

Lysosomes are highly dynamic, single membrane-bound compartments that are critical for maintaining cellular homeostasis. These organelles, which appear to vary between 200 nm–1 μm in diameter, originate from the maturation of endocytic vesicles and via the enrichment of newly synthesized lysosomal proteins in the trans-Golgi network. Lysosomes mediate the targeted degradation of intracellular and extracellular cargoes, including those trafficked by the endosomal and autophagic pathways, to produce monosaccharides, amino acids, and free fatty acids, among other molecules. However, recent discoveries have revealed additional lysosomal functions, including roles in nutrient and metabolic sensing, gene regulation, inflammation, membrane repair, and effects on the extracellular environment. Lysosomes are found throughout the cytoplasm, but may have a preferential distribution depending on their specific activity or cell state. For example, due to their origin, lysosomes predominantly localize to the perinuclear region, but also maintain this distribution under conditions of proteolytic stress. However, secretory lysosomes localize to the cell periphery and also appear to be less acidic (Keeling et al., 2018). Lysosomal positioning is also influenced by contact with the endoplasmic reticulum, mitochondria and the Golgi apparatus. Lysosomes are rarely stationary and are trafficked bidirectionally along microtubules, with anterograde and retrograde movements mediated by kinesin or dynein. While lysosomes are known to play a causative role in storage diseases, their broader dysfunction contributes to a variety of neurodegenerative diseases, including Alzheimer’s disease, frontotemporal dementia, age-related macular degeneration (AMD), amyotrophic lateral sclerosis, Parkinson’s disease, and Huntington’s disease, among others (Keeling et al., 2018; Malik et al., 2019). Retinal pigment epithelial (RPE) cells provide a unique model to study lysosomal biology and the consequences of their dysfunction. The RPE forms a highly specialized monolayer in the retina that is intimately associated with photoreceptors. Overlying photoreceptors shed outer segments (photoreceptor outer segments, POS) as part of the daily photoreceptor renewal, which are engulfed and trafficked for degradation in lysosomes of RPE cells. Here, we review our novel findings showing how disease-related pathways in the retina affect these organelles and how lysosomal defects may contribute to RPE damage as part of a clinically well-defined pathway of RPE atrophy leading to irreversible vision loss in AMD and other retinopathies.
The RPE monolayer is arranged in a mosaic-like pattern (referred to as ‘cobblestone morphology’) with each RPE cell providing a dense bed of apical microvilli that wrap around overlying POS. The monolayer forms a polarized epithelium, which is structurally and physiologically specialized to undertake a variety of activities that are integral to maintaining normal vision. One such function is the receptor-mediated internalization of POS, where ~10% of outer segments from each overlying photoreceptor cell are engulfed every 24 hours throughout life and trafficked via the phagosome/endosome and autophagy pathways for lysosomal degradation (Figure 1A). Here, their molecular components are recycled for reuse in the visual process (Keeling et al., 2018). Consequently, the RPE monolayer is considered to possess the highest proteolytic burden in the human body. Early studies in Rhesus monkey retinas showed each RPE cell supports between 39–45 photoreceptors. Depending on their relative position in the retina (central vs. peripheral), each RPE cell may ingest between 2000–4000 discs per day. Over 70 years, each RPE is estimated to process up to one billion POS (Keeling et al., 2018). However, using three-dimensional-reconstructed tissues from the central mouse retina, we recently showed that an RPE cell can support up to 216 photoreceptors (Keeling et al., 2020b). Hence, the proteolytic burden of the RPE monolayer is likely to be much greater than previously thought. With advancing age, the process of POS breakdown becomes less efficient, resulting in the accumulation of autofluorescent, non-degradable inclusion bodies termed lipofuscin