

Abstract
Pathogen detection from biological and environmental samples is important for global disease control. Despite advances in pathogen detection using deep learning, current algorithms have limitations in processing long genomic sequences. Through the deep cross-fusion of cross, residual and deep neural networks, we developed DCiPatho for accurate pathogen detection based on the integrated frequency features of 3-to-7 k-mers. Compared with the existing state-of-the-art algorithms, DCiPatho can be used to accurately identify distinct pathogenic bacteria infecting humans, animals and plants. We evaluated DCiPatho on both learned and unlearned pathogen species using both genomics and metagenomics datasets. DCiPatho is an effective tool for the genomic-scale identification of pathogens by integrating the frequency of k-mers into deep cross-fusion networks. The source code is publicly available at https://github.com/LorMeBioAI/DCiPatho.
Keywords: pathogen identification, K-mer frequency, metagenomics, deep cross-fusion networks
INTRODUCTION
Pathogens are major threats to human and animal health, as well as the environment. According to statistics from the World Health Organization, ~13 million children die from infectious diseases every year worldwide, accounting for 25.5% of the global total annual mortality rate [1]. Plant pathogen infections cause ~30% of crop yield losses globally, exacerbating the food crisis [2]. Therefore, rapid pathogen detection is of great significance for public health [3], food safety [4], animal health [5], plant quarantine [6] and environmental quality [7] studies from a One Health perspective [8].
Pathogen detection based on DNA sequencing is mainly categorized into taxonomy-dependent and taxonomy-independent approaches, which require appropriate computational methods [9]. The former approaches rely heavily on the type of pathogen database [10, 11]. Taxonomy-independent approaches identify pathogens directly from DNA sequences, omitting taxonomic assignment. In these approaches, the algorithms still must be trained on available references for accurate pathogen detection. For instance, machine learning tools, such as Pathogenicity Prediction for Bacterial Genomes (PaPrBaGs) [12] and Bacterial Pathogenicity Classification via Sparse-SVM (BacPaCS) [13], are currently applied for open-view pathogen detection using the precomputed databases of sequences and peptide features of a custom reference database. However, the performance of the above methods is often limited by advances in algorithms, the composition of pretrained databases and the fast evolution and emergence of novel pathogens [14].
Advanced deep learning models are state-of-the-art (SOTA) technologies to improve the performance of DNA sequence classification [15]. Existing models generally take k-mers as the basic processing unit [14, 16, 17]. The k-mer-based methods mainly fall into two categories [18–20]. One category is the frequency-based k-mer feature method. For example, the k-mer frequency was used as the key feature to classify transposable elements using a convolutional neural network (CNN) model with a stacked autoencoder [21, 22] to predict ncRNA–protein interactions [23] or using bidirectional long short-term memory (BiLSTM) [24]. The other category is k-mer encoding representation methods [19], in which k-mers are converted into a vector space, such as one-hot, word2vec, dna2vec, GloVe and bidirectional encoder representations from transformer (BERT) [25–28]. For example, one-hot encoding and k-mer embedding were used to identify chromatin regions using CNN and BI-LSTM [25] and YY1-mediated chromatin loops from DNA sequences using CNN [26]. In DeePaC [14], CNN and LSTM were used to build a model for pathogenicity classification. BERTax [16] based on BERTs was proposed to classify the kingdom, phylum and genus of DNA sequences. Both DeePaC [14] and BERTax [16] are read-based methods. Read-based encoding model prediction mainly relies on the voting outcomes of 300 –1500 bp short reads of gene sequences. Although deep learning methods have wider applicability and better generalization performance in DNA sequence analysis, there are still some shortcomings with respect to their use in pathogen identification. (i) The k-mers can be defined on arbitrary length sequences and constitute an unbiased, general and complete set of sequence features. However, a single frequency rather than the cooccurrence of several frequencies will sometimes cause information loss [24, 29, 30]. (ii) Current deep learning models such as CNN, attention and BERT require fixed length or short sequences as input and consist of sequence features with k-mer coding representation [26, 31]. (iii) If a mean over all the read-length subsequence predictions constitutes the final prediction, without considering the interaction between reads, some global context characteristics of pathogen genomes may be ignored [14, 16]. (iv) The hardware cost of a single iteration of the large language model is climbing higher. Overall, the application potential of current deep learning methods is severely limited by their insufficiency in handling whole genome sequences up to millions of bp.
Therefore, considering automatically learning feature interactions using deep learning networks [32] and extracting arbitrary lengths of k-mer frequencies, we proposed a deep neural network model that can be used to automatically combine k-mer frequency information to efficiently learn the crossover and nonlinear features to generate more effective models. Each layer of the cross network produces higher order interactions based on existing ones and keeps the interactions from previous layers. The model does not require manual feature engineering or an ergodic search and has a low computational cost [30, 32–35]. Our results show that DCiPatho outperforms the competing SOTA methods.
