

Abstract
The blood-brain barrier is the interface through which the brain interacts with the milieu and consists mainly of a sophisticated network of brain endothelial cells that forms blood vessels and selectively moves molecules inside and outside the brain through multiple mechanisms of transport. Although brain endothelial cell function is crucial for brain homeostasis, their role in neurodegenerative diseases has historically not been considered with the same importance as other brain cells such as microglia, astroglia, neurons, or even molecules such as amyloid beta, Tau, or alpha-synuclein. Alzheimer’s disease is the most common neurodegenerative disease, and brain endothelial cell dysfunction has been reported by several groups. However, its impairment has barely been considered as a potential therapeutic target. Here we review the most recent advances in the relationship between Alzheimer’s disease and brain endothelial cells commitment and analyze the possible mechanisms through which their alterations contribute to this neurodegenerative disease, highlighting their inflammatory phenotype and the possibility of an impaired secretory pattern of brain endothelial cells that could contribute to the progression of this ailment. Finally, we discuss why shall brain endothelial cells be appreciated as a therapeutic target instead of solely an obstacle for delivering treatments to the injured brain in Alzheimer’s disease.
Key Words: dementia, endothelial cells, neurodegeneration, neuroinflammation, neuronal death, paracellular transport, transcellular transport
Introduction
Although the blood-brain barrier (BBB) disruption has been demonstrated to contribute to Alzheimer’s disease (AD) progression, most of the designed treatments underestimate its relevance and do not consider the BBB.
For years, the BBB was thought to be a poorly permeable structure that hinders the free pass of molecules between the brain and the milieu. However, as its study continued, this concept has been redefined, and now a plethora of evidence indicates that more than a barrier itself, the BBB acts as a highly dynamic and selective portal through which several biomolecules pass (Profaci et al., 2020).
Unfortunately, the role of the BBB in AD has been undervalued, and other brain cells have gained more relevance in the development of new treatments. Nevertheless, relevant findings strongly indicate that the importance of the brain endothelial cells (BECs) is not less than other cell types since BECs of AD patients bear the highest number of expressed genes related to AD risk (Yang et al., 2022). This finding calls for further investigation on BECs not only related to their role in the pathogenesis of AD but also regarding the possibility of considering these cells as a therapeutic target.
In this review, we analyze and discuss the possible mechanisms through which BECs could promote and positively modulate AD progression by analyzing the mechanisms of their disruption and impairment of their secretory activity. Furthermore, we also discuss the potential therapeutic agents that could contribute to AD treatment by targeting BECs. Since BECs participate in the development of AD and are one of the main cells affected by this neurodegenerative disease, they could be one of the main targets for treating it.
Search Strategy
Studies cited in this review were published between 1987 and 2023, and were searched on the PubMed, Science Direct, and Web of Science databases using the following keywords: Alzheimer’s disease, blood brain barrier, blood brain barrier transport, neurodegeneration, brain endothelial cells, amyloid beta, brain endothelial cells treatment, tau, and neuroinflammation.
The Blood-Brain Barrier
The BBB is a multi-layered structure composed of a thick glycocalyx, pericytes, a basement membrane, astrocytic endfeet, microglia, and non-fenestrated brain blood vessels, which are formed by BECs that are linked with tight junctions. All these cells and structures form the neurovascular unit (Solár et al., 2022; Varatharaj and Galea, 2017; Figure 1A). Each of these components contributes to the correct function of the BBB, such as keeping an adequate microenvironment for optimal brain function, clearing cellular metabolites and synaptic material, and importing the required nutrients for brain maintenance.
Figure 1.
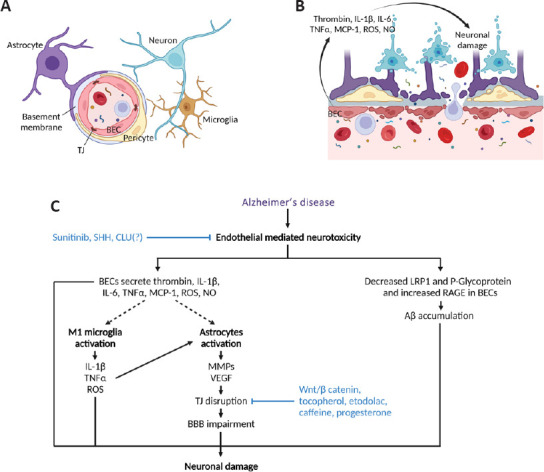
Impaired brain endothelial cells in Alzheimer’s disease.
(A) Elements of a healthy BBB include pericytes, astrocytes, basement membrane, microglia, and BECs closely linked by TJ. (B) In AD, BECs secrete proinflammatory factors that contribute to neuronal damage. (C) Diagram showing how endothelial-mediated neurotoxicity could be targeted in AD to decrease neuronal damage. Whether CLU(?) inhibits endothelial-mediated neurotoxicity remains to be determined. Aβ: Amyloid beta; AD: Alzheimer’s disease; BBB: blood-brain barrier; BEC: brain endothelial cell; CLU: clusterin; IL-1β: interleukin-1β; IL-6: interleukin-6; LRP1: low-density lipoprotein receptor-related protein 1; MCP-1: monocyte chemoattractant protein-1; MMPs: metalloproteinases; NO: nitric oxide; RAGE: receptor for advanced glycation endproducts; ROS: reactive oxygen species; SHH: sonic hedgehog; TJ: tight junction; TNFα: tumor necrosis factor α; VEGF: vascular endothelial growth factor; Wnt: wingless and Int-1. Created with BioRender.com.
Glycocalyx
The glycocalyx is a matrix enriched with proteoglycans, glycoproteins, and glycosaminoglycans. This structure covers the luminal surface of BECs, and its density is higher in the BBB compared to the glycocalyx of the peripheral endothelium (Profaci et al., 2020; Walter et al., 2021). The BBB glycocalyx modulates the immune response, coagulation processes, neuronal homeostasis, and gene expression. Therefore, its correct function is essential for maintaining the BBB integrity (Zhao et al., 2021).
Basal membrane
The basal membrane is an essential non-cellular component of the BBB which is classified as parenchymal basal membrane and endothelial basal membrane. The parenchymal basal membrane is secreted by astrocytes; meanwhile, the endothelial basal membrane is made by secretions of pericytes and BECs. Its biochemical and structural composition consists of a layer with a thickness between 50 and 150 nm, which is highly organized and mainly enriched with collagen IV, laminin, and perlecan. At a functional level, its relevance relies on its role as support for BECs in the BBB (Xu et al., 2018).
Astrocytic endfeet
Astrocytes are a subtype of glial cells that have the ability to regulate the passage of nutrients, including oxygen and glucose, depending on the energy requirements or demand of brain tissue; they are also capable of regulating the dilation of brain arterioles through the secretion of calcium-dependent vasoactive substances (Howarth, 2014). Astrocytes usually have a process called astrocytic endfeet through which these cells make contact with the basal membrane, while wrapping endothelial cells and pericytes (Profaci et al., 2020). The interaction between BECs and astrocytic endfeet is essential for the modulation of blood flow and cerebrospinal fluid in the brain parenchyma, and they stabilize the BBB integrity and barrier function (Howarth, 2014; Profaci et al., 2020).
Pericytes
Pericytes are multipotent cells present in all the vascularized tissues of the body (Liu et al., 2020). Within the brain, they are attached to the basal membrane of the vasculature, and they have important roles, such as modulation of angiogenic processes, permeability, blood flow, and traffic of molecules and immune cells through the BBB (Profaci et al., 2020). Moreover, pericytes have a role in the maintenance and survival of endothelial cells of the BBB through the secretion of factors such as platelet-derived growth factor, angiopoietin 1, transforming growth factor beta, and sphingosine 1 phosphate (Sharma et al., 2019). Pericytes are also involved in BBB permeability and neuroimmune response by increasing BBB leakiness, secretion of cytokines and chemokines, facilitating peripheral immune cell transport, and stimulating resident and recruited immune cells (Erickson and Banks, 2018). Pericytes induce the polarization of astrocytic feet, contributing to the maintenance of the interaction between astrocytic endfeet and BECs (Armulik et al., 2010). These properties make pericytes an essential component of the neurovascular unit.
