

Abstract
Overexpressed Aurora A, amplified centrosomes, and aneuploidy are salient features of estrogen-induced mammary preinvasive lesions and tumors in female August–Copenhagen Irish (ACI) rats. Intimately involved in these events are cyclins and their associated cyclin-dependent kinase (CDK) partners. Cyclin E1·CDK2 overexpression plays an important dual role in late G1/S phase of the cell cycle in cancer cells. It increases DNA replication providing growth advantage to cancer cells and facilitates aberrant centrosome duplication, generating chromosomal instability and aneuploidy leading to tumor development. Presented herein, a 24.0- and 45.0-fold elevation in cyclin E1 and CDK2 was found in 17β-estradiol (E2)-induced ACI rat mammary tumors (MTs), respectively. Cyclin E·CDK2 positive staining was confined to the large round cells found within focal dysplasias, ductal carcinomas in situ, and invasive MTs. Co-immunoprecipitation and in vitro kinase activity of these tumors revealed that these cell cycle entities are functional. When mammary tissue derived from untreated normal, E2-induced hyperplasia and primary tumors were normalized to cyclin E1 levels, low molecular weight (LMW) cyclin E1 forms (33- and 45-kDa) were detected in all of these tissue groups. Moreover, increasing concentrations of protease inhibitor in tissue lysates resulted in a marked reduction of LMW forms, indicating that the presence of cyclin E1 LMW forms can be markedly reduced. Significant increases in cyclin E1 mRNA (2.1-fold) were detected in primary ACI rat E2-induced breast tumors, and quantitative real-time polymerase chain reaction revealed a 20% amplification of the cyclin E1 gene (CCNE1). Collectively, these results support the involvement of cyclin E1·CDK2 in centrosome overduplication during each stage of E2-induced mammary tumorigenesis.
Keywords: Cyclin E1·CDK2, Estrogen, ACI rat, Breast cancer
Introduction
Breast cancer (BC) comprises about 30% of all cancers occurring in women in the USA [1]. In recent years, gene expression profiling has led to the identification of human BC subtypes with distinctive molecular features having both prognostic and clinical implications. Notably, luminal ductal A/B subtypes comprise about 73% of all female BC cases [2, 3]. Characteristic of these BC subtypes is the presence of estrogen receptor α (ERα), while others exhibit slightly lower frequencies of both ERα and progesterone receptor expression [4, 5]. The defining feature of human sporadic luminal A/B subtype BCs, but not limited to these BC subtypes, is chromosomal instability—aneuploidy [6, 7]. Initiating these molecular alterations in BC is the overexpression of Aurora A, a mitotic kinase oncoprotein, involved in precipitating centrosome amplification resulting in the mis-segregation of chromosomes, aneuploidy, and ultimately, tumor development [8–10]. These molecular changes occur in human ductal BC with frequencies between 80% and 94% [11, 12].
In BC, the overexpression of cyclin E1·cyclin-dependent kinase 2 (CDK2) plays an important dual role in late G1/S phase of the cancer cell cycle. The first is DNA replication initiated by c-myc overexpression via estrogen stimulation, providing a growth advantage to BC cells [13, 14]. The second is the promotion of aberrant centrosome duplication by the concomitant overexpression of cyclin E1·CDK2, also a consequence of estrogen action [15–18]. It has been shown that cyclin E1·CDK2 is localized to centrosomes [19]. Some of its target centrosomal proteins include nucleophosmin/B23, CP110, Mps1 kinase, and CG-NAP [20–23]. Importantly, evidence has shown that a single maternal centriole can rapidly generate multiple daughter centrioles during a single S phase and that overexpressed cyclin E1·CDK2 has an essential role in this process [19]. This event would account for the presence of either odd (i.e., [3, 5]) or even (i.e., [4, 6]) number of centrosomes within a single malignant breast cell. Direct evidence that centrosome amplification can initiate oncogenesis is gleaned from Drosophila cell lines, which exhibit extra centrosomes capable of generating metastatic tumors when transplanted into hosts [24].
To begin to assess the role of cyclin E·CDK2 overexpression in BC, we employed a murine/rodent model, the estrogen (E2)-induced mammary tumors (MTs) of the intact female August–Copenhagen Irish (ACI) rat. This model remarkably recapitulates the histopathological and molecular changes commonly seen in luminal ductal human BCs; that is, c-myc overexpression and amplification, Aurora A overexpression, centrosome amplification, chromosomal instability, and aneuploidy occurring in early focal dysplasias, ductal carcinomas in situ (DCISs), as well as in invasive frank carcinomas [25–27]. The detection of these molecular changes early in MT development indicates that they are likely equally important in both initiation and progression of ductal mammary cancer development.
The present report provides evidence that in mammary glands of intact female ACI rats, estrogen treatment drives the overexpression of cyclin E1·CDK2 in cells specifically confined to focal dysplasias, DCISs, and invasive breast tumors. Cyclin E1 mRNA levels are also markedly elevated in E2-elicited ACI rat preinvasive breast lesions and in primary tumors. These data are consistent with the notion that cyclin E1·CDK2 is involved in centrosome overduplication seen only in these early breast lesions and primary breast neoplasms.
