2-enamide (1y) as a substrate. The gram-scale synthesis of isoquinolone 3a was also carried out using 5 mol % of [Cp*Co(CO)I2] and 10 mol % of AgOAc with 74% yield (see the Supporting Information).
Finally, we have performed a few mechanistic experiments in order to understand the mechanism of the current reaction (Scheme 3). When the H/D exchange experiment was performed using CD3OD in TFE as a solvent, we did not observe any significant amount of deuterium incorporation at the ortho-position in the starting material (Scheme 3a). This might be due to the rapid deuterium exchange with the solvent TFE. Therefore, we have performed a similar reaction in 1,2-DCE using D2O as the D-source. This resulted in the incorporation of 78% of D incorporation at each ortho-position of [D]n-1a. This result led us to conclude that, in the current reaction, the C–H activation step is reversible. When the intermolecular competitive reaction was performed with N-chlorbenzamides having an electron-donating Me and an electron-withdrawing CF3 group at para-positions, it was found that the substrate having an electron-withdrawing CF3 group reacts preferably compared to the substrate having an electron-donating Me group (Scheme 3b). This observation can be concluded in terms of carboxylate-assisted C–H activation.19 In order to determine the nature of the C–H activation step, we have performed the KIE experiments. The KIE value obtained from the competitive experiment was found to be 3.5 (Scheme 3c).
Scheme 3. Mechanistic Findings.

Then, we performed parallel reactions at 30, 60, 90, and 120 min, which resulted in the KIE values of 3.0, 2.3, 2.1, and 2.1, respectively (Scheme 3c). These KIE values indicate that the C–H activation might be the rate-determining step.
Based on the mechanistic experiments and relevant reports,7,8 a plausible mechanism for the current [4 + 2] annulation reaction is depicted in Scheme 4. The catalytic cycle begins with the formation of catalytically active Co(III) species A by the reaction between [Cp*Co(CO)I2] and AgOAc. At the same time, NaOAc deprotonates the N-chlorobenzamide, which in turn undergoes ortho-metalation with A to form a five-membered cobaltacycle B. Subsequently, regioselective insertion of vinyl acetate into the cobaltacycle A led to the formation of a seven-membered cobaltacycle species C. The regioselectivity of the vinyl acetate insertion is governed by the weak chelation between the carbonyl oxygen of the vinyl acetate and the cobalt center.
Scheme 4. Plausible Mechanism.

The intermediate C upon migratory insertion results in the formation of a N–Cl isoquinolone D along with the generation of Cp*Co(I) species. The Cp*Co(I) get oxidizes back to Cp*Co(III) via oxidative addition into the N–Cl bond to form species E. This step is responsible for the redox-neutral nature of this reaction. The intermediate E upon acetate elimination and subsequent protodemetalation furnishes an intermediate F along with concomitant generation of catalytically active Co(III) species A. Finally, isoquinolone F upon base-mediated tautomerization leads to the formation of the desired isoquinolone 3.
Conclusions
In conclusion, an efficient redox-neutral protocol has been developed for the synthesis of isoquinolones starting from N-chlorobenzamides and vinyl acetate using an inexpensive Co(III) catalyst. The reaction utilizes vinyl acetate as a cheap and safe acetylene surrogate. The reaction can be performed at ambient temperature without the requirement of any inert conditions. Also, the reaction demonstrated an excellent functional group tolerance along with a broad scope and gram-scale applicability. The reaction can be extended for the synthesis of a pyridine derivative via vinylic C–H functionalization. The preliminary mechanistic studies revealed that the C–H activation proceeds via the base assistance, and it might be the rate-determining step.
Experimental Section
General Remarks
All commercial reagents and solvents were used without additional purification, unless otherwise stated. Column chromatography was performed on silica gel (100–200 mesh) using a suitable solvent system. 1H and 13C NMR spectra were recorded in DMSO-d6 at 500 MHz. High-resolution mass spectrometry (HRMS) spectra were recorded using electrospray ionization time-of-flight (ESI-TOF) techniques. The starting N-chlorobenzamides were prepared from commercially available carboxylic acids in 2 steps following the literature protocol.12 Vinyl acetate is commercially available and used as such without any further purification.
General Procedure for the Cp*Co(III)-Catalyzed C–H Functionalization of N-Chlorobenzamides with Vinyl Acetate
To a screw-capped seal tube vial with a Teflon stir bar was added N-chlorobenzamide 1 (0.6 mmol, 1.0 equiv), vinyl acetate 2 (3.0 mmol, 258.0 mg, 5.0 equiv), [Cp*Co(CO)I2] (10.0 mol %, 28.5 mg), AgOAc (20.0 mg, 20 mol %), NaOAc (73.8 mg, 1.5 equiv), and TFE (2,2,2-trifluoroethanol) (3.6 mL) under an air atmosphere. The reaction mixture was stirred at 30 °C for 12 h. Then, it was filtered through a short pad of celite, and the celite pad was washed with DCM (15 mL × 2). The solvent was removed using a rotary evaporator, and the residue was purified by silica gel column chromatography using n-hexane/EtOAc as an eluent to give the desired isoquinolone derivatives. The yields were calculated with respect to N-chlorobenzamides, which are the limiting agents.
Spectral Data of All Compounds
Isoquinolin-1(2H)-one (3a)7

