


Key Words: axonal degeneration, axonal spheroid formation, ionomycin, store-operated calcium entry, myelin, Nile red, peri-axonal swelling
Abstract
The formation of axonal spheroid is a common feature following spinal cord injury. To further understand the source of Ca2+ that mediates axonal spheroid formation, we used our previously characterized ex vivo mouse spinal cord model that allows precise perturbation of extracellular Ca2+. We performed two-photon excitation imaging of spinal cords isolated from Thy1YFP+ transgenic mice and applied the lipophilic dye, Nile red, to record dynamic changes in dorsal column axons and their myelin sheaths respectively. We selectively released Ca2+ from internal stores using the Ca2+ ionophore ionomycin in the presence or absence of external Ca2+. We reported that ionomycin dose-dependently induces pathological changes in myelin and pronounced axonal spheroid formation in the presence of normal 2 mM Ca2+ artificial cerebrospinal fluid. In contrast, removal of external Ca2+ significantly decreased ionomycin-induced myelin and axonal spheroid formation at 2 hours but not at 1 hour after treatment. Using mice that express a neuron-specific Ca2+ indicator in spinal cord axons, we confirmed that ionomycin induced significant increases in intra-axonal Ca2+, but not in the absence of external Ca2+. Periaxonal swelling and the resultant disruption in the axo-myelinic interface often precedes and is negatively correlated with axonal spheroid formation. Pretreatment with YM58483 (500 nM), a well-established blocker of store-operated Ca2+ entry, significantly decreased myelin injury and axonal spheroid formation. Collectively, these data reveal that ionomycin-induced depletion of internal Ca2+ stores and subsequent external Ca2+ entry through store-operated Ca2+ entry contributes to pathological changes in myelin and axonal spheroid formation, providing new targets to protect central myelinated fibers.
Introduction
Neurological recovery after central nervous system trauma is dependent on the location of impact, and the extent of primary and secondary injury (Kwon et al., 2004; Hachem and Fehlings, 2021; Hejrati and Fehlings, 2021). However, a precise mechanistic understanding of secondary white matter degeneration remains incomplete (Dumont et al., 2001; Sekhon and Fehlings, 2001; McDonald and Sadowsky, 2002). Ultrastructural and histological studies examining compressed or contused spinal cords in rodents and humans revealed several pathological changes in white matter. The most pronounced changes are myelin vesiculation, periaxonal swelling, focal axonal swellings referred to as axonal spheroids that form along the lengths of axons, and severed axonal terminals referred to as endbulbs or retraction clubs (Anthes et al., 1995; Tator and Koyanagi, 1997; Beirowski et al., 2010; Ek et al., 2012; Hill, 2017). These changes have obvious dire consequences to those that suffer an SCI as once axons are severed, they fail to regenerate, and thereby become a correlate of irremediable clinical disability.
Time-lapse two-photon excitation imaging of contused spinal cord axons in real-time has shown that the fate of dystrophic axons either remains stable, fragment, or spontaneously recovers (Williams et al., 2014; Horiuchi et al., 2015; Orem et al., 2020b; Rajaee et al., 2020). Although the precise molecular mechanism(s) that influence axonal fate after SCI remain poorly understood, a role for intra-axonal Ca2+ accumulation and clearance has been proposed (Balentine and Spector, 1977; Happel et al., 1981; LoPachin and Lehning, 1997; Ma, 2013; Williams et al., 2014; Orem et al., 2020b). In support, studies expressing genetically encoded Ca2+ indicators in axons subjected to SCI in vivo and ex vivo, have shown that almost all axons that become fragmented experience an initial Ca2+ rise, whereas axons that restore Ca2+ to normal levels spontaneously recover (Stirling et al., 2014a; Williams et al., 2014; Tang et al., 2015; Orem et al., 2017, 2020a, b).
Even though targeting external sources of Ca2+ entry including glutamate receptors, voltage-gated Ca2+ channels, and reversal of the Na+/Ca2+ exchanger are highly protective in models of anoxic/ ischemic white matter injury, they have largely failed to prevent axonal degeneration in models of SCI and following clinical trials (George et al., 1995; Stirling and Stys, 2010; Fern et al., 2014; Williams et al., 2014). Collectively, these results suggest that other routes of Ca2+ entry such as mechanical disruptions in the axolemma (Williams et al., 2014), release from intra-axonal Ca2+ stores (Stirling et al., 2014a; Orem et al., 2017, 2020a), or store-operated Ca2+ entry (SOCE) (Orem et al., 2020b) may contribute to “bystander” degeneration of axons after SCI. SOCE is a central mechanism to refill depleted Ca2+ stores by inducing Ca2+ entry through plasma membrane-localized Orai channels (Putney, 2011, 2018). Although excessive SOCE has been implicated in central nervous system trauma (Zhang et al., 2014; Rao et al., 2015; Orem et al., 2020b; Wang et al., 2021) and disease (Secondo et al., 2018; Kappel et al., 2019; Huang et al., 2020), the role of SOCE in central myelinated fiber injury remains unclear. Alternatively, combinations of the above routes of Ca2+ release/ entry, or other mechanisms may induce axonal degeneration, such as axonal transport interruption/ failure (Povlishock, 1992; Coleman and Perry, 2002; Williams et al., 2014; Rajaee et al., 2020) or autophagy (del Mar et al., 2015; Vahsen et al., 2020). Regardless of the source of intra-axonal Ca2+ overload, axonal damage is likely mediated through Ca2+-induced microtubule instability and Ca2+-dependent calpain activation causing the breakdown of vital axonal cytoskeletal elements (Wang et al., 2012).
To further our understanding of the source of Ca2+ that mediates axonal spheroid formation, we used our previously characterized ex vivo spinal cord model that allows precise perturbation of intra and extracellular sources of Ca2+ and time-lapse two-photon excitation imaging to track dynamic changes in axons and myelin in real-time (Okada et al., 2014; Stirling et al., 2014a; Orem et al., 2017, 2020b; Stivers et al., 2017). The aim of this study is to selectively release Ca2+ from the axoplasmic reticulum using the Ca2+ ionophore ionomycin in the presence or absence of extracellular Ca2+ to evaluate the effect of SOCE on axonal degeneration.
Methods
Animals
All experiments were conducted in accordance with the University of Louisville Institutional Animal Care and Use Committee (protocol# 19513, approved on August 12, 2021) adhering to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th ed, National Research Council, 2011). Adult 6–8-week-old, 18–25 g transgenic male or female hemizygous Thy1YFP+ (IMSR_JAX: 003782) purchased from The Jackson Laboratory (Bar Harbor, ME, USA) were bred in-house as described here (Okada et al., 2014; Stirling et al., 2014a; Orem et al., 2020b; Rajaee et al., 2020). Adult 6–8-week-old, 18–25 g transgenic male or female advillin-Cre+:Ai9fl/fl:Ai95fl/fl mice (ISMR_JAX: 032027, IMSR_JAX:007909, IMSR_Jax: 024105, respectively; The Jackson Laboratory) were bred in house as briefly described here (Orem et al., 2020a, b). The latter express tdTomato and the genetic Ca2+ indicator, GCaMP6f in sensory neurons and their axons specifically due to Cre-mediated recombination by the advillin-Cre driver mice. Genotyping confirmed the presence of the transgenes.
Ex vivo live spinal cord imaging model
The ex vivo spinal cord model used here has been explained in detail previously (Okada et al., 2014; Stirling et al., 2014a; Orem et al., 2017, 2020b; Stivers et al., 2017). Briefly, mice were euthanized with an injection of 200 mg/kg of sodium pentobarbital (50 mg/mL; Diamondback Drugs, Scottsdale, AZ, USA) or an overdose of ketamine (500 mg/kg; Convertus, Dublin, OH, USA) intraperitoneally. Mice were then intracardially perfused with ice cold, oxygenated (95% O2: 5% CO2; Welders Suppy, Louisville, KY, USA) low Ca2+ artificial cerebrospinal fluid (aCSF; in mM: NaCl 126, NaHCO3 26, KCl 3, NaH2PO4 1.25, MgSO4 2, CaCl2 0.1, and dextrose 10; Sigma-Aldrich, St. Louis, MO, USA) to keep the spinal cords viable and the cervical region of spinal cords were removed and placed in a customized imaging chamber (RC-27L Large Bath Chamber with Slice supports; Warner Instruments, Hamden, CT, USA). The perfusate was then slowly changed to oxygenated 2 mM Ca2+ aCSF (in mM: NaCl 126, NaHCO3 26, KCl 3, NaH2PO4 1.25, MgSO4 2, CaCl2 2, and dextrose 10; Sigma-Aldrich). The temperature of the aCSF was held constant at 37°C by an inline heater (Warner Instruments).
