

Abstract
Paragangliomas (PGLs) are rare neuroendocrine tumours. About 30–40 % of these tumours are mutated in one of the different susceptibility genes, including those encoding the different subunits of the succinate dehydrogenase, a complex involved both in the tricarboxylic acid cycle and in the oxygen transport chain. The aim of this work was to investigate whether SDHB mutations may account for alterations in cell metabolism and functions. Since human PGL cell lines are not available, we used the neuroblastoma cell line (SK-N-AS) stably transfected with the wild-type human SDHB or different SDHB-mutated constructs carrying some significant mutations found in our patients affected by PGLs. Similarly to succinate dehydrogenase (SDH)-mutated tumour cells, mutated SK-N-AS clones showed reduced SDH enzyme activity. All clones showed normal citrate synthase activity, reduced oxygen consumption and reduced carbonic anhydride production, thus demonstrating a decreased in mitochondrial metabolism. In two of the three mutated SK-N-AS, we also found an increase in HIF1α expression. Surprisingly and unexpectedly, in all the SDHB-mutated clones, we found a significant decrease in glucose uptake and in lactate culture medium concentration, suggesting also a decrease of cytosolic metabolism. Finally, we found that these energetic changes were associated to an increase in cell proliferation and migration. Overall, these data demonstrate that although SDHB mutations significantly downregulate both mitochondrial and cytoplasmic cellular metabolism, these mutations are associated to an upregulation of some cellular functions, such as growth rate and invasiveness.
Keywords: Neuroblastoma Cell Line, Ethylene Glycol Tetraacetic Acid, SDHB Mutation, SDHB Gene, Bismuth Subnitrate
Introduction
Paragangliomas (PGLs) are rare neuroendocrine tumours arising from neural crest-derived cells [1]. In the last two decades, it has been demonstrated that about 30–40 % of these tumours are mutated in one of the different susceptibility genes [reviewed in 2, 3]. These genes include those encoding the four subunits of the succinate dehydrogenase (SDHA, SDHB, SDHC and SDHD) [4–7] and SDHAF2, which is responsible for the flavination of the SDHA subunit [8]. Mitochondrial succinate dehydrogenase (SDH) is an enzyme involved in the tricarboxylic acid (TCA) cycle, where it catalyses the oxidation of succinate to fumarate, and in the mitochondrial electron transport chain. SDHA and SDHB are the two catalytic subunits, while SDHC and SDHD are the structural subunits, which anchor the complex into the inner mitochondrial membrane.
Heterozygous germ line mutations in SDHA have been shown to cause abdominal PGL [4], while germ line mutations in SDHB, SDHC and SDHD, and SDHAF2 genes are responsible for the occurrence of syndromes named PGL4, PGL3, PGL1 and PGL2, respectively. These syndromes are characterized by the occurrence of PGL at different sites in the body (head and neck, thorax, abdomen) [8–16]. In particular, it has been demonstrated that SDHB germ line mutations are very often associated to malignant PGLs.
We have recently demonstrated that in patients affected by PGLs, genetic defects in one of SDH-encoding genes significantly affect the complex expression and function [17]. Although the correlation between the onset of these tumours and genetic mutations has been well-established, very few information are available on the biochemical/functional modifications induced in the cells by SDH mutations. Such dynamic studies cannot be carried out in tumour tissues and need to be performed on living cells. Unfortunately, at present, SDH knockdown animal models or a human neural crest-derived cell line are not available. Therefore, to overcome this problem, we used the neuroblastoma cell line SK-N-AS to investigate whether SDHB mutations account for different functional and metabolic alterations in these cells after transfection.
The characterization and the comprehension of the cellular changes due to SDH mutations, that lead to tumour development, may constitute a first step to a therapeutic pharmacological approach for these aggressive tumours.
Materials and Methods
SDHB Cloning and Site-Direct Mutagenesis
RNA was extracted from tissue using the commercial RNeasy Mini extraction kit (Qiagen) following the manufacturer’s instruction. Full-length SDHB complementary DNA (cDNA) was synthesized from 0.5 μg total RNA samples as follows. RNA, preheated at 80 °C for 5 min, was reverse-transcribed in 10 μl of reaction buffer (50 mM Tris HCl, pH 8.3, 75 mM KCl, 15 mM MgCl2, 10 mM DTT and 500 mM dNTP) containing 10 pmol of specific primers and 100 units of Superscript II in the presence of 15 units of RNase at 42 °C for 50 min.
A 2-μl human cDNA coding of SDHB was first amplified using 0.2 mM of the following primers: 5′-GCTGGACGTCAGGAGCC-3′ (forward) and 5′-GCTCTGAGGTGGTTATAAATC-3′ (reverse), mixed with 2X GC Buffer I, 200 mM dNTP, 5 U TaKaRa LA Taq polymerase (TaKaRa Bio Inc.) in a final volume of 50 μl. After an initial step of 2 min at 95 °C, 40 cycles at 95 °C for 30 s, 58 °C for 30 s and 72 °C for 2 min were performed before a final extension at 72 °C for 7 min. Total amplified product was then analysed on a 1-% agarose ethidium bromide gel using DNA molecular weight (Roche, Indianapolis, USA). Once obtained, the cDNA has been cloned using TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA) following the manufacturer’s instruction.