Microglia
Microglia are innate resident cells of the central nervous system (CNS), derived from the mesoderm in embryonic stages (Ginhoux and Prinz, 2015). These cells are activated in response to pathogens, tissue damage, and toxic substances (Fatoba et al., 2020). When activated, microglia interact with the microvasculature, increasing the BBB’s permeability, producing reactive oxygen species, and promoting lymphocyte and monocyte migration (da Fonseca et al., 2014; Huang et al., 2021).
Brain endothelial cells
In the BBB, BECs are tightly joined by tight junctions formed by the proteins occludin, claudins 1, 3, and 5, cingulin, and zonula occludens ZO-1 and ZO-2. BECs interactions are also reinforced by adherent junctions mediated by cadherins and catenin. BECs have a key role in solute transport across the BBB by paracellular and transcellular mechanisms, and tight junctions prevent paracellular diffusion of large molecules, but low molecular mass solutes can cross the BBB through this route (Sharma et al., 2019). However, the overall consequences of tight junctions are a low permeability of BECs through the paracellular route. Furthermore, transcellular traffic in BECs is also decreased compared to other peripheral endothelial cells. Although its vesicular transport is not as active as other peripheral blood vessels (Profaci et al., 2020), recent evidence has demonstrated that macromolecule trafficking is more dynamic than previously thought since plasma proteins were found in the brain parenchyma after their systemic administration (Yang et al., 2020).
Transport Function of Brain Endothelial Cells
BECs function as a physical portal through which diverse molecules move between the milieu and the CNS; they harbor multiple transport mechanisms across their membrane. The mechanisms of transport employed by molecules are paracellular and transcellular transport.
Paracellular Transport
The intercellular space is the gap amid two BECs that has a width of approximately 1.4 and 1.8 nm. The transport of molecules through the intercellular space is called paracellular transport, and in the BBB, only small molecules can move across it due to the presence of tight junctions such as occludins, claudins, or junctional adhesion molecules along with associated proteins like ZO-1, ZO-2, ZO-3 or cingulin, that together restrict the pass of large molecules. Therefore, paracellular transport is almost nonexistent in the BBB (Yazdani et al., 2019; Profaci et al., 2020).
Transcellular Transport
The main mechanism of transport across the BBB is transcellular transport which is the traffic of molecules across the BEC membrane. Transcellular transport can be divided into three main types: passive diffusion, carrier-mediated transport, and transcytosis (Yazdani et al., 2019).
Passive diffusion
This mechanism occurs mainly with hydrophobic molecules, which due to their characteristics and molecular weight (less than 500 DA), are capable of crossing both the apical and basolateral domains of BECs (Yazdani et al., 2019).
Carrier mediated transport
Solute carriers transport their specific solutes by passive or active mechanisms; these solutes play vital roles in energy metabolism and the proper functioning of all CNS components. Among these solutes are carbohydrates, amino acids, nucleotides, inorganic ions, fatty acids, vitamins, amines, cholines, monocarboxylic acids, and hormones (Sweeney et al., 2019; Yazdani et al., 2019; Profaci et al., 2020).
Carriers that move molecules against their concentration gradient require energy provided by ATP hydrolysis. This mechanism of transport moves proinflammatory molecules, drugs, xenobiotics, and nucleosides outside the CNS (Sweeney et al., 2019).
ABC transporters employ ATP to transport solute across the cell membrane. They are multi-domain integrated membrane proteins (Pereira et al., 2018) with two nucleotide-binding domains involved in transportation, and two transmembrane domains, related to transfer across lipid membranes (Bakos and Homolya, 2007). Remarkably, ABC transporters are unidirectional, from the cytoplasm to the blood, and are expressed in endothelial cells.
Transcytosis
Transcytosis is a cellular process of moving macromolecules from one surface or domain of a polarized cell to another (Fung et al., 2018).
Receptors can mediate the movement of many proteins and large peptides through the BBB. The ligand-receptor interaction triggers an endocytic event which forms vesicles that envelope the ligand-receptor complex; afterward, the ligand passes across the BECs and, through an exocytic mechanism, is released in the opposite compartment of the cell (Abbott et al., 2010; Ayloo and Gu, 2019; Sweeney et al., 2019). This transport system is called receptor-mediated transcytosis (RMT). RMT is specific as it involves ligand-receptor interaction. This transport has been described for transferrin, insulin, growth factors, and lipoproteins, whose receptors are located or recruited in clathrin-coated pits that are enriched in the brain endothelium (Hervé et al., 2008; Pulgar, 2019; Sharma et al., 2019).
In contrast, absorptive-mediated transcytosis (AMT) involves the unspecific interaction of a ligand with a moiety at the luminal surface of endothelial cells which is driven by ionic forces. Several cationic proteins such as albumin, protamine, avidin, and histone use AMT to enter the brain parenchyma. Cationic molecules bind to the luminal and abluminal side of BECs through anionic sites formed by the glycocalyx components such as heparan sulfate and chondroitin sulfate. Some studies have demonstrated the participation of caveolae in AMT, however, caveolae-independent AMT can also occur, this was observed for the transport of synthetic peptides B, additionally, clathrin-coated pits in the luminal side of BECs are negatively charged, suggesting their interaction with cationic proteins (Hervé et al., 2008).
The two main mechanisms of transcytosis in BECs are clathrin and caveolae-mediated transcytosis. The main difference between them is the type of molecules that coat their vesicles which are clathrins and caveolins, respectively (Ayloo and Gu, 2019; Yazdani et al., 2019). Both mechanisms of transport are susceptible to changes derived from insults or aging.
Stroke alters the transport across BECs and promotes the breakdown of the BBB in an ordered manner. First, transcellular trafficking mediated by caveolae increases within hours with a modest leakage at the beginning of the insult; however, after almost a day of the insult, the increase of BBB permeability is evident, which suggests that the impairment of transcellular trafficking comes first before the alteration of paracellular transport (Knowland et al., 2014).
Remarkably, as individual ages, caveolae transport increases meanwhile, clathrin transport decreases (Yang et al., 2020). Therefore, it will be interesting to know whether the increased caveolae transcytosis is a conserved mechanism of brain injury that could also be present in neurodegenerative diseases such as AD.
The Blood-Brain Barrier in Alzheimer’s Disease
AD is a progressive neurodegenerative disorder and the leading cause of dementia worldwide (Deture and Dickson, 2019). Clinically, it begins with mild cognitive impairment that worsens over the years in a significant percentage of the patients that develop severe cognitive impairment, defects in visual space perception, and motor alterations with emotional and behavioral changes (Tarawneh and Holtzman, 2012). AD correlates with neuronal death and atrophy in the cerebral cortex and hippocampus. Senile plaques and neurofibrillary tangles are considered hallmarks of AD that are accompanied by neurodegeneration, loss of synapses, neuroinflammation, and oxidative stress (Nelson et al., 2009; Goel et al., 2022). Amyloid beta peptide (Aβ) is the main constituent of senile plaques; mutations in amyloid precursor protein (APP), presenilin 1 and 2 (PSEN1, PSEN2) induce Aβ aggregates in heritable AD (Petit et al., 2022). Amyloid plaques are related to degenerated nerve endings and are higher in aged individuals (Sadigh-Eteghad et al., 2015).