To visualize myelin, the lipophilic dye Nile Red (Cat# N1142; ThermoFisher Scientific, Waltham, MA, USA) was first dissolved in dimethylsulfoxide (VWR, Radnor, PA, USA) and used at a final concentration of 25 μM, then directly and briefly added to the perfusion chamber (custom built) in 5 μL volumes until clear labeling of myelin surrounding YFP+ axon profiles was obtained. Importantly, using this experimental perfusion system, the spinal cord remained viable up to 13 hours after isolation based on morphological observation of axons, oligodendroglia, and myelin as previously reported (Okada et al., 2014).
Zero Ca2+ aCSF
For zero Ca2+ aCSF experiments, baseline images were first collected in 2 mM Ca2+ aCSF, and then the perfusion buffer was slowly changed to oxygenated zero Ca2+ aCSF (in mM: NaCl 126, NaHCO3 26, KCl 3, NaH2PO4 1.25, MgSO4 2, MgCl2·6H2O 2, EGTA 1; Sigma-Aldrich) for 1 hour, after which zero Ca2+ aCSF baseline images were taken.
Pharmacological agents
Ionomycin calcium salt was purchased from Tocris Bioscience (Minneapolis, MN, USA) and used per the manufacturer’s recommendations. Ionomycin was dissolved in DSMO first to make a 2 mM stock concentration. Ionomycin was then dissolved in 2 mM Ca2+ aCSF at a final concentration of 1, 3, or 5 µM and perfused for 1 hour. Similarly, ionomycin was dissolved in zero Ca2+ aCSF for a final concentration of 3 µM and perfused for 1 hour. After 1 hour, the ionomycin buffer was washed out with either zero or normal 2 mM Ca2+ aCSF and images were collected every 15 minutes for an additional hour. To inhibit store-operated Ca2+ entry (SOCE), we pretreated spinal cords for 30 minutes with YM-58483 (an established blocker of SOCE; 500 nM; Tocris Bioscience) prior to ionomycin perfusion in aCSF. Thapsigargin, an irreversible Sarco-Endoplasmic Reticulum Ca2+ ATPase inhibitor, was purchased from AdipoGen Life Sciences (San Diego, CA, USA) and used per the manufacturer’s recommendations. Thapsigargin was dissolved in DMSO to make a 5 mM stock solution and further diluted in 2 mM Ca2+ aCSF to make a final concentration of 1 µM to pretreat the spinal cord for 60 minutes to empty calcium stores.
Microscopy
Time-lapse two-photon excitation images of fluorescently labeled spinal cords were obtained using a commercial A1RMP+ multiphoton confocal microscope (Nikon Instruments Inc., Melville, NY, USA) and Element’s software (Nikon Instruments Inc.). Briefly, the mid-cervical level of the spinal cords was aligned under a 25×, 1.1 NA objective and excited with a wavelength of 950 nm (~17 mW measured at the exit of the objective) for simultaneous imaging of Nile Red and YFP, or tdtomato and GCaMP6f (EGFP). The fluorescence emitted was filtered through appropriate dichroic and bandpass filters to isolate the fluorescent emission of the fluorophores as previously described (Okada et al., 2014).
Image analysis
Time-lapse image recordings (123.15 µm wide by 665.08 µm in length) of the cervical region of the gracile fasciculus were collected in z-stacks (2.0 µm/step size) at 1 hour after 2 mM Ca2+ aCSF perfusion, 1 hour after zero Ca2+ aCSF perfusion, then every 15 minutes for each treatment paradigm. The 4D image data was then imported into ImageJ (Version 1.52; NIH, Bethesda, MD, USA, RRID: SCR_003070; Schneider et al., 2012) for visualization and analysis. Axonal spheroids were defined as axonal swellings two times or greater than the width of the axon at baseline. The number of axonal spheroids was counted from four maximum intensity projections divided equally from superficial to deeper depths.
From the same images as above, myelin rings were defined as myelin swellings that were three times or greater than the width of the axon at baseline that either surrounded axonal spheroids or were empty rings. Similarly, periaxonal swelling was defined as myelin that had separated from its axon creating a periaxonal space between the myelin and its axon.
To assess changes in intra-axonal free Ca2+, 4D image data were opened in ImageJ as above, and axons (tdTomato+) were traced using the multipoint tool. The mean fluorescence value of the GCaMP6f signal was obtained for each axon normalized to the average background signal of the channel to obtain a true GCaMP6f signal in the axon. One hundred axons were measured at baseline and then averaged to obtain the average calcium signal in baseline axons. After ionomycin treatment, images were again analyzed, and axons were traced using the same method as described for the baseline. The calcium signal was measured and expressed as a ratio ΔF/F0 where the change in calcium signal from baseline (ΔF) over the initial calcium signal at baseline (F0). All images were coded and analyzed by individuals blind to treatment.
Statistical analysis
Data analysis was performed with Sigma Plot (version 11.0; Systat Software Inc., Palo Alto, CA, USA). Data were expressed as the mean ± SD. A one-way repeated measures analysis of variance and Holm-Sidak all pairwise multiple comparison test was used to determine if there were significant differences in axonal spheroids and myelin rings between treatment groups (n = 3–5 animals/group). A Spearman Rank Order Correlation was used to determine the strength of the relationship between periaxonal swelling and axonal spheroids (n = 8 time points from 4 animals per group). A Kruskal-Wallis one-way analysis of variance on Ranks and Student-Newman-Keuls method of multiple comparisons was used to determine if there were significant differences between intra-axonal Ca2+ levels between groups as the data failed normality (n = 100 axons/group from 4 animals per group. A P value less than 0.05 was considered significant.
Results
Ionomycin dose-dependently induces the axonal spheroid formation
To further our knowledge of the role of extracellular versus intracellular sources of Ca2+ in central myelinated fiber injury, we took advantage of the well-characterized Ca2+ ionophore, ionomycin, that dose-dependently releases Ca2+ from Ca2+ stores within the cell (Yoshida and Plant, 1992; Morgan and Jacob, 1994). To visualize axons and myelin simultaneously, we isolated spinal cords from Thy1YFP+ mice that express YFP in dorsal column axons and labeled myelin with the lipophilic fluorescent dye Nile red as previously described (Okada et al., 2014; Stirling et al., 2014a; Orem et al., 2017). We then imaged the ex vivo spinal cords in real-time using two-photon excitation microscopy.
In control conditions with normal 2 mM Ca2+ aCSF perfusate to mimic in vivo conditions, time-lapse recordings of the dorsal surface of the living spinal cord revealed long parallel aligned YFP+ axons with few axonal spheroids during the observation period (Figure 1A, F, and K). The addition of ionomycin to normal 2 mM Ca2+ aCSF perfusate for 60 minutes followed by 2 mM Ca2+ aCSF for an additional 60 minutes, dose-dependently induced a progressive increase in axonal spheroid formation defined as axonal swellings three times or greater than the parent axon. Spinal cords treated with 1 µM ionomycin aCSF induced a gradual but small number of axonal spheroids from 30 to 120 minutes of observation (Figure 1B–E). In contrast, spinal cords treated with 3 µM ionomycin aCSF perfusate showed a gradual but pronounced increase in axonal spheroids (Figure 1G–J). However, the 5 µM ionomycin aCSF-treated spinal cords (Figure 1L–O) revealed a massive increase in axonal spheroids that began at 30 minutes of ionomycin treatment resulting in almost complete axonal fragmentation by 60 minutes of perfusion.
Figure 1.

Ionomycin dose-dependently induces pronounced axonal spheroid and myelin “ring” formation.
Representative maximum intensity projections of ex vivo living cervical dorsal column axons (A–O; YFP+, grayscale) and their corresponding myelin sheath (a–o; Nile red, grayscale) captured with two-photon excitation microscopy. Axonal spheroids, intact axons, and myelin rings were identified with arrowheads, open arrowheads, and arrows, respectively (A–O, a–o). In baseline conditions (0’) of normal 2 mM Ca2+ artificial cerebrospinal fluid, axons appear normal (A, F, K) and ensheathed in myelin (a, f, k) with few if any signs of pathology. In contrast, the addition of the Ca2+ ionophore ionomycin (iono) to normal Ca2+ artificial cerebrospinal fluid for 60 minutes dose-dependently (1 µM, B–E; 3 µM, G–J; 5 µM, L–O) induces pronounced axonal spheroid formation) over time (′ refers to minutes). By 90 minutes of observation, few intact axons remained in the 3 and 5 µM ionomycin group (I, N), whereas most axons appeared normal with few spheroids in the 1 µM ionomycin group from 30 to 120 minutes of observation (B–E). Similarly, 1 µM ionomycin (iono) induced very little pathology in myelin at 60 (c) and 120 minutes (e) of observation. However, the majority of “myelin rings” surrounded axonal spheroids (compare arrows in e with corresponding arrowheads in E). In distinction, both 3 µM (h, j) and 5 µM ionomycin (m, o) induced pronounced formation of myelin rings that surrounded axonal spheroids at 60 and 120 minutes of observation. Scale bars: 25 µm.