MATERIALS AND METHODS
Dataset preparation
We downloaded the complete genomes of 32 927 bacteria from the Reference Sequence Database of the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/refseq) in June 2022. The labels of bacterial complete genomes were curated by comparison with the collected information of plant, animal and human bacterial pathogens from the literature and various database websites. Briefly, plant pathogens were obtained from the plant pathogenic bacteria list on the International Society of Plant Pathology Committee (https://www.isppweb.org); animal and human pathogens were obtained from BacPaCs [13] and bacterial_refseq_pathogens in SURPIrt-dist [36] and the pathogen database on the NCBI website (https://www.ncbi.nlm.nih.gov/pathogens/) and MBPD [37]. For the label of each genomes, it was double-checked in manual curation by searching of ‘the strain identifier’ AND (‘infection’ OR ‘diseases’). The complete genomes of the 32 927 bacteria were labelled pathogenic or nonpathogenic bacterial strains. Based on the genus level, 22 046 genomes were labelled pathogenic bacterial strains (1269 genera), and 10 882 genomes were labelled nonpathogenic bacterial strains (6568 genera). Multiple sequences of chromosomes and plasmids were included for the complete genome sequences. To obtain ample characteristics of the genomic sequences, we performed a series of processing steps on the initial complete genome sequences. We combined the internal sequences of chromosomes and plasmids in the complete genome of each strain into a single DNA sequence file and stored it as a fasta file while retaining only the ID information of the first sequence to match the Latin name of the strain. Finally, each strain ID corresponds to a long DNA sequence and unique species classification information. The DNA sequence lengths of the bacterial pathogens ranged from 1.1 to 11.6 million bp, with a median length of 3 783 317 bp. The nonpathogenic sequence lengths were distributed between 4.3 and 10.5 million bp (median length: 3 784 056 bp). The processed dataset is named BacRefSeq. To reduce the learning cost and hardware resource cost for beginners and other researchers, we selected a mini dataset named Mini-BacRefSeq from BacRefSeq. Mini-BacRefSeq comprises of 1506 complete genomes including 707 pathogenic bacterial strains (540 species, including animal, human and plant pathogens) and 799 nonpathogenic bacterial strains (687 species).
Overview of the framework
The proposed framework DCiPatho is outlined in Figure 1. Given the input of a genomic sequence, k-mer frequency features are first extracted. The features are introduced into a feature cross-fusion prediction network, where CrossNet, ResNet and DeepNet are connected in a side-by-side manner, higher order feature combinations are generated explicitly and implicitly and more nonlinear features are obtained. Then, the outputs of the three networks are combined in the combination layer, and finally, the combined features are scored in the sigmoid layer to obtain the pathogenicity prediction results. The detailed parameter optimization of the DCiPatho model is available in Supplementary 1.1 and Supplementary Table 1.
Figure 1.

Overview of DCiPatho. (A) Starting from known pathogen and nonpathogen sequences, we propose a vector representation and a deep learning architecture to train the models for pathogen classification. (B) Neural network architecture of the DCiPatho model. The k-mer frequency feature is initially fed into three network modules in parallel. The ResNet module is constructed from N residual units, which adds the original input feature after ReLU transformation. The DeepNet module is constructed from N numbers of fully connected feedforward neural networks. The CrossNet module is composed of poly-cross layers. Each output of the three modules is equipped with a ReLU activation function. Then, the outputs of the three modules are concatenated into a combination layer, which has multiple dense layers with a sigmoid activation function to obtain the prediction of pathogenic probabilities.
The k-mer frequency feature
The k-mer approach has been successfully used for genome sequence analysis in bioinformatics [38]. Therefore, we used k-mer frequencies to characterize gene sequence feature information, where we notated  as a sequence of length
as a sequence of length  , and
, and  {
{ }, where
}, where  . The k-mer is the subsequence of length
. The k-mer is the subsequence of length  in the sequence. For a sequence with a length of
in the sequence. For a sequence with a length of  , the k-mer frequency is calculated using the sliding window method with a step size of 1 and a window size of
, the k-mer frequency is calculated using the sliding window method with a step size of 1 and a window size of  . Sliding is performed in a step-by-step manner from the first to the
. Sliding is performed in a step-by-step manner from the first to the  position, and the sliding frame is moved one base position at a time until the entire genomic sequence becomes ergodic. The derived feature vector is denoted as
position, and the sliding frame is moved one base position at a time until the entire genomic sequence becomes ergodic. The derived feature vector is denoted as  {
{ }, where
}, where  is the original cumulative frequency of the corresponding feature and
is the original cumulative frequency of the corresponding feature and  is the total number of all possible k-mer feature frequencies. Since DNA has a double-stranded structure, each DNA sequence can be sequenced from either strand. Therefore, for a certain subsequence, the k-mer frequency can be combined with the occurrence frequency of its reverse complementary sequence. Considering that a palindrome sequence is the same as its reverse complementary sequence, when
is the total number of all possible k-mer feature frequencies. Since DNA has a double-stranded structure, each DNA sequence can be sequenced from either strand. Therefore, for a certain subsequence, the k-mer frequency can be combined with the occurrence frequency of its reverse complementary sequence. Considering that a palindrome sequence is the same as its reverse complementary sequence, when  is an odd number, the dimension of the feature vector
is an odd number, the dimension of the feature vector  can be simplified to the dimension of
can be simplified to the dimension of  , and when
, and when  is even, the dimension of the eigenvector can be simplified to
is even, the dimension of the eigenvector can be simplified to  . Therefore, we used the above modified k-mer frequency extraction method to extract the k-mer features of the DNA sequences. When
. Therefore, we used the above modified k-mer frequency extraction method to extract the k-mer features of the DNA sequences. When  values from 3 to 7 are selected, the corresponding feature dimensions are as follows:
values from 3 to 7 are selected, the corresponding feature dimensions are as follows:  .
.
The frequency feature information for K3, K4, K5, K6, K7 and K3–K7 in the BacRefSeq dataset was calculated and normalized using the k-mer frequency method as aforementioned. The performance of different k-mer sizes and their combinations was compared to obtain the optimal input of the k-mer features (Supplementary 1.2 and Supplementary Figure 1A).
Feature cross and fusion prediction network
A DCiPatho network consists of input, a combination of CrossNet, ResNet and DeepNet, and a scoring layer. The specific neural network structure is shown in Figure 1B. As shown in this figure, k-mer feature vectors are fed into CrossNet, and the elements of the feature vectors are computed interactively to obtain feature crosses. Implicit crosses are obtained using ResNet. In DeepNet, the original k-mer features are retained, which increases the nonlinear expression capability of the model. The model has both low-order and high-order cross terms, thereby providing a better model representation capability.