Materials and Methods
Animals and Treatment
All studies were conducted in intact 6–8-week female ACI rats purchased from Harlan Sprague Dawley Inc. (Indianapolis, IN, USA). They were housed in an AAALAC-accredited facility with an approved animal protocol. The rats were acclimated for 1 week prior to treatment, randomly distributed into control and treatment groups, and treated with a single pellet containing either 20 mg cholesterol alone or 3 mg of E2 plus 17 mg of cholesterol (Hormone Pellet Press, Leawood, KS, USA) implanted in the shoulder region for 4.0–6.0 months as previously described [19, 25, 26]. The E2 serum levels were as previously reported (123.5 ± 4.8 and 121.8 ± 3.0 pg/ml, at 4.0 and 6.0 months E2 treatment) [26].
Centrosome Amplification: Size and Number
Centrosome's size and number were analyzed by confocal microscopy of paraffin sections immunolabeled with a monoclonal antibody against the centrosomal protein γ-tubulin. Mammary tissues from age-matched untreated control and 6.0-month E2-treated ACI rats were analyzed (n = 5/group). Serial sections of MTs were immunolabeled with γ-tubulin and Hoechst 33342, to label the centrosomes and the nuclei, respectively [28, 29]. Determinations made were based on average values of centrosomes in at least four randomly selected fields of view. A minimum of 100 centrosomes were analyzed. The size of the epithelial cell centrosomes were normalized against centrosomes in nearby fibroblasts. This normalization process infers that the size of the centrosome fibroblasts is fairly consistent among animals of different species. In addition, it allows a fair comparison of centrosome labeling from different tissues immunolabeled at various times, and their image captured in separate sessions. Centrosomes were scored as amplified if they were larger in size and/or number than seen in normal, untreated rat mammary epithelial tissue. Centrosome area (size) was measured in maximum intensity projections of seven consecutive 0.5-µm optical sections. Centrosomes were scored as amplified in size when their measured area was more than two times the average seen in untreated mammary glands. A cut off two times larger was established to exclude centrosomes equivalent in size to late G2 centrosomes of normal tissues. Additionally, centrosomes were scored as amplified in number when a cluster of more than two centrosomes was associated with a single nucleus.
Western Blot Analysis
Mammary gland and MT lysates (50 μg protein) were prepared and analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, followed by immunoblotting as described previously [26]. Membranes were probed with cyclin E1 (M-20) and β-actin (I-19; Biotechnology, Santa Cruz, CA, USA) or CDK2 (2B6) antibodies (Neomarkers, Fremont, CA, USA). For detection, horseradish peroxidase-conjugated secondary antibodies were used, and protein expression was visualized using enhanced chemiluminescence (ECL Amersham, Piscataway, NJ, USA).
Immunoprecipitation and Kinase Assay
Proteins for immunoprecipitation (500 μg) were precleared with appropriate normal IgG and A/G agarose (Santa Cruz Biotechmology, Santa Cruz, CA, USA), incubated with 1 μg of cyclin E1 (M20) or cdk2 (M2) (Santa Cruz), for 2 h with constant mixing, followed by a 2-h incubation with 30 μL A/G agarose slurry. Normal IgG was used as a negative control. Pelleted beads were washed with phosphate buffer solution to remove unbound antibody and protein and immediately used for histone H1-kinase assays (Upstate USA, Inc., Charlottesville, VA, USA) following the manufacturer's protocols. Histone H1 protein was then subjected to SDS gel electrophoresis, dried in a gel dryer, exposed to phosphor plate, and visualized in a Molecular Dynamics Phosphor Imager (Amersham).
Histopathological Analysis
Hematoxylin and eosin staining was performed on paraffin-embedded sections (5–6 μm) as described previously [20]. The sections were examined for hyperplasia and early E2-induced neoplastic changes and identified as: (1) dysplastic foci, seen after 3.0- to 5.0-month E2 treatment, contained cells with large, light staining nuclei and prominent nucleoli, with or without secondary lumens; (2) large dsyplastic foci, after 4.0- to 5.0-month E2 treatment, were similar to the human designation for atypical ductal hyperplasia (ADH) [30] and characterized by large, evenly spaced nuclei arranged in a monotonous pattern which occasionally formed a rosette-like pattern around more evenly shaped secondary lumens; (3) DCIS in addition to the ADH features, had regular punched-out secondary lumens, with a cross sectional diameter of >2 mm. The majority of the DCISs were the cribiform type. Less frequent were comedo, papillary, and solid-type DCISs.
Immunohistochemistry
Five- to 6-μM formalin-fixed paraffin-embedded mammary glands were analyzed by immunohistochemistry as described previously [20]. Anti-cyclin E1 (M20) (Santa Cruz) was used, as well as anti-CDK2 (2B6) antibodies (Neomarkers, Fremont, CA, USA). Negative controls were performed in the absence of primary antibody. Sections were then incubated with a biotinylated anti-rabbit secondary antibody, and signal amplification was accomplished using Vector Laboratories Elite ABC reagent (Burlingame, CA, USA). To determine the frequency of cyclin E1 positive cells, a minimum of 200 cells were counted from age-matched cholesterol-treated mammary glands, E2-induced hyperplastic regions, and preneoplastic lesions.