The mutant constructs were generated by site-directed mutagenesis using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). SDHB wild-type gene cloned in the pcDNA3.1/V5-His-TOPO® TA plasmid was used as template. We generated three missense mutations (Table 1).
Table 1.
Neuroblastoma cell clones
| Clone | Variation | Type of mutation |
|---|---|---|
| pcDNA | Empty vector | No |
| SDHB | Wild type | No |
| SDHB |
c.302 G > A p.Cys101Tyr |
Missense |
| SDHB |
c.572 G > A p.Cys191Tyr |
Missense |
| SDHB |
c.487 T > C p.Ser163Pro |
Missense |
cDNA for wild-type SDHB subunit was cloned, and cDNAs corresponding to mutations were generated by site-directed mutagenesis on the wild-type sequence. Neuroblastoma cells were stably transfected with the different constructs. pcDNA and SDHB wt clones were used as controls
The corresponding modified primers used to generate mutated alleles are the following:
for p.Cys101Tyr
5′-CATCTGTGGCTCTTATGCAATGAACATCAA-3′ (forward)
5′-TTGATGTTCATTGCATAAGAGCCACAGATG-3′ (reverse)
for p.Cys191Tyr
5′-GCATTCTCTGTGCCTACTGTAGCACCAGCT-3′ (forward)
5′-AGCTGGTGCTACAGTAGGCACAGAGAATGC-3′ (reverse)
for p.Ser163Pro
5′-AAGAAGGATGAACCTCAGGAAGGCAAGCAG-3′ (forward)
5′-CTGCTTGCCTTCCTGAGGTTCATCCTTCTT-3′ (reverse).
In bold are the base change. For both wild type and each mutant, the sequence of the SDHB gene was verified by sequencing the entire encoding region.
Cell Transfection and Clone Selection
The neuroblastoma cell line SK-N-AS was grown in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10 % foetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C in 5 % CO2-humidified atmosphere, to about 90 % confluence and transfected with the different constructs using Lipofectamine 2000 (Invitrogen) following manufacturer’s instruction. Cells were split 48 h later, and the standard medium was replaced by medium containing 0.6 mg/ml G418. After 3 weeks of selection, DNA was purified from the different cell clones and verified by sequencing. Positive clones were grown in media with lower amount of G418 for maintenance (0.2 mg/ml).
Cell Homogenates and Lysates Preparation
Unless specified, all reagents were purchased from Sigma (Sigma-Aldrich, Milan, Italy). Homogenates were prepared as already described [18] with minor modifications. Briefly, the cell pellets were resuspended in a solution containing 120 mM KCl, 20 mM HEPES, 2 mM MgCl2, 1 mM ethylene glycol tetraacetic acid (EGTA) and 5 mg/ml BSA and homogenized in a hand-held homogenizer. The homogenate was centrifuged at 800g for 10 min at 4 °C, and the enzyme assays were performed on the supernatant. For Western blot, cell pellets were lysed in lysis buffer (containing 50 mM Tris HCl pH = 7.5, 120 mM NaCl, 1 mM EGTA, 6 mM EDTA, 15 mM Na4P2O7, 20 mM NaF, 1 % Triton X-100, protease inhibitor cocktail) homogenized in a hand-held homogenizer, incubated for 30 min on ice. Lysates were clarified by centrifugation at 10,000g for 15 min at 4 °C. Supernatants were quantified for protein content (Coomassie Blue reagent was from Bio-Rad, Hercules, CA, USA) [19]. All procedures were carried out on ice or at 4 °C.
SDH and Citrate Synthase Activity Measurement
Tissue homogenates (50 μg) were incubated in a phosphate buffer containing sodium azide; 2,6 dichlorophenolindophenol (DCPIP); sodium succinate and phenazine methosulfate. Complex II specific activity was evaluated by measuring the decrease in absorbance due to the oxidation of DCPIP at 600 nm [18]. CS activity was measured by incubating the homogenates (25 μg) in a Tris buffer, containing acetyl coenzyme A, 5,5′dithiobis(2-nitrobenzoic acid) and oxaloacetate, and measuring the increase in absorbance at 412 nm [18]. The enzymatic activities were evaluated by photometry using the VICTOR3 1420 Multilabel Counter (Packard Instruments, Perkin-Elmer).