Close observation of the response of individual axons over time revealed that axons treated with 3 µM ionomycin first formed axonal swellings along the length of the axon, followed by a gradual increase in size to form prolate spheroid shaped structures interspersed by thinning strands of the axon. These “football’-like structures then formed rounded axonal spheroids that were completely separated from each other (Figure 1F–J). Interestingly, axonal spheroids formed adjacent to thin axonal segments and rarely lined up with adjacent axonal spheroids from neighboring axons (e.g., arrowheads, Figure 1H) possibly due to space restrictions within the densely packed myelinated fibers. We also noted that large-caliber fibers appeared less sensitive to ionomycin than smaller-caliber fibers as only the large-caliber fibers remained at 120 minutes of observation (Figure 1H–J). Given the gradual but pronounced induction of axonal spheroids, we decided that 3 µM ionomycin aCSF was an appropriate dosage to assess molecular mechanisms of Ca2+-induced central myelinated fiber injury.
Ionomycin dose-dependently causes pronounced changes in myelin
We next assessed ionomycin-induced changes in the myelin sheath using the fluorescent lipophilic dye, Nile red in real time. Due to the high content of lipids in myelin, Nile red preferentially labels myelin in white matter and is therefore a useful tool to visualize myelin in living nervous tissue in health and disease (Okada et al., 2014; Stirling et al., 2014b; Chu et al., 2017; Orem et al., 2017; Grochmal et al., 2018; Teo et al., 2021). In control conditions, myelin appeared unremarkable as “railroad tracks” that ensheathed axons (Figures 1a, f, k, and 2A). In distinction, myelin changed from mostly parallel tracks in 1 µM ionomycin treated spinal cords (Figure 1c and e) to mostly bright concentric rings following perfusion with 3 µM ionomycin (Figure 1h and j) and 5 µM ionomycin aCSF (Figure 1m and o).
Figure 2.
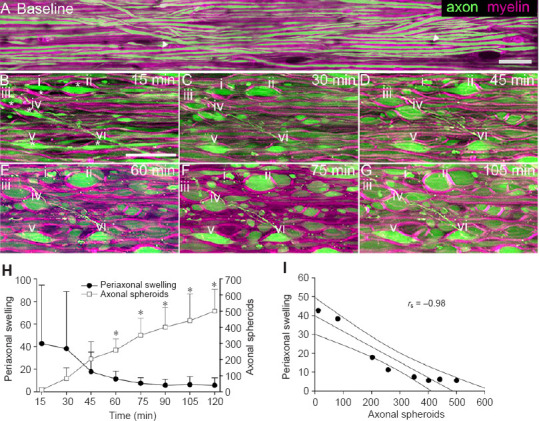
Time-lapse recordings reveal periaxonal swelling of myelin creates a physical space and mold for subsequent axonal spheroid formation.
(A–G) Representative maximum intensity projections of living spinal cord central myelinated fibers from time-lapse two-photon excitation microscopy (scale bars: 25 µm). (A) In baseline conditions of normal Ca2+ artificial cerebrospinal fluid (aCSF), dorsal column axons (green, YFP+) appear normal with few signs of pathology such as axonal spheroids or swellings. Myelin (magenta, Nile red) appears as “rail road” tracks surrounding axons as expected with few signs of pathology such as myelin vesicles, periaxonal swelling, or myelin rings. Small ~1 µm gaps in myelin appear for individual fibers at nodes of Ranvier as expected (arrowheads in A). (B) After 15 minutes of 3 µM ionomycin treatment, some fibers show periaxonal swelling of myelin (*) where the myelin separates from its axon, e.g. i–vi). (C–G) Time lapse recordings of regions of periaxonal swelling (i–vi) from 30 minutes to 45 minutes after ionomycin treatment reveal that axons swell within the “mold” created by separated myelin. These myelin “rings” surround the forming axonal spheroids giving them their characteristic shape over time (E–G). Interestingly, axonal spheroids rarely align between adjacent neighboring fibers but appear diagonally across from one another between adjacent fibers. (H) Quantification of regions of periaxonal swelling and axonal spheroid formation over time following 3 µM ionomycin treatment reveals that periaxonal swelling is an early event followed by axonal spheroids. Axonal spheroids and periaxonal swelling are expressed as mean ± SD with periaxonal swelling being normalized to baseline; repeated measures analysis of variance; *P < 0.05; n = 4. (I) Scatterplot of periaxonal swelling versus axonal spheroids. The best fit line and 95% confidence intervals are shown. The two variables are nearly perfectly negatively correlated. Spearman Rank Order Correlation; Correlation coefficient (rs) –0.976; P = 0.00000020; n = 8 time points from 4 ionomycin-treated spinal cords.
Of note, acute changes in myelin following 3 µM ionomycin treatment often began as periaxonal swellings, where the myelin separates away from the axon forming an enlarged periaxonal space (Figure 2B). Periaxonal swelling is rarely seen in normal aCSF control conditions (Figure 1A) and up to 13 hours of observation as previously reported (Okada et al., 2014). Within the regions of enlarged periaxonal space, some axons began to swell, and formed spheroids within the surrounding “mold” of myelin, giving the former their characteristic shape (Figure 2C–G). Quantification of these results reveals that periaxonal swelling is an early event after 3 µM ionomycin treatment followed by the gradual formation of axonal spheroids within the now enlarged periaxonal space (Figure 2H). Axonal spheroids significantly (repeated measures analysis of variance, P < 0.001) increased from 45 to 120 minutes after ionomycin treatment whereas periaxonal swelling decreased during this time interval. In support, we found a strong inverse correlation between periaxonal swelling and axonal spheroid formation (Spearman Rank Order Correlation; correlation coefficient –0.976; P = 0.00000020; Figure 2I). Collectively, our data show that ionomycin-induced Ca2+ release results in pathological changes to myelin prior to axons.
Both intra-axonal and extracellular sources of Ca2+ contribute to ionomycin-induced axonal spheroid formation
We next sought the route of Ca2+ that mediates ionomycin-induced axonal spheroid formation. Ionomycin can source Ca2+ from internal Ca2+ stores within the axon (i.e., axoplasmic reticulum) as supported by the known role of ionomycin in ER-mediated Ca2+ release and activation of T-cells (Chatila et al., 1989; Donnadieu et al., 1995). Ionomycin has also been shown to facilitate Ca2+ entry from extracellular sources and not by direct action across the plasmalemma, suggestive of channel-mediated entry (Morgan and Jacob, 1994). To determine the role of intra-axonal Ca2+ store release of Ca2+ versus external sources of Ca2+ we conducted 3 µM ionomycin aCSF experiments in the presence (2 mM Ca2+ aCSF) or absence of external Ca2+ (Ca2+ substituted with Mg2+ and the Ca2+ chelator EGTA). Following the acquisition of baseline images at 60 minutes after perfusion with aCSF only, 3 µM ionomycin was then perfused for 60 minutes in ionomycin-treated groups followed by perfusion of the relevant aCSF for an additional 60 minutes (e.g., 2 mM Ca2+ or zero Ca2+ aCSF). In control conditions of normal (2 mM) Ca2+ aCSF (Figure 3A–C, G) and zero Ca2+ aCSF only treated spinal cords (Figure 3D–F), few if any axonal spheroids were present over the period of observation. In zero Ca2+ aCSF-only preparations, a few axons experienced a small “sickle-shaped” disturbance over the 2 hours of observation but the axons remained intact suggestive that some external Ca2+ may be important to maintain the normal linear appearance of axons (Figure 3E and F). There was no significant difference in axonal spheroids between all treatment groups at baseline, 15, and 30 minutes following ionomycin application (repeated measures analysis of variance, P > 0.05, Figure 3V). At 45 minutes of ionomycin perfusion, there was a significant increase in axonal spheroids between the normal Ca2+ aCSF plus 3 µM ionomycin treated group versus normal Ca2+ aCSF only, and zero Ca2+ aCSF control groups (Figure 3V). However, there was not a significant difference (P = 0.42) between the normal Ca2+ aCSF plus 3 µM ionomycin group versus the zero Ca2+ aCSF plus 3 µM ionomycin group. This suggests that intra-axonal sources of Ca2+ may be important for initial spheroid formation (Figure 3V).
Figure 3.

Extracellular Ca2+ entry through SOCE contributes to ionomycin-induced axonal spheroid and myelin ring formation.