Interestingly, epidemiological studies associate AD with vascular risk factors (Breteler, 2000) since vascular dysregulation from the early stage of the disease contributes to its progression (Iturria-Medina et al., 2016). Aβ is toxic to the cerebral and peripheral endothelium, generating neurotoxicity since it causes endothelium-dependent vasoconstriction (Sutton et al., 1997). The accumulation of Aβ is also observed at the micro and macrovascular level, which promotes cerebral amyloid angiopathy, which culminates in severe cerebral vascular dysfunction (Saito et al., 2021). There is evidence that Tau pathology also causes cerebral vascular dysfunction in the hippocampus, which correlates with loss of the BBB integrity (Canepa and Fossati, 2021). Moreover, Tau can generate vascular dysfunction as neurofibrillary tangles formed by the deposition of phosphorylated Tau in the microvasculature disrupt the vessels and cause vascular inflammation (Cisternas et al., 2019). Finally, phosphorylated Tau is also abundant in perivascular spaces and may contribute to neurovascular pathology if its clearance is decreased (Canepa and Fossati, 2021).
Furthermore, current evidence also suggests that vascular dysfunction plays a significant role in AD. Senile plaques and neurofibrillary tangles may result from inadequate blood supply (Kelleher and Soiza, 2013). Furthermore, BECs may contribute to the formation of amyloid deposits around blood vessels because they express secretases that cleave the APP protein, which allows its accumulation (D’Uscio et al., 2017).
Remarkably, BECs are the cell type of the brain that expresses the largest amount of AD risk genes; some of them are associated with immune and inflammatory responses (Wang et al., 2012; Yang et al., 2022). Therefore, the altered BEC phenotype in AD suggests the participation of these cells in the onset and progression of the disease. In fact, there are multiple alterations in the endothelial cells, such as increased permeability, dysfunctional transport, and secretion of proinflammatory factors that are described below.
Although paracellular transport in the BBB is minimal in physiological conditions, multiple studies suggest that this process is impaired in AD. Recently it was demonstrated that AD brains displayed an abnormal distribution pattern of the tight junction protein Claudin 5 in blood vessels, thus suggesting a disruption of the BBB impermeability (Soto-Rojas et al., 2021a). This idea is supported by findings of plasma proteins in AD brains and imaging studies indicate that BBB disruption can be appreciated even in early AD patients (van de Haar et al., 2016; Sweeney et al., 2018).
BBB impairment in AD patients has been evidenced by gadolinium and protein infiltration in brain areas such as the hippocampus, prefrontal cortex, and entorhinal cortex (Alajangi et al., 2022; Kurz et al., 2022). In AD, BBB breakdown has been associated with PSEN1 mutations, apolipoprotein ε4 (APOE4) allele, metalloprotease-9 (MMP-9) expression, cyclophilin A, APP mutations, and Aβ aggregation (Alajangi et al., 2022).
BBB impairment alters permeability and clearance of substances, cell infiltration, and cerebral blood flow. MMPs are known to degrade tight junctions and basal membranes, causing BECs damage and detachment, pericyte loss, astrocytic endfeet detachment, and thus BBB damage (Knox et al., 2022). In contrast, brain damage activates microglia into proinflammatory M1 phenotype that secretes inflammatory mediators such as tumor necrosis factor α (TNF-α) and interleukin (IL)-1β and produces reactive oxygen species, which in turn activates astrocytes that secrete MMPs causing disruption of tight junctions and immune cells infiltration. Astrocytes also secrete vascular endothelial growth factor that downregulates occludin and claudin 5 and induces the proliferation and migration of BECs (Archie et al., 2021).
Furthermore, BBB damage in AD is widely associated with impaired Aβ clearance since the expression of low-density lipoprotein receptor-related protein 1, and P-glycoprotein transporters that remove Aβ from brain parenchyma is reduced in BECs, while receptor for advanced glycation endproducts (RAGE) that performs the opposite role is increased, thus promoting Aβ deposition in the brain (Dib et al., 2021; Gong and Jia, 2022; Kurz et al., 2022).
Recent findings show that insulin receptors (IR) are more expressed in BECs than in brain parenchyma (Yang et al., 2022; Leclerc et al., 2023), suggesting a crucial role of the endothelium in the transport of insulin and glucose through the pathway initiated by this hormone. Furthermore, IR activation occurs mainly in BECs instead of brain parenchyma. However, in an AD rodent model, this activation is not observed, thus suggesting a dysfunctional insulin signaling in this neurodegenerative disease (Leclerc et al., 2023). It remains to be studied whether in AD the IR remain internalized in BECs, or whether they activate alternative insulin signaling pathways. Recently, it was reported that insulin transport across BECs is mediated by clathrin in vitro. Since aging shifts clathrin-mediated transport to caveolae-mediated transport in BECs, this change could impair insulin signaling and contribute to AD onset (Yang et al., 2020; Pemberton et al., 2022).It was also demonstrated that the transport of insulin in the hypothalamus is mediated by caveolae (Pemberton et al., 2022); thus, the insulin transport might depend on the brain regions where endocytosed. These results encourage further investigation of the mechanisms of insulin transport across the BBB in the different areas of the central nervous system.
In 1999, it was discovered that endothelial cells from AD patients promote neurotoxicity. These findings gave rise to the endothelial-mediated neurotoxicity hypothesis, which proposes that BECs produce soluble factors capable of harming or killing neurons (Grammas et al., 1999). Multiple studies have demonstrated that in AD, BECs secrete proinflammatory factors such as thrombin, IL-1β, IL-6, TNF-α or monocyte chemoattractant protein-1 that could contribute directly to neuronal damage (Figure 1B and C; Grammas and Ovase, 2001; Yin et al., 2010; Grammas et al., 2014). Furthermore, BECs also secrete molecules that induce neuronal damage, such as reactive oxygen species or nitric oxide, which are overproduced in BECs from AD patients (Grammas, 2000).
Additionally, proinflammatory factors also act on BECs (Versele et al., 2022). Therefore, it is likely that BECs are susceptible to peripheral inflammation and, in turn, respond to peripheral cytokines that could modify their phenotype. In fact, BECs can secrete proinflammatory factors in response to systemic inflammation, altering brain function (Skelly et al., 2013). Interestingly, AD patients display elevated peripheral inflammatory cytokines (Tan et al., 2007) which could activate BECs and stimulate them to secrete proinflammatory factors inside the brain and promote neurodegeneration.
Altogether, this information indicates a central role of BECs in AD pathophysiology. Notwithstanding, BECs have historically not been considered with the same importance as other cell types and molecules to develop therapeutic strategies against this neurodegenerative condition.
Targeting the Blood-Brain Barrier as a Potential Treatment for Alzheimer’s Disease
Since 2003, the Food and Drug Administration has not approved new treatments for AD. The BBB is a limitation for the emergence of new therapies given that the vast majority of small molecules and biological drugs do not cross it. Therefore, several approaches are aimed at increasing its permeability and penetration into the cerebral parenchyma (Pardridge, 2020). Although BBB damage in AD has been evidenced in several studies in human and animal models, most drugs designed for AD treatment have shown poor effectiveness due to their inability to enter the CNS.
Pharmacological treatment of AD has been focused on cholinergic transmission, Aβ and Tau pathologies, neuroinflammation, and cholesterol metabolism (Nehra et al., 2022). Only small molecules such as acetylcholinesterase inhibitors (198–380 Da) and memantine (179 Da) can enter the brain parenchyma but do not significantly improve AD symptoms. Aβ secretases inhibitors and neurotrophins such as nerve growth factor and brain-derived neurotrophic factor have been evaluated without success since their high molecular weight or polarity results in low CNS penetration. Anti-Aβ antibodies have been proposed as a promising alternative for AD treatment. However, only aducanumab has shown brain penetration and beneficial effects in Aβ plaque reduction, but this effect was associated with BBB disruption since this drug causes cerebral microhemorrhages and vascular edema, which positively correlate with Aβ decrease (Pardridge, 2020).