Western Blot Analysis
Samples of clarified lysates (30 μg of proteins) were separated by SDS/PAGE and transferred onto PVDF (Immobilon, Millipore, MA, USA), as previously described [19]. Bound antibodies were detected using ECL reagents (Immobilon, Millipore, MA, USA) and analysed with a Bio-Rad ChemiDoc Imaging System for dedicated chemiluminescent image acquisition (Imaging and Analysis Software by Bio-Rad, Quantity One). The polyclonal anti-SDHB was from Atlas Antibodies (Atlas Antibodies, Stockholm, Sweden), and the monoclonal anti-subunit IV of complex IV was from Invitrogen (Molecular Probes, Inc., Eugene, OR, USA). The monoclonal anti-hypoxia-inducible factor (HIF)1α was from BD Transduction Laboratories (BD Biosciences, NJ, USA). The polyclonal anti-VDAC1, the polyclonal anti-actin and all the secondary antibodies, such as the anti-rabbit, the anti-mouse and the anti-goat immunoglobulin G conjugated to horseradish peroxidase, were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Transmission Electron Microscopy
The different neuroblastoma cell clones were pelleted by centrifugation, fixed in 4 % glutaraldehyde, post-fixed in 1 % osmium tetroxide and embedded in Epon 812. Ultra-thin sections were stained with uranyl acetate and alkaline bismuth subnitrate and examined under a JEM 1010 electron microscope (Jeol, Tokyo, Japan) at 80 kV. Micrographs were taken at ×12,000 and ×50,000 final magnification using a MegaView III digital camera (SIS-Soft Imaging System, Munster, Germany). The first were used for count for point determination of mitochondrial amount, and the latter ones were used for analysis of mitochondrial and internal cristae volumes. Briefly, to analyse the number of mitochondria, five cytoplasmic fields for single clone were chosen at random and a count by point was performed. Areas of mitochondria and areas of cristae were measured using iTEM image analysis software (SIS).
Glucose Uptake Measurement
Glucose transport was assayed essentially as previously described [20]. Briefly, to determine glucose uptake, cells were seeded in six-well plates and serum-starved in DMEM with low glucose (1 g/l) overnight. 2-deoxy-glucose uptake was evaluated in a buffered solution (140 mM NaCl, 20 mM HEPES/Na, 2.5 mM MgSO4, 1 mM CaCl2 and 5 mM KCl, pH 7.4) containing 0.5 μCi/ml [3H]deoxy-glucose. Glucose uptake was allowed at 37 °C for 10 min. Cells were subsequently washed with cold phosphate-buffered saline (PBS) and lysed with 0.1 M NaOH. Incorporated radioactive was assayed by liquid scintillation counting.
Oxygen Consumption Analysis
Quantitation of oxygen consumption by the SDHB wild-type and the SDHB-mutated neuroblastoma clone cells was conducted by means of the Oxygraph system (Hansatech Instruments, Norfolk, UK). Cells (300,000) were loaded in the chamber containing 300 μl of DMEM with glutamine 2 mM and 10 % foetal calf serum. Oxygen consumption was monitored for 5 min at 37 °C.
Detection of Released CO2 by Radioactive Glucose
The production of 14CO2 was measured from the D-[U-14C]glucose oxidation as previously described [21]. Briefly, cells were plated in six-well dishes, serum deprived for 24 h and then incubated with the medium added with 0.2 μCi/ml of D-[U-14C]glucose. Each dish was covered with a disc of Whatman paper. The paper was wetted with 100 μl of phenylethylamine-methanol (1:1) to trap the produced CO2. After incubation, 200 μl of 4M H2SO4 was added to the wells. Finally, after 1 h at 37 °C, the papers were removed and transferred to scintillation vials for radioactive measurements, by scintillation counting (Tri-Carb 2800TR Liquid Scintillation Analyzer, Perkin-Elmer). Resulted values were normalized on protein concentration.
Lactate Concentration Measurement
Cells were seeded in six-well plates, let them grown to confluence, washed twice with PBS and culturing media replaced with fresh one. After 24 h, media were collected and centrifuged at 16,000×g, 10 min at 4 °C, and the supernatants were transferred in clean tubes. Lactate was measured in the cultured media with Lactate Assay kit (Source Bioscience Life Sciences) according to the manufacturer’s instruction.
Proliferation Assay
To evaluate [3H]thymidine incorporation, cells at 50 % confluence and previously serum-starved for 24 h were challenged with 1 % FCS for 24 h. [3H]thymidine (0.5 μCi/well) was added for the last 2 h of incubation. Cells were washed twice in ice-cold phosphate-buffered saline (PBS) before the addition of 500 μl of 10 % trichloroacetic acid for 30 min at 4 °C. Washed twice with 250 μl of 5 % trichloroacetic acid, the cells were then lysed in 0.25 N NaOH (500 μl/well) for 1 h at 37 °C. Incorporation of [3H]thymidine was measured by scintillation counting (Tri-Carb 2800TR Liquid Scintillation Analyzer, Perkin-Elmer) [20].
Wound-Healing Migration Assay
Confluent cells were serum-starved for 24 h, and the dishes were scored with a sterile 200 μl micropipette tip and photographed. Thereafter, cells were treated with 1 % serum. After 24 h, the wounds were photographed again to visualize incoming cells [22]. Briefly, to analyse the wound healings, we measured the pixels number in which cells were absent, both at T = 0 and T = 24 h, and the difference was expressed as percentage of healing. For each clone, experiments were repeated four times.
Statistical Analysis
Statistical analysis was performed using Student’s t test, and p < 0.05 was considered significant. Data are reported as means ± SD of at least three experiments with similar results.