White solid (73.2 mg, 84%); eluent (50–60% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.27 (s, 1H), 8.19 (d, J = 7.9 Hz, 1H), 7.72–7.65 (m, 1H), 7.63 (d, J = 7.4 Hz, 1H), 7.50–7.43 (m, 1H), 7.17 (d, J = 7.0 Hz, 1H), 6.54 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.9, 137.9, 132.3, 128.9, 126.7, 126.3, 126.2, 126.1, 104.6.
6-Methylisoquinolin-1(2H)-one (3b)20

Yellow solid (61.0 mg, 64%); eluent (70–80% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.14 (brs, 1H), 8.06 (d, J = 8.2 Hz, 1H), 7.42 (s, 1H), 7.29 (dd, J = 8.2, 1.3 Hz, 1H), 7.13 (d, J = 6.7, 1H), 6.45 (d, J = 7.1 Hz, 1H), 2.41 (s, 3H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.6, 142.1, 137.6, 128.8, 127.5, 126.5, 125.5, 123.8, 104.2, 21.0.
6-(tert-Butyl)isoquinolin-1(2H)-one (3c)20

Yellow solid (78.5 mg, 65%); eluent (50–60% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.15 (brs, 1H), 8.10 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 7.54 (dd, J = 8.7, 1.8 Hz, 1H), 7.13 (m, 1H), 6.53 (d, J = 7.1 Hz, 1H), 1.32 (s, 9H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.6, 155.0, 137.8, 128.7, 126.4, 124.1, 123.9, 122.0, 104.9, 34.7, 30.8.
6-Phenylisoquinolin-1(2H)-one (3d)20

White solid (109.0 mg, 82%); eluent (60–65% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.27 (brs, 1H), 8.24 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 1.6 Hz, 1H), 7.84–7.73 (m, 3H), 7.52 (t, J = 7.6 Hz, 2H), 7.44 (t, J = 7.4 Hz, 1H), 7.25–7.15 (m, 1H), 6.62 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.6, 143.7, 139.2, 138.4, 129.3, 129.0, 128.2, 127.4, 127.0, 125.0, 124.9, 123.9, 104.8.
6-Fluoroisoquinolin-1(2H)-one (3e)7

White solid (68.3 mg, 70%); eluent (50–60% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.32 (brs, 1H), 8.22 (dd, J = 8.9, 5.9 Hz, 1H), 7.48 (dd, J = 10.1, 2.6 Hz, 1H), 7.31 (td, J = 8.9, 2.7 Hz, 1H), 7.26–7.19 (m, 1H), 6.53 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 164.4 (d, JC–F = 248.9 Hz), 161.2, 140.4 (d, JC–F = 10.6 Hz), 130.5, 130.2 (d, JC–F = 10.2 Hz), 123.0, 114.8 (d, JC–F = 23.6 Hz), 110.9 (d, JC–F = 21.8 Hz), 104.2; 19F NMR (470 MHz, DMSO-d6) δ −107.4.
6-Chloroisoquinolin-1(2H)-one (3f)20

White solid (86.2 mg, 80%); eluent (80–85% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.38 (brs, 1H), 8.15 (d, J = 8.6 Hz, 1H), 7.78 (d, J = 1.7 Hz, 1H), 7.48 (dd, J = 8.5, 1.9 Hz, 1H), 7.23 (d, J = 7.1 Hz, 1H), 6.53 d, J = 7.2 Hz, 1H; 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.2, 139.4, 137.2, 130.5, 128.9, 126.3, 125.2, 124.6, 103.6.
6-Bromoisoquinolin-1(2H)-one (3g)7

Yellow solid (80.7 mg, 60%); eluent (100% ethyl acetate); 1H NMR (500 MHz, DMSO-d6) δ 11.27 (s, 1H), 8.08 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 1.8 Hz, 1H), 7.61 (dd, J = 8.4, 2.0 Hz, 1H), 7.26–7.15 (m, 1H), 6.52 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.2, 139.5, 130.4, 129.0, 128.9, 128.2, 126.2, 124.8, 103.4.
6-Iodoisoquinolin-1(2H)-one (3h)7

Yellow solid (73.3 mg, 45%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.36 (brs, 1H), 8.13 (m, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.78 (dd, J = 8.3, 1.2 Hz, 1H), 7.27–7.13 (m, 1H), 6.49 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.5, 139.5, 134.7, 134.5, 130.1, 128.5, 125.1, 103.3, 100.5.
6-Nitroisoquinolin-1(2H)-one (3i)17