BECs microvasculature expresses the transferrin receptor 1, hence this receptor has been tested for drug delivery into the brain via transcytosis. However, transferrin transport across brain endothelial cells has been controversial, despite transferrin transcytosis was demonstrated in rats by the presence of 125I-labelled transferrin in the brain parenchyma, it was mainly accumulated in the brain endothelium, this evidence was supported by the use of a radiolabeled antibody against transferrin receptor ([125I]OX26) which was found mainly in BECs and extravascular [125I]OX26 was observed only in periventricular neurons (Fishman et al., 1987; Moos and Morgan, 2001). Moreover, the Divalent metal transporter 1 (DMT1), which transports iron either bound or unbound to transferrin, was detected in primary cultures of BECs, thus suggesting the transport of iron through this transporter and not by transferrin-mediated transcytosis. However, the absence of transferrin transcytosis cannot be completely assumed since several works reported the absence of the DMT1 transporter, in brain endothelial cells. Taking together this information, two mechanisms for brain iron transport have been proposed, transferrin-mediated endocytosis through DMT1, and transferrin transcytosis across BECs without DMT1 participation (Skjørringe et al., 2015; Johnsen et al., 2019).
TfR-mediated transport has been tested as a therapeutic approach. Several types of TfR ligands have been employed such as antibodies, liposomes, and nanoparticles. One of the most evaluated strategies is the use of antibodies against TfR conjugated with neuroprotective drugs such as BDNF or epidermal growth factor, with positive results in preclinical models, although with a very low percentage of brain uptake (Johnsen et al., 2019). Interestingly, it was reported that the reduced affinity between the antibody and TfR enhances transcytosis and antibody uptake in the brain parenchyma, this was assessed by designing an antibody with low affinity for TfR and high affinity for beta-secretase 1 secretase, observing its accumulation in the mouse brain and the reduction of Aβ with a single systemic dose (Yu et al., 2011).
Transcytosis-mediated delivery of gold nanoparticles (AuNP) was also probed using a monoclonal antibody for mouse transferrin receptors. AuNPs coupled to transferrin antibody were injected in the caudal vein of mice and followed at 10 minutes, 30 minutes, 2.5 hours, and 24 hours after administration. AuNPs were observed in the brain at all temporal times increasing gradually with time and within different localizations. Some were located at the luminal membrane of endothelial cells in blood capillaries, other AuNPs were in the wall of clathrin-coated pits of the luminal membrane and inside of lysosomal and non-lysosomal vesicles. Only a few particles were found at the basal membrane of the endothelium, however, no AuNP particles were observed beyond the basal membrane of brain capillaries (Cabezón et al., 2015).
The fusion of the TNF receptor 2 extracellular domain with the heavy chain of a monoclonal antibody against IR or transferrin receptor has been developed to mediate TNF receptor 2 brain entry by RMT and reduce neuroinflammation by sequestering TNF-α. RMT strategy was also applied with the erythropoietin receptor to promote neurite outgrowth. Moreover, the RMT delivery system increased brain uptake to 1–3% ID/brain (Pardridge, 2020). These proposals have a high potential and shall be explored in depth.
IR-mediated transcytosis has been also employed for brain drug delivery. Preclinical studies developed humanized anti-IR antibodies conjugated with an anti-Aβ antibody, iduronate 2-sulfatase, arylsulfatase, and N-sulfoglucosamine sulfohydrolase showing a brain uptake between 1–3% ID/100 g tissue (Pulgar, 2019). However, brain insulin transport by transcytosis is controversial. A recent work showed that insulin receptor blockade or its deficiency did not affect insulin transport across BBB in mice (Rhea et al., 2018).
Several studies have shown that increasing the function of ABC transporters, such as ABCB1, ABCA1, ABCG2, and ABCC1, has beneficial effects in AD models, promoting the clearing of Aβ peptides (Abuznait and Kaddoumi, 2012).
ABCB1 is highly expressed in neurocapillary endothelial cells and has lower concentrations in neurons, astrocytes, and pericytes. ABCB1 mediates Aβ efflux, probably facilitating the transfer of endocytosed Aβ from the endothelial cells to plasma. ABCB1 also inhibits the penetrability of soluble Aβ, transported by RAGE, from the apical to the basolateral side of capillary endothelial cells (Behl et al., 2021).
ABCA1 is expressed in astrocytes, microglial cells, neurons, and brain capillary endothelial cells (Fujiyoshi et al., 2007) and, importantly, regulates the efflux of phospholipids and cholesterol from microglial cells and astrocytes, increasing the lipidation of apoE (Wolf et al., 2012), altering APP processing, and facilizing Aβ degradation (Behl et al., 2021). ABCA1 expression decreases Aβ formation, secretion, and deposition (Terwel et al., 2011). Conversely, the absence of ABCA1 decreased lipidation and expression of apoE and increased insoluble Aβ (Koldamova et al., 2005).
ABCG2 is abundant in the luminal membrane of endothelial cells of the nervous system and multiple organs (Tachikawa et al., 2005). It also engages in Aβ transport, impeding Aβ entry into the brain (Do et al., 2012) and maintains the BBB structure. ABCG2 expression is enhanced in neurons of patients with AD and inhibits oxidative stress and inflammatory response by interacting with the cell signaling of the nuclear factor κB (Abuznait and Kaddoumi, 2012).
ABCC1 (multidrug resistance protein 1) is expressed on the luminal side of the BBB and also in neurons, pericytes, microglia, and astrocytes (Wolf et al., 2012). Knock-out mice have high Aβ in the brain, but the enzymes that produce Aβ are normal, suggesting a role in Aβ clearance (Krohn et al., 2011). ABCC1 participates in the endocytosis of Aβ facilitated by lipoprotein receptor-related protein 1 (Kanekiyo and Bu, 2014). The role of ABC transporters in Aβ clearance and its notable expression in BECs make them a good therapeutic target for AD treatment.
The inhibition of RAGE is a possible approach to decrease the effects of its activation and inflammation.
Anti-RAGE antibody inhibits inflammation, cytokines, and RAGE expression. It also prevents monocyte infiltration, thus avoiding pro-inflammatory responses and neurotoxicity. BBB permeability is the most critical obstacle for using anti-RAGE antibodies (Kozyrev et al., 2020).
FPS-ZM1 is a small inhibitor that blocks the binding of the V-domain and Aβ1–42 (Deane et al., 2012) and inhibits the activation of microglia by advanced glycation endproducts, oxidative stress, and inflammation.
Azeliragon (PF-04494700) is a small molecule that antagonizes the RAGE pathway by inhibiting the binding of the extracellular domain of RAGE to Aβ1–42, with a reduction in neuroinflammation, levels of Aβ1–42 and inflammatory cytokines (Burstein et al., 2018). However, a phase III study discovered that it did not exhibit a beneficial effect (2-Year Extension Study of Azeliragon in Subjects with Alzheimer’s Disease (STEADFAST Extension) - Full Text View - ClinicalTrials.gov, n.d.).
Several compounds, such as tranilast, emetine, aminopyrimidine derivatives, and phenyl benzoxazoles with effects in Aβ clearance, can have an effect and can be tested (Singh and Agrawal, 2022).
BECs receptors and transporters reduce Aβ deposition and could be valuable targets for therapeutic agents, and more research is needed to explore this possibility (Zhang et al., 2022). Most of the studies consider the BBB as an obstacle for drug delivery to the CNS in AD patients. However, few of them consider the BBB not only as a delivery barrier but also as a therapeutic target. Therefore, modest emphasis has been made on targeting BECs to treat AD.
