


Abstract
Target search models of DNA-binding proteins in cells typically consider search mechanisms that include 3D diffusion and 1D sliding, which can be characterized by single-molecule tracking on DNA. However, the finding of liquid droplets of DNA and nuclear components in cells cast doubt on extrapolation from the behavior in ideal non-condensed DNA conditions to those in cells. In this study, we investigate the target search behavior of DNA-binding proteins in reconstituted DNA-condensed droplets using single-molecule fluorescence microscopy. To mimic nuclear condensates, we reconstituted DNA-condensed droplets using dextran and PEG polymers. In the DNA-condensed droplets, we measured the translational movement of four DNA-binding proteins (p53, Nhp6A, Fis and Cas9) and p53 mutants possessing different structures, sizes, and oligomeric states. Our results demonstrate the presence of fast and slow mobility modes in DNA-condensed droplets for the four DNA-binding proteins. The slow mobility mode capability is correlated strongly to the molecular size and the number of DNA-binding domains on DNA-binding proteins, but only moderately to the affinity to single DNA segments in non-condensed conditions. The slow mobility mode in DNA-condensed droplets is interpreted as a multivalent interaction mode of the DNA-binding protein to multiple DNA segments.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Sequence-specific DNA-binding proteins are required to search for and bind to their target sequences embedded within long genomic DNA molecules. To seek out targets of ∼20 bp within ∼109 bp genomes in a physiologically relevant timescale, four search mechanisms have been proposed: 1D sliding, hopping/jumping, 3D diffusion, and intersegmental transfer (1–8). DNA-binding proteins move along single DNA molecules via 1D sliding with continuous contact to DNA or with hopping/jumping, to read the DNA sequence and recognize specific targets. When proteins dissociate from DNA, 3D diffusion in solution can alter the regions of DNA scanned. Proteins can transfer between two adjacent DNA segments through an intermediate bound to the two DNA segments using intersegmental transfer. Theoretical studies suggest that the integration of multiple search mechanisms can facilitate the target search (9–12). For example, the tumor suppressor p53, used as one of the model DNA-binding proteins in this study, has been experimentally proven to possess all four search mechanisms by tracking the proteins on DNA at the single-molecule level (13–21). In general, 3D diffusion is the common search strategy for DNA-binding proteins, whereas the choice of other search mechanisms varies across these proteins. Nhp6A, the second model used in this study, readily slides along DNA, whereas Fis and Cas9, the third and fourth models used in this study, are mostly stationary and show 1D sliding by a small fraction of bound proteins (22–26). Both Nhp6A and Fis exhibit intersegmental transfer (27). The target search behavior of these DNA-binding proteins in cells was also measured and interpreted using these search mechanisms (22,26,28–30). However, the recent finding of liquid droplet (condensate) formation in cells cast doubt on the extrapolation from the behavior in a non-condensed DNA condition to that in cells.
Recent accumulated data suggest that genomic DNA and nuclear components undergo liquid-liquid phase separation (LLPS) to form liquid droplets, and this process can regulate the transcriptional activity at a level that is not achieved under dispersed conditions. Nucleated transcriptional condensates amplified gene expression (31), and droplets of engineered transcription factors formed at target promoters increased gene expression (32). On the other hand, disruption of long-distance association of DNA segments through nuclear condensates decreased transcriptional activity (33). Furthermore, uptake of the p53 transcription factor into 53BP1 droplets enhanced target gene activation (34). Also, p53 drove nuclear speckle association of p53 target genes and boosted RNA expression (35). Considering these data, we should take into account the liquid droplet formation of DNAs in the understanding of the target search mechanism of DNA-binding proteins in cells. In DNA-condensed droplets, DNA-binding proteins may contact not only single DNA segments, but also simultaneously to multiple DNA regions located nearby, as observed in molecular dynamics simulations of p53 (36,37). In contrast, the proposed search mechanisms, with the exception of intersegmental transfer, do not consider these multivalent interactions of DNA-binding protein to multiple DNA segments.