Yellow solid (77.6 mg, 68%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.62 (d, J = 2.4 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 8.18 (dd, J = 8.7, 2.3 Hz, 1H), 7.40–7.29 (m, 1H), 6.81 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 160.8, 149.7, 138.6, 131.3, 129.6, 129.0, 121.8, 119.7, 104.6.
6-(Trifluoromethyl)isoquinolin-1(2H)-one (3j)7

White solid (94.6 mg, 74%); eluent (80–85% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.50 (brs, 1H), 8.35 (d, J = 8.4 Hz, 1H), 8.09 (s, 1H), 7.72 (dd, J = 8.3, 0.8 Hz, 1H), 7.30 (d, J = 7.1 Hz, 1H), 6.70 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.0 (s), 138.1 (s), 132.1 (q, J = 31.7 Hz), 130.6 (s), 128.4 (s), 128.1 (s), 123.8 (q, J = 272.9 Hz), 123.5 (d, J = 3.4 Hz), 121.8 (d, J = 2.0 Hz), 104 (s); 19F NMR (470 MHz, DMSO-d6) δ −61.5.
Methyl 1-Oxo-1,2-dihydroisoquinoline-6-carboxylate (3k)17

Yellow solid (59.7 mg, 49%); eluent (100% ethyl acetate); 1H NMR (500 MHz, DMSO-d6) δ 11.41 (brs, 1H), 8.28 (m, 2H), 7.96 (dd, J = 8.4, 1.5 Hz, 1H), 7.24 (m, 1H), 6.70 (d, J = 7.1 Hz, 1H), 3.91 (s, 3H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 165.7, 161.2, 137.8, 132.7, 129.9, 128.8, 127.7, 127.3, 125.6, 104.6, 52.4.
7-Methylisoquinolin-1(2H)-one (3l)20
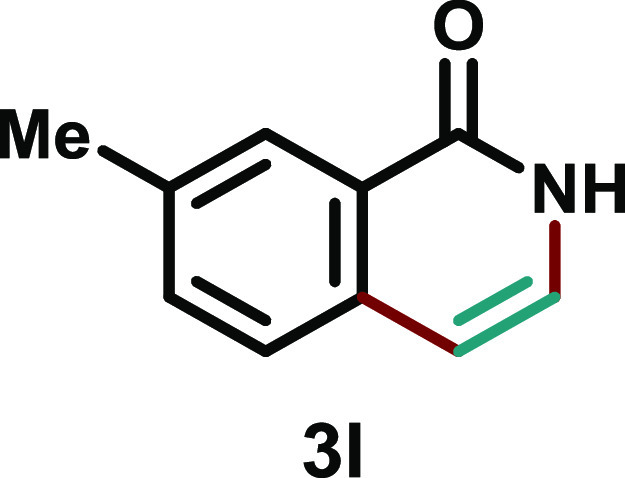
Yellow solid (50.6 mg, 53%); eluent (70–80% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.10 (brs, 1H), 7.99 (s, 1H), 7.51 (dt, J = 8.0, 4.8 Hz, 2H), 7.08 (d, J = 7.1 Hz, 1H), 6.49 (d, J = 7.1 Hz, 1H), 2.42 (s, 3H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.7, 135.7, 135.5, 133.5, 127.9, 126.1, 126.0, 104.4, 20.9 (one carbon is missing in the aromatic region due to the overlap).
7-Methoxyisoquinolin-1(2H)-one (3m)8

Yellow solid (44.1 mg, 42%); eluent (100% ethyl acetate); 1H NMR (500 MHz, DMSO-d6) δ 11.20 (brs, 1H), 7.66–7.51 (m, 2H), 7.31 (dd, J = 8.5, 2.8 Hz, 1H), 7.04 (d, J = 6.8 Hz, 1H), 6.51 (d, J = 7.1 Hz, 1H), 3.85 (s, 3H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.5, 157.9, 131.8, 128.0, 127.3, 126.4, 122.1, 107.1, 1045, 55.3.
7-Nitroisoquinolin-1(2H)-one (3n)

Yellow solid (77.6 mg, 68%); eluent (70–75% ethyl acetate in hexane); m.p. 220–222 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.77 (brs, 1H), 8.87 (s, 1H), 8.43 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.49–7.39 (m, 1H), 6.72 (d, J = 6.7 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.2, 145.1, 142.9, 133.5, 128.1, 126.2, 125.7, 122.7, 104.1; HRMS (ESI)m/z calcd. for C9H7N2O3 [M + H]+: 191.0457, found: 191.0485.
7-Chloroisoquinolin-1(2H)-one (3o)17

White solid (65.7 mg, 61%); eluent (100% ethyl acetate); 1H NMR (500 MHz, DMSO-d6) δ 11.31 (brs, 1H), 8.12 (d, J = 1.7 Hz, 1H), 7.70 (d, J = 2.1 Hz, 2H), 7.19 (d, J = 7.1 Hz, 1H), 6.56 (d, J = 7.1 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 160.7, 136.5, 132.4, 130.8, 129.5, 128.5, 127.2, 125.6, 104.0.
7-Bromoisoquinolin-1(2H)-one (3p)17