Feature optimization consists of the following three parts
CrossNet
In CrossNet, explicit k-mer feature crosses are obtained directly. The cross network consists of cross layers, where each layer obeys the following equation:
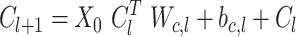 |
(1) |
where l = 1, 2, … , NC. Cl and Cl + 1 are the outputs of the lth and l + 1th cross layers, respectively; C0 is X0 and Wc,l and bc,l are the connection parameters between these two layers. All of the variables in the above equation are column vectors. The output of each layer is the output of the previous layer and the feature crossing.
The unique structure of CrossNet allows the degrees of cross features to increase with the depth of the layer, with one layer of CrossNet providing a maximum of two-dimensional cross features, two layers providing a maximum of three-dimensional cross features and so on.
Therefore, CrossNet can be used to efficiently learn the cross combination of gene k-mer features in a parameter-sharing manner by controlling the number of stacked layers, thereby avoiding manual feature engineering.
ResNet
A multilayer perceptron (MLP) is the main structure in ResNet, and a multilayer residual network is used as a specific implementation of MLP compared with the standard neural network, with the perceptron as the basic unit. Through adequate implicit cross combination of the various dimensions of the feature vector by the multilayer residual network, the model can capture the information within the nonlinear and combinatorial features.
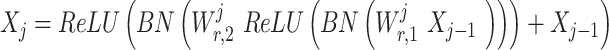 |
(2) |
where j = 1,2, … ,NR; ReLU is the rectified linear unit; xj-1 is the input to the residual network at layer j; Xj is the output; Wjr,1 is the first connection layer weight at layer j and Wjr,2 is the second connection layer weight. BN is obtained by normalizing the features in each dimension in a batch of data by subtracting them from the mean and dividing by the standard deviation. The new scaled feature values have a mean of 0 and a variance of 1.
DeepNet
In DeepNet, which is a fully connected feedforward neural network, the original sequence features are dimensionally reduced, and a cross combination of high-dimensional nonlinear features is learned. The output values of each layer are as follows:
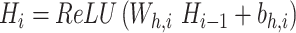 |
(3) |
where I = 1, 2, … , NF; H0 uses X0; Hi is the output of layer I; Wh,i is the fully connected network connection weight; and bh,i is the bias value.
The network modules of the ResNet, CrossNet and DeepNet networks were designed by an ablation study to optimize the module structure for the pathogen classification model of DCiPatho (Supplementary 1.3 and Supplementary Figure 1B).
Feature concatenation and a scoring layer
Finally, the outputs from CrossNet, ResNet and DeepNet are concatenated as sequential feature representations, and feature dimensionality reduction is then achieved through multiple fully connected layers. Pathogenicity prediction is a dichotomous problem i.e. output pathogenicity and nonpathogenicity are obtained by a logit scoring layer.
The feature 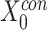 is a combination of cross features and dimensionality reduction features:
is a combination of cross features and dimensionality reduction features:
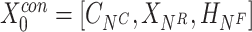 |
(4) |
Feature  is then passed through
is then passed through  fully connected layers for dimensionality reduction. The final output is
fully connected layers for dimensionality reduction. The final output is  :
:
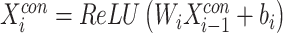 |
(5) |
where  ; and
; and  and
and  are the neural weights and bias value of layer
are the neural weights and bias value of layer  , respectively.
, respectively.
The logit layer classification formula is as follows:
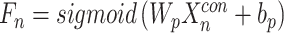 |
(6) |
The probability of the final output pathogenicity  is distributed between 0 and 1.
is distributed between 0 and 1.  is the scoring layer connection weights, and
is the scoring layer connection weights, and  is the bias value.
is the bias value.
This model uses BCELoss as the loss function, and  and
and  denote the loss of the nth sample and total loss, respectively, as follows:
denote the loss of the nth sample and total loss, respectively, as follows:
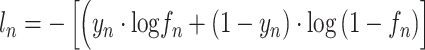 |
(7) |
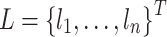 |
(8) |
where  and
and  are the true and predicted labels of the nth sample, respectively.
are the true and predicted labels of the nth sample, respectively.
Benchmarking
To benchmark our method against the SOTA deep learning methods and machine learning methods in pathogen prediction, we compared BERTax [16], DeePaC [14], DeepTE [21], PaPrBaG [12] and BacPaCS [13], EC-DFR [39], PlasClass [40], XGBoost and AdaBoost from the scikit-learn library [41] with DCiPatho on the BacRefSeq dataset. For a fair comparison, each model was trained and optimized to obtain the corresponding optimal hyperparameters. The detailed parameter settings and the implementations can be found in Supplementary 1.4.
Evaluation metrics
In this study, we evaluated the comprehensive performance of the DCiPatho network and other artificial intelligence methods for classifying pathogenic sequences using the evolution metric of accuracy (ACC), matthews correlation coefficient (MCC), F1 score, area under curve (AUC), precision-recall curve (PRC) and receiver operating characteristic curve (ROC) in previous studies [42, 43].
RESULTS
Comparison between DCiPatho and other methods on the BacRefSeq dataset
DCiPatho was first benchmarked against the baseline deep learning methods and machine learning methods, including BERTax [16], DeePaC [14], DeepTE [21], PaPrBaG [12] and BacPaCS [13], EC-DFR [39], PlasClass [40], XGBoost and AdaBoost in pathogen prediction on the BacRefSeq dataset (Table 1 and Supplementary Table 2). To guarantee the independence of the test set and enable a robust evaluation, we employed the 5-fold cross-validation approach to partition the training, validation and testing sets of the BacRefSeq dataset. In each fold, 80% of the data was utilized for training, 10% for validation and parameter tuning, whereas the remaining 10% was used for testing the model’s performance. We evaluated our model on the test set and reported the average metrics, and the evaluation results are presented in Table 1 and Supplementary Table 2. More results with other popular deep learning methods, such as Bi-LSTM, Attention and Transformer, can be found in the Supplementary 1.5. The PRC and ROC curves of the best models can be seen in Figure 2A and B. We found that the DCiPatho network showed the highest ACC, AUC, F1 and MCC scores among all the models. In the ROC and PRC curves, DCiPatho had the best performance, with ROC and PRC values of 98.2 and 99.2%, respectively. Compared with the DNA representation of k-mer frequency, the results of sequence-based encoding were not ideal. Therefore, DCiPatho is a better way to obtain genome features, which can better solve the problem of long genome sequence representation than read-sequence-based encoding. The results show that DCiPatho can be used to effectively differentiate between pathogenic and nonpathogenic bacteria.