In this study, we investigated the target search behavior of DNA-binding proteins in DNA-condensed droplets reconstituted in vitro in a chamber (mimicking droplets in cells) using single-molecule fluorescence microscopy. We confirmed that single-molecule fluorescence microscopy can be used to track molecules in liquid droplets of proteins (38,39). In this study, we applied this method to monitor the behavior of DNA-binding proteins in DNA-condensed droplets. To reconstitute liquid droplets of DNA for single-molecule measurements, we used the LLPS of two polymers, dextran and PEG (40) and the uptake of a long DNA into dextran-rich droplet phases (41) (Figure 1A). In the reconstituted droplets of DNAs, we measured the dynamics of four DNA-binding proteins: p53, Nhp6A, Fis and Cas9 (Figure 1A). The four proteins were chosen as models, because their target search mechanisms on dispersed DNAs were well characterized, as described above. In addition, the data of these model proteins with different structures, sizes, and oligomeric states can be used to determine the molecular factors that govern the target search mechanism in DNA-condensed droplets. Our results show the presence of fast and slow mobility modes in DNA-condensed droplets for each of the four DNA-binding proteins. The slow mobility mode capability was correlated to the molecular size or the number of DNA-binding domains, rather than to the affinity to single DNA segments in non-condensed conditions, suggesting multivalent interactions of DNA-binding proteins to multiple DNA segments.
Figure 1.

Molecular uptake of DNA and DNA-binding proteins into Dextran-rich droplets. (A) Schematic diagram of the in vitro DNA-LLPS mimicking system for single-molecule measurements of DNA-binding proteins (left) and DNA-binding protein structures used in this study (right). In the left panel, a dextran-rich droplet is formed by mixing dextran and PEG, which recruits λ DNA used in this study. Individual DNA-binding proteins, which are recruited by condensed DNAs in the droplets, are tracked using single-molecule fluorescence microscopy (dashed arrows). In the light panel, colored structures except for grey denote the domains (or regions) that can interact to DNA. PDB codes used for Nhp6A and Fis are 1LWM and 3IV5, respectively. Schematic structures are presented for p53 and Cas9. (B) DIC and fluorescent images of FITC-labeled dextran or sytox green-intercalated λ DNA in dextran and PEG. (C) DIC and fluorescent images of Alexa488- or Atto488-labeled DNA-binding proteins in 4.1 nM λ DNA of droplets. Protein sample concentrations were set to 100 nM. Scale bars in panel (B) and (C) were 40 μm.
MATERIALS AND METHODS
Protein samples
For p53 samples, we prepared the p53 tetramer as well as the NCT, TC and dimer mutants, as described previously (15,17,21,38,42). For the p53 tetramer, a thermostable and cysteine-modified human p53 mutant (C124A, C135V, C141V, W146Y, C182S, V203A, R209P, C229Y, H233Y, Y234F, N235K, Y236F, T253V, N268D, C275A, C277A and K292C) was used (15). The tetrameric TC mutant corresponds to residues 293–393 of the polypeptide with an additional N-terminal cysteine (15). The tetrameric NCT mutant corresponds to residues 1–363 (17) and the dimer mutant contains the L344A mutation (38,42). The gene of p53-CTD(R/KtoA) mutant (K370A, K372A, K373A, R379A, K381A, K382A and K386A) was generated by cloning the mutated DNA fragments to the p53 vector cleaved by Kpn I and PpuM I with In-Fusion HD Cloning Kit (Takara). This mutant was expressed and purified as described above.
The Escherichia coli Fis Q21C psuedo-wildtype, Fis Q21C R85A DNA-binding defective mutant, and Saccharomyces cerevisiae Nhp6A 2-Cys mutant (containing Cys at residue 2 and the C-terminal end) were expressed and purified without tags, as described previously (23,24,26). Deactivated Cas9 (dCas9: M1C, D10A, C80S, H840A, and C574S) from Streptococcus pyogenes was expressed and purified fused to a soluble maltose binding protein (MBP) tag at the N terminus (dCas9–MBP) or purified without the MBP tag using TEV protease (25). ST-dCas9-MBP, which is dCas9–MBP fused to residues 2–16 of Nhp6A (VTPREPKKRTTRKKK) at the C-terminus, was prepared as described previously (25).
