

One of the most enigmatic problems in biomedical research surrounds the phenomenon that neurodegenerative diseases target specific cell types and brain regions. This is difficult to explain because the proteins that cause them are widely expressed, often highest in resistant regions. This mystery is further complicated by the fact that some disease-causing proteins are associated with multiple diseases. For example, the protein alpha-synuclein forms toxic aggregates in multiple diseases including Parkinson’s disease, dementia with Lewy bodies, and multiple systems atrophy, whereas the protein TDP43 can cause amyotrophic lateral sclerosis or frontotemporal lobar degeneration (Alegre-Abarrategui et al., 2019; Schweingruber and Hedlund, 2022). Alzheimer’s disease is associated with aggregates from two proteins, the amyloid precursor protein (APP) and Tau. Familial forms of Alzheimer’s disease are often associated with mutations in APP and enzymes that process APP. In contrast, mutations in Tau are not linked to Alzheimer’s disease but instead to frontal temporal dementia, whereas non-mutated Tau is also associated with several other neurodegenerative diseases (Carroll et al., 2021). Huntington’s disease is one of the few types of neurodegenerative disease that is strictly genetic. It is caused by an abnormally long polyglutamine tract in the Huntingtin protein, and the longer the tract the earlier the disease emerges. Remarkably, very long polyglutamine tracts also cause a wider distribution of affected brain regions, presenting with different clinical signs. The prion protein (PrP) also causes multiple, distinct diseases, collectively called prion diseases (Jackson, 2014).
Prion diseases come in three basic forms. The most infamous form involves infection from an exogenous source, such as contaminated food or medical procedures. The infectious agents, called prions, consist mostly of aggregated PrP (Prusiner, 1982; Bueler et al., 1993; Brandner et al., 1996). Although these acquired forms are the main cause of prion disease in farmed and wild animals, they account for only 1% of all human prion diseases. A more common form of prion disease in humans, accounting for about 15% of all cases, is caused by mutations in the gene encoding PrP. This form is especially fascinating because it can be separated into three subclasses of diseases known as fatal familial insomnia (FFI), genetic Creutzfeldt-Jakob disease (gCJD), and Gerstmann-Sträussler-Scheinker syndrome, where each is caused by specific mutations and has specific neuropathological changes and clinical signs (Jackson, 2014). The final form of prion disease is sporadic, which simply means there is no mutation and no evidence of infection. In each of these basic forms of prion disease, the amount, distribution, and shapes of PrP aggregates can vary tremendously. Furthermore, other hallmarks of prion disease, namely the distribution and density of spongiform degeneration and the concentration of infectivity, can also vary greatly, from barely detectable to highly abundant. The age at disease onset also varies greatly. This is easy to understand in acquired and sporadic prion diseases because the disease-triggering events may happen at any age, but not for genetic prion diseases, where the mutation is present throughout life. For example, the two most common mutations, D178N causing FFI and E200K causing gCJD, have ages at onset ranging from 12 to 89 for FFI and 31 to 92 for gCJD (Minikel et al., 2019). Interestingly, the D178N mutation can also cause gCJD when in cis with the M129V substitution. Therefore, the mammalian brain can express mutant PrP for decades without overt abnormalities and this wide variation in human prion diseases may also exist in experimental models.
To understand the molecular pathways acting in neurodegenerative diseases researchers often employ mouse models. In fact, some of the earliest mouse models of any neurodegenerative disease were prion disease models since they could be established before the discovery of genes linked to neurodegenerative diseases and the development of methods for the genetic engineering of mice. Thus, mouse models of acquired prion diseases became highly studied in the early days of neurodegenerative disease research.
Perhaps the most intensively studied mouse model of all prion diseases is the model of acquired prion disease developed at the Rocky Mountain Laboratories (RML) in the US. It was originally derived from a goat with scrapie prion disease, and subsequently passaged tens of times through multiple mammalian species, until the final RML strain of mouse-adapted scrapie prion disease was established. After multiple decades of passaging, the most virulent, fastest-killing prions were selected.