Table 1.
Comparing the performance of DCiPatho to nine advanced machine learning / deep learning methods on identifying the complete genome of pathogens
| Methods | ACC (%) | MCC (%) | AUROC (%) | F1 Score (%) |
|---|---|---|---|---|
| PlasClass | 93.11 ± 1.05 (bc) | 83.52 ± 0.83 (c) | 96.73 ± 0.36 (b) | 94.21 ± 1.14 (b) |
| XGBoost | 92.78 ± 0.48 (bc) | 83.81 ± 0.69 (bc) | 96.93 ± 0.56 (b) | 94.18 ± 1.10 (b) |
| AdaBoost | 77.87 ± 2.27 (e) | 43.99 ± 1.65 (f) | 83.41 ± 0.98 (e) | 84.91 ± 1.21 (d) |
| PaPrBaG | 93.95 ± 0.32 (ab) | 87.25 ± 0.61 (a) | 96.54 ± 0.44 (b) | 94.79 ± 0.93 (ab) |
| BacPaCS | 66.12 ± 1.39 (f) | 47.14 ± 1.01 (e) | 65.80 ± 0.63 (f) | 73.77 ± 1.06 (e) |
| DeePac (ResNet) | 84.44 ± 1.09 (d) | 63.23 ± 1.02 (d) | 95.12 ± 0.43 (bc) | 87.95 ± 1.01 (c) |
| DeepTE | 91.88 ± 0.66 (c) | 83.25 ± 1.02 (c) | 95.90 ± 0.70 (bc) | 95.12 ± 0.81 (bc) |
| EC-DFR | 92.74 ± 0.49 (bc) | 85.41 ± 1.19 (b) | 90.82 ± 0.62 (d) | 94.19 ± 0.85 (b) |
| Bertax | 85.83 ± 1.32 (d) | 70.03 ± 1.21 (d) | 92.10 ± 1.02 (c) | 90.10 ± 0.96 (c) |
| DCiPatho | 95.14 ± 0.28 (a) | 88.52 ± 0.81 (a) | 98.49 ± 0.39 (a) | 96.41 ± 0.20 (a) |
Notes: For genome sequences, we used the BacRefSeq test dataset. Different lower case letters in parentheses indicate a significant difference among the mean of the evaluation metric of different models by the analysis of variance (ANOVA) by post hoc Tukey test at the 5% level of significance (P < 0.05, n = 5). In contrary, the difference between the means of each evaluation metric is not statistically significant for the models with the same letter. The best performance of evaluated models is highlighted in bold.
Figure 2.

Benchmarking the performance of the DCiPatho model using the BacRefSeq dataset. (A) ROC curves of DCiPatho and other methods. (B) PRC curves of DCiPatho and other methods.
Performance of DCiPatho using the PATRIC dataset
We compared the prediction accuracy of DCiPatho to the nine SOTA methods on a new pathogen dataset of PATRIC to further evaluate the performance of the DCiPatho model in novel pathogen identification. PATRIC has a total of 878 pathogenic sequences belonging to 179 genera with a median genome sequence length of 4 940 713 bp. DCiPatho showed the best accuracy, with ~8 and 11% higher accuracy than BacPaCS, DeepTE and BERTax, respectively, which are advanced deep learning methods (Figure 3A). However, the prediction accuracy of DCiPatho was 82.24%, which is not as high as that on the BacRefSeq test set (95.21%). This could be explained by the fact that only 114 pathogen species were shared between the BacRefSeq and PATRIC datasets (Figure 3B). On the PATRIC dataset, the DCiPatho model effectively identified the pathogen species present (100% median accuracy) in the BacRefSeq dataset compared with those absent (70.86% median accuracy) from the training set i.e. novel pathogen species (Figure 3C). For unlearned pathogens, 45 of 149 species (40%) were identified well by the DCiPatho model, with a prediction accuracy of >80% (Figure 3D). This suggests that DCiPatho outperformed other models in terms of generalized performance and accurately identified novel, unknown and unlearned pathogen species.
Figure 3.

Evaluation of DCiPatho on the PATRIC dataset. (A) Benchmark of different classifiers in comparison with DCiPatho on the PATRIC dataset. (B) Difference in bacterial composition between the BacRefSeq and PATRIC datasets. (C) Prediction performance of PATRIC for present and absent pathogens in the BacRefSeq dataset.