within lysosomes and other membrane-bound vesicles of RPE cells (Ng et al., 2008; Keeling et al., 2018). Histological analysis of aged donor eye tissues revealed that by the 8th decade of life, ~20% of the RPE cytoplasm was filled with such aggregates. Although other post-mitotic cells also accumulate lipofuscin with age, the excess accumulation of lipofuscin containing specific retinoids in RPE is associated with common blinding conditions such as AMD as well as rare/hereditary retinopathies including Stargardt disease (Keeling et al., 2018). The importance of efficient POS clearance to RPE function was highlighted by findings demonstrating a ~50% reduction in lysosomal enzyme activity upon lipofuscin exposure. Lipofuscin also caused the permeabilization of lysosomes. A major retinoid of RPE lipofuscin is N-retinylidine-N-retinylethanolamine, a pyridinium bis-retinoid and photo-inducible free radical generator, which impairs POS breakdown. Modification of POS by lipid peroxidation, which generates malondialdehyde and 4-hydroxynonenal, also impairs their degradation in lysosomes (Keeling et al., 2018). Of note, the acidic environment of lysosomes provides ideal conditions for the accumulation of the highly pathogenic Alzheimer’s-linked amyloid beta (Aβ) proteins (Lee et al., 2022). This is of interest, as histopathological analyses of donor eye tissues show high levels of Aβ which naturally deposits near RPE cells as a consequence of age as well as in advanced AMD (Lynn et al., 2017). The autofluorescent properties of RPE inclusion bodies can be non-invasively assessed in patients by fundus autofluorescence imaging, which in effect provides some insights into the biochemical constituents within these cells. Confocal microscopy analysis of RPE cells in donor retinas showed different phenotypes of granules based on their autofluorescence properties, size and composition, which may be correlated with different types/populations of overlying photoreceptors (Bermond et al., 2020). Regions of the retina where RPE cells have atrophied can thus be visualized as dark patches, associated with the loss of overlying photoreceptors in patients with geographic atrophy AMD. By contrast, the margins of such atrophic lesions show abnormal autofluorescence, often preceding cell death (Holz et al., 2007). Fundus autofluorescence imaging is therefore widely utilized and is an accepted clinical end-point and a gold standard for assessing RPE pathology. Collectively, this evidence highlights the critical role played by lysosomes, the impairment of which is directly linked with RPE dysfunction and a major pathway associated with developing blindness.
Figure 1.
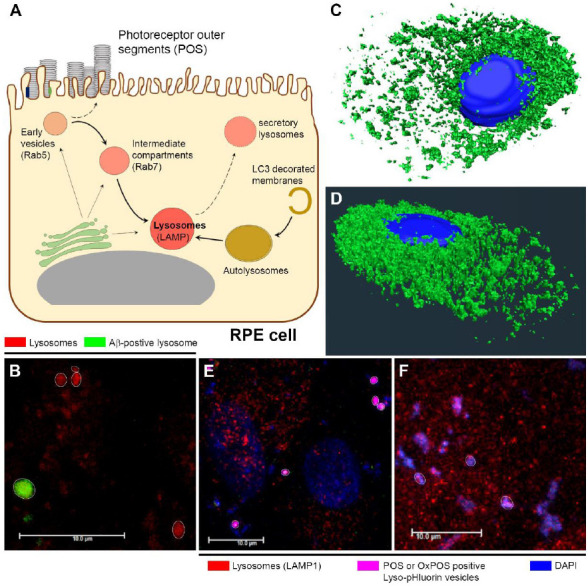
Cargo trafficking in RPE cells and the fate of lysosomes in retinopathies.
(A) Schematic illustrating the receptor-mediated internalization and subsequent trafficking of POS cargos in the phagosome/endosome and autophagy pathways of RPE cells. Key trafficking compartments are indicated by specific markers. Early and mature lysosomes are labeled with LAMP1 and LAMP2 respectively. Pathways of recycling endosomes as well as secretory lysosomes are shown along dashed arrows, whilst links between various trafficking vesicles with the Golgi are also indicated. (B) Confocal immunofluorescence image showing lysosomes co-localized with Aβ in green, which appear enlarged compared to lysosomes without Aβ cargos (red). (C, D) The LysoTracker™ labeled lysosomal population in RPE cells rendered in three-dimensional, showing their predominantly perinuclear clustering with fewer vesicles distributed peripherally which may be involved in secretion. (E) Confocal immunofluorescence images showing Lyso-pHluorin positive vesicles co-localized with POS or (F) OxPOS cargos to report intraluminal lysosomal pH in RPE cells. Lysosomes discernible as single organelles in the focal plane are shown highlighted by a white circle. Scale bars: 10 μm. Aβ: Amyloid beta; DAPI: 4′,6-diamidino-2-phenylindole; LAMP: lysosome-associated membrane protein; LC3: microtubule-associated protein 1A/1B-light chain 3; OxPOS: oxidized photoreceptor outer segments; POS: photoreceptor outer segments; Rab: Ras-related protein; RPE: retinal pigment epithelium. Unpublished data.