Yellow solid (84.7 mg, 63%); eluent (100% ethyl acetate); 1H NMR (500 MHz, DMSO-d6) δ 11.32 (brs, 1H), 8.27 (d, J = 2.1 Hz, 1H), 7.83 (dd, J = 8.6, 2.3 Hz, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.21 (d, J = 7.0 Hz, 1H), 6.55 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 160.5, 136.7, 134.9, 129.5, 128.7, 128.5, 127.5, 118.8, 103.9.
6,7-Dimethylisoquinolin-1(2H)-one (3q) and 5,6-Dimethylisoquinolin-1(2H)-one (3q′)17

White solid (46.8 mg, 45%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.03 (s, 1H), 7.93 (s, 1H), 7.40 (s, 1H), 7.11–7.00 (m, 1H), 6.42 (d, J = 7.1 Hz, 1H), 2.34 (s, 6H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.6, 141.7, 136.0, 135.2, 127.9, 126.6, 126.2, 124.2, 104.1, 19.6, 19.4.
8-Fluoroisoquinolin-1(2H)-one (3r)17

White solid (40.9 mg, 41%); eluent (80–85% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.24 (brs, 1H), 7.65 (td, J = 7.9, 4.9 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.17 (m, 2H), 6.52 (dd, J = 7.2, 2.2 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 161.6 (d, JC–F = 261.1 Hz), 159.3 (d, JC–F = 2.7 Hz), 140.9, 133.4 (d, JC–F = 9.9 Hz), 130.2, 122.3, 114.9 (d, JC–F = 5.4 Hz), 112.8 (d, JC–F = 21.2 Hz), 104.0; 19F NMR (470 MHz, DMSO-d6) δ −111.2.
Benzo[g]isoquinolin-1(2H)-one (3u)20

Yellow solid (64.4 mg, 55%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.02 (brs, 1H), 8.87 (s, 1H), 8.20–8.13 (m, 2H), 8.02 (d, J = 8.4 Hz, 1H), 7.66–7.61 (m, 1H), 7.58–7.52 (m, 1H), 7.11 (dd, J = 7.2, 5.7 Hz, 1H), 6.64 (d, J = 7.2 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 162.2, 134.8, 133.8, 130.9, 129.2, 128.0, 127.8, 127.4, 125.7, 124.9, 123.9, 104.5 (one carbon is missing in the aromatic region due to the overlap).
Thieno[2,3-c]pyridin-7(6H)-one (3v)7

Yellow solid (50.8 mg, 56%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.47 (brs, 1H), 8.02 (d, J = 4.9 Hz, 1H), 7.37 (d, J = 4.9 Hz, 1H), 7.26 (d, J = 6.8 Hz, 1H), 6.71 (d, J = 6.9 Hz, 1H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 158.4, 146.1, 133.6, 130.1, 129.1, 124.9, 102.0.
3-Methylpyridin-2(1H)-one (3w)
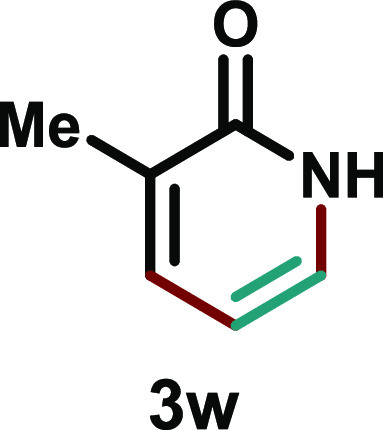
Yellow solid (40.6 mg, 62%); eluent (80–90% ethyl acetate in hexane); m.p. 144–146 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.42 (brs, 1H), 7.29–7.25 (m, 1H), 7.19 (dd, J = 6.7, 1.6 Hz, 1H), 6.06 (t, J = 6.6 Hz, 1H), 1.96 (s, 3H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 162.7, 137.6, 132.3, 128.5, 104.6, 16.3; HRMS (ESI)m/z calcd. for C6H8NO [M + H]+: 110.0606, found: 110.0649.
3-Benzylpyridin-2(1H)-one (3x)21