The endothelial-mediated neurotoxicity hypothesis, together with the cumulative evidence of increased BBB leakiness in AD, suggests that BECs are a promising therapeutic target. Preclinical studies demonstrated that targeting the proinflammatory phenotype of BECs with the multikinase inhibitor Sunitinib decreased TNF-α, IL1-β, thrombin, and Aβ expression in BECs and improved cognitive functions in AD murine models (Grammas et al., 2014). Moreover, the plasma-derived protein clusterin also decreased the proinflammatory phenotype of BECs in another AD rodent model (de Miguel et al., 2021), suggesting that there are proteins and drugs capable of restoring the altered BECs in AD. Interestingly, these effects could be promoted without the need to cross the BBB; thus, it could be highly feasible to target BECs by systemic drug delivery. This information suggests that BECs are a potential target to treat AD that needs to be further explored.
There are other potential therapeutic agents with possibilities of producing their beneficial effects in the brain without crossing the BBB. One of them is the Gap27 antibody, which inactivates connexins after binding to them and therefore inhibiting neuroinflammation. It was demonstrated that Gap27 inhibited reactive gliosis in an inflammation model based on lipopolysaccharide exposure. Since reactive gliosis is a hallmark of neurodegenerative diseases such as AD (Fakhoury, 2018), it is possible that blocking connexin activity at the luminal side of BECs with Gap27 could represent a feasible therapeutic strategy to treat these ailments (de Bock et al., 2022).
Targeting caveolae transport mechanisms in BECs of AD patients could also represent a potential therapeutic strategy. It has been proposed that inhibiting caveolae-mediated endocytosis with the statin atorvastatin could aid in the treatment of AD since its blockade could increase the transforming growth factor beta signaling (Fessel, 2020). Remarkably, statin treatment correlates with a decrease of AD risk (Jeong et al., 2021) and its potential to treat AD should be further explored.
Another interesting approach to identifying possible therapeutic strategies is the study of molecules involved in the development of the BBB. Recent studies have demonstrated that components of the Wingless and Int-1 (Wnt) signaling, which are involved in BBB development, are decreased in an AD rodent model. Remarkably, the activation of Wnt/β catenin signaling restored the levels of Claudin-5, ZO-1, and the glucose transporter Glut1, thus decreasing BBB leakage induced by Aβ (Wang et al., 2022). These results suggest that the molecules that modulate the development of the BBB could restore BECs in neurodegenerative conditions. Another morphogen that participates in BBB development is Sonic Hedgehog (SHH) (Alvarez et al., 2011). Curiously, SHH can reduce proinflammatory cytokines on BECs such as IL-8 or monocyte chemoattractant protein-1, which are increased in BECs of AD patients (Grammas and Ovase, 2001; Grammas et al., 2006; Alvarez et al., 2011). This information suggests that SHH could be studied as a potential therapeutic agent to restore the BBB in AD; nevertheless, there is contrasting evidence with respect to its role in this neurodegenerative disease that should be considered when interpreting the results of SHH on BECs and AD (Yang et al., 2021). In this regard, other morphogens should be explored in the near future as potential candidates to improve BEC integrity and function as a therapeutic approach to treat AD by targeting cerebral vasculature.
Significantly, paracellular transport in BECs is also impaired in AD (Soto-Rojas et al., 2021b). Addressing this impairment by restoring tight junction proteins could provide promising therapeutic approaches for this neurodegenerative disease (Sharma et al., 2022). Mice carrying the APOE4 allele display a conspicuous leakage at the BBB that is accompanied by an overexpression of the proinflammatory cytokine CypA mainly in pericytes; since these BBB alterations were associated with neural dysfunction, the restoration of BBB leakage through anti-inflammatory agents could be used as a therapeutic strategy for AD treatment. Notably, the treatment with cyclosporine decreased BBB permeability and the activity of MMP-9. The action of cyclosporine was proposed to be on MMP-9 derived from pericytes (Bell et al., 2012); whether cyclosporine could also act on BECs to decrease thrombin or fibrin in brain parenchyma is unknown. In contrast, inhibition of the mammalian target of rapamycin signaling directly in BECs with rapamycin improved BBB integrity by upregulating tight junction proteins and decreasing their permeability to fibrinogen (van Skike et al., 2018), however, whether mammalian target of rapamycin inhibition also restores the secretory phenotype of BECs remains to be seen. Other studies using tocopherol and etodolac also improved BBB integrity by increasing its impermeability and restoring ZO-1 and Claudin 5 levels (Elfakhri et al., 2019). However, the authors did not perform behavioral tests and did not evaluate whether these molecules also had a direct effect on the secretory pattern of BECs.
Caffeine restores BBB integrity in an AD mice model by decreasing BBB leakage and increasing the expression of ZO-1. Additionally, cognitive impairments were attenuated with this treatment. Interestingly, there was a reduction of thrombin and proinflammatory cytokines such as TNF-α and IL-1β in mice brains (Kim et al., 2022). This evidence also suggests that not only is the integrity of the BBB restored but also the secretory phenotype of BECs since the decrease of thrombin, TNF-α and IL-1β in the brain could also be derived from a decreased production or transport of them from BECs given that the microvasculature releases these molecules in AD (Grammas and Ovase, 2001; Yin et al., 2010). Remarkably, rivaroxaban, an inhibitor of the Xa coagulation factor, had similar effects on the expression of these neurotoxic factors in the same animal model, either in brain parenchyma or BECs (Kim et al., 2022).
Interestingly, it was observed that progesterone reduces MMP-9 levels and increases the expression of claudins 1 and 5, improving BBB integrity; moreover, progesterone decreases oxidative stress and inflammation, having a protecting role in CNS and although this hormone could target BECs, it remains to be determined (Alajangi et al., 2022).
Viral vectors have been proposed for gene delivery to the CNS, adeno-associated viruses (AAV) are considered suitable because of their safety and long-term expression in CNS. Modifications of AAV capsid improve its crossing through the BBB, such as AAV-BR1 capsid which mediates the selective transduction of mouse brain and spinal cord microvasculature, or AAV-PHP.eB based on Cre recombinase for the specific targeting of neurons that showed a transduction efficiency of 69% and 55% in cortical and striatal neurons, respectively, after its systemic administration in mice (Chan et al., 2017; Körbelin et al., 2016).
Finally, stem cell treatment also shows significant results at the preclinical and clinical stages (Brody et al., 2022; López-Ornelas et al., 2022; Gonçalves et al., 2023). The most employed in preclinical and clinical models are mesenchymal stem cells and neural stem cells (López-Ornelas et al., 2022). Interestingly, phase I clinical trials with mesenchymal stem cells, which are immunomodulatory, demonstrated an increase of anti-inflammatory cytokines in serum samples of AD patients (Gao et al., 2016; Brody et al., 2022), however, whether this treatment also had an effect on BECs by modulating their inflammatory profile was not determined and should be explored in the future.
Conclusion
New technologies and research approaches have shed light on the cell biology of BECs, which are now recognized to have a key role in brain physiology and the pathophysiology of neurodegenerative diseases. Although emerging literature has highlighted the relevance of BBB alteration to promote or maintain AD, there is not enough focus on strategies for repairing BBB impairments, and most of the studies consider the BBB as an obstacle to deliver drugs rather than as a therapeutic target.
Throughout the years most of the therapeutic strategies for treating neurodegenerative diseases have been focused on improving the passing of candidate drugs across the BBB to reach brain parenchyma, however, there is a lack of attention on the healing of the impaired elements of the BBB in AD, such as BECs which are one of the most altered cells in this ailment. This approach has been poorly tested and BECs should now be considered in the design of new treatments in the future. The more the BECs and other components of the BBB are considered as targets to treat AD and other neurodegenerative diseases, the higher the odds a potential treatment will have to thrive through clinical trials.
Additional file: Open peer review report 1 (79.1KB, pdf) .