Later, following the discovery of the gene encoding PrP and the invention of genetic engineering methods, the ability to study genetic models emerged. Mouse models based on randomly integrated transgenes have dominated the neurodegenerative disease field because of their ease of generation and because their high expression levels typically enhance neurological disease. However, overexpression, which is not a feature of natural animal or human prion diseases, creates confounders. During synthesis, PrP passes through the endoplasmic reticulum and Golgi apparatus and finally resides at the cell surface. Therefore, overexpressed PrP creates an unnatural processing, chaperoning, and degradation burden on these and other organelles and displaces molecules that should otherwise occupy those spaces. These issues are then confounded when a mutation is added: when disease emerges in such mutant models, how much of the effect is due to the overexpression problems, and how much to the mutation? Another issue is that spatial expression patterns differ between different transgenic lines that have integrated into random locations in the genome (Kaczmarczyk and Jackson, 2015). The spatial expression pattern variability creates another confounder: when different phenotypes emerge from transgenes with different sequences, how much of this difference is due to the spatial expression pattern differences, and how much to the sequence differences? These issues from over-expression and variable spatial expression patterns can be avoided with knock-in mice where all variants are expressed from the gene’s native location.
Even though knock-in models avoid these two serious issues, they are rarely used because of two important limitations. First, they are much more difficult to engineer, although CRISPR/Cas-based tools are lowering this barrier. Second, they typically develop a mild disease. Indeed, to enhance disease in APP knock-in mice, alleles were engineered to carry three or four mutations, but the mice were only mildly affected, with a normal lifespan (Saito et al., 2014). To accelerate disease in Huntington’s disease knock-in mice, they were engineered to carry mutations severe enough to cause disease in children (Lin et al., 2001). Nonetheless, these models also have a mild phenotype, without a shortened lifespan. Similarly, PrP knock-in models bearing single mutations also have mild phenotypes (Jackson et al., 2009, 2013). Importantly, despite having mild phenotypes, knock-in models have been useful for discovering molecular pathways in pre-onset stages of neurodegenerative disease.
In two recent studies, cell type-specific molecular responses were analyzed for mouse models of acquired and genetic prion diseases. The first employed the RML model in wild-type mice (Kaczmarczyk et al., 2022), while the second employed two knock-in models expressing the mouse equivalent of the D178N and E200K mutations (Bauer et al., 2022). Notably, all models and controls expressed PrP from the same, endogenous location in the genome, avoiding the confounders described above. To discover molecular responses, epitope-tagged ribosomes were expressed in specific cell types using RiboTag mice (Figure 1). From brain homogenates of diseased or control RiboTag mice, the epitope-tagged ribosomes were immunopurified, and the attached mRNAs, representing the genome-wide pool of translating mRNAs (translatome), were analyzed with next-generation sequencing methods. There were some technical differences between the two studies, such as different Illumina sequencing platforms, but the overall similarities, including the same mouse genetic background, enable a basic comparison of results.
Figure 1.
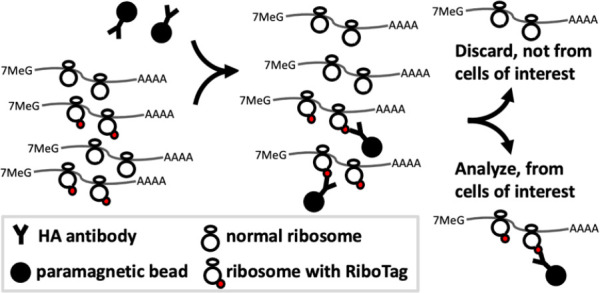
Capture of mRNA from specific cell types with RiboTag.
Paramagnetic beads labeled with an antibody specific to the HA (hemagglutinin) epitope are added to homogenates (left) made from tissues in which RiboTag-labeled ribosomes (HA epitope fused to ribosomal protein l22) are expressed in a cell type of interest. The HA antibody::bead complexes capture the RiboTag protein (middle), and those ribosomes and their associated mRNAs are purified away from ribosomes and mRNAs from undesired cell types (right).