Brown solid (72.1 mg, 65%); eluent (80–90% ethyl acetate in hexane); 1H NMR (500 MHz, DMSO-d6) δ 11.40 (brs, 1H), 7.25–7.17 (m, 5H), 7.16–7.11 (m, 2H), 6.06 (t, J = 6.6 Hz, 1H), 3.67 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 162.2, 140.2, 138.0, 133.1, 132.1, 128.8, 128.3, 125.9, 104.8, 35.5.
Acknowledgments
The authors thank SERB, New Delhi, India for financial support in the form of Start-up Research Grant (File No: SRG/2020/000288; IITM/SERB/ABP/306). They also thank IIT Mandi for the Seed-Grant funding (IITM/SG/ABP/76). T.R., A.G., and Y.N.A. thank the Ministry of Human Resources Development (MHRD), India, for the research fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02352.
Accession Codes
CCDC 2239457 and 2237759 contain the supporting crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/datarequest/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: +44 1223 336033.
Author Contributions
§ T.R., A.G., and Y.N.A. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Matsui T.; Sugiura T.; Nakai H.; Iguchi S.; Shigeoka S.; Takada H.; Odagaki Y.; Nagao Y.; Ushio Y. Novel 5-HT3 Antagonists. Isoquinolinones and 3-Aryl-2-Pyridones. J. Med. Chem. 1992, 35, 3307–3319. 10.1021/jm00096a001. [DOI] [PubMed] [Google Scholar]; b Rigby J. H.; Maharoof U. S. M.; Mateo M. E. Studies on the Narciclasine Alkaloids: Total Synthesis of (+)- Narciclasine and (+)-Pancratistatin. J. Am. Chem. Soc. 2000, 122, 6624–6628. 10.1021/ja000930i. [DOI] [Google Scholar]; c Hudlicky T.; Rinner U.; Gonzalez D.; Akgun H.; Schilling S.; Siengalewicz P.; Martinot T. A.; Pettit G. R. Total Synthesis and Biological Evaluation of Amaryllidaceae Alkaloids: Narciclasine, Ent-7-Deoxypancratistatin, Regioisomer of 7-Deoxypancratistatin, 10b-Epi-Deoxypancratistatin, and Truncated Derivatives. J. Org. Chem. 2002, 67, 8726–8743. 10.1021/jo020129m. [DOI] [PubMed] [Google Scholar]; d Pettit G. R.; Meng Y.; Herald D. L.; Graham K. A. N.; Pettit R. K.; Doubek D. L. Isolation and Structure of Ruprechstyril from Ruprechtia Tangarana. J. Nat. Prod. 2003, 66, 1065–1069. 10.1021/np0300986. [DOI] [PubMed] [Google Scholar]; e Asano Y.; Kitamura S.; Ohra T.; Itoh F.; Kajino M.; Tamura T.; Kaneko M.; Ikeda S.; Igata H.; Kawamoto T.; Sogabe S.; Matsumoto S.; Tanaka T.; Yamaguchi M.; Kimura H.; Fukumoto S. Discovery, Synthesis and Biological Evaluation of Isoquinolones as Novel and Highly Selective JNK Inhibitors (2). Bioorg. Med. Chem. 2008, 16, 4699–4714. 10.1016/j.bmc.2008.02.028. [DOI] [PubMed] [Google Scholar]; f Sunderland P. T.; Woon E. C. Y.; Dhami A.; Bergin A. B.; Mahon M. F.; Wood P. J.; Jones L. A.; Tully S. R.; Lloyd M. D.; Thompson A. S.; Javaid H.; Martin N. M. B.; Threadgill M. D. 5-Benzamidoisoquinolin-1-Ones and 5-(ω-Carboxyalkyl)Isoquinolin-1-Ones as Isoform-Selective Inhibitors of Poly(ADP-Ribose) Polymerase 2 (PARP-2). J. Med. Chem. 2011, 54, 2049–2059. 10.1021/jm1010918. [DOI] [PubMed] [Google Scholar]
- a Allison J. B. Biological Evaluation of Proteins. Physiol. Rev. 1955, 35, 664–700. 10.1152/physrev.1955.35.3.664. [DOI] [PubMed] [Google Scholar]; b Briet N.; Brookes M. H.; Davenport R. J.; Galvin F. C. A.; Gilbert P. J.; Mack S. R.; Sabin V. Synthesis of Novel Substituted Isoquinolones. Tetrahedron 2002, 58, 5761–5766. 10.1016/S0040-4020(02)00573-2. [DOI] [Google Scholar]; c Konovalenko A. S.; Shablykina O. V.; Shablykin O. V.; Moskvina V. S.; Brovarets V. S. 1H-Isochromene-1-Ones and Isoquinoline-1(2H)-Ones with Carbonyl Group in Position 3: Features of Synthetic Approaches and Transformation. Arkivoc 2023, 2022, 79–112. 10.24820/ark.5550190.p011.861. [DOI] [Google Scholar]
- Hua R. Isoquinolone Syntheses by Annulation Protocols. Catalysts 2021, 11, 620–687. 10.3390/catal11050620. [DOI] [Google Scholar]