Acknowledgments:
We thank the academic workshop Comunicación entre las células del Sistema nervioso y su implicación en enfermedades neurológicas of Universidad Nacional Autónoma de México for the academic support to Alejandro Rodríguez-Oviedo. We also thank B.A. Sharon Lezama (Excelsior University, Albany, NY, USA) for revising the paper.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement: The data are available from the corresponding author on reasonable request.
Open peer reviewer: Erik J. Behringer, Loma Linda University, USA.
P-Reviewer: Behringer EJ; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Abuznait AH, Kaddoumi A. Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci. 2012;3:820–831. doi: 10.1021/cn300077c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alajangi HK, Kaur M, Sharma A, Rana S, Thakur S, Chatterjee M, Singla N, Jaiswal PK, Singh G, Barnwal RP. Blood-brain barrier:emerging trends on transport models and new-age strategies for therapeutics intervention against neurological disorders. Mol Brain. 2022;15:49. doi: 10.1186/s13041-022-00937-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 5.Archie SR, al Shoyaib A, Cucullo L. Blood-brain barrier dysfunction in CNS disorders and putative therapeutic targets:an overview. Pharmaceutics. 2021;13:1779. doi: 10.3390/pharmaceutics13111779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 7.Ayloo S, Gu C. Transcytosis at the blood-brain barrier. Curr Opin Neurobiol. 2019;57:32–38. doi: 10.1016/j.conb.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakos É, Homolya L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1) Pflugers Arch. 2007;453:621–641. doi: 10.1007/s00424-006-0160-8. [DOI] [PubMed] [Google Scholar]
- 9.Behl T, Kaur I, Sehgal A, Kumar A, Uddin MS, Bungau S. The interplay of ABC transporters in Aβtranslocation and cholesterol metabolism:implicating their roles in Alzheimer's disease. Mol Neurobiol. 2021;58:1564–1582. doi: 10.1007/s12035-020-02211-x. [DOI] [PubMed] [Google Scholar]
- 10.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breteler MMB. Vascular risk factors for Alzheimer's disease:an epidemiologic perspective. Neurobiol Aging. 2000;21:153–160. doi: 10.1016/s0197-4580(99)00110-4. [DOI] [PubMed] [Google Scholar]
- 12.Brody M, Agronin M, Herskowitz BJ, Bookheimer SY, Small GW, Hitchinson B, Ramdas K, Wishard T, McInerney KF, Vellas B, Sierra F, Jiang Z, Mcclain-Moss L, Perez C, Fuquay A, Rodriguez S, Hare JM, Oliva AA, Jr, Baumel B. Results and insights from a phase I clinical trial of Lomecel-B for Alzheimer's disease. Alzheimers Dement. 2022;19:261–273. doi: 10.1002/alz.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer's disease. J Prev Alzheimers Dis. 2018;5:149–154. doi: 10.14283/jpad.2018.18. [DOI] [PubMed] [Google Scholar]
- 14.Cabezón I, Manich G, Martín-Venegas R, Camins A, Pelegrí C, Vilaplana J. Trafficking of gold nanoparticles coated with the 8D3 anti-transferrin receptor antibody at the mouse blood-brain barrier. Mol Pharm. 2015;12:4137–4145. doi: 10.1021/acs.molpharmaceut.5b00597. [DOI] [PubMed] [Google Scholar]
- 15.Canepa E, Fossati S. Impact of Tau on neurovascular pathology in Alzheimer's disease. Front Neurol. 2021;11:573324. doi: 10.3389/fneur.2020.573324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cisternas P, Taylor X, Lasagna-Reeves CA. The amyloid-Tau-neuroinflammation axis in the context of cerebral amyloid angiopathy. Int J Mol Sci. 2019;20:6319. doi: 10.3390/ijms20246319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.da Fonseca ACC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, Lima FRS. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362. doi: 10.3389/fncel.2014.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bock M, de Smet M, Verwaerde S, Tahiri H, Schumacher S, van Haver V, Witschas K, Steinhäuser C, Rouach N, Vandenbroucke RE, Leybaert L. Targeting gliovascular connexins prevents inflammatory blood-brain barrier leakage and astrogliosis. JCI Insight. 2022;7:e135263. doi: 10.1172/jci.insight.135263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Miguel Z, Khoury N, Betley MJ, Lehallier B, Willoughby D, Olsson N, Yang AC, Hahn O, Lu N, Vest RT, Bonanno LN, Yerra L, Zhang L, Saw NL, Fairchild JK, Lee D, Zhang H, McAlpine PL, Contrepois K, Shamloo M, et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature. 2021;600:494–499. doi: 10.1038/s41586-021-04183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ, Thiyagarajan M, Zarcone T, Fritz G, Friedman AE, Miller BL, Zlokovic BV. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener 2019. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dib S, Pahnke J, Gosselet F. Role of ABCA7 in human health and in Alzheimer's disease. Int J Mol Sci. 2021;22:4603. doi: 10.3390/ijms22094603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Do TM, Noel-Hudson MS, Ribes S, Besengez C, Smirnova M, Cisternino S, Buyse M, Calon F, Chimini G, Chacun H, Scherrmann JM, Farinotti R, Bourasset F. ABCG2- and ABCG4-mediated efflux of amyloid-βpeptide 1-40 at the mouse blood-brain barrier. J Alzheimers Dis. 2012;30:155–166. doi: 10.3233/JAD-2012-112189. [DOI] [PubMed] [Google Scholar]
- 25.D'Uscio LV, He T, Katusic ZS. Expression and processing of amyloid precursor protein in vascular endothelium. Physiology (Bethesda) 2017;32:20–32. doi: 10.1152/physiol.00021.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elfakhri KH, Abdallah IM, Brannen AD, Kaddoumi A. Multi-faceted therapeutic strategy for treatment of Alzheimer's disease by concurrent administration of etodolac and α-tocopherol. Neurobiol Dis. 2019;125:123–134. doi: 10.1016/j.nbd.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson MA, Banks WA. Neuroimmune axes of the blood-brain barriers and blood-brain interfaces:bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. 2018;70:278–314. doi: 10.1124/pr.117.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fakhoury M. Microglia and astrocytes in Alzheimer's disease:implications for therapy. Curr Neuropharmacol. 2018;16:508–518. doi: 10.2174/1570159X15666170720095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatoba O, Itokazu T, Yamashita T. Microglia as therapeutic target in central nervous system disorders. J Pharmacol Sci. 2020;144:102–118. doi: 10.1016/j.jphs.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Fessel J. Caveolae, CD109, and endothelial cells as targets for treating Alzheimer's disease. Alzheimers Dement (N Y) 2020;6:e12066. doi: 10.1002/trc2.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fishman JB, Rubin JB, Handrahan JV, Connor JR, Fine RE. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J Neurosci Res. 1987;18:299–304. doi: 10.1002/jnr.490180206. [DOI] [PubMed] [Google Scholar]
- 32.Fujiyoshi M, Ohtsuki S, Hori S, Tachikawa M, Terasaki T. 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J Neurochem. 2007;100:968–978. doi: 10.1111/j.1471-4159.2006.04240.x. [DOI] [PubMed] [Google Scholar]
- 33.Fung KYY, Fairn GD, Lee WL. Transcellular vesicular transport in epithelial and endothelial cells:Challenges and opportunities. Traffic. 2018;19:5–18. doi: 10.1111/tra.12533. [DOI] [PubMed] [Google Scholar]