Reverse Transcriptase PCR (RT-PCR)
All reagents for reverse transcriptase (RT)-polymerase chain reaction (PCR) were from Invitrogen Corp., Carlsbad, CA, USA. First strand synthesis was carried out following the manufacturer's protocol for M-MLV RT with 5 μg of total RNA and 250 ng of random primer. cDNA was then amplified by recombinant Taq DNA polymerase and gene specific primers for cyclin E1 (FWD 5-AGAGCAGCAGGGGTCTACAA-3′; REV 5-CCATGGCCTCCTTAACTTCA-3′; 196 bp amplicon) or β-actin (FWD 5-ATGGTGGGTATGGGTCAGAA-3′; REV 5-TCCATATCGTCCCAGTTGGT-3′; 119 bp amplicon) for 30 cycles (95°C/45 s, 60°C/45 s, and 72°C/45 s). PCR amplicons were separated by 1–2% agarose gel electrophoresis and visualized with ethidium bromide. Densitometry was performed using a Bio-Rad (Hercules, CA, USA) Gel Doc EQ.
DNA Isolation and Quantitative Real-Time PCR (Q-PCR) for Gene Amplification
Cholesterol-treated mammary glands and MTs from ten individual rats were quickly frozen in liquid nitrogen, and DNA extracted by the LiCl method described by Gemmell and Akiyama [31]. Genomic DNA was precipitated with two volumes of ethanol, washed with 70% ethanol, air dried, and resuspended in Tris/EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). DNA integrity was evaluated by agarose gel electrophoresis. Aliquots of 50 ng genomic DNA from control mammary glands and MTs were amplified using Platinum SYBR Green quantitative real-time (Q)-PCR SuperMix UDG (Invitrogen Corporation, Carlsbad, CA, USA) and gene specific primers (same as RT-PCR) for cyclin E1 and β-actin in a 7300 real-time PCR System (Applied Biosystems, Foster City, CA, USA). Myc gene amplification was used as a positive control (FWD 5-CAAGAGGTGCCATGTCTCTACTCA-3′; REV 5-CAGCTGGATAGTCCTTCCTTGTG-3′; 74 bp amplicon). The baseline was set automatically, and the threshold Ct value was measured during the exponential phase of the amplification.
Statistical Analyses
One-way analysis of variance with Dunn or Tukey post hoc tests was used for statistical evaluation. Values are expressed as mean ± standard error. Statistical significance was assumed when p < 0.05 was obtained.
Results
Amplification of Centrosomes
Excessive centrosome duplication is characteristic of E2-induced MTs in female ACI rats as evaluated by confocal microscopy of paraffin-embedded sections immunolabeled with a monoclonal antibody against the centrosomal protein γ-tubulin (Fig. 1). Compared to adjacent normal hyperplasia, in E2-induced primary MT, 88–93% of the cells exhibited centrosome amplification, both in terms of size (volume) and number (p < 0.001). In comparison, only a 7% increase in centrosome number was seen in ductal cells without atypia. Otherwise, normal hyperplastic mammary tissue adjacent to E2-induced MTs exhibited a normal centrosome staining distribution as that observed in untreated normal cycling mammary cells.
Fig. 1.
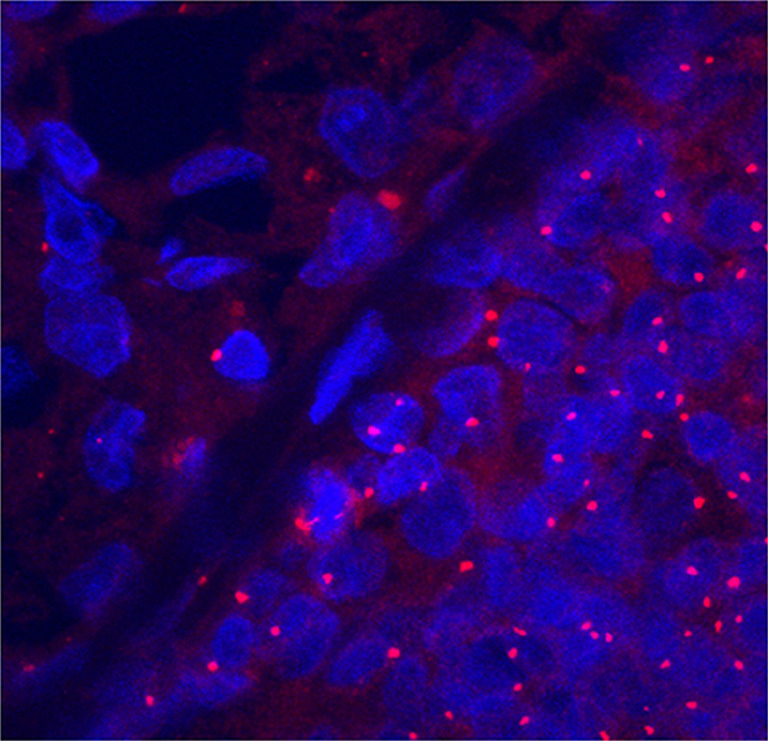
Centrosome amplification in E2-induced mammary tumors (MTs). Centrosomes and nuclei were observed by confocal microscopy of sections labeled with an antibody against γ-tubulin (red) and DNA dye Hoechst 33342 (blue) in areas corresponding to hematoxylin/eosin stained sections. In E2-induced MT cells (lower right), the centrosomes were consistently amplified in both size and number compared to adjacent normal hyperplasia (upper left).