Results
SDHB Mutations Induce a Decrease in SDH Activity and an Increase in HIF1α Expression Without Impairing Protein Expression and Mitochondria Number and Morphology
To assess if different SDHB mutations might confer different biochemical and/or functional characteristics to neuroblastoma cells, we cloned the wild-type (wt) human SDHB gene into pcDNA3.1/TOPO vector. Subsequently, we generated SDHB mutant constructs by site-directed mutagenesis. The missense mutations have been chosen among those found in our cohort of patients affected by PGLs (Table 1).
The neuroblastoma cell line (SK-N-AS) was screened for all the SDH subunit genes and found wild type. Subsequently, the line was stably transfected with the wild-type human SDHB or the different SDHB mutant constructs. The presence of the SDHB wild-type or mutated sequences in the different SK-N-AS clones was verified by sequencing the entire SDHB encoding region purified from the different cell populations. We then evaluated the SDH activity of the different cell clones and found a significant decrease, ranging from 27 to 47 %, in all the SDHB-mutated clones compared to the cells overexpressing the SDHB wild-type sequence (control; Fig. 1a). The expression of the complex, as evaluated by Western blot analysis, did not correlate with the results of the enzymatic assay. In fact, we did not find any difference in the SDHB expression levels among the clones (Fig. 1b).
Fig. 1.

Succinate dehydrogenase (SDH) and citrate synthase (CS) activities and mitochondrial protein expression in SDHB-mutated neuroblastoma clones. The histogram represents a the SDH and c CS activities expressed as the percentage of the activity measured in cell homogenates. All the mutated clones showed a significantly decreased SDH activity. CS activities were comparable among all the samples. Bars are the means of five independent experiments (each of them conducted in duplicate samples) ± SD, *p < 0.05 compared to controls (pcDNA and wt-transfected clones). b Total protein lysates were assessed for mitochondrial proteins such as SDHB, voltage-dependent anion channel (VDAC) and subunit IV of the cytochrome c oxidase (COX) by Western blot analysis. Blot is representative of five independent experiments, and actin immunoblot has been used as loading control. d Densitometric analysis of Western blot bands of SDHB, performed using imaging and analysis software by Bio-Rad (Quantity One), showed no differences in protein expression in all the different clones. Bars are the means of five independent experiments
In order to evaluate if the expression of mutated SDHB could somehow affect the activity of other mitochondrial enzymes, we also analysed the activity of the citrate synthase (CS) in the different clones. This enzyme is present in the mitochondrial matrix and is involved in the TCA cycle. Differently from SDH, the activity of CS was found comparable among all the samples (Fig. 1c). Also, the expression of other mitochondrial proteins, such as the voltage-dependent anion channel (VDAC), a protein located on the mitochondrial outer membrane, and the cytochrome c oxidase (COX), which is an enzyme of the respiratory chain located in the inner mitochondrial membrane, resulted unchanged in the different neuroblastoma cell clones (Fig. 1b). These results suggest that the expression of different SDHB mutations did not induce, in our model, a generalized impairment of the mitochondria, but altered the SDH activity specifically.
It is known that alterations of SDH enzymatic activity are coupled to increased levels of HIF1α [23]. As expected, in the mutated clones, we found an increase in HIF1α expression (Fig. 2a). At densitometric analysis, the increase was found statistically significant in the two clones expressing the missense mutations, p.Cys101Tyr and p.Cys191Tyr (Fig. 2b).
Fig. 2.

HIF1α expression in SDHB-mutated neuroblastoma clones. a Total protein lysates were assessed for HIF1α by Western blot analysis. Blot is representative of three independent experiments, and actin immunoblot has been used as loading control. b The histogram represent the densitometric analysis of Western blot bands, performed using imaging and analysis software by Bio-Rad (Quantity One). Bars are the means of three independent experiments ± SD, *p < 0.05 compared to controls (wt-transfected clones)
It has been demonstrated that in PGLs, SDH mutations result not only in a destabilization of the complex II, with loss of the enzymatic activity, but also in an abnormal mitochondrial morphology [24]. In order to confirm the data obtained by the Western blot analysis suggesting no alteration in the mitochondria and to check their morphology, we used the electron microscopy approach. As shown in Fig. 3, and confirmed by using the count-for-point method (Table 2), both the mitochondrial shape and amount were found comparable in all the samples. Moreover, the analysis of the area ratios showed that also the internal mitochondrial membranes forming the cristae were similar in all the neuroblastoma clones (Table 2).
Fig. 3.

Transmission electron microscopy of ultra-thin sections of the different neuroblastoma cell clones examined under a JEM 1010 electron microscope. Micrographs were taken at ×12,000 final magnification using a MegaView III digital camera (SIS-Soft Imaging System, Munster, Germany). Images are representative of five cytoplasmic fields for single clone
Table 2.