For GFP, the negatively charged (−30) mutant (62936; Addgene) with a N-terminal 6 × His tag was prepared (43). The GFP mutant was expressed using BL21 Star (DE3) cells in LB medium by 0.5 mM IPTG at 25ºC for 20 h after OD600 of the culture reached 0.6. The cells were lysed by sonication in 20 mM Tris, 1.5 M NaCl, a protease inhibitor cocktail (cOmplete, Mini; Sigma-Aldrich), 10 mM MgCl2, and 10 U/mL DNase at pH 8.0. The supernatant was collected after centrifugation. The GFP mutant was purified using a HisTrap column (HisTrap FF crude; Cytiva) in a solution containing 20 mM Tris, 1.5 M NaCl, and 100 or 250 mM imidazole (pH 8.0). Purity was confirmed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The sample was stored at –80ºC.
Labeling with fluorophores
For single-molecule measurements of DNA-binding proteins, the proteins except for GFP were labeled with Atto488 (ATTO-TEC) or Alexa488 (Thermo Fisher) using maleimido chemistry and then purified using a cation exchange chromatography (23,24,26,38). For single-molecule measurements of λ DNA in droplets, λ DNA (NEW ENGLAND BioLabs, Inc.) was annealed with Atto488-labeled DNA (5′-AGGTCGCCGCCC[Atto488]-3′, Sigma-Aldrich) and then purified using a NucleoSpin gDNA Clean-up kit (Takara).
Titration experiments
The fluorescence anisotropy of the 6-FAM-labeled DNA was measured with increasing the concentration of non-labeled proteins including p53 dimer mutant (L344A), Fis, Nhp6A, and dCas9–MBP at 25°C using a fluorescence spectrometer (FP-6500; JASCO Co., Tokyo, Japan) with a home-built autorotating polarizer (15). Non-labeled proteins were manually titrated into a solution containing 5 nM 6-FAM-labeled dsDNA, 20 mM HEPES, 0.5 mM EDTA, 1 mM DTT, 0.5 mg/mL BSA, and 50 mM KCl, and 1 mM MgCl2 at pH 7.9 (17). The non-specific DNA duplex used in this study was composed of 5′-6-FAM-AATATGGTTTGAATAAAGAGTAAAGATTTG-3′ and its non-labeled complementary sequence (Sigma-Aldrich). Titration curves were fitted using the following equations based on a one-to-one binding model:
 |
(1) |
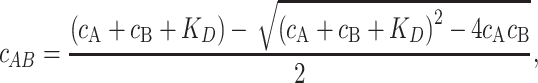 |
(2) |
where robs, rA, rAB, KD, cA and cB indicate the observed anisotropy, anisotropy of free molecule A, anisotropy of the complex of molecules A and B, dissociation constant, the total concentration of molecule A, and total concentration of molecule B, respectively. The analysis was conducted using the Igor software.
Sample solution preparation for measurements
For dextran-rich droplet formation, we used the solution containing 20 mM HEPES, 1 mM EDTA, 150 mM KCl, 5 wt% PEG (PEG6000; MW 7300–9300; Wako Pure Chemical Ind.), 5 wt% dextran (MW 180 000–210 000; Wako Pure Chemical Ind.), and 100 nM FITC-dextran (average MW 40 000) at pH 7.4. The sample solution was prepared using 20 or 30 wt% PEG and dextran stock solutions, and the droplet formation was triggered by adding PEG stock solution into the solution containing the components except for PEG. For uptake of DNA into dextran-rich droplets, we used the solution containing 20 mM HEPES, 1 mM EDTA, 150 mM KCl, 5 wt% PEG, 5 wt% dextran, 1 nM λ DNA, and 5 μM sytox green (Thermo Fisher Scientific) at pH 7.4. For uptake of the labeled proteins into DNA-rich droplets, we used the solution containing 20 mM HEPES, 1 mM EDTA, 150 mM KCl, 5 wt% PEG, 5 wt% dextran, 1.3 nM λ DNA, and 100 nM labeled proteins at pH 7.4. For single-molecule measurements of the labeled proteins and GFP into DNA-rich droplets, we used the solution containing 20 mM HEPES, 1 mM EDTA, 150 mM KCl, 5 wt% PEG, 5 wt% dextran, 1.3–10.4 nM λ DNA, and 1.5–2.0 nM labeled proteins at pH 7.4. The stock solution of λ DNA was replaced with distilled water using NucleoSpin gDNA Clean-up kit for all cases and concentrated to be 32 nM using vacuum-centrifugal evaporator (MV-100; TOMY) for preparing 10.4 nM λ DNA solution. The λ DNA concentrations in dextran-rich droplets were estimated to be enhanced 3.1-fold by measuring the volume fraction of dextran-rich phase after centrifuge and by assuming the uptake of all λ DNAs into it. The viscosity of DNA droplets was estimated to be 150–160 cP by calculating the ratio of the diffusion coefficient of GFP in dilute solution (44) to the average value measured in DNA droplets. The viscosity did not depend on DNA concentrations, suggesting that high concentration of dextran in droplets was the primary determinant due to its excluded volume effect. The viscosity of DNA droplets was close to the minimum range of viscosity reported for biomolecular droplets (45). After the preparation of sample solutions, we immediately performed the subsequent microscopic measurements until the droplet size became extremely large.