Cyclin E1:CDK2 Expression During E2-Elicited Breast Oncogenesis
Because of the invariability of chromosomal instability and aneuploidy seen in ACI rat preinvasive lesions and MTs, believed to arise as a consequence of centrosome overduplication, it seemed pertinent to evaluate the possible involvement of cyclin E1·CDK2 in mammary oncogenesis. Cyclin E1 protein expression was assessed in mammary glands from age-matched cholesterol-treated controls, 4.0-month E2-treated, and primary E2-induced MTs by Western blot analysis. A significant, 3.3-fold, increase in cyclin E1 protein expression was seen after 4.0 months of E2 treatment, a time period of early DCIS occurrence. In MT samples, cyclin E1 expression was markedly increased 24.0-fold compared to age-matched cholesterol-treated controls. The binding partner of cyclin E1, CDK2, was also significantly elevated, 27.0- and 45.0-fold, in mammary glands after 4.0 months of E2 treatment and in E2-induced MTs, respectively (Fig. 2a, b).
Fig. 2.

Western blot analysis of cyclin E1 and CDK2 (a, b), in vivo complex formation between cyclin E1 and CDK2 (c), and in vitro kinase activity (d). a Representative western blots of individual, age-matched, untreated control mammary glands (C1, C2), 4.0-month E2-treated mammary glands (E4-1 and E4-2), and primary tumors (T1, T2) (n = 6). b Protein expression was measured using a Molecular Dynamics PDSI with ImageQuant software and graphed as fold increase. Cyclin E1 expression increased 3.3- and 24.0-fold, after 4.0-month E2-treatment and in E2-induced tumors, respectively; while CDK2 expression increased 27.0- and 45.0-fold, respectively. β-actin was used as a loading control. c Cyclin E1 and CDK2 immunoprecipitated from E2-induced tumors and subjected to western blot analysis to determine the presence of CDK2 protein. Cyclin E1 co-immunoprecipitated with CDK2, but did not with normal rabbit IgG used as negative control. d Cyclin E1 immunoprecipitates subjected to in vitro kinase assays as described in the text. Kinase activities were higher in mammary tumors (T) than in untreated control mammary glands. Data represent the mean ± standard error. Statistical significance was determined by t test. *p < 0.04 vs control, **p < 0.001 vs control. The dotted line indicates the mean of control untreated age-matched samples.
Cyclin E1 Immunoprecipitation and Kinase Assay
To determine whether the elevated levels of cyclin E1 protein expression were functionally active in MTs, co-immunoprecipitation and kinase assay studies were performed using histone H1 as a substrate. Specific antibodies to cyclin E1 and CDK2 were used to immunoprecipitate their respective proteins in MT samples and subjected to western blot analysis with antibodies against CDK2. Direct association between cyclin E1 and CDK2 was observed (Fig. 2c). However, no binding of cyclin E1 to CDK4 was detected (data not shown). To assess the functionality of the cyclin E1·CDK2 complexes, immunoprecipitated cyclin E1 protein from control age-matched cholesterol-treated and E2-induced MT samples was subjected to an in vitro kinase assay utilizing [32P]-ATP and histone H1 as a substrate. Kinase activity in E2-induced MTs was significantly elevated when compared to age-matched cholesterol-treated controls (Fig. 2d).
Cyclin E1 Low Molecular Weight Forms
Low molecular weight (LMW) forms of cyclin E1 protein, 33- to 45-kDa, have been associated with high tumor grade and genomic instability in human BC [32]. To determine whether LMW cyclin E1 forms are unique to malignant breast cells, Western blot analysis of control, 4.0-month E2-treated, and E2-induced MTs were evaluated for the presence of LMW cyclin E1 forms. When the samples were normalized to total protein, multiple LMW forms were detected in MT samples containing high protein levels (Fig. 3a). However, when samples were normalized to cyclin E1 levels, similar banding patterns were present in control mammary tissue, E2-treated hyperplasia, and MTs with long (30 min) exposure times. To ascertain whether LMW cyclin E1 forms were generated by postnecropsy protein degradation, higher concentrations of a cocktail of protease inhibitors (PIs) were tested during the protein extraction of E2-induced MT samples using one of the following four lysis buffers: 0× without PIs, 1× containing the standard concentration of PIs, and 5.0× or 7.5× containing 5.0 or 7.5 times standard PI concentrations, respectively. When the protein extraction procedure was conducted in the absence of PIs, the presence of the full-length cyclin E1 was markedly lower, and the 45-kDa form intensified, compared to standard PI levels, resulting in a ratio of 0.54 (Fig. 3b). When cyclin E1 protein was isolated with 1× standard lysis buffer, the full-length cyclin E1 was markedly elevated, and the 45 kDa form declined with a ratio of 3.5. Significantly, protein extraction using the 5.0× and 7.5× lysis buffers increased the ratio of full-length vs negligible levels of the 45-kDa cyclin E1 form with ratios of 12.8 and 16.5, respectively. No other LMW forms were visible at shorter exposure times; however, these forms were faintly detected at prolonged exposure times (data not shown).
Fig. 3.