- 34.Gao F, Chiu SM, Motan DAL, Zhang Z, Chen L, Ji HL, Tse HF, Fu QL, Lian Q. Mesenchymal stem cells and immunomodulation:current status and future prospects. Cell Death Dis. 2016;7:e2062. doi: 10.1038/cddis.2015.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginhoux F, Prinz M. Origin of microglia:current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015;7:a020537. doi: 10.1101/cshperspect.a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goel P, Chakrabarti S, Goel K, Bhutani K, Chopra T, Bali S. Neuronal cell death mechanisms in Alzheimer's disease:an insight. Front Mol Neurosci. 2022;15:409. doi: 10.3389/fnmol.2022.937133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonçalves R de J, Vasques J, da Silva-Junior A, Gubert F, Mendez-Otero R. Mesenchymal stem cell- and extracellular vesicle-based therapies for Alzheimer’s disease:progress, advantages, and challenges. Neural Regen Res. 2023;18:1645–1651. doi: 10.4103/1673-5374.361546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong M, Jia J. Contribution of blood-brain barrier-related blood-borne factors for Alzheimer's disease vs. vascular dementia diagnosis:A pilot study. Front Neurosci. 2022;16:1292. doi: 10.3389/fnins.2022.949129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grammas P, Moore P, Weigel PH. Microvessels from Alzheimer's disease brains kill neurons in vitro. Am J Pathol. 1999;154:337–342. doi: 10.1016/S0002-9440(10)65280-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grammas P. A damaged microcirculation contributes to neuronal cell death in Alzheimer's disease. Neurobiol Aging. 2000;21:199–205. doi: 10.1016/s0197-4580(00)00102-0. [DOI] [PubMed] [Google Scholar]
- 41.Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels in Alzheimer's disease. Neurobiol Aging. 2001;22:837–842. doi: 10.1016/s0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- 42.Grammas P, Samany PG, Thirumangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer's disease microvessels:implications for disease pathogenesis. J Alzheimers Dis. 2006;9:51–58. doi: 10.3233/jad-2006-9105. [DOI] [PubMed] [Google Scholar]
- 43.Grammas P, Martinez J, Sanchez A, Yin X, Riley J, Gay D, Desobry K, Tripathy D, Luo J, Evola M, Young A. A new paradigm for the treatment of Alzheimer's disease:targeting vascular activation. J Alzheimers Dis. 2014;40:619–630. doi: 10.3233/JAD-2014-132057. [DOI] [PubMed] [Google Scholar]
- 44.Hervé F, Ghinea N, Scherrmann JM. CNS delivery via adsorptive transcytosis. AAPS J. 2008;10:455–472. doi: 10.1208/s12248-008-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howarth C. The contribution of astrocytes to the regulation of cerebral blood flow. Front Neurosci. 2014;8:103. doi: 10.3389/fnins.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption:effects and mechanisms. CNS Neurosci Ther. 2021;27:36–47. doi: 10.1111/cns.13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Alzheimer's Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeong SM, Shin DW, Yoo TG, Cho MH, Jang W, Lee J, Kim SY. Association between statin use and Alzheimer's disease with dose response relationship. Sci Rep. 2021;11:15280. doi: 10.1038/s41598-021-94803-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnsen KB, Burkhart A, Thomsen LB, Andresen TL, Moos T. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 2019;181:101665. doi: 10.1016/j.pneurobio.2019.101665. [DOI] [PubMed] [Google Scholar]
- 50.Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-βclearance in Alzheimer's disease. Front Aging Neurosci. 2014;6:93. doi: 10.3389/fnagi.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kelleher RJ, Soiza RL. Evidence of endothelial dysfunction in the development of Alzheimer's disease:Is Alzheimer's a vascular disorder? Am J Cardiovasc Dis. 2013;3:197. [PMC free article] [PubMed] [Google Scholar]
- 52.Kim S, Moon GJ, Kim HJ, Kim DG, Kim J, Nam Y, Sharma C, Leem E, Lee S, Kim KS, Ha CM, McLean C, Jin BK, Shin WH, Kim DW, Oh YS, Hong CW, Kim SR. Control of hippocampal prothrombin kringle-2 (pKr-2) expression reduces neurotoxic symptoms in five familial Alzheimer's disease mice. Br J Pharmacol. 2022;179:998–1016. doi: 10.1111/bph.15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–617. doi: 10.1016/j.neuron.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knox EG, Aburto MR, Clarke G, Cryan JF, O'Driscoll CM. The blood-brain barrier in aging and neurodegeneration. Mol Psychiatry. 2022;27:2659–2673. doi: 10.1038/s41380-022-01511-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 56.Körbelin J, Dogbevia G, Michelfelder S, Ridder DA, Hunger A, Wenzel J, Seismann H, Lampe M, Bannach J, Pasparakis M, Kleinschmidt JA, Schwaninger M, Trepel M. A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. EMBO Mol Med. 2016;8:609–625. doi: 10.15252/emmm.201506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kozyrev N, Albers S, Yang J, Prado VF, Prado MAM, Fonseca GJ, Jane Rylett R, Dekaban GA. Infiltrating hematogenous macrophages aggregate around β-amyloid plaques in an age- and sex-dependent manner in a mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2020;79:1147–1162. doi: 10.1093/jnen/nlaa093. [DOI] [PubMed] [Google Scholar]
- 58.Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Brüning T, Plath AS, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J. Cerebral amyloid-βproteostasis is regulated by the membrane transport protein ABCC1 in mice. J Clin Invest. 2011;121:3924–3931. doi: 10.1172/JCI57867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurz C, Walker L, Rauchmann BS, Perneczky R. Dysfunction of the blood-brain barrier in Alzheimer's disease:Evidence from human studies. Neuropathol Appl Neurobiol. 2022;48:e12782. doi: 10.1111/nan.12782. [DOI] [PubMed] [Google Scholar]
- 60.Liu Q, Yang Y, Fan X. Microvascular pericytes in brain-associated vascular disease. Biomed Pharmacother. 2020;121:109633. doi: 10.1016/j.biopha.2019.109633. [DOI] [PubMed] [Google Scholar]
- 61.López-Ornelas A, Jiménez A, Pérez-Sánchez G, Rodríguez-Pérez CE, Corzo-Cruz A, Velasco I, Estudillo E. The impairment of blood-brain barrier in Alzheimer's disease:challenges and opportunities with stem cells. Int J Mol Sci. 2022;23:10136. doi: 10.3390/ijms231710136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leclerc M, Bourassa P, Tremblay C, Caron V, Sugère C, Emond V, Bennett DA, Calon F. Cerebrovascular insulin receptors are defective in Alzheimer's disease. Brain. 2023;146:75–90. doi: 10.1093/brain/awac309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moos T, Morgan EH. Restricted transport of anti-transferrin receptor antibody (OX26) through the blood-brain barrier in the rat. J Neurochem. 2001;79:119–129. doi: 10.1046/j.1471-4159.2001.00541.x. [DOI] [PubMed] [Google Scholar]
- 64.Nehra G, Bauer B, Hartz AMS. Blood-brain barrier leakage in Alzheimer's disease:From discovery to clinical relevance. Pharmacol Ther. 2022;234:108119. doi: 10.1016/j.pharmthera.2022.108119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease:a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pardridge WM. Treatment of Alzheimer's disease and blood-brain barrier drug delivery. Pharmaceuticals. 2020;13:394. doi: 10.3390/ph13110394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pemberton S, Galindo DC, Schwartz MW, Banks WA, Rhea EM. Endocytosis of insulin at the blood-brain barrier. Front Drug Deliv. 2022;2:32. doi: 10.3389/fddev.2022.1062366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pereira CD, Martins F, Wiltfang J, da Cruz E Silva OAB, Rebelo S. ABC transporters are key players in Alzheimer's disease. J Alzheimers Dis. 2018;61:463–485. doi: 10.3233/JAD-170639. [DOI] [PubMed] [Google Scholar]