Representative maximum intensity projections captured using two-photon excitation microscopy of living spinal cord axons ex vivo (YFP+, grayscale, A–R). In baseline conditions of normal 2 mM Ca2+ artificial cerebrospinal fluid (aCSF; A, G, and M) or zero Ca2+ aCSF (D and J), axons appear normal with few if any axonal spheroids present. In the absence of ionomycin (iono), perfusion of spinal cords with normal Ca2+ (A–C) or zero Ca2+ aCSF (D–F) does not induce a significant change in axonal spheroids over time (from 0 to 120 minutes, V). The addition of ionomycin (3 µM) for 60 minutes following baseline recordings profoundly increases axonal spheroid formation in normal Ca2+ aCSF treated spinal cords evident at 45 minutes of ionomycin treatment and up to 120 minutes of observation (H, I, P, and V). In contrast, the absence of external Ca2+ significantly reduces ionomycin-induced axonal spheroid formation beginning at 75 minutes of observation (K, L, Q, and V). Importantly, 30 minutes of pretreatment with the store-operated calcium entry inhibitor YM58483, significantly reduced ionomycin-induced axonal spheroid formation from 60 minutes to 120 minutes of observation (N, O, R, and V). Myelin staining with Nile red (grayscale, S, T, and U) reveals that ionomycin in normal Ca2+ aCSF (S) induces myelin ring formation (e.g., arrows) that often contain axonal spheroids. Both zero Ca2+ aCSF (T) and YM58483 (U) significantly reduce myelin ring formation (W) at 120 minutes versus 2 mM Ca2+ aCSF plus ionomycin treated spinal cords, however, pretreatment of YM58483 significantly reduced myelin ring formation from 60 to 120 minutes of observation. Compare normal myelin (arrowheads in U) with myelin rings (arrows, U). Scale bars: 25 µm. Axonal spheroids and myelin rings are expressed as mean ± SD per timepoint and significance is established using a repeated measures analysis of variance with the Holm-Sidak method of multiple comparisons (V and W). (V) Ca2+ aCSF only vs. Ca2+ aCSF + ionomycin (iono): 75–120 minutes *P < 0.001; 45, 60 minutes *P < 0.01. 0 (zero) Ca aCSF only vs. Ca2+ + iono: 75–120 minutes #P < 0.001; 45, 60 minutes #P < 0.01. Ca2+ aCSF + iono vs. 0 Ca2+ aCSF + iono: 75–120 minutes ψP < 0.001. Ca2+ aCSF + iono vs. Ca2+ aCSF + iono + YM (YM58483): 60 minutes φP < 0.05; 75–90 minutes φP < 0.01; 105–120 minutes φP < 0.001. (W) Ca2+ aCSF + iono vs. 0 Ca2+ aCSF + iono at 120 minutes **P < 0.01; and Ca2+ aCSF + iono + YM at 60 minutes **P < 0.01; 120 minutes ***P < 0.001, n = 3–5 animals/group.
In distinction, at 75 minutes of perfusion in the presence or absence of external Ca2+, there was a significant decrease (P < 0.001) in axonal spheroids in zero Ca2+ aCSF plus 3 µM ionomycin group (Figure 3J–L, V) compared with normal Ca2+ aCSF plus 3 µM ionomycin (Figure 3G–I, V). A significant difference between these two treatment groups remained up to 120 minutes of observation (Figure 3P, Q, and V). Together these results clearly support a role for externally sourced Ca2+ in ionomycin-induced axonal spheroid formation.
YM 58483, a potent inhibitor of store-operated-calcium entry, reduces ionomycin-induced axonal spheroid formation
Given that some axonal spheroids formed acutely when perfused with zero Ca2+ aCSF plus ionomycin perfusate, and the known role of ionomycin-induced Ca2+ release from internal stores (Imboden and Weiss, 1987), we reasoned that an intra-axonal source of Ca2+ may play an initial role followed by a pronounced role for extracellular Ca2+. The main Ca2+ store in axons is the axoplasmic reticulum, and both RyR and IP3R are known to release Ca2+ from the ER/axoplasmic reticulum and contribute to secondary degeneration following SCI (Stirling et al., 2014a; Orem et al., 2017, 2020a). Once Ca2+ is released from the ER/axoplasmic reticulum, store-operated Ca2+ entry (SOCE) is the main pathway to restore Ca2+ stores with extracellular Ca2+ (Smyth et al., 2010; Orem et al., 2020b). We, therefore, hypothesized that SOCE mediates ionomycin-induced axonal spheroid formation.
To test this hypothesis, we pretreated spinal cords for 30 minutes with YM58483 (500 nM), a potent and well-known inhibitor of SOCE, in normal Ca2+ aCSF and then induced axonal spheroid formation using 3 µM ionomycin. As shown in Figure 3, pretreatment with the SOCE inhibitor reduced axonal spheroid formation versus normal Ca2+ and zero Ca2+ aCSF plus ionomycin treatment groups (Figure 3M–O, R). Quantification of these results showed that inhibiting SOCE with YM58483 significantly (P < 0.001) reduced axonal spheroid formation (YM58483) compared with vehicle control treated spinal cord preparations (normal aCSF plus 3 µM ionomycin) from 60 to 120 minutes of observation (Figure 3V). These results support a role for SOCE in axonal spheroid formation induced by ionomycin.
SOCE inhibition reduces myelin ring formation
To determine if SOCE inhibition protected myelin after ionomycin application, we quantified the number of myelin rings with a diameter 3 times that of normal-appearing myelin. As shown in Figure 3, in control conditions at baseline there was no significant difference (P > 0.05) in myelin rings between any of the treatment groups (Figure 3W). In contrast, 3 µM ionomycin in the presence of normal Ca2+ aCSF induced a pronounced increase in myelin rings that was lessened in zero Ca2+ aCSF plus ionomycin (Figure 3S and T) and greatly reduced following pretreatment with YM58483 in normal Ca2+ aCSF plus ionomycin (Figure 3S and U). Quantification of these results showed that pretreatment with YM58483 significantly (P < 0.001) reduced myelin ring formation (YM58483) compared with vehicle control treated spinal cord preparations (normal aCSF plus 3 µM ionomycin) that continued to 120 minutes of observation (Figure 3W). In distinction, perfusion of spinal cords in zero Ca2+ aCSF in the presence of ionomycin significantly (P = 0.007) reduced myelin ring formation compared to normal Ca2+ plus ionomycin at 120 minutes of observation. These results suggest a role for external Ca2+ entry through SOCE in myelin ring formation.
Calcium store depletion prevents ionomycin-induced axonal spheroid and myelin ring formation.
To further confirm that ionomycin-induced axonal spheroid formation mechanistically involves Ca2+ release from internal Ca2+ stores and subsequent SOCE, we hypothesized that depleting Ca2+ stores prior to ionomycin treatment would be highly protective as no stored Ca2+ would be available to be released in axons. To test this hypothesis, we pretreated spinal cords with thapsigargin, a specific and potent inhibitor of sarco/ER Ca2+ ATPase (SERCA) pumps and well-characterized Ca2+ store depleting agent (Lytton et al., 1991), in the presence or absence of ionomycin. As shown in Figure 4, there was no significant difference (P > 0.05) in axonal spheroids between the normal Ca2+ aCSF plus thapsigargin treated group versus normal Ca2+ aCSF alone group during 2 hours of observation (Figure 4A–D, K, see also Figure 3V). Importantly, pretreatment of thapsigargin in normal Ca2+ aCSF completely prevented ionomycin-induced axonal spheroid formation (see Figure 4I and compare Figure 4F–H, K with Figure 3V). Similar to the results for axonal spheroids, Ca2+ store depletion prevented the ionomycin-induced myelin ring formation, and the mean number of myelin rings was not significantly different from normal Ca2+ aCSF controls (compare Figure 4E, J, L with Figure 3W). Together, these results confirm that Ca2+ release from internal stores is the likely mechanism that initiates ionomycin-induced axonal spheroid and myelin ring formation.
Figure 4.

Ca2+ store depletion prevents ionomycin-induced axonal spheroid and myelin ring formation.
Representative maximum intensity projections were captured using two-photon excitation microscopy of living spinal cord tissue ex vivo. Axons (YFP+, A–D and F–I) in green and myelin stained with Nile Red (D, E, I, J) in magenta. In baseline conditions, 2 mM Ca2+ artificial cerebrospinal fluid (aCSF), (A, F) axons appear intact with few swellings or spheroids. To deplete Ca2+ stores, thapsigargin (Tg, 1 µM) was added to normal Ca2+ aCSF. Importantly, Ca2+ store depletion did not induce axonal spheroids for up to 120 min of observation (B–E, K). Pretreatment of thapsigargin followed by ionomycin (iono; 3 µM) also did not induce significant changes in axonal spheroids (G–I, K, compare with Figure 3G–I, V). A composite of axons and myelin channels (D, I) show normal ensheathment of the axons and some periaxonal swelling (separation of myelin from axons, arrows in D, I). Ionomycin-induced myelin ring formation was also attenuated (E, J, L, compare with Figure 3S and W). Scale bars: 25 µm. (K, L) Axonal spheroids and myelin rings are expressed as mean ± SD per time point and statistical significance was determined using repeated measures analysis of variance, n = 3–4 per group. No significant difference (P > 0.05) was found between normal Ca2+ aCSF only (same data from Figure 3 for comparison only), normal Ca2+ aCSF plus Tg, and normal Ca2+ aCSF plus Tg plus 3 µM ionomycin.