High transferability of DCiPatho to metagenomes
To evaluate the transferability of DCiPatho from genome to metagenome prediction, we assessed the performance of DCiPatho on metagenome-assembled genomes (MAGs) from the gut microbiome of hospitalized adults [44]. A total of 665 high-quality MAGs (19 pathogen and 56 nonpathogen species, completeness >90% and contamination <5%) were filtered using CheckM [45] via the lineage-specific workflow. We found that the mean prediction accuracy on MAGs was only 62.31%, which is unsatisfactory using the pretrained DCiPatho model on the BacRefSeq dataset based on complete genomes. We suspect that the inaccuracy could be attributed to significant differences in sequence types and species composition between the BacRefSeq and MAG datasets. Therefore, we de novo trained the DCiPatho model on the MAG dataset to identify pathogen potential for metagenomics using the pipeline method shown in Figure 1. Remarkably, the average prediction accuracy of de novo-trained DCiPatho in pathogen identification drastically increased from 62.31 (using the pretrained DCiPatho model) to 95.45% with an MCC of 86.40%, a ROC of 99.17% and an F1 score of 89.28% (Figure 4A). Interestingly, the de novo-trained DCiPatho model shows excellent performance in identifying the pathogen species both absent and present in the BacRefSeq dataset, with median accuracies of 93.38 and 99.81%, respectively (Figure 4B). In detail, 16 of 19 pathogen species (82.2%) were identified by the de novo DCiPatho model with an accuracy of >85% (Figure 4C). This suggests that the DCiPatho model has superior performance on diverse dataset types for both genome and metagenome sequencing.
Figure 4.

High performance of the de novo DCiPatho model of MAGs based on metagenomic sequences. (A) Prediction accuracy of de novo DCiPatho on pathogen and nonpathogen MAGs. (B) Difference in de novo DCiPatho prediction of the pathogen MAGs present and absent in the BacRefSeq dataset. (C) Details of model performance in predicting the pathogenic potential of each pathogen MAG.
Feature representations and contribution analysis learned by DCiPatho
To examine the effectiveness of the feature representation learned by the trained DCiPatho model, we applied a two-level feature visualization strategy to visualize this feature representation. The details of the feature visualization can be found in Supplementary 2. We visualized abstract features in the input layer, ensemble layer and two dense layers of the trained DCiPatho model. Figure 5A shows the feature representations of four different layers for the model trained on the BacRefSeq dataset. The raw dataset representation is shown first. The pathogenic data points are mixed with the nonpathogenic data points. After the ResNet layer of our model, the pathogenic data points were gradually separated from the nonpathogenic data points. Based on the feature representations learned by the combination layer of the model, we can see that the data points are almost split into two parts. After the dense layer of the model, the data points seem to be more clearly divided into two groups, and the specificity in the 2D space becomes larger, which indicates a greater degree of discrimination. Thus, DCiPatho can effectively learn important feature representations.
Figure 5.

Feature representations and contribution analysis. (A) The t-SNE visualization analysis of different layers. (B) The pie chart shows the proportion of different k-mer sizes contributing to the order of magnitude, as reflected by the top 10, top 100, top 1000 and top 10 000. (C) Violin plots of the contribution of different k-mer sizes.
To further explain different k-mer vectors, we explored the impact of different k-mer sizes on the prediction results of DCiPatho. Since we extracted k-mers with varying lengths of K3-K7 in feature extraction, we divided the features into five groups according to the k-mer size. Then, we used Captum [46] to quantify the degree of pathogenic contribution of different k-mer sizes. In looking at each group, we visualized the distribution of importance from the top 10 to the top 10 000 shown in Figure 5B. In conclusion, we found that among the top 10 most essential feature contributions, only K6 and K7 are included, and as the k-mer size increases, the maximum value of the contribution degree in the model increases monotonically. This phenomenon proves our hypothesis because the larger the k-mer size is, the lower the frequency of the individual k-mer, and the more unique and representative they are. As shown in Figure 5C, we generated violin plots to show the contribution of the corresponding k-mer size with respect to the model, with the middle line representing the average and the lines at both ends representing the extreme values. Except for K3, the overall shape and distribution of various sizes are similar (the quartiles are very close), but there are more outliers in K7; as the size increases, the maximum value of the contribution degree also increases.
Comparative analysis of training and prediction times for DCiPatho and SOTA models
To further investigate the computational efficiency of DCiPatho, we compared the training time of DCiPatho to the nine SOTA models on the BacRefSeq dataset and the prediction time on the PATRIC dataset under the same hardware environment. Our analysis revealed that DCiPatho is highly flexible and capable of running smoothly on general computers. Furthermore, DCiPatho demonstrated significantly shorter prediction times of 3.8 s compared with the other deep learning models we tested, as shown in Supplementary 1.6 and Supplementary Table 4. These results indicate that DCiPatho offers not only superior performance but also improved computational efficiency, making it a highly promising tool for predicting pathogenicity from bacterial genomic data. However, it should be noted that the PATRIC dataset used for our analysis contained only 878 sequences, leading to most methods achieving a prediction time of <10 s. However, considering the possibility of future pretrained DCiPatho being used on larger datasets, our results suggest that DCiPatho is a more lightweight and high-performing option, as it was 2.97/1.6/4.28 (from 1.6 to 4.28) times faster than the other deep learning models we tested.
DISCUSSION
We proposed a novel deep learning model, DCiPatho, for the rapid, accurate and unbiased diagnosis of DNA-based pathogens. The key advances underlying our model include () accurate genomic scale classification at the Mb level of DNA sequences based on the enhanced features of k-mer frequencies with deep cross-fusion networks, (b) detection across a broad range of pathogenic bacteria infecting humans, animals and plants, (c) compatibility with both known and unknown and unlearned pathogen sequences and (d) a dual-use model of genomic and metagenomic DNA sequences from WGS and mNGS platforms. Importantly, we found that DCiPatho can accurately predict the potential of pathogens. The great power of DCiPatho can be observed, especially for species that did not appear in the training set. In addition, high accuracy and F1 scores were obtained, particularly for identification on the test set of BacRefSeq (mean accuracy = 95.21%) and metagenomic DNA (mean accuracy = 95.45%). Furthermore, 40% of the unlearned novel pathogen species in the PATRIC test dataset can be well predicted by our model with a prediction accuracy of >80%. Although the accuracy was similar or superior to that of the current pathogen detection tools [12–14, 47], the overall performance of DCiPatho is superior in general, with higher MCC and F1 scores.