Western blot analysis of cyclin E1 in protein-normalized (a) and cyclin-E1-normalized (b) proteins from control mammary gland (c), 4.0-month E2-treated mammary gland (E4), and primary tumor (T). a Low molecular weight (LMW) cyclin E1 forms were visible only in protein-normalized tumor tissue samples after enhanced chemiluminescence exposure time. When the same samples were normalized to cyclin E1 levels, similar LMW bands (24, 35, 40, and 45 kDa) were present in all treatment groups. b Cyclin E1 protein extracted with the standard lysis buffer, 1×, revealed a doublet of 50 and 45 kDa. In the absence of protease inhibitors, 0×, the same tumor tissue exhibited a diminished full-length cyclin E1 protein and prominent 45-kDa form. In the presence of excess protease inhibitor, 5 and 7.5×, there was a shift to the full-length cyclin E1.
Cyclin E1·CDK2 Expression in Preinvasive Lesions and MTs
Focal dysplasias, most frequently seen after 3.0 to 4.5 months of E2 treatment, consisted of cells with large round nuclei, two to three times larger than those seen in normal adjacent hyperplasia. A small fraction of cells from the resting ducts of cholesterol-treated mammary glands (Fig. 4a) and normal hyperplasia from E2-treated glands (Fig. 4b) exhibited scattered and weak nuclear cyclin E protein expression, 0.7 ± 0.4% and 1.02 ± 0.2%, respectively. In focal dysplasias (Fig. 4c), DCIS, both cribiform/comedo type commonly seen between 4.0 and 4.5 months of E2 treatment (Fig. 4d, e), and MTs (Fig. 4f) all exhibited a dramatic increase in the number of cyclin E1-stained cells residing in these preinvasive as well as invasive primary ACI rat MTs induced by E2. The 16- to 17-fold increase in cyclin E1 stained cells is consistent with its indicated involvement in centrosome amplification seen at these stages [14]. The temporal proximity of these events to centrosome amplification is noteworthy. Serial sections of E2-treated ACI rat mammary glands were also stained for CDK2. Cells in focal dysplasias (Fig. 5a), DCISs, both cribiform/comedo type (Fig. 5b), and invasive ductal MTs (Fig. 5c, d) exhibited marked CDK2 staining. In comparison, adjacent normal hyperplasia (Fig. 5a) exhibited modicum or no detectable CDK2 stained cells. The intensity and temporal overexpression of cyclin E1·CDK2 in preinvasive lesions, both focal dysplasias and DCISs, coincides with the marked elevation in oncoprotein Aurora A mitotic kinase [14].
Fig. 4.

Representative cyclin E immunohistochemistry (a–f) of E2-induced rat mammary premalignant lesions and mammary tumors. Weak positive nuclear staining for cyclin E1 was detected in cells from age-matched, untreated controls (a) and 4.0-month E2-treated normal hyperplasia (b). Focal dysplasia (c), ductal carcinoma in situ (d, e), and invasive ductal carcinoma (f) exhibited strong nuclear staining. Magnification: ×20 and ×40 for inserts.
Fig. 5.

Representative CDK2 immunohistochemistry (a–d) of E2-induced mammary premalignant lesions and mammary tumors (MTs). Intense CDK2 positive stained cells were present in E2-induced (a) focal dysplasia, (b) comedo/cribiform ductal carcinoma in situ, and in MT at low (c, ×20) and high magnification (d, ×40).
Cyclin E1 mRNA Expression
Cyclin E1 mRNA expression was determined by semiquantitative RT-PCR analysis in 4.0-month E2-treated mammary gland samples from female ACI rats and primary MTs (Fig. 6). Compared to age-matched cholesterol-treated controls, a moderate rise in cyclin E1 mRNA was detected after 4.0 months (1.46-fold) of treatment and a significant increase (2.1-fold) in E2-induced MTs.
Fig. 6.

Expression of cyclin E1 mRNA by semiquantitative reverse transcriptase-polymerase chain reaction. Top: a representative membrane showing amplified cyclin E1 (196 bp) and β-actin (119 bp) cDNA transcripts subjected to 1–2% agarose gel electrophoresis in untreated mammary glands (C1-2), E2-treated mammary glands (E4 1-2), and E2-induced mammary tumors (MTs) (T1-2). Bottom: cyclin E1 expression increased 1.46-fold after 4.0-month E2-treatment and 2.08-fold in E2-induced MTs. Densitometric values for cyclin E1 were normalized against β-actin, and fold increases were represented as the mean ± standard error (n = 6). Statistical significance was determined by one-way analysis of variance with a Tukey post hoc test, *p < 0.05 vs control. The dotted line indicates the mean of control untreated age-matched samples.
Q-PCR Analysis of Cyclin E1 Gene
Q-PCR analysis of cyclin E1 genomic DNA was performed on ten cholesterol-treated age-matched controls and an equal number of primary E2-induced MTs to assess gene copy number. The threshold for amplification was set at 1.8-fold based on the variability of Q-PCR in measuring gene copy number [33]. Moreover, 1.8-fold is at least two standard deviations relative gene copy number above the average gene copy number observed in control mammary gland. The amplification of c-myc was used as a positive control. In comparison to cyclin D1 (0.80 ± 0.5) or D3 (0.73 ± 0.05), cyclin E1 was significantly amplified in 20% of the E2-induced MTs examined (Fig. 7) compared to age-matched cholesterol-treated mammary glands.
Fig. 7.

Amplification of cyclins D1, D3, and E1 genes by quantitative real-time polymerase chain reaction. Relative quantification of gene copy number was determined by normalization to the β-actin gene. The assigned threshold for amplification was 1.8-fold over β-actin, control DNA from untreated mammary glands. Cyclin E1 was amplified in 20% of the E2-tumors tested, but D1 and D3 genes were not amplified. Known c-myc amplification was used as a positive control. The dotted line indicates the mean of control untreated age-matched samples.