To unravel the mechanisms involved in the proteolytic processing of POS and other cargos and gain insights into how these processes become impaired, we exploited an in vitro RPE cell model, which facilitates experimental studies, including the use of powerful microscopes to capture dynamic trafficking events in living cells. Mature RPE monolayers that are pigmented and which structurally and physiologically recapitulate the in situ RPE, were fed isolated POS. Following an approach to maximize binding whilst minimizing their premature/piecemeal internalization, POS molecules become synchronously engulfed in a receptor-mediated (tyrosine-protein kinase Mer and αvβ5 integrin) manner to be trafficked in the phagosome/endosome and autophagy pathways. The major early, intermediate and late trafficking compartments were labeled using Ras-related protein (Rab)5, Rab7, lysosomal-associated membrane protein (LAMP)1 and LAMP2 as well as microtubule-associated protein 1A/1B-light chain 3 (LC3B) to obtain temporal information on POS trafficking in unprecedented detail. POS cargos were visualized by three-dimensional-rendered confocal immunofluorescence microscopy, whilst an algorithm objectively quantified the extent of POS co-localization in each compartment. Dynamic events, including the responses of lysosomes to POS and other cargos, were captured by live/confocal imaging. Ultrastructural changes to trafficking vesicles alongside evidence of gradual POS degradation were reported by electron microscopy, which corroborated confocal data from living and fixed RPE monolayers.
POS molecules were trafficked via early-intermediate compartments with little evidence of degradation. However, co-localization to lysosomes near the nucleus and the basolateral membrane showed evidence of cargo breakdown alongside the formation of double-membrane phagophores and co-localization with LC3B-positive bodies (Keeling et al., 2019). Overall, POS degradation occurred along the apical-basal axis of RPE cells. Despite efficient proteolysis, some elements of POS appeared to persist within late compartments for at least 72 hours after their internalization, at least in culture (Keeling et al., 2020a). Interestingly, some LAMP-positive lysosomes were also positive for Rab7, perhaps indicative of transition states in trafficking vesicles. Although healthy RPE cells systematically trafficked POS cargos, we observed sizable time intervals within each step, alongside evidence of shuttling by other means to LC3B-positive membranes, perhaps via non-canonical autophagy, indicating a degree of flexibility which we speculate may contribute to RPE resilience with age and disease. This hypothesis was supported by subsequent findings showing markedly diminished POS levels in Rab5 and Rab7 compartments and premature POS trafficking to LAMP1/2 vesicles, correlated with enlarged lysosomes under conditions of oxidative stress. By contrast, impaired membrane fusion induced by Bafilomycin A1 resulted in failed vesicle maturation with evidence of POS being sequestered in early compartments alongside diminished trafficking to lysosomes (Keeling et al., 2019). This was reflected in changes to the dimensions of affected compartments with significant increases to the size of POS-positive early-intermediate vesicles alongside smaller lysosomes compared to their equivalents in healthy RPE. Under conditions of oxidative stress and dysregulated autophagy, we also observed the premature formation of autophagosomes. Our data were consistent with enlarged and increased autophagosomes reported in cultured RPE from donor AMD tissues. Furthermore, under both insults, the sizes of lysosomes without cargos remained normal, suggesting only POS-carrying organelles were affected, even under disease-linked conditions (Keeling et al., 2019). Our work thus unraveled the mechanisms by which incorrectly processed POS, whether by rapid trafficking to bypass upstream processing events or by being sequestered in early-intermediate compartments without sufficient co-localization to lysosomes, can contribute to the formation of insoluble inclusion bodies in RPE cells. Once formed, these aggregates cannot be removed via transport outside the