In this work, we challenged and resolved the current difficulty of machine learning classification on the Mb length of long DNA sequences at the full genomic scale. Currently, there are excellent tools such as DeePaC, which can be used to predict pathogenic potential based on the genomic sequences of bacteria, viruses and fungi [14, 48, 49] with prediction accuracies ranging from 87.8 to 95.0%. Generally, one-hot encoding and/or dna2vec sequence feature types or BERTax, whereas the BERT pretrained models consume considerable computational resources and may ignore global features in the genome to some extent [12–14]. The k-mer frequencies better account for global features. To avoid the influence of coupling relationships between sequences on the prediction results, we deployed the ResNet, CrossNet and DeepNet deep cross-fusion networks to train the classifier based on the combination of features from K3 to K7. Indeed, another issue of the k-mer-based classifier is determining the appropriate k-mer size to obtain a good trade-off between computational complexity and feature information. Several works have shown that short k-mers are sufficient to provide effective informative features [25, 50, 51]. Long k-mers may affect the model performance based on the number of uninformative k-mers and lead to heavy computational resource costs [52]. Thus, we developed a pipeline for feature extraction and combination on the frequency of different k-mers in the DCiPatho framework. Our assessment also showed that the combination k-mer features outperformed any feature inputs of a single k-mer from K3 to K7.
In the benchmark test, DCiPatho achieved the best performance in terms of the evaluation metrics in comparison with other classification models. XGBoost and AdaBoost are current advanced machine learning models [53], PlasClass, PaPrBaG, BacPaCS and EC-DFR are currently excellent bioinformatics classifiers, and DeePaC, BERTax and DeepTE are deep learning models. Another reason to start with the frame algorithms of the above tools is that they are more suitable for de novo model training and fair comparison based on the combination feature of k-mers of genomic scale DNA sequences. Furthermore, the deep cross-fusion networks for our DCiPatho model outperformed the individual and dual networks of ResNet, CrossNet and DeepNet.
Uncertainty in labels can cause serious problems with respect to large errors in model evaluation and the loss of important features, and it is crucial to accurately and consistently define human pathogens. However, these are not simple tasks [14]. Several tools are already available in the field of human pathogenic bacteria, and DCiPatho attempts to classify human, animal and plant pathogens at the genomic level. However, more research is needed to explore this issue. Examples include the use of confident learning [54] (finding and learning with label errors in the dataset) and the construction of multivariate classification models for animal, plant, human and zoonotic pathogens. Finally, there are other studies that could be considered to identify pathogenic bacteria not based on the amplicon level, metabolism level or transcriptome level.
There are still challenges and difficulties at this period, including classification at the transcriptome level and attempting to extract more advanced sequence-based features on the extremely large sequence of the entire genome. In addition, more research on enhancing the performance of DCiPatho on the 16S dataset and other gene sequencing methods is needed.
CONCLUSIONS
We developed a deep cross-fusion network model of DCiPatho for the genome-scale identification of human, animal and plant pathogens. We carefully designed and investigated the k-mer combination feature and network structure. We found that DCiPatho can be enhanced by cross features to identify pathogens in t-SNE feature visualization. DCiPatho achieves SOTA results and a shorter prediction time. The performance of the k-mer frequency cross feature is better than that of the k-mer encoding feature in the whole genome level pathogen prediction. DCiPatho is also easily extendable to WGS genomics to mNGS metagenomics data and might also be used as a general workflow for the construction of deep cross-fusion network architectures beyond pathogen detection at the long DNA sequence level. We anticipate a broad application of DCiPatho for open-view diagnosis in clinical, agricultural, fishery and veterinary settings in the current health era. For future work, we plan to build a long sequence deep learning feature representation and classification model on larger scale dataset.
Key Points
DCiPatho proposed a deep cross-fusion network for genome-scale pathogen detection.
DCiPatho can be used to extract cross features of k-mer frequency.
DCiPatho achieves SOTA results based on low computational power. We also demonstrate the capability of feature cross-combination compared with general feature engineering techniques.
DCiPatho can be used to detect a broad range of pathogenic bacteria infecting humans, animals and plants.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Ye Tao for valuable discussions and comments.
Author Biographies
Gaofei Jiang is associated professor at Nanjing Agricultural University (gjiang@njau.edu.cn)
Jiaxuan Zhang is a postgraduate student at Nanjing Agricultural University (andr3i@stu.njau.edu.cn)
Yaozhong Zhang is a PhD candidate at Nanjing Agricultural University (yaozhongzyz@stu.njau.edu.cn)
Xinrun Yang is a PhD candidate at Nanjing Agricultural University (xinrunyang@stu.njau.edu.cn)
Tingting Li is a postgraduate student at Nanjing Agricultural University (tingtingli@njau.edu.cn)
Ningqi Wang is a PhD candidate at Nanjing Agricultural University (wangnq@njau.edu.cn)
Xingjian Chen is a PhD candidate at City University of Hong Kong (xingjchen3-c@my.cityu.edu.hk)
Fang-Jie Zhao is professor at Nanjing Agricultural University (fangjie.zhao@njau.edu.cn)
Zhong Wei is professor at Nanjing Agricultural University (weizhong@njau.edu.cn)
Yangchun Xu is professor at Nanjing Agricultural University (ycxu@njau.edu.cn)
Qirong Shen is professor at Nanjing Agricultural University (shenqirong@njau.edu.cn)
Wei Xue is professor at Nanjing Agricultural University (xwsky@njau.edu.cn)
Contributor Information
Gaofei Jiang, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Jiaxuan Zhang, College of Artificial Intelligence, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Yaozhong Zhang, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Xinrun Yang, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Tingting Li, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Ningqi Wang, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Xingjian Chen, Department of Computer Science, City University of Hong Kong, Hong Kong 999077, China.