Analysis of mitochondria
| Clone | % of mitochondria | % of mitochondria areas (ma) | % of internal cristae areas (ica) | ma/ica ratio |
|---|---|---|---|---|
| pcDNA | 17 ± 0.2 | 100 ± 24 | 100 ± 15 | 6 |
| SDHB wt | 18 ± 2 | 101 ± 22 | 101 ± 13 | 5.9 |
| SDHB p.Cys101Tyr | 17 ± 3 | 91 ± 24 | 99 ± 16 | 5.5 |
| SDHB p.Cys191Tyr | 19 ± 4 | 124 ± 22 | 104 ± 7 | 7.2 |
| SDHB p.Ser163Pro | 18 ± 3 | 129 ± 21 | 119 ± 18 | 6.5 |
Briefly, to analyse the number of mitochondria, five cytoplasmic fields for single clone were chosen at random, and a count by point was performed. Areas of mitochondria and areas of cristae were measured using iTEM image analysis software (SIS). Mitochondrial swelling/disruption was estimated by the ratio between the surface area and overall areas of cristae
SDHB Mutations Affect Cell Metabolism in All Neuroblastoma Clones
It is well-known that the TCA cycle sustains the oxidative phosphorylation carried out by the mitochondrial respiratory chain, ending in oxygen reduction and ATP production. Thus, we measured mitochondrial oxygen consumption in each neuroblastoma clone, and we found that in all the SDHB-mutated cells, oxygen consumption was significantly lower than that in the wild-type ones in agreement with the finding of the decreased SDH activity (Fig. 4a, b). In order to understand the energy status of these cells, we also measured the CO2 production. In keeping with the oxygen consumption data, the CO2 levels were slightly but significantly lower in all the SDH-mutated clones (18 to 22 %, Fig. 3c) than in the wild-type ones.
Fig. 4.
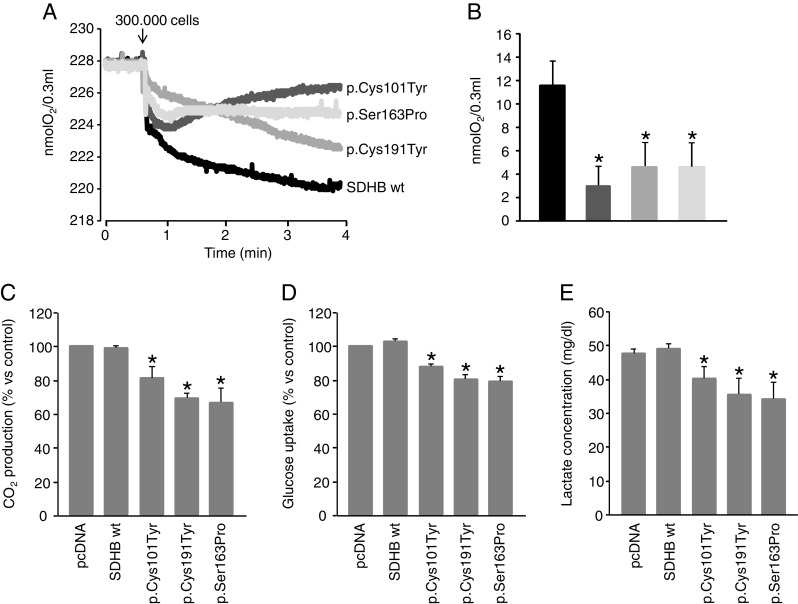
Study of O2 consumption, CO2 production, glucose uptake and lactate secretion in SDHB-mutated neuroblastoma clones. a Representative traces of oxygen consumption measured by means of the Oxygraph system (Hansatech Instruments, Norfolk, UK). Cells were loaded in the chamber containing 300 μl of DMEM + 10 % FCS. Oxygen consumption was monitored for about 4 min at 37 °C. b Oxygen consumption was significantly decreased in all the mutated clones (the black bar is the control, dark grey is p.Cys101Tyr, the grey is p.Cys191Tyr and light grey is p.Ser163Pro). Bars are the means of three independent experiments ± SD, *p < 0.05 compared to control (wt clone). c Basal CO2 levels were measured in cells serum-starved for 24 h and then incubated with the medium containing 0.2 μCi/ml D-[U-14C]glucose. CO2 amounts were slightly, but significantly lower in all the SDH-mutated clones (18 to 22 %, Fig. 3c) than in the wild-type ones. Bars are the means of three independent experiments, performed in duplicate, ± SD. *p < 0.05 compared to control (pcDNA and wt clones). d Glucose uptake was measured in cells serum-starved in DMEM with low glucose (1 g/l) overnight. 2-deoxy-glucose uptake was allowed at 37 °C for 10 min. Incorporated radioactive was assayed by liquid scintillation counting. All the mutated clones showed a slight, but significant decrease in glucose uptake. Bars are the means of three independent experiments (each of them conducted in triplicate samples) ± SD, *p < 0.05 compared to controls (pcDNA and wt). e Culturing media were replaced with fresh ones once cells reached 100 % confluence. After 24 h, media were collected and centrifuged, and the supernatants were transferred in clean tubes. Lactate was immediately measured in the cultured media with Lactate Assay kit (Source Bioscience Life Sciences) according to the manufacturer’s instruction. Again, the media of all the mutated clones showed a slight, but significant decrease in lactate concentration. Bars are the means of three independent experiments ± SD, *p < 0.05 compared to control (pcDNA and wt)
Cancer cells undergo deep changes in their metabolism. In fact, tumour cells undergo aerobic glycolysis, also called Warburg effect, coupled with increased glucose uptake due to incomplete glucose oxidation, thus promoting the production of glycolytic intermediates, needed to increase cancer biomass [25, 26]. To ascertain whether the different SDHB mutations might induce different effect on glycolysis, we measured the glucose uptake after having starved the cells overnight in the absence of serum and at low glucose concentration. Unexpectedly, we observed that glucose uptake was slightly, but significantly decreased in all the mutated clones. These decrements were ranging from 13 to 21 % (Fig. 4d). To address the possibility that the inversion of the Warburg phenotype could be dependent on a preferential lactate versus glucose uptake, we measured the lactate levels in the culturing medium. Indeed, the concentration of lactate was significantly lower in the media of all the mutated clones (16 to 30 %, Fig. 4e), suggesting an increase in the utilization of this substrate.