- a Song G.; Chen D.; Pan C. L.; Crabtree R. H.; Li X. Rh-Catalyzed Oxidative Coupling between Primary and Secondary Benzamides and Alkynes: Synthesis of Polycyclic Amides. J. Org. Chem. 2010, 75, 7487–7490. 10.1021/jo101596d. [DOI] [PubMed] [Google Scholar]; b Mochida S.; Umeda N.; Hirano K.; Satoh T.; Miura M. Rhodium-Catalyzed Oxidative Coupling/Cyclization of Benzamides with Alkynes via C-H Bond Cleavage. Chem. Lett. 2010, 39, 744–746. 10.1246/cl.2010.744. [DOI] [Google Scholar]; c Hyster T. K.; Rovis T. Rhodium-Catalyzed Oxidative Cycloaddition of Benzamides and Alkynes via C-H/N-H Activation. J. Am. Chem. Soc. 2010, 132, 10565–10569. 10.1021/ja103776u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Patureau F. W.; Glorius F. Oxidizing Directing Groups Enable Efficient and Innovative C–H Activation Reactions. Angew. Chem., Int. Ed. 2011, 50, 1977–1979. [DOI] [PubMed] [Google Scholar]; b Mo J.; Wang L.; Liu Y.; Cui X. Transition-Metal-Catalyzed Direct C–H Functionalization under External-Oxidant-Free Conditions. Synthesis 2015, 47, 439–459. 10.1055/s-0034-1379890. [DOI] [Google Scholar]; c Wang H.; Huang H. Transition-Metal-Catalyzed Redox-Neutral and Redox-Green C–H Bond Functionalization. Chem. Rec. 2016, 1807–1818. 10.1002/tcr.201500274. [DOI] [PubMed] [Google Scholar]; d Wang Z.; Xie P.; Xia Y. Recent Progress in Ru(II)-Catalyzed C–H Activations with Oxidizing Directing Groups. Chin. Chem. Lett. 2018, 29, 47–53. 10.1016/j.cclet.2017.06.018. [DOI] [Google Scholar]; e Aher Y. N.; Mondal B.; Pawar A. B.. Cp*Co(III)-Catalyzed C–H Functionalization Mediated by Oxidizing Directing Groups toward the Synthesis of Heterocycles. In Handbook of CH-Functionalization; John Wiley & Sons, Ltd, 2022; pp 1–28. [Google Scholar]
- a Wu J.; Cui X.; Chen L.; Jiang G.; Wu Y. Palladium-Catalyzed Alkenylation of Quinoline- N -Oxides via C–H Activation under External-Oxidant-Free Conditions. J. Am. Chem. Soc. 2009, 131, 13888–13889. 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]; b Guimond N.; Gouliaras C.; Fagnou K. Rhodium(III)-Catalyzed Isoquinolone Synthesis: The N-O Bond as a Handle for C-N Bond Formation and Catalyst Turnover. J. Am. Chem. Soc. 2010, 132, 6908–6909. 10.1021/ja102571b. [DOI] [PubMed] [Google Scholar]; c Ng K.-H.; Chan A. S. C.; Yu W.-Y. Pd-Catalyzed Intermolecular Ortho -C–H Amidation of Anilides by N-Nosyloxycarbamate. J. Am. Chem. Soc. 2010, 132, 12862–12864. 10.1021/ja106364r. [DOI] [PubMed] [Google Scholar]; d Tan Y.; Hartwig J. F. Palladium-Catalyzed Amination of Aromatic C-H Bonds with Oxime Esters. J. Am. Chem. Soc. 2010, 132, 3676–3677. 10.1021/ja100676r. [DOI] [PubMed] [Google Scholar]; e Rakshit S.; Grohmann C.; Besset T.; Glorius F. Rh(III)-Catalyzed Directed C-H Olefination Using an Oxidizing Directing Group: Mild, Efficient, and Versatile. J. Am. Chem. Soc. 2011, 133, 2350–2353. 10.1021/ja109676d. [DOI] [PubMed] [Google Scholar]
- Webb N. J.; Marsden S. P.; Raw S. A. Rhodium(III)-Catalyzed C–H Activation/Annulation with Vinyl Esters as an Acetylene Equivalent. Org. Lett. 2014, 16, 4718–4721. 10.1021/ol502095z. [DOI] [PubMed] [Google Scholar]
- Dana S.; Sureshbabu P.; Giri C. K.; Baidya M. Ruthenium(II)-Catalyzed C–H Activation/Annulation of Aromatic Hydroxamic Acid Esters with Enamides Leading to Aminal Motifs. Eur. J. Org. Chem. 2021, 2021, 1385–1389. 10.1002/ejoc.202001632. [DOI] [Google Scholar]