In the first study, to identify pre-onset and onset time points, brain activity was serially measured with electroencephalography (EEG) as the disease progressed (Kaczmarczyk et al., 2022). When the EEG showed differences between diseased and control mice, neuropathological changes were also present but behavioral changes had not yet developed. This happened at week 18 of a 23-week disease course and was considered the disease onset stage. At week 10, diseased and control mice had identical EEG profiles and showed no neuropathological or behavioral changes; it was therefore considered the pre-onset stage.
To study cell type-specific responses, homogenates were made from RiboTag mouse brain hemispheres that had the olfactory bulb removed. Analyses of the translatomes at the disease onset stage revealed massive but similar changes in all cell types, even though the mice looked healthy. At the pre-onset stage, cells made specific responses. For example, glutamatergic neurons, representing most of the excitatory neurons, had 38 differentially expressed genes (DEGs) involving cytoskeleton components and regulators of the cytoskeleton, including Arc (Kaczmarczyk et al., 2022). In contrast, GABAergic neurons, representing most inhibitory neurons, had 83 DEGs, including most genes of the core circadian rhythm pathway. Surprisingly, parvalbumin neurons, a large subset of GABAergic neurons previously reported to be highly vulnerable to many prion diseases, showed essentially no response with only 3 DEGs. Furthermore, somatostatin (SST) neurons, another GABAergic subset, had only 1 DEG, indicating that the circadian rhythm-related changes seen in the broad GABAergic population were from a narrow subset that includes neither the parvalbumin nor SST subpopulations. Interestingly, astrocytes responded with a reduced expression of many ribosomal and mitochondrial proteins, making up most of the 139 DEGs. Therefore, despite the lack of EEG, behavioral and neuropathological changes at the pre-onset timepoint, certain cells made specific, coordinated responses while others barely responded at all.
The study of FFI and gCJD mice also employed EEG analyses to build on the previous characterizations (Jackson et al., 2009, 2013). Both models had very mild EEG changes at 21 months of age (Bauer et al., 2022), leaving unchanged the notion from previous analyses that the age at onset is around 16 months. The pre-onset stage chosen for the translatome analysis was 9 months, which was estimated to be similar to the pre-onset stage in the RML model study.
In contrast to the first study, the second study did not include astrocytes. Furthermore, in the second study, the olfactory bulb and cerebellum were separated from the rest of the brain (considered the cerebrum) and the cerebellum and cerebrum were analyzed separately.
Like in the RML model, in the FFI and gCJD models, parvalbumin neurons showed little response, with only 2 or 3 DEGs in each model. However, in contrast to the RML model, the translatomes of the FFI and gCJD models revealed robust responses in SST neurons, with 684 and 153 DEGS, respectively. Some of the notable pathways these DEGs were associated with included translation and ribosome biogenesis, actin cytoskeleton, and Rho GTPases which regulate the actin cytoskeleton, most of which were upregulated. Further analyses indicated the mammalian target of rapamycin pathway was the signature response in SST neurons in both diseases. This pathway was not detected in any cell type in pre-onset RML model data. This similar response in both genetic models was surprising because they are clinically distinct and cause the most severe damage in different brain regions, the thalamus in FFI and the hippocampus in gCJD. Interestingly, these regions are in the cerebrum and enriched in glutamatergic neurons, but the corresponding samples purified from cerebral glutamatergic neurons showed a relatively mild response with only 3 and 11 DEGs, respectively. In both models, GABAergic neurons had the second strongest response, with 47 DEGs in the cerebrum and 28 in the cerebellum of FFI mice, and 14 DEGs in the cerebrum and 64 in the cerebellum of gCJD mice. Despite the similar total number of GABAergic DEGs as in the RML model, the DEGs in the FFI and gCJD models were unrelated to the circadian rhythm but instead to metal binding, T cell response, and synapses. Importantly, the refined experimental approach in the second study, where the cerebellum was separated from the cerebrum, should have enhanced the detection of pathways originally detected in the RML model if they were also present in the genetic models. These and other comparisons lead to the overall conclusion that, at pre-onset disease stages, FFI and gCJD models are surprisingly similar to each other but quite different from the RML model.