Proliferation and Migration is Increased in SDHB-Mutated Cells
The effects of the different mutations on cell growth rate and motility were evaluated in all the transfected cell types.
Cell proliferation was assessed by measuring [3H]thymidine incorporation, as an index of DNA synthesis. Thymidine incorporation was measured in the different clones challenged with 1 % of serum. As shown in Fig. 5, two of the three cell clones expressing the missense mutations, p.Cys101Tyr and p.Cys191Tyr, showed a statistically significant increased proliferation in comparison to controls.
Fig. 5.

Effects of SDHB mutations on proliferation of neuroblastoma clones. Labelled thymidine incorporation was measured in 24 h serum-starved neuroblastoma clones, at approximately 50 % confluence, treated with 1 % FCS for 24 h. Data are means of three independent experiments performed in triplicate. *p < 0.05 compared to controls (pcDNA and wt clones)
The wound-healing migration assay was planned to measure cell migration potential. Confluent cells were scraped with a sterile micropipette tip, challenged with 1 % serum and photographed at different times to visualize cell progression [22]. When compared to controls, all the mutated clones showed a significantly increased migration (Fig. 6).
Fig. 6.
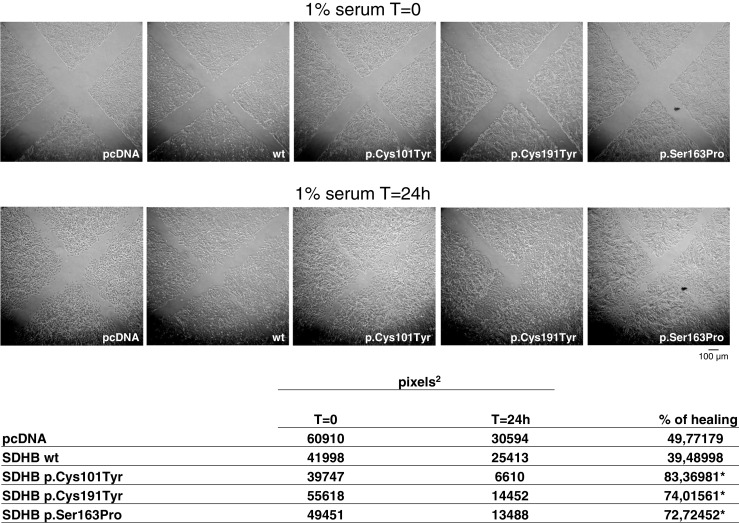
Effects of SDHB mutations on migration of neuroblastoma clones. Wound-healing migration assay: confluent and serum-starved cells were scratched with a micropipette tip and photographed (time T = 0). Cells were challenged with 1 % FCS, and after 24 h, the wounds were photographed again (time T = 24 h) to visualize the incoming cells. The experiment was repeated four times with similar results. Cell-free pixels, representing the wounds, were measured both at T = 0 and T = 24 h. The difference, expressed as percentage, represents the healing. In the figure are shown the representative pixels values, for each clone; experiments were repeated four times. *p < 0.05 compared to controls (pcDNA and wt-transfected clones)
Discussion
In the past few years, the correlation between SDH mutations and the onset of PGLs have been very well-established. Moreover, by studying tumour transcription profile, the intracellular pathways involved in tumour pathogenesis have been highlighted. Indeed, it has been demonstrated that SDHx-mutated tumours show a transcription profile associated with “pseudo-hypoxic” signalling [27]. In fact, succinate accumulation inhibits HIF degradation causing an activation of the hypoxia-induced angiogenesis. Also, von Hippel-Lindau (VHL) protein is involved in HIF degradation thus explaining the pseudo-hypoxic profiles found in VHL-mutated tumours. On the other hand, RET- and NF1-mutated PGLs are associated with abnormal activation of RAS/RAF/MAPK and PI3K/AKT/mTOR intracellular pathways, involved in cell proliferation and differentiation [28, 29]. Although SDH is a key enzyme involved in both the TCA and the oxidative phosphorylation, data on the consequences of SDH mutations on cellular metabolism are not yet available. In this paper, for the first time, we report the effects of different SDHB mutations on cellular metabolism. In a previous work, we demonstrated that SDHx-mutated tumours show a significant decrease in SDH activity and complex expression [17]. In the cell model used in this paper, we found a significant and specific decrease in SDH activity in all the SDHB-mutated clones but we could not reveal any differences in the complex expression. This finding may have different explanations: (a) the SDHB subunit is overexpressed in our clones and, although the half-life of the mutated protein is much shorter than that of the wt one [30], the protein levels might be still high, so that, small differences in protein levels might not be detectable by Western blot analysis; (b) the difference between the mutated and the wt isoforms is limited to a single amino acid, and this difference might not be detected by the antibody we used. In any case, also in view of the increased HIF1α expression we found, we believe that the significant decrease in SDH activity found in the mutated clones suggests that in our cultured cells, there is a dominant effect of the mutations similar to that observed in our patients, thus making our cell model suitable for evaluating the functional consequences of an impaired SDH activity similar to that found in SDH-mutated tumour tissue.