- a Gandeepan P.; Müller T.; Zell D.; Cera G.; Warratz S.; Ackermann L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]; b Carvalho R. L.; De Miranda A. S.; Nunes M. P.; Gomes R. S.; Jardim G. A. M.; Da Silva E. N. On the Application of 3d Metals for C-H Activation toward Bioactive Compounds: The Key Step for the Synthesis of Silver Bullets. Beilstein J. Org. Chem. 2021, 17, 1849–1938. 10.3762/bjoc.17.126. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Singh R.; Sathish E.; Gupta A. K.; Goyal S. 3d-Transition Metal Catalyzed C–H to C–N Bond Formation: An Update. Tetrahedron 2021, 100, 13247 10.1016/j.tet.2021.132474. [DOI] [Google Scholar]; d Ilies L. C–H Activation Catalyzed by Earth-Abundant Metals. Bull. Chem. Soc. Jpn. 2021, 94, 404–417. [Google Scholar]; e Mandal R.; Garai B.; Sundararaju B. Weak-Coordination in C–H Bond Functionalizations Catalyzed by 3d Metals. ACS Catal. 2022, 12, 3452–3506. 10.1021/acscatal.1c05267. [DOI] [Google Scholar]
- For recent reviews on Cp*Co(III)-catalysis: see,; a Chirila P. G.; Whiteoak C. J. Recent Advances Using [Cp*Co(CO)I2] Catalysts as a Powerful Tool for C-H Functionalisation. Dalton Trans. 2017, 46, 9721–9739. 10.1039/C7DT01980G. [DOI] [PubMed] [Google Scholar]; b Wang S.; Chen S. Y.; Yu X. Q. C-H Functionalization by High-Valent Cp*Co(III) Catalysis. Chem. Commun. 2017, 53, 3165–3180. 10.1039/C6CC09651D. [DOI] [PubMed] [Google Scholar]; c Prakash S.; Kuppusamy R.; Cheng C.-H. Cobalt-Catalyzed Annulation Reactions via C–H Bond Activation. ChemCatChem 2018, 10, 683–705. 10.1002/cctc.201701559. [DOI] [Google Scholar]; d Ghorai J.; Anbarasan P. Developments in Cp*CoIII-Catalyzed C–H Bond Functionalizations. Asian J. Org. Chem. 2019, 8, 430–455. 10.1002/ajoc.201800452. [DOI] [Google Scholar]; e Loup J.; Dhawa U.; Pesciaioli F.; Wencel-Delord J.; Ackermann L. Enantioselective C–H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem., Int. Ed. 2019, 58, 12803–12818. 10.1002/anie.201904214. [DOI] [PubMed] [Google Scholar]; f Bhaduri N.; Pawar A. B. Redox-Neutral C–H Annulation Strategies for the Synthesis of Heterocycles via High-Valent Cp*Co(III) Catalysis. Org. Biomol. Chem. 2023, 21, 3918–3941. 10.1039/D3OB00133D. [DOI] [PubMed] [Google Scholar]
- a Sivakumar G.; Vijeta A.; Jeganmohan M. Cobalt-Catalyzed Cyclization of N-Methoxy Benzamides with Alkynes Using an Internal Oxidant through C–H/N–O Bond Activation. Chem. - Eur. J. 2016, 22, 5899–5903. 10.1002/chem.201600471. [DOI] [PubMed] [Google Scholar]; b Chavan L. N.; Gollapelli K. K.; Chegondi R.; Pawar A. B. Cp*Co(III)-Catalyzed C–H Functionalization Cascade of N -Methoxyamides with Alkynedione for the Synthesis of Indolizidines. Org. Lett. 2017, 19, 2186–2189. 10.1021/acs.orglett.7b00904. [DOI] [PubMed] [Google Scholar]; c Sen M.; Mandal R.; Das A.; Kalsi D.; Sundararaju B. Cp*CoIII-Catalyzed Bis-Isoquinolone Synthesis by C–H Annulation of Arylamide with 1,3-Diyne. Chem. - Eur. J. 2017, 23, 17454–17457. 10.1002/chem.201704155. [DOI] [PubMed] [Google Scholar]; d Dey A.; Rathi A.; Volla C. M. R. Cobalt(III)-Catalyzed [4+2] Annulation of Heterobicyclic Alkenes by sp2 C–H Activation. Asian J. Org. Chem. 2018, 7, 1362–1367. 10.1002/ajoc.201800251. [DOI] [Google Scholar]; e Lerchen A.; Knecht T.; Koy M.; Daniliuc C. G.; Glorius F. A General Cp*Co III -Catalyzed Intramolecular C–H Activation Approach for the Efficient Total Syntheses of Aromathecin, Protoberberine, and Tylophora Alkaloids. Chem. - Eur. J. 2017, 23, 12149–12152. 10.1002/chem.201702648. [DOI] [PubMed] [Google Scholar]
- Yu X.; Chen K.; Guo S.; Shi P.; Song C.; Zhu J. Direct Access to Cobaltacycles via C–H Activation: N -Chloroamide-Enabled Room-Temperature Synthesis of Heterocycles. Org. Lett. 2017, 19, 5348–5351. 10.1021/acs.orglett.7b02632. [DOI] [PubMed] [Google Scholar]
- Yu X.; Chen K.; Wang Q.; Zhang W.; Zhu J. Co(III)-Catalyzed N-Chloroamide-Directed C–H Activation for 3,4-Dihydroisoquinolone Synthesis. Org. Chem. Front. 2018, 5, 994–997. 10.1039/C7QO01124E. [DOI] [Google Scholar]