- 69.Petit D, Fernández SG, Zoltowska KM, Enzlein T, Ryan NS, O'Connor A, Szaruga M, Hill E, Vandenberghe R, Fox NC, Chávez-Gutiérrez L. Aβprofiles generated by Alzheimer's disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Molecular Psychiatry. 2022;27:2821–2832. doi: 10.1038/s41380-022-01518-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease:Important unanswered questions. J Exp Med. 2020;217:e20190062. doi: 10.1084/jem.20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pulgar VM. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front Neurosci. 2019;12:1019. doi: 10.3389/fnins.2018.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rhea EM, Rask-Madsen C, Banks WA. Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol. 2018;596:4753–4765. doi: 10.1113/JP276149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta:a crucial factor in Alzheimer's disease. Med Princ Pract. 2015;24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saito S, Tanaka M, Satoh-Asahara N, Carare RO, Ihara M. Taxifolin:a potential therapeutic agent for cerebral amyloid angiopathy. Front Pharmacol. 2021;12:130. doi: 10.3389/fphar.2021.643357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharma B, Luhach K, Kulkarni GT. In vitro and in vivo models of BBB to evaluate brain targeting drug delivery. Brain targeted drug delivery systems:a focus on nanotechnology and nanoparticulates. In: Gao H, Gao X, editors. New York: Academic Press; 2019. pp. 53–101. [Google Scholar]
- 76.Sharma C, Woo H, Kim SR. Addressing blood-brain barrier impairment in Alzheimer's disease. Biomedicines. 2022;10:742. doi: 10.3390/biomedicines10040742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh H, Agrawal DK. Therapeutic potential of targeting the receptor for advanced glycation end products (RAGE) by small molecule inhibitors. Drug Dev Res. 2022;83:1257–1269. doi: 10.1002/ddr.21971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skelly DT, Hennessy E, Dansereau MA, Cunningham C. A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, [corrected] TNF-αand IL-6 challenges in C57BL/6 mice. PLoS One. 2013;8:e69123. doi: 10.1371/journal.pone.0069123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skjørringe T, Burkhart A, Johnsen KB, Moos T. Divalent metal transporter 1 (DMT1) in the brain:implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci. 2015;8:19. doi: 10.3389/fnmol.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Solár P, Zamani A, Lakatosová K, Joukal M. The blood-brain barrier and the neurovascular unit in subarachnoid hemorrhage:molecular events and potential treatments. Fluids Barriers CNS. 2022;19:29. doi: 10.1186/s12987-022-00312-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soto-Rojas LO, Campa-Córdoba BB, Harrington CR, Salas-Casas A, Hernandes-Alejandro M, Villanueva-Fierro I, Bravo-Muñoz M, Garcés-Ramírez L, de La Cruz-López F, Ontiveros-Torres MÁ, Gevorkian G, Pacheco-Herrero M, Luna-Muñoz J. Insoluble vascular amyloid deposits trigger disruption of the neurovascular unit in Alzheimer's disease brains. Int J Mol Sci. 2021a;22:3654. doi: 10.3390/ijms22073654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soto-Rojas LO, Pacheco-Herrero M, Martínez-Gómez PA, Campa-Córdoba BB, Apátiga-Pérez R, Villegas-Rojas MM, Harrington CR, de la Cruz F, Garcés-Ramírez L, Luna-Muñoz J. The neurovascular unit dysfunction in Alzheimer's disease. Int J Mol Sci. 2021b;22:2022. doi: 10.3390/ijms22042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sutton ET, Hellermann GR, Thomas T. beta-amyloid-induced endothelial necrosis and inhibition of nitric oxide production. Exp Cell Res. 1997;230:368–376. doi: 10.1006/excr.1996.3440. [DOI] [PubMed] [Google Scholar]
- 84.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier:from physiology to disease and back. Physiol Rev. 2019;99:21–78. doi: 10.1152/physrev.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem. 2005;95:294–304. doi: 10.1111/j.1471-4159.2005.03369.x. [DOI] [PubMed] [Google Scholar]
- 87.Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, Benjamin EJ, Au R, Kiel DP, Wolf PA, Seshadri S. Inflammatory markers and the risk of Alzheimer disease:the Framingham study. Neurology. 2007;68:1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 88.Tarawneh R, Holtzman DM. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med. 2012;2:a006148. doi: 10.1101/cshperspect.a006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JÅ, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-αexpression for microglial Aβphagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Hofman PAM, Verhey FRJ, Backes WH. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology. 2016;281:527–535. doi: 10.1148/radiol.2016152244. [DOI] [PubMed] [Google Scholar]
- 91.van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, Lechleiter JD, Galvan V. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer's disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol. 2018;314:H693–703. doi: 10.1152/ajpheart.00570.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017;60:1–12. doi: 10.1016/j.bbi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 93.Versele R, Sevin E, Gosselet F, Fenart L, Candela P. TNF-αand IL-1βmodulate blood-brain barrier permeability and decrease amyloid-βpeptide efflux in a human blood-brain barrier model. Int J Mol Sci. 2022;23:10235. doi: 10.3390/ijms231810235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walter FR, Santa-Maria AR, Mészáros M, Veszelka S, Dér A, Deli MA. Surface charge, glycocalyx, and blood-brain barrier function. Tissue Barriers. 2021;9:1904773. doi: 10.1080/21688370.2021.1904773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Q, Huang X, Su Y, Yin G, Wang S, Yu B, Li H, Qi J, Chen H, Zeng W, Zhang K, Verkhratsky A, Niu J, Yi C. Activation of Wnt/β-catenin pathway mitigates blood-brain barrier dysfunction in Alzheimer's disease. Brain. 2022;145:4474–4488. doi: 10.1093/brain/awac236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang S, Qaisar U, Yin X, Grammas P. Gene expression profiling in Alzheimer's disease brain microvessels. J Alzheimers Dis. 2012;31:193–205. doi: 10.3233/JAD-2012-120454. [DOI] [PubMed] [Google Scholar]
- 97.Wolf A, Bauer B, Hartz AMS. ABC transporters and the Alzheimer's disease enigma. Front Psychiatry. 2012;3:54. doi: 10.3389/fpsyt.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu L, Nirwane A, Yao Y. Basement membrane and blood-brain barrier. Stroke Vasc Neurol. 2018;4:78–82. doi: 10.1136/svn-2018-000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang AC, Stevens MY, Chen MB, Lee DP, Stähli D, Gate D, Contrepois K, Chen W, Iram T, Zhang L, Vest RT, Chaney A, Lehallier B, Olsson N, du Bois H, Hsieh R, Cropper HC, Berdnik D, Li L, Wang EY, et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature. 2020;583:425–430. doi: 10.1038/s41586-020-2453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, Toland A, Calcuttawala K, Shin H, Pálovics R, Shin A, Wang EY, Luo J, Gate D, Schulz-Schaeffer WJ, Chu P, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer's risk. Nature. 2022;603:885–892. doi: 10.1038/s41586-021-04369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang C, Qi Y, Sun Z. The role of sonic hedgehog pathway in the development of the central nervous system and aging-related neurodegenerative diseases. Front Mol Biosci. 2021;8:711710. doi: 10.3389/fmolb.2021.711710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yazdani S, Jaldin-Fincati JR, Pereira RVS, Klip A. Endothelial cell barriers:Transport of molecules between blood and tissues. Traffic. 2019;20:390–403. doi: 10.1111/tra.12645. [DOI] [PubMed] [Google Scholar]
- 103.Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer's disease. Am J Pathol. 2010;176:1600–1606. doi: 10.2353/ajpath.2010.090406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3:84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- 105.Zhang YL, Wang J, Zhang ZN, Su Q, Guo JH. The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer's disease. Neural Regen Res. 2022;17:2355–2363. doi: 10.4103/1673-5374.335829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao F, Zhong L, Luo Y. Endothelial glycocalyx as an important factor in composition of blood-brain barrier. CNS Neurosci Ther. 2021;27:26–35. doi: 10.1111/cns.13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.