Differential interference contrast (DIC) and fluorescence microscopy for ensemble measurements
We used the DIC detection mode of inverted microscope (IX-73; Olympus, Tokyo, Japan) equipped with a microscopic objective (LUCPlanFL N, NA = 0.7, 60×) and a color-sensitive camera (DP74; Olympus) (46). Light sources (U-LH100L and U-HGLGPS; Olympus) were used for DIC and fluorescence detection, respectively. The excitation wavelength was 470–495 nm, and the fluorescence at 510–550 nm was detected. The sample solution was cast on a coverslip (Matsunami Glass) and covered with a glass slide (Matsunami Glass). The coverslip and slide glass were cleaned with ethanol and 5 M KOH and coated with 0.5% 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer (Lipidureμ-CM5206; NOF Corp.) in ethanol before use (47). DIC and fluorescence images were obtained at 21–22°C. The uptake ratio of DNA-binding proteins inside to outside DNA droplets was calculated from the ratios of the fluorescence intensities inside to outside DNA droplets.
Single-molecule measurements
Single-molecule measurements were conducted following the method described in our previous papers with some modifications (38,39). The sample solutions were cast on a coverslip and covered with a glass slide (Matsunami Glass) using 20-μm thick double-sided tape. The coverslip and slide glass were cleaned with a solution containing 30% H2O2, 28% NH3 and H2O at a 1:1:1 ratio before use. To reduce fluorescent sample adsorption, the coverslip and slide glass were coated with 0.5% MPC polymer in ethanol before use. An inverted fluorescence microscope (IX-73; Olympus) with a total internal reflection fluorescence unit (IX3RFAEVAW; Olympus) was used (23,24). The objective lens (NA = 1.49) was illuminated using a 488-nm laser with highly inclined thin illumination (HILO) geometry. The fluorescence collected by the objective lens was detected using an EM-CCD camera (iXon Ultra 888; Andor). The laser power was in the range of 5–10 mW. Images were recorded at time intervals of 50 ms after reducing the number of observable molecules in DNA-rich droplets by photobleaching for 1–2 min at 21–22°C. The fluorescent spots of single molecules were tracked from sequential images using the ImageJ software with the plugin ‘Particle track and analysis’. To minimize the detection of molecules in PEG phase, we analyzed only a selected rectangle area that included DNA-rich droplets in the tracking. We selected trajectories with at least six consecutive points, and MSDs were calculated from all pairs of two-dimensional positions of a molecule at each time interval for all trajectories using our in-house program with some modifications (15,24,26). The MSD plots were fitted by 4Dt + 4σ2, where D and σ denote diffusion coefficient of molecules and spatial resolution, respectively. For diffusion analysis of each molecule, we used initial five displacements of trajectories (trajectories longer than 6 points were truncated) and calculated apparent diffusion coefficient (D*) values of individual molecules as described by Stracy et al. (48,49). The distribution of D* values was fit with the following equation:
 |
(3) |
where Aj, n and Dj* denote the amplitude of the jth mode, the number of steps in the trajectory and the diffusion coefficient of the jth mode, respectively. We used n = 5. The standard error for each D* bin was estimated using a bootstrap method with 1000 replicates and was used as the fitting weight. The fittings were performed using Igor. These analyses were done using data sets obtained in single or two independent experiments, and 5–54 droplets were analysed for each condition to minimize the difference of individual droplets. For the data of p53 in 4.1 nM DNA, the difference of percentages and diffusion coefficients for the two modes were within 1% and 0.02 μm2/s, respectively, in two independent experiments. Furthermore, the reliability of the obtained parameters was tested by comparing those obtained using the displacement distribution fitting method without taking into account the bias of molecules escaping from illumination thickness (50). For the data of p53 in 4.1 nM DNA, the difference of percentages and diffusion coefficients for two modes were within 1% and 0.08 μm2/s, respectively.