- a Muniraj N.; Prabhu K. R. Cobalt(III)-Catalyzed [4+2] Annulation of N-Chlorobenzamides with Maleimides. Org. Lett. 2019, 21, 1068–1072. 10.1021/acs.orglett.8b04117. [DOI] [PubMed] [Google Scholar]; b Ramesh B.; Jeganmohan M. Cobalt(III)-Catalyzed Redox-Neutral [4+2]-Annulation of N-Chlorobenzamides/Acrylamides with Alkylidenecyclopropanes at Room Temperature. Chem. Commun. 2021, 57, 3692–3695. 10.1039/D1CC00654A. [DOI] [PubMed] [Google Scholar]; c Wu Z.; Zheng Q.; Lv G.; Lai R.; Hu Y.; Hai L.; Wu Y. Cobalt(III)-Catalyzed C–H Activation/Annulation Cascade Reaction of N-Chlorobenzamides with 2-Acetylenic Ketones at Room Temperature. Synthesis 2022, 54, 3289–3297. 10.1055/a-1794-1314. [DOI] [Google Scholar]; d Ramesh B.; Jeganmohan M. Cobalt(III)-Catalyzed Regio- and Chemoselective [4 + 2]-Annulation of N-Chlorobenzamides/Acrylamides with 1,3-Dienes at Room Temperature. J. Org. Chem. 2022, 87, 5713–5729. 10.1021/acs.joc.2c00072. [DOI] [PubMed] [Google Scholar]; e Yadav S. K.; Jeganmohan M. Cobalt(III)-Catalyzed Regioselective [4 + 2]-Annulation of N-Chlorobenzamides with Substituted Alkenes. J. Org. Chem. 2022, 87, 13073–13088. 10.1021/acs.joc.2c01588. [DOI] [PubMed] [Google Scholar]
- a Pawar A. B.; Agarwal D.; Lade D. M. Cp*Co(III)-Catalyzed C-H/N-N Functionalization of Arylhydrazones for the Synthesis of Isoquinolines. J. Org. Chem. 2016, 81, 11409–11415. 10.1021/acs.joc.6b02001. [DOI] [PubMed] [Google Scholar]; b Lade D. M.; Pawar A. B. Cp*Co(III)-Catalyzed Vinylic C–H Bond Activation under Mild Conditions: Expedient Pyrrole Synthesis via (3 + 2) Annulation of Enamides and Alkynes. Org. Chem. Front. 2016, 3, 836–840. 10.1039/c6qo00108d. [DOI] [Google Scholar]; c Pawar A. B.; Lade D. M. Cobalt(III)-Catalyzed C–H Halogenation of 6-Arylpurines: Facile Entry into Arylated, Sulfenylated and Alkoxylated 6-Arylpurines. Org. Biomol. Chem. 2016, 14, 3275–3283. 10.1039/c5ob02640g. [DOI] [PubMed] [Google Scholar]; d Aher Y. N.; Pawar A. B. Cp*Co(III)-Catalyzed C-H Amination/Annulation Cascade of Sulfoxonium Ylides with Anthranils for the Synthesis of Indoloindolones. Chem. Commun. 2021, 57, 7164–7167. 10.1039/D1CC02817K. [DOI] [PubMed] [Google Scholar]
- a Aher Y. N.; Lade D. M.; Pawar A. B. Cp*Ir(III)-Catalyzed C-H/N-H Functionalization of Sulfoximines for the Synthesis of 1,2-Benzothiazines at Room Temperature. Chem. Commun. 2018, 54, 6288–6291. 10.1039/C8CC03288B. [DOI] [PubMed] [Google Scholar]; b Lade D. M.; Aher Y. N.; Pawar A. B. Cp*Ir(III)-Catalyzed C–H/O–H Functionalization of Salicylaldehydes for the Synthesis of Chromones at Room Temperature. J. Org. Chem. 2019, 84, 9188–9195. 10.1021/acs.joc.9b01139. [DOI] [PubMed] [Google Scholar]
- Murugan S. J.; Jeganmohan M. Cp*Co(III)-Catalyzed Regioselective [4 + 2]-Annulation of N-Chlorobenzamides with Vinyl Acetate/Vinyl Ketones. J. Org. Chem. 2023, 88, 1578–1589. 10.1021/acs.joc.2c02640. [DOI] [PubMed] [Google Scholar]
- We observed around 5-6% of the coresspoding benzamide (non-separable) along with the isoquinolone derivatives 3g and 3h.
- Ackermann L. Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- Sun R.; Yang X.; Li Q.; Xu K.; Tang J.; Zheng X.; Yuan M.; Fu H.; Li R.; Chen H. Divergent Synthesis of Isoquinolone and Isocoumarin Derivatives by the Annulation of Benzoic Acid with N-Vinyl Amide. Org. Lett. 2019, 21, 9425–9429. 10.1021/acs.orglett.9b03638. [DOI] [PubMed] [Google Scholar]
- Kruse L. I.; Kaiser C.; DeWolf W. E.; Finkelstein J. A.; Frazee J. S.; Hilbert E. L.; Ross S. T.; Flaim K. E.; Sawyer J. L. Some Benzyl-Substituted Imidazoles, Triazoles, Tetrazoles, Pyridinethiones, and Structural Relatives as Multisubstrate Inhibitors of Dopamine.Beta.-Hydroxylase. 4. Structure-Activity Relationships at the Copper Binding Site. J. Med. Chem. 1990, 33, 781–789. 10.1021/jm00164a051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.