Discussion
We previously reported that during E2-induced mammary oncogenesis in intact female ACI rats, employing relatively low serum estrogen levels, cyclin D1 expression was substantial in normal hyperplastic mammary cells elicited by estrogen treatment and endogenous cycling levels of progesterone [34]. Additionally, cyclin D1, albeit widely expressed in adjacent hyperplasia, was found more uniformly and further elevated in cells of preinvasive lesions and in E2-induced MTs. While a loss of growth regulation, exemplified by aberrant cyclin D1 overexpression, is a common integral occurrence in BC cell proliferation, a more egregious incipient event is the precipitation of centrosome overduplication and subsequent generation of chromosomal instability and aneuploidy; without affecting DNA replication. This occurs when DNA replication and centrosome duplication cycles, normally highly coordinated, are uncoupled, as seen in papilloma cells derived from p53-null mice [34]. The uncoupling of these two critical cycles leads to centrosome hyperamplification in these cells. In ACI rat E2-treated mammary glands, overexpression of cyclin D3 was specifically confined to the larger cells within focal dysplasias, DCISs (see Journal cover), and primary invasive MT; however, it was not detected in mammary cells of adjacent normal hyperplasia [34]. Therefore, unlike cyclins D1, as well as cyclins A2 and B (Li and Li, unpublished data), only cyclins D3·CDK4 and E1·CDK2, shown herein, are solely overexpressed in mammary cells of preinvasive lesions and primary MTs elicited by E2 in ACI rats, but not detected in adjacent hyperplasia. It has been suggested that cyclin D2 and D3 possess nonredundant functions which are not directly related to DNA synthesis and hence cell proliferation [28, 35]. Moreover, there is evidence that excessive activation at CDK4 forming complexes with D cyclins can promote aberrant centrosome duplication [29]. This is consistent with a number of human cancers which exhibit appreciable cyclin D3 overexpression associated with either centrosome aberrations or aneuploidy [36, 37]. In a study of 83 human BC specimens, largely ductal, moderate to strong levels of cyclin D3 expression were reported in 37% of the samples examined [38]. Comparable increases in cyclin E1 expression in BC have been reported [39, 40]. However, it should be noted that less is known concerning the involvement of CDK4/CDK6, both kinases which form complexes with the D cyclin family members, particularly cyclin D2 and D3, in the centrosome duplication cycle [15].
Cyclin E1 LMW forms have been associated with elevated cyclin E1·CDK2 activity, poor prognosis in women with BC, and considered potent predictors of survival [30]. However, our findings in ACI rat E2-induced MTs provide evidence that the presence of such LMW cyclin E1 forms are independent of oncogenesis and dependent on the relative protein concentration of cyclin E1 in the tissue. Our results are consistent with a previous study indicating that in human BCs, LMW cyclin E1 forms are not unique to malignant cells, but the result of excessively high protein levels [45], and the presence of tumor-specific proteases that promote the degradation of cyclin E1 protein [31]. In the studies presented herein, we have shown that in E2-induced ACI rat MTs, most of the LMW cyclin E1 forms were generated after protein extraction and dependent on the level of the protease inhibitor cocktail. Moreover, the presence of cyclin E1 LMW forms in normal cycling mammary tissue, at higher protein concentrations, indicates that these forms are not the sole propriety of breast tumor tissue.
The overexpression of cyclin E1 protein in E2-induced MTs is due in part to mRNA overexpression and to cyclin E1 gene (CCNE1) amplification. However, only a modest but significant rise in cyclin E1 mRNA and amplification of CCNE1 were detected. Although mRNA levels may not be commensurate with protein expression levels in E2-induced MTs, such discrepancies have also been found in human BC cell lines [41] and may be partially explained by a defect in cyclin E1 degradation [41, 42]. While CCNE1 is amplified in 20% of E2-induced ACI rat MTs, human BC cell lines and primary breast tumors exhibit a similar gene amplification frequency of CCNE1 [41, 43, 44].
Collectively, these results support the contention that the highly overexpressed cyclins E1·CDK2 and D3·CDK4 in E2-treated ACI rat mammary cells, confined to preinvasive breast lesions and frank tumors, act in concert to facilitate centrosome overduplication leading to chromosomal instability and aneuploidy, without affecting DNA replication. Further studies are needed to ascertain the relative importance of these two cyclins and their respective kinases in altering the centrosome cycle, the generation of amplified centrosomes, and eventual chromosomal instability and aneuploidy found in all stages of estrogen-driven oncogenesis in the ACI rat.
Acknowlegements
The authors acknowledge the expert clerical assistance of Ms. Lauren Overton from the University of Kansas Medical Center, Department of Pharmacology.
Disclosures
None.
Abbreviations
- BC
Breast cancer
- CCNE1
Cyclin E1 gene
- CDK
Cyclin-dependent kinase
- DCIS
Ductal carcinoma in situ
- E2
17β-estradiol
- ERα
Estrogen receptor α
- LMW
Low molecular weight
- MT
Mammary tumor
- PR
Progesterone receptor
- SE
Standard error
Footnotes
Grant support
This study was supported by the Susan G. Komen for the Cure Grant number BCTR42906 to J.J. Li.