RESULTS
In vitro DNA droplet system for single-molecule analysis of DNA-binding proteins
To mimic LLPS of DNAs in cells, we used the mixture of dextran and PEG polymers, which form dextran-rich droplets (40) (Figure 1A). Dextran-rich droplets can efficiently recruit long DNA molecules (41), resulting in DNA-rich droplets (Figure 1A). The target search dynamics of DNA-binding proteins inside such DNA droplets could be observed using single-molecule fluorescence microscopy (Figure 1A).
As a proof of concept, we investigated the formation of dextran-rich droplets in a solution containing 150 mM KCl at pH 7.4 (near-physiological condition) in the presence of dextran and PEG. DIC imaging demonstrated the formation of spherical droplets, and fluorescent images of dextran labeled with a fluorescent dye FITC, confirming the dextran-rich droplet formation (Figure 1B, left). Next, we prepared DNA-rich droplets in order to investigate the target search dynamics of DNA-binding proteins. We used λ DNA that is a 48.5 kb non-specific substrate for the proteins studied here except for Fis (51,52). Fluorescent images with the DNA intercalator sytox green demonstrated that λ DNAs were efficiently recruited into dextran-rich droplets, resulting in DNA-condensed droplet formation (Figure 1B, right). We further investigated the localization of DNA-binding proteins into DNA droplets. We tested four DNA-binding proteins with different structures, sizes, and oligomeric states: p53, Nhp6A, Fis and Cas9 (Figure 1A). p53 is homo-tetramer of 393 residues with folded and disordered DNA-binding domains. Nhp6A is monomer of 93 residues with a folded HMGB DNA-binding domain and a disordered DNA-binding region. Fis subunits contain 98 residues that fold into an interwrapped compact dimer containing two helix-turn-helix DNA-binding domains. Cas9, clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein, is monomer of 1368 residues, and its multiple folded domains interact with DNA. For Cas9, dCas9-MBP, which is deficient for DNA cleavage and is fused to the maltose binding protein, was used in the absence of guide RNA (25). Fluorescent images at the ensemble level (100 nM labeled proteins) demonstrated that all four proteins labeled with the fluorescent dye Alexa488 or Atto488 were highly localized into DNA droplets (Figure 1C). The uptake ratios of these proteins inside to outside DNA droplets were 4.8 ± 0.4 for p53, 4.3 ± 0.3 for Nhp6A, 6.9 ± 0.8 for Fis, and 12 ± 1 for dCas9–MBP. The uptake level of these DNA-binding proteins, except dCas9–MBP, was significantly reduced in the absence of DNA (Supplementary Figure S1). These results demonstrate that non-specific DNA-binding activity enables or assists recruitment of these DNA-binding proteins into DNA-condensed droplets.
A slow mobility mode in DNA-condensed droplets was observed for four DNA-binding proteins
To clarify the target search dynamics of labeled DNA-binding proteins in non-labeled DNA droplets, we conducted single-molecule tracking measurements. We used fluorescence microscopy with highly inclined and laminated optical sheet (HILO) illumination (38,39) (Figure 2A). For single-molecule detection, the concentrations of labeled DNA-binding proteins were reduced to 1.5–2.0 nM. We first measured the dynamics of p53–Alexa488 (Supplementary movie S1). Many bright mobile spots were observed inside the droplets. We tracked the centers of the bright spots inside the droplets (Figure 2B). Translational dynamic properties were analyzed using mean square displacement (MSD) plots obtained from the traces. The MSD plots revealed a linear relationship with the time interval, indicating that p53 molecules diffused inside the DNA droplets (Figure 2C). As DNA concentration increased, the slope of MSD plots decreased, reflecting reduced diffusion of p53. To further examine this behavior, we plotted distribution histograms of the apparent diffusion coefficients D* obtained from single-molecule trajectories (Figure 2D). The histograms were fitted with the sum of two theoretical distributions with two average diffusion coefficients. 39 ± 1% of p53 molecules partitioned into a slow mobility mode (DS = 0.085 ± 0.001 μm2/s) and 61 ± 1% into a fast mobility mode (DF = 0.518 ± 0.009 μm2/s) in 4.1 nM DNA. The percentage of the slow mobility mode increased to 72 ± 2% in 33 nM DNA, reducing the average diffusion observed in Figure 2C.