cell. We also investigated modifications to POS including those that promote their aggregation within cells. UV irradiation of isolated POS converts regularly-arranged membranes into an osmiophilic electron-dense mass (OxPOS). These co-localized to enlarged lysosomes and autophagy bodies, resulting in up to a doubling of vesicle size (Keeling et al., 2020a). Whilst POS breakdown was correlated with diminishing luminal content and increasing distance from the apical RPE surface, OxPOS showed no evidence of degradation for at least 72 hours (the duration of the experiment) following their internalization. Impaired proteolysis was also indirectly quantified by recapitulating the parameters of fundus autofluorescence imaging in a research-grade/laboratory confocal system for use in cultured cells, which to our knowledge was the first such attempt. Whilst autofluorescence readouts of POS-fed monolayers produced no marked changes, supporting prior evidence of efficient cargo degradation in healthy RPE (Keeling et al., 2018, 2019), a single OxPOS pulse resulted in autofluorescence levels that were 3–5 fold greater than unfed/control or POS exposed cultures (Keeling et al., 2020a), demonstrating that a clinical biomarker of RPE health can be reproduced in vitro.
In further studies, we investigated potential effects of Aβ in lysosomes of RPE using our in vitro cell model. We observed oligomeric Aβ1–42 to rapidly enter RPE cells, resulting in swollen lysosomes (Figure 1B). Aβ continued to accumulate in lysosomes over a prolonged period, peaking at 24 hours following initial exposure (Lynn et al., 2021). There was a rapid and substantial burst of lysosomal cathepsin B in response, which succeeded in eliminating only a small proportion of Aβ. By contrast, a smaller and more sustained cathepsin B response successfully cleared POS cargos, revealing contrasting lysosomal responses to different cargo types. The effects of Aβ exposure on RPE physiology were evaluated by subsequently feeding cultures with a synchronized POS pulse, which showed > 20% fewer POS-positive lysosomes at timeframes that were critical for POS degradation (Lynn et al., 2021). Chronic Aβ exposure could therefore result in proteolytically deficient RPE, where insufficient function of lysosomes could eventually contribute to the accumulation of pathogenic inclusion bodies within cells; a novel pathway through which Aβ can contribute to RPE dysfunction in aged/AMD retinas. Our work also showed that RPE cells contain a sizable lysosomal population, likely in the hundreds, which is consistent with reported numbers in other cells (Keeling et al., 2018), but may be higher given the cell’s exceptionally high proteolytic burden (Figure 1C and D). Outstanding questions remain on the importance of intraluminal acidity and the intracellular localization of lysosomes in RPE cells. To address these we investigated the intraluminal acidity/pH of these organelles. N-retinylidine-N-retinylethanolamine is reported to impair the activity of the vATPase transporter which acidifies lysosomes, resulting in the accumulation of POS (Bergmann et al., 2004). Our recent findings demonstrate that intraluminal lysosomal pH becomes significantly less acidic upon co-localization with either POS or OxPOS (Figure 1E and F). However, POS is effectively degraded in lysosomes by healthy RPE cells, whereas OxPOS, which is resistant to degradation, becomes sequestered in these compartments (Keeling et al., 2019, 2020a). Therefore, impaired lysosomal acidification may be an early event, likely preceding the well-documented pathogenic effects of N-retinylidine-N-retinylethanolamine. In this respect, lysosomal defects in RPE may share similarities with failed acidification of autolysosomes reported in neurons of Alzheimer’s mouse models (Lee et al., 2022). Our findings thus provide further mechanistic insights into how lysosomes in RPE cells become dysfunctional, showing similarities with lysosomal dysfunction reported in other types of neurodegenerative conditions.
This work was supported by PhD Studentships from the Macular Society, Biotechnology and Biological Sciences Research Council (BBSRC) SoCoBio-DTP and funding from the Gift of Sight Appeal.