Ionomycin treatment increases intra-axonal free Ca2+ derived from both internal and external sources.
To confirm that ionomycin treatment increases Ca2+ levels directly in axons, we generated advilin-Cre+/–:Ai9fl/fl:Ai95fl/fl transgenic mice that express the fluorescent protein tdTomato (Ai9) and the genetic-encoded Ca2+ indicator, GCaMP6f (Ai95), in DRG neurons (advil-Cre driver) that project their axons in the superficial dorsal columns of the spinal cord. Utilizing this approach, we were able to simultaneously record changes in axoplasmic free Ca2+ (GCaMP6f) specifically in axons (tdTomato+) following the addition of ionomycin. Live two-photon excitation recordings of ex vivo spinal cord axons revealed that in baseline 2 mM Ca2+ aCSF and zero Ca2+ aCSF only conditions, Ca2+ was barely detected in tdTomato+ axons as expected up to 2 hours of observation (Figure 5A, D, and G–I). In contrast, the addition of 3 µM ionomycin to normal Ca2+ aCSF significantly (P < 0.05) increased intra-axonal Ca2+ levels compared to normal Ca2+ and zero Ca2+ aCSF controls at both one hour and two hours of observation (Figure 5B, E, and G–I). However, when ionomycin was added to zero Ca2+ aCSF, intra-axonal Ca2+ levels were significantly (P < 0.05) less than in the presence of normal Ca2+ plus ionomycin at both 1 hour and 2 hours of observation. These data suggest that in normal Ca2+ aCSF plus ionomycin treated spinal cords, the rise in intra-axonal Ca2+ also includes Ca2+ entry from the extracellular space that is prevented in zero Ca2+ aCSF plus ionomycin conditions (Figure 5C, F, and G–I). In support of Ca2+ release from internal stores, quantification of Ca2+ changes in axons revealed that in zero Ca2+ aCSF plus ionomycin conditions there was a significant (P < 0.05) rise in free Ca2+ levels compared with both normal and zero Ca2+ aCSF controls (Figure 5D and F–I). Importantly, axonal spheroids were always associated with an increased intra-axonal Ca2+ level in normal Ca2+ aCSF plus ionomycin (Figure 5E1) and in zero Ca2+ plus ionomycin conditions (Figure 5F1). Together, these results confirm that ionomycin treatment increases intra-axonal Ca2+ levels from both internal and external sources that result in axonal spheroid formation.
Figure 5.

Ionomycin increases axoplasmic free Ca2+ levels derived from both internal and external sources subsequent to axonal spheroid formation.
Representative maximum intensity projections captured using two-photon excitation microscopy of living spinal cord axons ex vivo (tdTomato, magenta A–C) and the corresponding genetic Ca2+ indicator GCaMP6f (D–F). (A) At 1 hour of observation in normal Ca2+ artificial cerebrospinal fluid (aCSF) only conditions, axons appear normal (tdTomato, magenta) with low levels of GCaMP6F as indicated by the heatmap (D). (B) However, following 1 hour of 3 µM ionomycin (iono) treatment in normal Ca2+ aCSF, axons and axonal spheroids (arrows) contained increased levels of Ca2+ as shown in E. Similarly, in zero Ca2+ aCSF plus ionomycin conditions, axons and axonal spheroids still contained elevated levels of Ca2+ but the intensity of the Ca2+ signal was reduced. D1, E1, F1 are higher magnification images of the boxed area in D, E, F respectively. Scale bar: 25 µm. (G) Representative traces of Ca2+ changes in axons over time in normal Ca2+ aCSF, zero Ca2+ aCSF and ionomycin in the presence of normal or zero Ca2+ conditions. As revealed in the box and whisker plots, quantification of axonal Ca2+ changes normalized to baseline (delta F/F0) are shown in H and I at 1 hour and 2 hours, respectively. In normal Ca2+ aCSF conditions in the presence of ionomycin (Ca2+ + iono), there was a significant increase (p < 0.001) in the median axonal Ca2+ levels vs. normal Ca2+ aCSF only (Ca2+), zero Ca2+ aCSF only (0 Ca2+), and zero Ca2+ aCSF + ionomycin (0 Ca2+ + iono) at 1 hour (H) and 2 hours (I) of observation. There was also a significant increase in axonal Ca2+ levels in zero Ca2+ aCSF + ionomycin (0 Ca2+ + iono) versus normal Ca2+ aCSF only (Ca), and zero Ca2+ aCSF only at both time points. Data are expressed as median (line within the box), 25th, 75th percentile (bottom and top of the box) and 5th, 95th percentile (bottom and top of the error bars). Outliers are shown as open circles. Significance was established using a Kruskal-Wallis analysis of variance on Ranks and Student-Newman-Keuls (SNK) multiple comparisons. *P < 0.05, Ca2+ + iono vs. Ca2+, 0 Ca2+, and 0 Ca2+ + iono groups; #P < 0.05, zero Ca2+ + iono vs. Ca2+ and zero Ca2+ only groups. n = 100 axons per group from 4 animals per group.
Discussion
The precise molecular mechanisms that cause central myelinated fiber degeneration remain poorly understood, however, a central role for pathological Ca2+ in axonal degeneration has been established (Stirling and Stys, 2010; Lingor et al., 2012; Ma, 2013; Maxwell, 2015; Yong et al., 2021). In contrast, very little is known about living myelin and the source(s) of Ca2+ or other mediators in myelin injury remains unclear (Balentine, 1988; Micu et al., 2006; Tsutsui and Stys, 2013; Hamilton et al., 2016; Fern and Matute, 2019; Warnock et al., 2020). To further our knowledge of the source of Ca2+ that mediates myelin and axonal degeneration, we used the well-characterized Ca2+ ionophore, ionomycin, to promote Ca2+ release from internal stores and time-lapse two-photon excitation imaging to document dynamic changes in myelin and axons in real time.
Here, utilizing Nile red as a vital dye to visualize dynamic changes in living myelin, we reveal that ionomycin causes central myelin to separate from its axon, i.e., periaxonal swelling, where the periaxonal space enlarges over time. Second, we reveal that periaxonal swelling often precedes axonal spheroid formation. In support, time-lapse recordings show that within the enlarged periaxonal spaces, axons began to swell and form spheroids, the latter identical to those described following contusive SCI in vivo (Williams et al., 2014; Rajaee et al., 2020). Interestingly, their characteristic shape was “molded” and confined within the disrupted axo-myelinic domains. As spheroids grew within these regions, the axonal segments between the spheroids thinned, myelin collapsed, and the now detached spheroids were often encased in myelin that appeared as “rings” in 2D optical cross-sections. It is unlikely at this stage of central myelinated fiber degeneration that these changes are reversible. Third, we uncover that these acute pathological changes in axons and their myelin sheaths depend initially on internal sources of Ca2+ as store depletion prior to ionomycin treatment abolished ionomycin-induced pathological changes in axons and myelin. Lastly, we show that the major route for external Ca2+ in ionomycin-mediated central myelinated fiber pathology is through SOCE, revealing a potential therapeutic target to protect myelinated fibers.
Of particular interest in the current study was the spatiotemporal relationship between periaxonal swelling and subsequent axonal spheroid formation. Here, we found that periaxonal swelling was often the first morphological change detected following ionomycin-induced Ca2+ release. Given the importance of an intact axo-myelinic interface for myelinated fiber physiology (Cohen et al., 2020), and glial-mediated axonal trophic support (Mot et al., 2018), disruption of these vital domains will likely have profound effects on impulse propagation, fidelity, and axonal survival.
As the axo-myelinic interface remains intact with few if any disruptions in control conditions ex vivo up to 2 hours and up to 13 hours ex vivo in normal Ca2+ aCSF (Okada et al., 2014), it is reasonable to suggest that the ionomycin-induced pathology occurs initially at the level of the myelin sheath. Our results are in agreement with previous ultrastructural studies that have exposed excised rat peripheral nerve or dorsal columns to calcium ionophores and reported similar Ca2+ induced alterations in the myelin sheath including vesicular degeneration of the outermost facing myelin exposed to the external space, paranodal vesiculation, and enlarged periaxonal space (Schlaepfer, 1977; Smith et al., 1985; Smith and Hall, 1994). Furthermore, as Ca2+-dependent proteases have been isolated from purified myelin and mediate cleavage of key structural proteins, the observed pathological changes in myelin could be induced by calpain-mediated degradation (Banik et al., 1985) as well as myelinolytic Ca2+-dependent phospholipases (Smith and Hall, 1988; Lopez-Vales et al., 2008; Shi et al., 2011; Warnock et al., 2020).