Nowadays, it is well-known that mitochondria are dynamic organelles that can change in number and morphology. Mitochondrial integrity is maintained through a continuous process by which mitochondria divide and fuse forming a dynamic interconnecting network undergoing remarkable structural changes both during apoptosis and under oxidative stress. According to the very limited data of the literature, PGL cells from SDHD mutation carriers are reported to present an increased number of swollen mitochondria [24]. By electron microscopy and Western blot analysis, we could demonstrate that, in our experimental conditions, both the number and the morphology of mitochondria are not impaired. Also in this case, the discrepancy with data obtained from tumour tissues might be due to the presence of the wt SDHB protein that is probably sufficient not to drastically impair mitochondrial homeostasis. In spite of that, the metabolic functions of the SDHB-mutated clones resulted significantly changed. Reducing equivalents, coming from the TCA, are necessary for the respiratory chain to work properly. As expected, oxygen consumption was significantly decreased in all the clones, confirming that a compromised SDH activity influences the reduction of oxygen to water. In line with these results, we also found a decreased CO2 production, suggesting a reduced glucose decarboxylation and an increased HIF1α expression in two clones. These results are in agreement with a recent study in which the authors demonstrated the presence of differences in determinants of energy metabolism in PGLs [31]. In particular, the authors reported that in all SDH-mutated tumours, they found an accumulation of succinate associated with a very low content of ATP/ADP/AMP.
In contrast, the data on glucose uptake and lactate production were somehow surprising. In fact, our hypothesis was that a reduction in SDH activity might be associated to a marked Warburg effect causing a shift from a complete glycolysis to an aerobic glycolysis, with an increased glucose uptake and lactate production. On the contrary, we found a reduced glucose uptake and a reduced lactate concentration in the medium of all the clones. Recently, Fiaschi and colleagues [32] demonstrated that there was a metabolic reprogramming of prostate cancer cells, which gradually became independent from glucose use, while increasing lactate uploading and consumption. In our experimental conditions, the decreased lactate concentration in the medium, coupled to a decreased glucose uptake, suggests a shift in metabolite consumption and energy metabolism in the different mutated clones, similarly to the prostate cancer cells.
In our study, this reprogramming in energy production, induced by SDHB mutations, is associated to differences in cell proliferation and migration, as demonstrated by the increased thymidine incorporation and wound-healing assays. Although not experimentally proved, it is reasonable to hypothesize that these increases in proliferation and migration are consequences of the mutated metabolism and not the other way round, that is an adaptation of the metabolism to an increased cell division rate or migration. It is worth noting that the two mutated clones presenting a significant increase in cell growth are the same, presenting also a significant increase in HIF1α expression, thus suggesting a direct link between cell growth and hypoxia.
To the best of our knowledge this is the first study evaluating the metabolic and functional cell modifications induced by SDHB mutations. We are well aware that our experimental model may not closely resemble the in vivo conditions of SDHB-mutated tumour cells, but it, nevertheless, allowed us to dynamically evaluate the functional consequences of an impaired SDH activity at the cellular level. Such a resulted decrease is associated to a shift in energy metabolism, cell proliferation and migration.
An improved comprehension of the cellular mechanisms leading to tumour development may constitute the first step for a therapeutic pharmacological approach aimed at limiting the biological aggressiveness of the SDHB-mutated PGLs.
Acknowledgments
This work has been supported by Fondazione Cassa di Risparmio di Pistoia e Pescia (Prot. 2010.0278), Ministery grant on Rare diseases (10FF11) and Istituto Toscano Tumori (ITT, Prot. AOOGRT/325462/Q.80.110) to M.M.
Conflicts of Interest
The authors do not have any conflict of interest to disclose.