Figure 2.

Single-molecule tracking of DNA-binding proteins in DNA-condensed droplets. (A) Schematic diagram of single-molecule tracking of fluorescent DNA-binding proteins in a DNA droplet. HILO illumination minimizes background fluorescence, enabling single-molecule detection of DNA-binding proteins (pink) in the DNA (light blue) droplet. (B) Typical trajectories of DNA-binding protein molecules in DNA droplets. Typical trajectories of single molecules over 250 ms (red) are overlaid in time-averaged fluorescent images (white and black). Scale bars are 10 μm. (C) Mean square displacement (MSD) plots of the labelled DNA-binding proteins in DNA droplets. Errors of MSD plots denote the standard errors. Straight lines show the best-fit linear functions for the MSD data. (D) Distributions of average diffusion coefficients of individual molecules of the DNA-binding proteins. Black dashed curves represent the best fitted curves by Equation (3) with two components. Colored solid curves denote each of the two components.
To test the generality of slow mobility mode in DNA droplets, we examined the translational dynamics of other DNA-binding proteins labeled with Atto488: Nhp6A, Fis, and dCas9–MBP. Like p53, the three proteins showed a linear relation of MSD plots against the time interval, confirming their diffusion in DNA droplets (Figure 2C). As the DNA concentration of droplets increased, the slopes of MSD plots of the three proteins decreased. Furthermore, slow mobility modes were found in the D* distributions and their percentages were increased with increasing DNA concentration, similar to the p53 case (Figure 2D). The slow mobility mode percentage of the small proteins (Nhp6A and Fis) was lower than that of the large proteins (p53 and dCas9–MBP) in 4.1 nM DNA, and became saturated and close to that of the large proteins in 33 nM DNA (Figure 3A). Furthermore, the D values of the fast mobility mode decreased to a small degree for p53 and Fis or did not show any systematic change for Nhp6A and dCas9–MBP, as DNA concentration increased (Figure 3B). The D values of the fast mobility mode for DNA-binding proteins distributed in a similar range but with a small variation. Therefore, we conclude that the slow mobility mode in DNA droplets is commonly observed for the four DNA-binding proteins and that the slow mobility mode percentage varies with the specific protein and with DNA concentration.
Figure 3.

Diffusion mode properties of DNA-binding proteins in DNA droplets as a function of DNA concentration of droplets. (A) DNA-concentration dependence of slow mobility mode percentages of four DNA-binding proteins. (B) DNA-concentration dependence of diffusion coefficients for the fast mobility mode of four DNA-binding proteins. In panels A and B, errors denote the fitting errors.
To interpret the two mobility modes of DNA-binding proteins, we measured the behaviour of Atto488-labeled λ DNA in droplets. The MSD plots showed a linear relationship against the time interval in 4.1 nM DNA, supporting that DNA is retained in liquid state in droplets (Supplementary Figure S2A). In the D* distribution of DNA, the peak was located at ∼0.1 μm2/s and the population decreased by ∼1.0 μm2/s (Supplementary Figure S2B). In 33 nM DNA, a similar result was obtained (Supplementary Figure S2C, D). The D value of the two mobility modes of DNA-binding proteins ranged within the D* distribution of DNA, suggesting the possibility that these DNA-binding proteins in the two mobility modes diffuse in DNA droplets while interacting with DNA transiently or for extended time. The slow mode percentage of DNA-binding proteins became larger with increasing DNA concentration (Figure 2D), whereas the peak height of DNA in the D* distribution did not depend on DNA concentration (Supplementary Figure S2B, D).