At disease onset, the RML model had thousands of DEGs in all cell types, with similar pathways between them. The disease onset stage was not examined for the genetic models, which is unfortunate because it would be useful to know if the responses in each cell type also converge and if they resemble the responses in the RML model. Such a finding could support the notion that a therapy developed for acquired prion disease could also be applied to genetic prion diseases. However, a therapy targeting a specific pathway may be ineffective when used at disease onset, when there are thousands of DEGs and lots of distinct pathways affected, some of which may also need to be modulated. In such a scenario, multiple therapies may be needed to control specific pathways to change the disease course.
To conclude, as described at the beginning of this perspective for other proteins such as Tau and alpha-synuclein, PrP should be viewed as causing multiple, distinct diseases, especially when comparing genetic and acquired forms. The significance of this thinking is that therapies that are effective on acquired prion disease may target changes that do not exist in genetic prion diseases and thus may not work for them.
I would like to thank members of our lab for their constructive feedback.
This work was supported by Wallenberg Center for Molecular Medicine, the Knut and Alice Wallenberg foundation, and the King Gustaf and Queen Victoria foundation (to WSJ).
Additional file: Open peer review report 1 (82.2KB, pdf) .
Footnotes
P-Reviewer: De Cecco E; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
Open peer reviewer: Elena De Cecco, University of Zurich, Switzerland.
References
- 1.Alegre-Abarrategui J, Brimblecombe KR, Roberts RF, Velentza-Almpani E, Tilley BS, Bengoa-Vergniory N, Proukakis C. Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. 2019;138:681–704. doi: 10.1007/s00401-019-02010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer S, Dittrich L, Kaczmarczyk L, Schleif M, Benfeitas R, Jackson WS. Translatome profiling in fatal familial insomnia implicates TOR signaling in somatostatin neurons. Life Sci Alliance. 2022;5:e202201530. doi: 10.26508/lsa.202201530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 4.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 5.Carroll T, Guha S, Nehrke K, Johnson GVW. Tau post-translational modifications:potentiators of selective vulnerability in sporadic Alzheimer's disease. Biology (Basel) 2021;10:1047. doi: 10.3390/biology10101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson WS. Selective vulnerability to neurodegenerative disease:the curious case of Prion Protein. Dis Model Mech. 2014;7:21–29. doi: 10.1242/dmm.012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63:438–450. doi: 10.1016/j.neuron.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson WS, Borkowski AW, Watson NE, King OD, Faas H, Jasanoff A, Lindquist S. Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proc Natl Acad Sci U S A. 2013;110:14759–14764. doi: 10.1073/pnas.1312006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaczmarczyk L, Jackson WS. Astonishing advances in mouse genetic tools for biomedical research. Swiss Med Wkly. 2015;145:w14186. doi: 10.4414/smw.2015.14186. [DOI] [PubMed] [Google Scholar]
- 10.Kaczmarczyk L, Schleif M, Dittrich L, Williams RH, Koderman M, Bansal V, Rajput A, Schulte T, Jonson M, Krost C, Testaquadra FJ, Bonn S, Jackson WS. Distinct translatome changes in specific neural populations precede electroencephalographic changes in prion-infected mice. PLoS Pathog. 2022;18:e1010747. doi: 10.1371/journal.ppat.1010747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 12.Minikel EV, Vallabh SM, Orseth MC, Brandel JP, Haïk S, Laplanche JL, Zerr I, Parchi P, Capellari S, Safar J, Kenny J, Fong JC, Takada LT, Ponto C, Hermann P, Knipper T, Stehmann C, Kitamoto T, Ae R, Hamaguchi T, et al. Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology. 2019;93:e125–134. doi: 10.1212/WNL.0000000000007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 14.Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 15.Schweingruber C, Hedlund E. The cell autonomous and non-cell autonomous aspects of neuronal vulnerability and resilience in amyotrophic lateral sclerosis. Biology (Basel) 2022;11:1191. doi: 10.3390/biology11081191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.