References
- 1.The American Cancer Society . Cancer facts & figures 2008. New York: The American Cancer Society; 2008. [Google Scholar]
- 2.Yang XR, Sherman ME, Rimm DL, Lissowska J, Brinton LA, Peplonska B, Hewitt SM, Anderson WF, Szeszenia-Dabrowska N, Bardin-Mikolajczak A, et al. Differences in risk factors for breast cancer molecular subtypes in a population-based study. Cancer Epidemiol Biomarkers Prev. 2007;16:439–443. doi: 10.1158/1055-9965.EPI-06-0806. [DOI] [PubMed] [Google Scholar]
- 3.Surveillance, Epidemiology, and End Results (SEER) Oncogene. 2008 Feb 21; 27(9):1253-1262. Program (www.seercancer.gov) SEER*Stat, Database: Incidence- SEER17 Reqs Limited-Use, Nov. 2006 Sub (2000-2004) Linked to County, Attributes- Total US 1969-2004 Counties, National Cancer Institute, DCCPS, Surveillance, Research Program, Cancer Statistics Branch, released April 2007. Based on the November 2006 submission
- 4.Alberts SR, Ingle JN, Roche PR, Cha SS, Wold LE, Farr GH, Jr, Krook JE, Wieand HS. Comparison of estrogen receptor determinations by a biochemical ligand-binding assay and immunohistochemical staining with monoclonal antibody ER1D5 in females with lymph node positive breast carcinoma entered on two prospective clinical trials. Cancer. 1996;78:764–772. doi: 10.1002/(SICI)1097-0142(19960815)78:4<764::AID-CNCR12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.Li CI, Daling JR, Malone KE. Incidence of invasive breast cancer by hormone receptor status from 1992 to 1998. J Clin Oncol. 2003;21:28–34. doi: 10.1200/JCO.2003.03.088. [DOI] [PubMed] [Google Scholar]
- 6.Arnerlov C, Emdin SO, Cajander S, Bengtsson NO, Tavelin B, Roos G. Intratumoral variations in DNA ploidy and s-phase fraction in human breast cancer. Anal Cell Pathol. 2001;23:21–28. doi: 10.1155/2001/430674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiesener B, Hauser-Kronberger CE, Zipperer E, Dietze O, Menzel C, Hacker GW. p34cdc2 in invasive breast cancer: relationship to DNA content, Ki67 index and c-erbB-2 expression. Histopathology. 1998;33:522–530. doi: 10.1046/j.1365-2559.1998.00500.x. [DOI] [PubMed] [Google Scholar]
- 8.Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22:451–464. doi: 10.1023/A:1023789416385. [DOI] [PubMed] [Google Scholar]
- 9.Marumoto T, Zhang D, Saya H. Aurora-A—a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 10.Li JJ, Li SA. Mitotic kinases: the key to duplication, segregation, and cytokinesis errors, chromosomal instability, and oncogenesis. Pharmacol Ther. 2006;111:974–984. doi: 10.1016/j.pharmthera.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Kimura M, Matsunaga K, Fukada D, Mori H, Okano Y. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999;59:2041–2044. [PubMed] [Google Scholar]
- 12.Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Roger I, Solomon DL, Sewing A, Land H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene. 1997;14:2373–2381. doi: 10.1038/sj.onc.1201197. [DOI] [PubMed] [Google Scholar]
- 14.Butt AJ, Caldon CE, McNeil CM, Swarbrick A, Musgrove EA, Sutherland RL. Cell cycle machinery: links with genesis and treatment of breast cancer. Adv Exp Med Biol. 2008;630:189–205. doi: 10.1007/978-0-387-78818-0_12. [DOI] [PubMed] [Google Scholar]
- 15.Fukasawa K. P53, cyclin-dependent kinase and abnormal amplification of centrosomes. Biochim Biophys Acta. 2008;1786:15–23. doi: 10.1016/j.bbcan.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol. 1999;9:429–432. doi: 10.1016/S0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson RL, Maller JL. Cyclin E-dependent localization of MCM5 regulates centrosome duplication. J Cell Sci. 2008;121:3224–3232. doi: 10.1242/jcs.034702. [DOI] [PubMed] [Google Scholar]
- 19.Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene. 2007;26:6280–6288. doi: 10.1038/sj.onc.1210456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/S0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell. 2002;3:339–350. doi: 10.1016/S1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- 22.Fisk HA, Winey M. The mouse Mps1p-like kinase regulates centrosome duplication. Cell. 2001;106:95–104. doi: 10.1016/S0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura T, Takahashi M, Kim HS, Mukai H, Ono Y. Centrosome-targeting region of CG-NAP causes centrosome amplification by recruiting cyclin E-cdk2 complex. Genes Cells. 2005;10:75–86. doi: 10.1111/j.1365-2443.2005.00816.x. [DOI] [PubMed] [Google Scholar]
- 24.Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JJ, Papa D, Davis MF, Weroha SJ, Aldaz CM, El-Bayoumy K, Ballenger J, Tawfik O, Li SA. Ploidy differences between hormone- and chemical carcinogen-induced rat mammary neoplasms: comparison to invasive human ductal breast cancer. Mol Carcinog. 2002;33:56–65. doi: 10.1002/mc.10022. [DOI] [PubMed] [Google Scholar]