The delay in ionomycin-induced axonal changes may result from limited accessibility of ionomycin to the axon initially, as the surface of the axon is 99% ensheathed in myelin. However, once the axo-myelinic interface is disrupted, this may facilitate increased access of ionomycin from the extracellular space to the periaxonal space. This may also explain why myelinated fibers with greater diameter are often refractory to ionomycin-induced axonal changes versus smaller diameter fibers and require increased concentrations of ionomycin to induce axonal damage.
Regardless of the precise route of access to the axon, how does ionomycin mechanistically induce pathological changes in axons? We predict that ionomycin initially induces the release of Ca2+ from internal stores. In support, thapsigargin-mediated Ca2+ store depletion was completely protective and prevented ionomycin-induced axonal spheroid formation. Furthermore, a maneuver that is widely used to bypass T-cell receptor-mediated activation uses ionomycin to release Ca2+ from internal stores (Imboden and Weiss, 1987; Yoshida and Plant, 1992; Morgan and Jacob, 1994). The resultant decrease in stored Ca2+ then drives entry of Ca2+ but not from simple diffusion across the plasmalemma but from SOCE (Imboden and Weiss, 1987; Morgan and Jacob, 1994). Axons are also known to store Ca2+ primarily in the axoplasmic reticulum, an extension of the ER that is continuous along the entire length of the axon (Lindsey and Ellisman, 1985; Ozturk et al., 2020). Therefore, ionomycin could induce axonal Ca2+ stores to release Ca2+ into the axoplasm driving replenishment by SOCE. Excessive SOCE would then provide the necessary external Ca2+ to drive axonal spheroid formation. In agreement, we and others have shown that targeting RyR and IP3R, key Ca2+ release channels on the ER and axoplasmic reticulum, as well as SOCE, decreases secondary axonal degeneration following SCI (Stirling et al., 2014a; Orem et al., 2017, 2020b), ischemia (Ouardouz et al., 2003; Tu et al., 2020), and TBI (Hou et al., 2015; Rao et al., 2015; Liang et al., 2019). Here, we mechanistically link Ca2+ store depletion and SOCE that collectively causes pathological Ca2+ accumulation in both myelin and axons.
Similarly, Ca2+ oscillations have been recorded in myelin (Baraban et al., 2018) and increased Ca2+ accumulation occurs in myelin following injury (Micu et al., 2007). Furthermore, oligodendrocytes express all RyR and IP3R (Haak et al., 2001; Ruiz et al., 2010), and RyR-mediated Ca2+ release from internal Ca2+ stores contributes to excitotoxicity (Ruiz et al., 2010). Therefore, it is plausible that the same mechanistic processes described above supporting a role for SOCE in axonal spheroid formation could also cause pathological changes in myelin. In support, glial Ca2+ signaling involves the release of Ca2+ from the ER and replenishment through SOCE (Papanikolaou et al., 2017). Whereas aberrant SOCE has been shown to contribute to cuprizone-induced demyelination (Wang et al., 2015), and EAE (Schuhmann et al., 2010). Whether noncompact myelin contains a similar endomembrane system as axons remains to be determined; however, mitochondria are also found in myelin (Rinholm et al., 2016) and could contribute to Ca2+ buffering/release in myelin. Further work is necessary to determine the molecular mechanisms of Ca2+ signaling dynamics in myelin in health and disease. We also cannot rule out indirect effects on other cell types such as microglia and astrocytes that through excessive Ca2+ signaling, could release toxic inflammatory molecules. Future experiments could utilize fluorescently tagged ionomycin to further elucidate its distribution within the spinal cord.
Our ex vivo spinal cord model facilitates direct observation of the dynamic response of central myelinated fibers to injury over time with a high spatial resolution (Okada et al., 2014; Stirling et al., 2014b; Orem et al., 2017). Furthermore, this model is advantageous as it restricts the delivery of drugs to the isolated spinal cord and thereby prevents systemic effects on other organ systems. Moreover, it allows the manipulation of the perfusate bathing the cord to precisely control levels of external Ca2+ or other aCSF constituents that is not practical in vivo. The pathological changes in myelin and axons shown here also mirror what has been observed following clinically relevant SCI models in vivo including periaxonal swelling, myelin vesiculation, terminal endbulb formation, and spheroid formation following SCI in vivo (Balentine, 1978; Anthes et al., 1995).
This study has some limitations that should be noted such as lack of blood-derived elements, chemical versus mechanical injury-induced axonal spheroid formation, and the length of time ex vivo models remain viable. The model’s excised nature and removal from the blood supply prevents a peripheral immune response and other blood-derived elements from potentially contributing to the axo-myelinic pathology. While a limiting factor in regard to comparing our findings directly to in vivo outcomes, our model, as described above, does permit inspection of SCI in a more controlled environment that still mirrors observations in in vivo studies. The viability of ex vivo spinal transplants is limited to only several hours after spinal cord isolation while secondary white matter degeneration occurs over a protracted period of time in vivo. Future studies can remedy this limitation with an in vivo model which can be observed for weeks following SCI.
In summary, we have provided evidence that supports a novel role for SOCE in mediating pathological Ca2+ accumulation in both myelin injury and axonal spheroid formation. Further understanding of key SOCE effector molecules and their role in white matter injury may lead to novel targets to protect vital axo-myelinic domains and preserve white matter function after injury.
Acknowledgments:
We thank Starlynn Okada, Jamie Seo, Tarah Lancaster, Behrad Bakhtiari, and Kelsey Littrel (Kentucky Spinal Cord Injury Research Center University of Louisville, School of Medicine, Louisville, KY, USA) for technical assistance, genotyping, and animal care.
Footnotes
Funding: This work was supported by NIH-NINDS NS092680 (to DPS).
Conflicts of interest: The authors declare no competing financial interests.
Data availability statement: All relevant data are within the paper.
C-Editors: Zhao M, Liu WJ; S-Editor: Li CH; L-Editors: Li CH, Song LP; T-Editor: Jia Y
References
- 1.Anthes DL, Theriault E, Tator CH. Characterization of axonal ultrastructural pathology following experimental spinal cord compression injury. Brain Res. 1995;702:1–16. doi: 10.1016/0006-8993(95)01028-6. [DOI] [PubMed] [Google Scholar]
- 2.Balentine JD. Pathology of experimental spinal cord trauma. II. Ultrastructure of axons and myelin. Lab Invest. 1978;39:254–266. [PubMed] [Google Scholar]
- 3.Balentine JD. Spinal cord trauma:in search of the meaning of granular axoplasm and vesicular myelin. J Neuropathol Exp Neurol. 1988;47:77–92. doi: 10.1097/00005072-198803000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Balentine JD, Spector M. Calcification of axons in experimental spinal cord trauma. Ann Neurol. 1977;2:520–523. doi: 10.1002/ana.410020612. [DOI] [PubMed] [Google Scholar]
- 5.Banik NL, McAlhaney WW, Hogan EL. Calcium-stimulated proteolysis in myelin:evidence for a Ca2+-activated neutral proteinase associated with purified myelin of rat CNS. J Neurochem. 1985;45:581–588. doi: 10.1111/j.1471-4159.1985.tb04026.x. [DOI] [PubMed] [Google Scholar]
- 6.Baraban M, Koudelka S, Lyons DA. Ca (2+) activity signatures of myelin sheath formation and growth in vivo. Nat Neurosci. 2018;21:19–23. doi: 10.1038/s41593-017-0040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beirowski B, Nogradi A, Babetto E, Garcia-Alias G, Coleman MP. Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J Neuropathol Exp Neurol. 2010;69:455–472. doi: 10.1097/NEN.0b013e3181da84db. [DOI] [PubMed] [Google Scholar]
- 8.Chatila T, Silverman L, Miller R, Geha R. Mechanisms of T cell activation by the calcium ionophore ionomycin. J Immunol. 1989;143:1283–1289. [PubMed] [Google Scholar]