The authors wish to thank Dr. David A. Tumbarello (University of Southampton, UK) for scrutiny of the manuscript. We also thank Dr. Savannah A. Lynn (University of Southampton, UK) for images of Aβ co-localization in lysosomes, as well as Mr. Charles Ellis and Dr. David S. Chatelet (University of Southampton, UK) for generating three-dimensional-rendered lysosomes in RPE cells. Professor Andrew J. Lotery (University of Southampton, UK) and Dr. David A. Tumbarello are acknowledged for co-supervision of the work.
Footnotes
C-Editors: Zhao M, Sun Y, Yu J; T-Editor: Jia Y
References
- 1.Bergmann M, Schütt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:562–564. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]
- 2.Bermond K, Wobbe C, Tarau IS, Heintzmann R, Hillenkamp J, Curcio CA, Sloan KR, Ach T. Autofluorescent granules of the human retinal pigment epithelium:phenotypes, intracellular distribution, and age-related topography. Invest Ophthalmol Vis Sci. 2020;61:35. doi: 10.1167/iovs.61.5.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143:463–472. doi: 10.1016/j.ajo.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 4.Keeling E, Lotery AJ, Tumbarello DA, Ratnayaka JA. Impaired cargo clearance in the retinal pigment epithelium (RPE) underlies irreversible blinding diseases. Cells. 2018;7:16. doi: 10.3390/cells7020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keeling E, Chatelet DS, Johnston DA, Page A, Tumbarello DA, Lotery AJ, Ratnayaka JA. Oxidative stress and dysfunctional intracellular traffic linked to an unhealthy diet results in impaired cargo transport in the retinal pigment epithelium (RPE) Mol Nutr Food Res. 2019;63:e1800951. doi: 10.1002/mnfr.201800951. [DOI] [PubMed] [Google Scholar]
- 6.Keeling E, Culling AJ, Johnston DA, Chatelet DS, Page A, Tumbarello DA, Lotery AJ, Ratnayaka JA. An in-vitro cell model of intracellular protein aggregation provides insights into RPE stress associated with retinopathy. Int J Mol Sci. 2020a;21:6647. doi: 10.3390/ijms21186647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keeling E, Chatelet DS, Tan NYT, Khan F, Richards R, Thisainathan T, Goggin P, Page A, Tumbarello DA, Lotery AJ, Ratnayaka JA. 3D-reconstructed retinal pigment epithelial cells provide insights into the anatomy of the outer retina. Int J Mol Sci. 2020b;21:8408. doi: 10.3390/ijms21218408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, Chan H, Bouchet-Marquis C, Bleiwas C, Berg MJ, Huo C, Peddy J, Pawlik M, Levy E, Rao M, Staufenbiel M, Nixon RA. Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Aβin neurons, yielding senile plaques. Nat Neurosci. 2022;25:688–701. doi: 10.1038/s41593-022-01084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynn SA, Keeling E, Munday R, Gabha G, Griffiths H, Lotery AJ, Ratnayaka JA. The complexities underlying age-related macular degeneration:could amyloid beta play an important role? Neural Regen Res. 2017;12:538–548. doi: 10.4103/1673-5374.205083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynn SA, Johnston DA, Scott JA, Munday R, Desai RS, Keeling E, Weaterton R, Simpson A, Davis D, Freeman T, Chatelet DS, Page A, Cree AJ, Lee H, Newman TA, Lotery AJ, Ratnayaka JA. Oligomeric Aβ(1-42) Induces an AMD-like phenotype and accumulates in lysosomes to impair RPE function. Cells. 2021;10:413. doi: 10.3390/cells10020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malik BR, Maddison DC, Smith GA, Peters OM. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019;12:100. doi: 10.1186/s13041-019-0504-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng KP, Gugiu B, Renganathan K, Davies MW, Gu X, Crabb JS, Kim SR, Rózanowska MB, Bonilha VL, Rayborn ME, Salomon RG, Sparrow JR, Boulton ME, Hollyfield JG, Crabb JW. Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics. 2008;7:1397–1405. doi: 10.1074/mcp.M700525-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]