References
- 1.Mannelli M, Castellano M, Schiavi F, Filetti S, Giacchè M, Mori L, Pignataro V, Bernini G, Giachè V, Bacca A, Biondi B, Corona G, Di Trapani G, Grossrubatscher E, Reimondo G, Arnaldi G, Giacchetti G, Veglio F, Loli P, Colao A, Ambrosio MR, Terzolo M, Letizia C, Ercolino T, Opocher G, Italian Pheochromocytoma/Paraganglioma Network Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94:1541–1547. doi: 10.1210/jc.2008-2419. [DOI] [PubMed] [Google Scholar]
- 2.Welander J, Soderkvust P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18:R253–R276. doi: 10.1530/ERC-11-0170. [DOI] [PubMed] [Google Scholar]
- 3.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- 4.Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 7.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 8.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linnoila RI, Keiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168–1180. doi: 10.1016/0046-8177(90)90155-X. [DOI] [PubMed] [Google Scholar]
- 10.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 11.Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, van Heerden J, Lteif AN, Young WF., Jr Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006;118:1109–1117. doi: 10.1542/peds.2005-2299. [DOI] [PubMed] [Google Scholar]
- 12.Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with SDHB-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- 13.Schiavi F, Boedeker CC, Bausch B, Peçzkowska M, Gomez CF, Strassburg T, Pawlu C, Buchta M, Salzmann M, Hoffmann MM, Berlis A, Brink I, Cybulla M, Muresan M, Walter MA, Forrer F, Välimäki M, Kawecki A, Szutkowski Z, Schipper J, Walz MK, Pigny P, Bauters C, Willet-Brozick JE, Baysal BE, Januszewicz A, Eng C, Opocher G, Neumann HP, European-American Paraganglioma Study Group Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 14.Mannelli M, Ercolino T, Giachè V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44:586–587. doi: 10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C, European-American Paraganglioma Study Group Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 16.Simi L, Sestini R, Ferruzzi P, Gaglianò MS, Gensini F, Mascalchi M, Guerrini L, Pratesi C, Pinzani P, Nesi G, Ercolino T, Genuardi M, Mannelli M. Phenotype variability of neural crest derived tumours in six Italian families segregating the same founder SDHD mutation Q109X. J Med Genet. 2005;42:e52. doi: 10.1136/jmg.2004.030353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rapizzi E, Ercolino T, Canu L, Giaché V, Francalanci M, Pratesi C, Valeri A, Mannelli M. Mitochondrial function and content in pheochromocytoma/paraganglioma of succinate dehydrogenase mutation carriers. Endocr Relat Cancer. 2012;19:261–269. doi: 10.1530/ERC-11-0263. [DOI] [PubMed] [Google Scholar]
- 18.Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assay of the respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 19.Rapizzi E, Taddei ML, Fiaschi T, Bruni P, Chiarugi P. Sphingosine-1phosphate increases muscle glucose uptake through a redox-dependent trans-activation of insulin receptor. Cell Mol Life Sci. 2009;66:3207–3218. doi: 10.1007/s00018-009-0106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rapizzi E, Donati C, Cencetti F, Nincheri P, Bruni P. Sphingosine 1-phosphate differentially regulates serum-induced proliferative response in C2C12 reserve cells and myoblasts. Mol Cell Biochem. 2008;314:193–199. doi: 10.1007/s11010-008-9780-y. [DOI] [PubMed] [Google Scholar]
- 21.Fiaschi T, Buricchi F, Cozzi G, Matthias S, Parri M, Raugei G, Ramponi G, Chiarugi P. Redox-dependent and ligand-independent trans-activation of insulin receptor by globular adiponectin. Hepatology. 2007;46:130–139. doi: 10.1002/hep.21643. [DOI] [PubMed] [Google Scholar]
- 22.Parri M, Buricchi F, Taddei ML, Giannoni E, Raugei G, Ramponi G, Chiarugi P. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J Biol Chem. 2005;280:34008–34018. doi: 10.1074/jbc.M502879200. [DOI] [PubMed] [Google Scholar]
- 23.Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 24.Douwes Dekker PB, Hogendoorn PC, Kuipers-Dijkshoorn N, Prins FA, van Duinen SG, Taschner PE, van der Mey AG, Cornelisse CJ. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J Pathol. 2003;201:480–486. doi: 10.1002/path.1461. [DOI] [PubMed] [Google Scholar]
- 25.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 26.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Favier J, Brière JJ, Burnichon N, Rivière J, Vescovo L, Benit P, Giscos-Douriez I, De Reyniès A, Bertherat J, Badoual C, Tissier F, Amar L, Libé R, Plouin PF, Jeunemaitre X, Rustin P, Gimenez-Roqueplo AP. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4(9):e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Califano D, Rizzo C, D’Alessio A, Colucci-D’Amato GL, Cali G, Bartoli PC, Santelli G, Vecchio G, de Franciscis V. Signaling through Ras is essential for ret oncogene-induced cell differentiation in PC12 cells. J Biol Chem. 2000;275:19297–19305. doi: 10.1074/jbc.M905866199. [DOI] [PubMed] [Google Scholar]
- 29.Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 30.Yang C, Matro JC, Huntoon KM, Ye DY, Huynh TT, Fliedner SM, Breza J, Zhuang Z, Pacak K. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012;26:4506–4516. doi: 10.1096/fj.12-210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao JU, Engelke UF, Rodenburg RJ, Wevers RA, Pacak K, Eisenhofer G, Qin N, Kusters B, Goudswaard AG, Lenders JW, Hermus AR, Mensenkamp AR, Kunst HP, Sweep FC, Timmers HJ. Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clin Cancer Res. 2013;19:3787–3795. doi: 10.1158/1078-0432.CCR-12-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–5140. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]