- 9.Chu TH, Cummins K, Sparling JS, Tsutsui S, Brideau C, Nilsson KPR, Joseph JT, Stys PK. Axonal and myelinic pathology in 5xFAD Alzheimer's mouse spinal cord. PLoS one. 2017;12:e0188218. doi: 10.1371/journal.pone.0188218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen CCH, Popovic MA, Klooster J, Weil MT, Mobius W, Nave KA, Kole MHP. Saltatory conduction along myelinated axons involves a periaxonal nanocircuit. Cell. 2020;180:311–322. e315. doi: 10.1016/j.cell.2019.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman MP, Perry VH. Axon pathology in neurological disease:a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- 12.del Mar N, von Buttlar X, Yu AS, Guley NH, Reiner A, Honig MG. A novel closed-body model of spinal cord injury caused by high-pressure air blasts produces extensive axonal injury and motor impairments. Exp Neurol. 2015;271:53–71. doi: 10.1016/j.expneurol.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donnadieu E, Bismuth G, Trautmann A. The intracellular Ca2+concentration optimal for T cell activation is quite different after ionomycin or CD3 stimulation. Pflugers Arch. 1995;429:546–554. doi: 10.1007/BF00704160. [DOI] [PubMed] [Google Scholar]
- 14.Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS. Acute spinal cord injury, part I:pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24:254–264. doi: 10.1097/00002826-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Ek CJ, Habgood MD, Dennis R, Dziegielewska KM, Mallard C, Wheaton B, Saunders NR. Pathological changes in the white matter after spinal contusion injury in the rat. PLoS One. 2012;7:e43484. doi: 10.1371/journal.pone.0043484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fern R, Matute C. Glutamate receptors and white matter stroke. Neurosci Lett. 2019;694:86–92. doi: 10.1016/j.neulet.2018.11.031. [DOI] [PubMed] [Google Scholar]
- 17.Fern RF, Matute C, Stys PK. White matter injury:Ischemic and nonischemic. Glia. 2014;62:1780–1789. doi: 10.1002/glia.22722. [DOI] [PubMed] [Google Scholar]
- 18.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grochmal J, Teo W, Gambhir H, Kumar R, Stratton JA, Dhaliwal R, Brideau C, Biernaskie J, Stys PK, Midha R. A novel approach to 32-channel peripheral nervous system myelin imaging in vivo, with single axon resolution. J Neurosurg. 2018;130:163–171. doi: 10.3171/2017.6.JNS17239. [DOI] [PubMed] [Google Scholar]
- 20.Haak LL, Song LS, Molinski TF, Pessah IN, Cheng H, Russell JT. Sparks and puffs in oligodendrocyte progenitors:cross talk between ryanodine receptors and inositol trisphosphate receptors. J Neurosci. 2001;21:3860–3870. doi: 10.1523/JNEUROSCI.21-11-03860.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hachem LD, Fehlings MG. Pathophysiology of spinal cord injury. Neurosurg Clin N Am. 2021;32:305–313. doi: 10.1016/j.nec.2021.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton NB, Kolodziejczyk K, Kougioumtzidou E, Attwell D. Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature. 2016;529:523–527. doi: 10.1038/nature16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Happel RD, Smith KP, Banik NL, Powers JM, Hogan EL, Balentine JD. Ca2+-accumulation in experimental spinal cord trauma. Brain Res. 1981;211:476–479. doi: 10.1016/0006-8993(81)90976-8. [DOI] [PubMed] [Google Scholar]
- 24.Hejrati N, Fehlings MG. A review of emerging neuroprotective and neuroregenerative therapies in traumatic spinal cord injury. Curr Opin Pharmacol. 2021;60:331–340. doi: 10.1016/j.coph.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Hill CE. A view from the ending:Axonal dieback and regeneration following SCI. Neurosci Lett. 2017;652:11–24. doi: 10.1016/j.neulet.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Horiuchi H, Oshima Y, Ogata T, Morino T, Matsuda S, Miura H, Imamura T. Evaluation of injured axons using two-photon excited fluorescence microscopy after spinal cord contusion injury in YFP-H line mice. Int J Mol Sci. 2015;16:15785–15799. doi: 10.3390/ijms160715785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou PF, Liu ZH, Li N, Cheng WJ, Guo SW. Knockdown of STIM1 improves neuronal survival after traumatic neuronal injury through regulating mGluR1-dependent Ca(2+) signaling in mouse cortical neurons. Cell Mol Neurobiol. 2015;35:283–292. doi: 10.1007/s10571-014-0123-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang AS, Tong BCK, Wu AJ, Chen X, Sreenivasmurthy SG, Zhu Z, Liu J, Su C, Li M, Cheung KH. Rectifying attenuated store-operated calcium entry as a therapeutic approach for Alzheimer's disease. Curr Alzheimer Res. 2020;17:1072–1087. doi: 10.2174/1567205018666210119150613. [DOI] [PubMed] [Google Scholar]
- 29.Imboden JB, Weiss A. The T-cell antigen receptor regulates sustained increases in cytoplasmic free Ca2+through extracellular Ca2+influx and ongoing intracellular Ca2+mobilization. Biochem J. 1987;247:695–700. doi: 10.1042/bj2470695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kappel S, Borgstrom A, Stoklosa P, Dorr K, Peinelt C. Store-operated calcium entry in disease:Beyond STIM/Orai expression levels. Semin Cell Dev Biol. 2019;94:66–73. doi: 10.1016/j.semcdb.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451–464. doi: 10.1016/j.spinee.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Liang Y, Tong F, Zhang L, Zhu L, Li W, Huang W, Zhao S, He G, Zhou Y. iTRAQ-based proteomic analysis discovers potential biomarkers of diffuse axonal injury in rats. Brain Res Bull. 2019;153:289–304. doi: 10.1016/j.brainresbull.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Lindsey JD, Ellisman MH. The neuronal endomembrane system. III. The origins of the axoplasmic reticulum and discrete axonal cisternae at the axon hillock. J Neurosci. 1985;5:3135–3144. doi: 10.1523/JNEUROSCI.05-12-03135.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lingor P, Koch JC, Tönges L, Bähr M. Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 2012;349:289–311. doi: 10.1007/s00441-012-1362-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LoPachin RM, Lehning EJ. Mechanism of calcium entry during axon injury and degeneration. Toxicol Appl Pharmacol. 1997;143:233–244. doi: 10.1006/taap.1997.8106. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Vales R, Navarro X, Shimizu T, Baskakis C, Kokotos G, Constantinou-Kokotou V, Stephens D, Dennis EA, David S. Intracellular phospholipase A(2) group IVA and group VIA play important roles in Wallerian degeneration and axon regeneration after peripheral nerve injury. Brain. 2008;131:2620–2631. doi: 10.1093/brain/awn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 38.Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol Dis. 2013;60:61–79. doi: 10.1016/j.nbd.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maxwell WL. Development of concepts in the pathology of traumatic axonal and traumatic brain injury. Brain neurotrauma:molecular, neuropsychological, and rehabilitation aspects. In: Kobeissy FH, editor. Boca Raton (FL): CRC Press; 2015. [Google Scholar]
- 40.McDonald JW, Sadowsky C. Spinal-cord injury. Lancet. 2002;359:417–425. doi: 10.1016/S0140-6736(02)07603-1. [DOI] [PubMed] [Google Scholar]
- 41.Micu I, Ridsdale A, Zhang L, Woulfe J, McClintock J, Brantner CA, Andrews SB, Stys PK. Real-time measurement of free Ca2+changes in CNS myelin by two-photon microscopy. Nat Med. 2007;13:874–879. doi: 10.1038/nm1568. [DOI] [PubMed] [Google Scholar]
- 42.Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- 43.Morgan AJ, Jacob R. Ionomycin enhances Ca2+influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem J. 1994;300(Pt 3):665–672. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mot AI, Depp C, Nave KA. An emerging role of dysfunctional axon-oligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin Neurosci. 2018;20:283–292. doi: 10.31887/dcns.2018.20.4/amot. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Research Council (2011) Guide for the Care and Use of Laboratory Animals. 8th ed. Washington, DC: The National Academies Press; [Google Scholar]
- 46.Okada SL, Stivers NS, Stys PK, Stirling DP. An ex vivo laser-induced spinal cord injury model to assess mechanisms of axonal degeneration in real-time. J Vis Exp. 2014:e52173. doi: 10.3791/52173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orem BC, Rajaee A, Stirling DP. IP3R-mediated intra-axonal Ca(2+) release contributes to secondary axonal degeneration following contusive spinal cord injury. Neurobiol Dis. 2020a;146:105123. doi: 10.1016/j.nbd.2020.105123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orem BC, Partain SB, Stirling DP. Inhibiting store-operated calcium entry attenuates white matter secondary degeneration following SCI. Neurobiol Dis. 2020b;136:104718. doi: 10.1016/j.nbd.2019.104718. [DOI] [PubMed] [Google Scholar]