Origin of the slow mobility mode in DNA-condensed droplets
The slow mobility mode lasted for at least 250 ms (corresponding to 5 data points) in DNA droplets, which may reflect long-lived complexes between DNA-binding proteins and DNA. An alternative interpretation of the slow mobility mode is physical trapping of DNA-binding proteins within the condensed DNA network. To test these possibilities, we measured the dynamics of GFP, which lacks DNA-binding activity, in the DNA droplets. We used the −30 charged mutant of GFP (43) in order to minimize non-specific electrostatic interactions to DNA. The slopes of MSD plots of GFP were not significantly dependent on DNA concentration of the droplets, which differed from that of the DNA-binding proteins (Supplementary Figure S3A). The D* distribution analysis demonstrated that the slow mobility mode was still observed for GFP, but the percentage of the slow mobility mode was ≤ 9% in all DNA concentrations tested (Supplementary Figure S3B, C). This fraction presumably reflects physical trapping of DNA-binding proteins within condensed DNAs. In contrast, the DNA-binding proteins exhibited 37∼60% of the slow mobility modes in 16 nM DNA (Supplementary Figure S4A). The gap between GFP and DNA-binding proteins implies that the majority of the slow mobility mode is due to formation of long-lived complexes between DNA-binding proteins and DNA in DNA droplets.
To confirm the interaction between DNA-binding proteins and DNA in the slow mobility mode, we investigated the salt concentration dependence of translational mobility of p53 in DNA droplets. When the salt concentration increased from 50 mM to 500 mM KCl, the slope of the MSD plots increased (Figure 4A). In addition, the slow mobility mode percentage decreased from 47 ± 1% in 50 mM KCl to 25 ± 1% in 500 mM KCl, whereas the fast mobility mode percentage increased (Figure 4B,C). These data support that the weakened electrostatic interaction upon elevating salt concentration destabilizes the slow mobility mode. The remaining percentage of slow mobility mode in 500 mM KCl may include the physical, but non-electrostatic trapping within condensed DNAs. Furthermore, the D value of the fast mobility mode increased from 0.37 ± 0.01 μm2/s in 50 mM KCl to 0.56 ± 0.01 μm2/s in 500 mM KCl (Figure 4C). This suggests that high salt concentration suppresses the transient binding fraction of p53 to DNA, promoting the free diffusion fraction of p53 in the fast mobility mode. Taken together, the slow mobility mode reflects the formation of long-lived complex between DNA-binding protein and DNA as well as the physical trapping in condensed DNA.
Figure 4.

Single-molecule tracking of p53 in DNA-condensed droplets in different salt concentrations. (A) Mean square displacement (MSD) plots of p53 in DNA droplets (4.1 nM) in varying salt concentrations. Errors denote the standard errors. Straight lines show the best-fit linear functions for the MSD data. (B) Distributions of average diffusion coefficients of individual molecules of p53. Black dashed curves represent the best fitted curves by Equation (3) with two components. Colored solid curves denote each of the two components. (C) Salt-concentration dependence of diffusion coefficients of the fast mobility mode and the fast mobility mode percentage. Errors denote the fitting errors.
Since some DNA-binding proteins show a preferential backward motion by interacting with DNA confined in cells (53–56), we next examined the distribution of angles between consecutive translocations at 50 ms in DNA droplets. In the distribution of p53, the population of backward angles near 180° was ∼2-fold larger than that of the other angle region for all DNA concentrations (Supplementary Figure S5A). To identify which mode possesses this backword motion preference, we examined the relationship between the angle and the average displacement of two consecutive steps (Supplementary Figure S5B). The plot densities of backward angle were larger than those of the other angles in the short displacement region (<0.1 μm) corresponding to the slow mobility mode. Furthermore, the backward angle preference in the slow mobility mode was observed for other DNA-binding proteins (Supplementary Figure S6). In contrast, GFP, as a control, did not show any angle preference (Supplementary Figure S6). These results support that molecules in the slow mobility mode interact with or are trapped by DNA that is confined within the droplets.