Fang-Jie Zhao, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Zhong Wei, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Yangchun Xu, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Qirong Shen, Jiangsu Provincial Key Laboratory for Organic Solid Waste Utilization, Laboratory of Bio-interactions and Crop Health, Jiangsu Collaborative Innovation Center for Solid Organic Waste Resource Utilization, National Engineering Research Center for Organic-based Fertilizers, Joint International Research Laboratory of Soil Health, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
Wei Xue, College of Artificial Intelligence, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China.
FUNDING
This work was supported by the National Natural Science Foundation of China [grant number 42090062 to G.J. and F.J.Z., 42090064 to Q.S., 42007038 to G.J., 42277113 to Z.W., and 31972504 to Y.X.], the Fundamental Research Funds for the Central Universities (grant number KYT2023001 to Z.W., XUEKEN2023039 to Q.S. and XUEKEN2023044 to W.R.), and the Natural Science Foundation of Jiangsu Province (SBK2023030230 to G.J.), and the China National Tobacco Corporation [grant number 110202101047(LS-07) to G.J.], and Jiangxi Branch of China National Tobacco Corporation [grant number 2021.01.010 to Z.W.].
DATA AVAILABILITY
All code is publicly available at https://github.com/LorMeBioAI/DCiPatho. The dataset of BacRefSeq, mini-BacRefSeq, PATRIC and MAGs are available at https://zenodo.org/record/7571307.
AUTHORS’ CONTRIBUTIONS
Conceptualization: G.J., W.X., Z.W.; Resources: G.J., W.X.; Methodology: G.J., J.Z., X.Y., Y.Z., N.W., W.X.; Data curation: G.J., J.Z., X.Y., Y.Z.,T.L., N.W.; Formal analysis: G.J., J.Z., X.Y., Y.Z., T.L., N.W., W.X.; Funding acquisition: G.J., Z.W., Y.X., Q.S., W.X.; Investigation: G.J., J.Z., X.Y., Y.Z., N.W., W.X.; Project administration: G.J., Z.W., W.X.; Supervision: G.J., Z.W., W.X.; Visualization: G.J., J.Z., X.Y., Y.Z., Writing—original draft: G.J., W.X.; Writing—review & editing: G.J., W.X., J.Z., X.Y., Y.Z., Y.X., Q.S., Z.W.
REFERENCES
- 1. Hasanzad M, Sarhangi N, Aghaei Meybodi HR, et al. Precision medicine in non communicable diseases. Int J Mol Cell Med 2019;8:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Savary S, Willocquet L, Pethybridge SJ, et al. The global burden of pathogens and pests on major food crops. Nat Ecol Evol 2019;3(3):430–9. [DOI] [PubMed] [Google Scholar]
- 3. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol 2019;14:319–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saravanan A, Kumar PS, Hemavathy RV, et al. Methods of detection of food-borne pathogens: A review. Environ Chem Lett 2021;19(1):189–207. [Google Scholar]
- 5. Vidic J, Manzano M, Chang C-M, Jaffrezic-Renault N. Advanced biosensors for detection of pathogens related to livestock and poultry. Vet Res 2017;48(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong A-Y, Wang Z, Huang J-J, et al. Bioinformatic tools support decision-making in plant disease management. Trends Plant Sci 2021;26:953–67. [DOI] [PubMed] [Google Scholar]
- 7. Regan JF, Makarewicz AJ, Hindson BJ, et al. Environmental monitoring for biological threat agents using the autonomous pathogen detection system with multiplexed polymerase chain reaction. Anal Chem 2008;80:7422–9. [DOI] [PubMed] [Google Scholar]
- 8. Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018;19:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li N, Cai Q, Miao Q, et al. High-throughput metagenomics for identification of pathogens in the clinical settings. Small Methods 2021;5:2000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miao J, Han N, Qiang Y, et al. 16SPIP: A comprehensive analysis pipeline for rapid pathogen detection in clinical samples based on 16S metagenomic sequencing. BMC Bioinformatics 2017;18:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalantar KL, Carvalho T, de Bourcy CFA, et al. IDseq: An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020;9:giaa111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deneke C, Rentzsch R, Renard BY. PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data. Sci Rep 2017;7:39194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barash E, Sal-Man N, Sabato S, et al. BacPaCS-bacterial pathogenicity classification via sparse-SVM. Bioinformatics 2019;35:2001–8. [DOI] [PubMed] [Google Scholar]
- 14. Bartoszewicz JM, Seidel A, Rentzsch R, et al. DeePaC: Predicting pathogenic potential of novel DNA with reverse-complement neural networks. Bioinformatics 2020;36:81–9. [DOI] [PubMed] [Google Scholar]
- 15. Mo Z, Zhu W, Sun Y, et al. One novel representation of DNA sequence based on the global and local position information. Sci Rep 2018;8:7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mock F, Kretschmer F, Kriese A, et al. Taxonomic classification of DNA sequences beyond sequence similarity using deep neural networks. Proc Natl Acad Sci U S A 2022;119:e2122636119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oh M, Zhang L. DeepMicro: deep representation learning for disease prediction based on microbiome data. Sci Rep 2020;10:6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H, Cimen E, Singh N, et al. Deep learning for plant genomics and crop improvement. Curr Opin Plant Biol 2020;54:34–41. [DOI] [PubMed] [Google Scholar]
- 19. He Y, Shen Z, Zhang Q, et al. A survey on deep learning in DNA/RNA motif mining. Brief Bioinform 2021;22:bbaa229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Talukder A, Barham C, Li X, et al. Interpretation of deep learning in genomics and epigenomics. Brief Bioinform 2021;22:bbaa177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan H, Bombarely A, Li S. DeepTE: A computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 2020;36:4269–75. [DOI] [PubMed] [Google Scholar]
- 22. Nakano FK, Mastelini SM, Barbon S, et al. Improving hierarchical classification of transposable elements using deep neural networks. In: 2018 International Joint Conference on Neural Networks (IJCNN). International Neural Network Society, Rio de Janeiro, RJ, Brazil, 2018, pp. 1–8.