- 26.Li JJ, Weroha SJ, Lingle WL, Papa D, Salisbury JL, Li SA. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc Natl Acad Sci USA. 2004;101:18123–18128. doi: 10.1073/pnas.0408273101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li JJ, Li SA. Deciphering the conundrum of estrogen-driven breast cancer: Aurora kinase deregulation. In: Melmed SRH, Chanson P, Christen Y, editors. Hormonal control of cell cycle. Heidelberg: Springer; 2007. pp. 49–62. [Google Scholar]
- 28.Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle. 2005;4:330–338. [PubMed] [Google Scholar]
- 29.Nelsen CJ, Kuriyama R, Hirsch B, Negron VC, Lingle WL, Goggin MM, Stanley MW, Albrecht JH. Short term cyclin D1 overexpression induces centrosome amplification, mitotic spindle abnormalities, and aneuploidy. J Biol Chem. 2005;280:768–776. doi: 10.1074/jbc.M407105200. [DOI] [PubMed] [Google Scholar]
- 30.Page DL, Dupont WD. Anatomic markers of human premalignancy and risk of breast cancer. Cancer. 1990;66:1326–1335. doi: 10.1002/1097-0142(19900915)66:14+<1326::AID-CNCR2820661405>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 31.Spruck C, Sun D, Fiegl H, Marth C, Mueller-Holzner E, Goebel G, Widschwendter M, Reed SI. Detection of low molecular weight derivatives of cyclin E1 is a function of cyclin E1 protein levels in breast cancer. Cancer Res. 2006;66:7355–7360. doi: 10.1158/0008-5472.CAN-05-3240. [DOI] [PubMed] [Google Scholar]
- 32.Akli S, Zheng PJ, Multani AS, Wingate HF, Pathak S, Zhang N, Tucker SL, Chang S, Keyomarsi K. Tumor-specific low molecular weight forms of cyclin E induce genomic instability and resistance to p21, p27, and antiestrogens in breast cancer. Cancer Res. 2004;64:3198–3208. doi: 10.1158/0008-5472.CAN-03-3672. [DOI] [PubMed] [Google Scholar]
- 33.Bodin L, Beaune PH, Loriot MA. Determination of cytochrome P450 2D6 (CYP2D6) gene copy number by real-time quantitative PCR. J Biomed Biotechnol. 2005;3:248–253. doi: 10.1155/JBB.2005.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weroha SJ, Li SA, Tawfik O, Li JJ. Overexpression of cyclins D1 and D3 during estrogen-induced breast oncogenesis in female ACI rats. Carcinogenesis. 2006;27:491–498. doi: 10.1093/carcin/bgi278. [DOI] [PubMed] [Google Scholar]
- 35.Wang X-J, Greenhalgh DA, Jiang A, He D, Zhong L, Brinkley BR, Toop DR. Analysis of centrosome abnormalities and angiogenesis in epidermal-target p53172H mutant and p53-knockout mice after chemical carcinogenesis: evidence for a gain of function. Mol Carcinogenesis. 1998;23:185–192. doi: 10.1002/(SICI)1098-2744(199811)23:3<185::AID-MC7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Neben K, Tews B, Wrobel G, Hahn M, Kokocinski F, Giesecke C, Krause U, Ho AD, Kramer A, Lichter P. Gene expression patterns in acute myeloid leukemia correlate with centrosome aberrations and numerical chromosome changes. Oncogene. 2004;23:2379–2384. doi: 10.1038/sj.onc.1207401. [DOI] [PubMed] [Google Scholar]
- 37.Russell A, Thompson MA, Hendley J, Trute L, Armes J, Germain D. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene. 1999;18:1983–1991. doi: 10.1038/sj.onc.1202511. [DOI] [PubMed] [Google Scholar]
- 38.Bartkova J, Zemanova M, Bartek J. Abundance and subcellular localisation of cyclin D3 in human tumours. Int J Cancer. 1996;65:323–327. doi: 10.1002/(SICI)1097-0215(19960126)65:3<323::AID-IJC8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 39.Keyomarsi K, O'Leary N, Molnar G, Lees E, Fingert HJ, Pardee AB. Cyclin E, a potential prognostic marker for breast cancer. Cancer Res. 1994;54:380–385. [PubMed] [Google Scholar]
- 40.Scott KA, Walker RA. Lack of cyclin E immunoreactivity in non-malignant breast and association with proliferation in breast cancer. Br J Cancer. 1997;76:1288–1292. doi: 10.1038/bjc.1997.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enders GH. Cyclins in breast cancer: too much of a good thing. Breast Cancer Res. 2002;4:145–147. doi: 10.1186/bcr439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Courjal F, Louason G, Speiser P, Katsaros D, Zeillinger R, Theillet C. Cyclin gene amplification and overexpression in breast and ovarian cancers: evidence for the selection of cyclin D1 in breast and cyclin E in ovarian tumors. Int J Cancer. 1996;69:247–253. doi: 10.1002/(SICI)1097-0215(19960822)69:4<247::AID-IJC1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 44.Schraml P, Bucher C, Bissig H, Nocito A, Haas P, Wilber K, Seelig S, Kononen J, Mihatsch MJ, Dirnhofer S, et al. Cyclin E overexpression and amplification in human tumours. J Pathol. 2003;200:375–382. doi: 10.1002/path.1356. [DOI] [PubMed] [Google Scholar]
- 45.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]