- 49.Orem BC, Pelisch N, Williams J, Nally JM, Stirling DP. Intracellular calcium release through IP3R or RyR contributes to secondary axonal degeneration. Neurobiol Dis. 2017;106:235–243. doi: 10.1016/j.nbd.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 50.Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J, Stys PK. Depolarization-induced Ca2+release in ischemic spinal cord white matter involves L-type Ca2+channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 51.Ozturk Z, O'Kane CJ, Perez-Moreno JJ. Axonal endoplasmic reticulum dynamics and its roles in neurodegeneration. Front Neurosci. 2020;14:48. doi: 10.3389/fnins.2020.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papanikolaou M, Lewis A, Butt AM. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct Funct. 2017;222:2993–3005. doi: 10.1007/s00429-017-1380-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Povlishock JT. Traumatically induced axonal injury:pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- 54.Putney JW. Origins of the concept of store-operated calcium entry. Front Biosci (Schol Ed) 2011;3:980–984. doi: 10.2741/202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Putney JW. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv Biol Regul. 2018;68:88–96. doi: 10.1016/j.jbior.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajaee A, Geisen ME, Sellers AK, Stirling DP. Repeat intravital imaging of the murine spinal cord reveals degenerative and reparative responses of spinal axons in real-time following a contusive SCI. Exp Neurol. 2020;327:113258. doi: 10.1016/j.expneurol.2020.113258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rao W, Zhang L, Peng C, Hui H, Wang K, Su N, Wang L, Dai SH, Yang YF, Chen T, Luo P, Fei Z. Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim Biophys Acta. 2015;1852:2402–2413. doi: 10.1016/j.bbadis.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Rinholm JE, Vervaeke K, Tadross MR, Tkachuk AN, Kopek BG, Brown TA, Bergersen LH, Clayton DA. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia. 2016;64:810–825. doi: 10.1002/glia.22965. [DOI] [PubMed] [Google Scholar]
- 59.Ruiz A, Matute C, Alberdi E. Intracellular Ca2+release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010;1:e54. doi: 10.1038/cddis.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlaepfer WW. Vesicular disruption of myelin simulated by exposure of nerve to calcium ionophore. Nature. 1977;265:734–736. doi: 10.1038/265734a0. [DOI] [PubMed] [Google Scholar]
- 61.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ:25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schuhmann MK, Stegner D, Berna-Erro A, Bittner S, Braun A, Kleinschnitz C, Stoll G, Wiendl H, Meuth SG, Nieswandt B. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol. 2010;184:1536–1542. doi: 10.4049/jimmunol.0902161. [DOI] [PubMed] [Google Scholar]
- 63.Secondo A, Bagetta G, Amantea D. On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front Mol Neurosci. 2018;11:87. doi: 10.3389/fnmol.2018.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sekhon LH, Fehlings MG. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine (Phila Pa 1976) 2001;26:S2–12. doi: 10.1097/00007632-200112151-00002. [DOI] [PubMed] [Google Scholar]
- 65.Shi Y, Sun W, McBride JJ, Cheng JX, Shi R. Acrolein induces myelin damage in mammalian spinal cord. J Neurochem. 2011;117:554–564. doi: 10.1111/j.1471-4159.2011.07226.x. [DOI] [PubMed] [Google Scholar]
- 66.Smith KJ, Hall SM, Schauf CL. Vesicular demyelination induced by raised intracellular calcium. J Neurol Sci. 1985;71:19–37. doi: 10.1016/0022-510x(85)90034-6. [DOI] [PubMed] [Google Scholar]
- 67.Smith KJ, Hall SM. Peripheral demyelination and remyelination initiated by the calcium-selective ionophore ionomycin:in vivo observations. J Neurol Sci. 1988;83:37–53. doi: 10.1016/0022-510x(88)90018-4. [DOI] [PubMed] [Google Scholar]
- 68.Smith KJ, Hall SM. Central demyelination induced in vivo by the calcium ionophore ionomycin. Brain. 1994;117(Pt 6):1351–1356. doi: 10.1093/brain/117.6.1351. [DOI] [PubMed] [Google Scholar]
- 69.Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stirling DP, Stys PK. Mechanisms of axonal injury:internodal nanocomplexes and calcium deregulation. Trends Mol Med. 2010;16:160–170. doi: 10.1016/j.molmed.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stirling DP, Cummins K, Wayne Chen SR, Stys P. Axoplasmic reticulum Ca(2+) release causes secondary degeneration of spinal axons. Ann Neurol. 2014a;75:220–229. doi: 10.1002/ana.24099. [DOI] [PubMed] [Google Scholar]
- 72.Stirling DP, Cummins K, Mishra M, Teo W, Yong VW, Stys P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain. 2014b;137:707–723. doi: 10.1093/brain/awt341. [DOI] [PubMed] [Google Scholar]
- 73.Stivers NS, Pelisch N, Orem BC, Williams J, Nally JM, Stirling DP. The toll-like receptor 2 agonist Pam3CSK4 is neuroprotective after spinal cord injury. Exp Neurol. 2017;294:1–11. doi: 10.1016/j.expneurol.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 74.Tang P, Zhang Y, Chen C, Ji X, Ju F, Liu X, Gan WB, He Z, Zhang S, Li W, Zhang L. In vivo two-photon imaging of axonal dieback, blood flow, and calcium influx with methylprednisolone therapy after spinal cord injury. Sci Rep. 2015;5:9691. doi: 10.1038/srep09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483–492. doi: 10.3171/jns.1997.86.3.0483. [DOI] [PubMed] [Google Scholar]
- 76.Teo W, Caprariello AV, Morgan ML, Luchicchi A, Schenk GJ, Joseph JT, Geurts JJG, Stys PK. Nile Red fluorescence spectroscopy reports early physicochemical changes in myelin with high sensitivity. Proc Natl Acad Sci U S A. 2021;118:e2016897118. doi: 10.1073/pnas.2016897118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsutsui S, Stys PK. Metabolic injury to axons and myelin. Exp Neurol. 2013;246:26–34. doi: 10.1016/j.expneurol.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 78.Tu CC, Wan BY, Zeng Y. STIM2 knockdown protects against ischemia/reperfusion injury through reducing mitochondrial calcium overload and preserving mitochondrial function. Life Sci. 2020;247:116560. doi: 10.1016/j.lfs.2019.116560. [DOI] [PubMed] [Google Scholar]
- 79.Vahsen BF, Ribas VT, Sundermeyer J, Boecker A, Dambeck V, Lenz C, Shomroni O, Caldi Gomes L, Tatenhorst L, Barski E, Roser AE, Michel U, Urlaub H, Salinas G, Bahr M, Koch JC, Lingor P. Inhibition of the autophagic protein ULK1 attenuates axonal degeneration in vitro and in vivo, enhances translation, and modulates splicing. Cell Death Differ. 2020;27:2810–2827. doi: 10.1038/s41418-020-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang H, Liu S, Tian Y, Wu X, He Y, Li C, Namaka M, Kong J, Li H, Xiao L. Quetiapine inhibits microglial activation by neutralizing abnormal STIM1-mediated intercellular calcium homeostasis and promotes myelin repair in a cuprizone-induced mouse model of demyelination. Front Cell Neurosci. 2015;9:492. doi: 10.3389/fncel.2015.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang J, Zhao W, Wang X, Gao H, Liu R, Shou J, Yan J. Enhanced store-operated calcium entry (SOCE) exacerbates motor neurons apoptosis following spinal cord injury. Gen Physiol Biophys. 2021;40:61–69. doi: 10.4149/gpb_2020040. [DOI] [PubMed] [Google Scholar]
- 82.Wang JT, Medress ZA, Barres BA. Axon degeneration:molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Warnock A, Toomey LM, Wright AJ, Fisher K, Won Y, Anyaegbu C, Fitzgerald M. Damage mechanisms to oligodendrocytes and white matter in central nervous system injury:The Australian Context. J Neurotrauma. 2020;37:739–769. doi: 10.1089/neu.2019.6890. [DOI] [PubMed] [Google Scholar]
- 84.Williams PR, Marincu BN, Sorbara CD, Mahler CF, Schumacher AM, Griesbeck O, Kerschensteiner M, Misgeld T. A recoverable state of axon injury persists for hours after spinal cord contusion in vivo. Nat Commun. 2014;5:5683. doi: 10.1038/ncomms6683. [DOI] [PubMed] [Google Scholar]
- 85.Yong Y, Hunter-Chang S, Stepanova E, Deppmann C. Axonal spheroids in neurodegeneration. Mol Cell Neurosci. 2021;117:103679. doi: 10.1016/j.mcn.2021.103679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoshida S, Plant S. Mechanism of release of Ca2+from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. J Physiol. 1992;458:307–318. doi: 10.1113/jphysiol.1992.sp019419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang M, Song JN, Wu Y, Zhao YL, Pang HG, Fu ZF, Zhang BF, Ma XD. Suppression of STIM1 in the early stage after global ischemia attenuates the injury of delayed neuronal death by inhibiting store-operated calcium entry-induced apoptosis in rats. Neuroreport. 2014;25:507–513. doi: 10.1097/WNR.0000000000000127. [DOI] [PubMed] [Google Scholar]