- 23. Peng C, Han S, Zhang H, et al. RPITER: a hierarchical deep learning framework for ncRNA–protein interaction prediction. Int J Mol Sci 2019;20:E1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Zhao Y, Gong W, et al. EDLMFC: An ensemble deep learning framework with multi-scale features combination for ncRNA–protein interaction prediction. BMC Bioinformatics 2021;22:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Min X, Zeng W, Chen N, et al. Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics 2017;33:i92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dao F-Y, Lv H, Zhang D, et al. DeepYY1: A deep learning approach to identify YY1-mediated chromatin loops. Brief Bioinform 2021;22:bbaa356. [DOI] [PubMed] [Google Scholar]
- 27. Andreopoulos WB, Geller AM, Lucke M, et al. Deeplasmid: Deep learning accurately separates plasmids from bacterial chromosomes. Nucleic Acids Res 2022;50:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma Y, Guo Z, Xia B, et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat Biotechnol 2022;40:921–31. [DOI] [PubMed] [Google Scholar]
- 29. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 30. Wang R, Fu B, Fu G, et al. Deep & cross network for ad click predictions. In: Proceedings of the ADKDD’17. Association for Computing Machinery, Nova Scotia, NS, Canada, 2017, pp. 1–7.
- 31. Marx V. Method of the year: Long-read sequencing. Nat Methods 2023;20:6–11. [DOI] [PubMed] [Google Scholar]
- 32. Guo H, Tang R, Ye Y, et al. DeepFM: a factorization-machine based neural network for CTR prediction. arXiv 2017; 1703.04247.
- 33. Cheng H-T, Koc L, Harmsen J, et al. Wide & deep learning for recommender systems. In: Proceedings of the 1st Workshop on Deep Learning for Recommender Systems. Association for Computing Machinery, Boston, BOS, USA, 2016, pp. 7–10.
- 34. Lian J, Zhou X, Zhang F, et al. xDeepFM: Combining explicit and implicit feature interactions for recommender systems. In: Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. Association for Computing Machinery, London, LON, United Kingdom, 2018, pp. 1754–63.
- 35. Sun Y, Pan J, Zhang A, et al. FM2: Field-matrixed factorization machines for recommender systems. Proc Web Conf 2021;2021:2828–37. [Google Scholar]
- 36. Gu W, Deng X, Lee M, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med 2021;27:115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang X, Jiang G, Zhang Y, et al. MBPD: A multiple bacterial pathogen detection pipeline for one health practices, iMeta 2023;2:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fiannaca A, La Rosa M, Rizzo R, et al. A k-mer-based barcode DNA classification methodology based on spectral representation and a neural gas network. Artif Intell Med 2015;64:173–84. [DOI] [PubMed] [Google Scholar]
- 39. Lin W, Wu L, Zhang Y, et al. An enhanced cascade-based deep forest model for drug combination prediction. Brief Bioinform 2022;23:bbab562. [DOI] [PubMed] [Google Scholar]
- 40. Pellow D, Mizrahi I, Shamir R. PlasClass improves plasmid sequence classification. PLoS Comput Biol 2020;16:e1007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: Machine learning in python. J Mach Learn Res 2011;12:2825–30. [Google Scholar]
- 42. Kha Q-H, Ho Q-T, Le NQK. Identifying SNARE proteins using an alignment-free method based on multiscan convolutional neural network and PSSM profiles. J Chem Inf Model 2022;62:4820–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kha Q-H, Tran T-O, Nguyen T-T-D, et al. An interpretable deep learning model for classifying adaptor protein complexes from sequence information. Methods 2022;207:90–6. [DOI] [PubMed] [Google Scholar]
- 44. Siranosian BA, Brooks EF, Andermann T, et al. Rare transmission of commensal and pathogenic bacteria in the gut microbiome of hospitalized adults. Nat Commun 2022;13:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parks DH, Imelfort M, Skennerton CT, et al. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 2015;25:1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kokhlikyan N, Miglani V, Martin M, et al. Captum: A unified and generic model interpretability library for PyTorch. arXiv 2020;2009.07896.
- 47. Bartoszewicz JM, Genske U, Renard BY. Deep learning-based real-time detection of novel pathogens during sequencing. Brief Bioinform 2021;22:bbab269. [DOI] [PubMed] [Google Scholar]
- 48. Bartoszewicz JM, Seidel A, Renard BY. Interpretable detection of novel human viruses from genome sequencing data. NAR Genom Bioinform 2021;3:lqab004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bartoszewicz JM, Nasri F, Nowicka M, et al. Detecting DNA of novel fungal pathogens using ResNets and a curated fungi-hosts data collection. Bioinformatics 2022;38:ii168–74. [DOI] [PubMed] [Google Scholar]
- 50. Shen Z, Bao W, Huang D-S. Recurrent neural network for predicting transcription factor binding sites. Sci Rep 2018;8:15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trabelsi A, Chaabane M, Ben-Hur A. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics 2019;35:i269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marçais G, DeBlasio D, Kingsford C. Asymptotically optimal minimizers schemes. Bioinformatics 2018;34:i13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zou Q, Liu Q. Advanced machine learning techniques for bioinformatics. IEEE/ACM Trans Comput Biol Bioinform 2019;16:1182–3. [Google Scholar]
- 54. Northcutt C, Jiang L, Chuang I. Confident learning: estimating uncertainty in dataset labels. J Artif Intell Res 2021;70:1373–411. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code is publicly available at https://github.com/LorMeBioAI/DCiPatho. The dataset of BacRefSeq, mini-BacRefSeq, PATRIC and MAGs are available at https://zenodo.org/record/7571307.