The number of DNA-binding domains of p53 affects slow and fast mobility modes in DNA droplets
The data for the four DNA-binding proteins in Figure 3A demonstrated that large DNA-binding proteins have a greater tendency to form the slow mobility mode within DNA droplets, especially in low DNA concentrations. Since the large DNA-binding proteins used in this study possess multiple DNA-binding domains or regions (Figure 1A), we posit that these DNA-binding proteins can simultaneously interact with multiple DNA segments located nearby in the slow mobility mode. To investigate how the number of DNA-binding domains of p53 affects the slow mode formation, we measured NCT and TC mutants of p53, which lack the C-terminal domain and N-terminal plus core domains, respectively, and maintain the tetramer (14,17,21), as well as the dimer-forming mutant (L344A) of p53 (57). The C-terminal and core domains of p53 are disordered and folded DNA-binding domains, respectively. The number of DNA-binding domains is reduced from 8 in the full-length p53 tetramer to 4 in the mutants. The three mutants showed a reduction of MSD plot slopes with elevating DNA concentration of droplets (Figure 5A), consistent with that of full-length p53. The D* distribution analysis of individual molecules demonstrated that the slow mobility mode percentages of these mutants increased in particular in 33 nM DNA (Figure 5B). In addition, the percentages of the slow mobility mode of these mutants were smaller than that of the full-length p53 tetramer in different DNA concentrations (Figure 5C). The slow mobility mode percentages of NCT and dimer mutants increased significantly in the range between 16 nM and 33 nM, and the slow mobility mode percentage of the TC mutant increased above 8.2 nM DNA. In contrast, the slow mobility mode percentage of the full-length p53 tetramer increased in the range between 4.1 and 33 nM. These results support that the slow mobility mode formation is suppressed by reducing the DNA-binding domains of p53. Furthermore, the similar results between NCT and TC mutants emphasize the importance of the number of DNA-binding domains of p53, regardless of domain composition, on the slow mobility mode formation.
Figure 5.

Single-molecule tracking of p53 mutants lacking a DNA-binding domain or with different oligomeric states in DNA-condensed droplets. (A) Mean square displacement (MSD) plots of NCT, TC, and dimer (L344A) mutants of p53 in DNA droplets. Errors denote the standard errors. Straight lines show the best-fit linear functions for the MSD data. NT, core, Tet, and CT represent the N-terminal, core, tetramerization, and C-terminal domains of p53, respectively. D represents the dimer mutation in the Tet domain. Green and yellow boxes represent folded regions, whereas the other colors (purple, black, and pink) represent disordered regions. (B) Distributions of average diffusion coefficients of individual molecules of NCT, TC and dimer (L344A) mutants of p53 in different DNA concentrations. Black dashed curves represent the best fitted curves by Equation (3) with two components. Colored solid curves denote each of the two components. (C, D) DNA-concentration (of droplets) dependence of slow mobility mode percentage (C) and diffusion coefficient of fast mobility mode (D). Errors denote the fitting errors.
The D value of the fast mobility mode for these mutants was larger than that for the full-length p53 tetramer (Figure 5D). This reflects that the deletion of the core and C-terminal domains from full-length p53 tetramers weakens the transient interaction to DNA in the fast mode, consistent with the weaker DNA-binding ability for NCT and TC mutants of p53 than that of full-length p53 (17). In addition, these mutations reduce the molecular size of p53, which enhances the diffusion of the fast mode in the absence of DNA interactions. Taken together, the reduced number of DNA-binding domains of p53 reduces the molecular surface size that can interact with DNA (assuming proportionality with the number of DNA-binding domains), thereby suppressing the percentage of molecules in the slow mobility mode and the transient association to DNA in the fast mobility mode.
Relationship of the slow mobility mode percentage in DNA-condensed droplets with molecular parameters of DNA-binding proteins
We examined the relationship between the molecular size and the slow mobility mode percentage for the four DNA-binding proteins and p53 mutants (Figure 6A and Supplementary Figure S4A). As the molecular size of DNA-binding proteins increased, the slow mode percentage increased in 4.1 nM DNA (r = 0.95 and P = 0.001 for closed symbol data). As the DNA concentration of droplets increased to 33 nM, the slow mobility mode percentages increased for all cases and almost saturated to 43–72% irrespective of their molecular sizes. In particular, the percentage of the slow mobility mode of small DNA-binding proteins was largely enhanced to the level of large ones in the high DNA concentration. Our results emphasize that large DNA-binding proteins are inclined to form the long-lived complex with condensed DNA in a low concentration of DNA, consistent with their large effective sizes (or surface areas) for interacting to DNA. Furthermore, the slow mobility mode percentage was strongly correlated with the number of DNA-binding domains in 4.1 nM DNA (r = 0.92 and P = 0.003 for closed symbol data; Figure 6B). As the DNA concentration of droplets increased, the correlation with the number of DNA-binding domains was smaller (Supplementary Figure S4B).
Figure 6.

Relationship of slow mobility mode percentages of DNA-binding proteins in DNA-condensed droplets to their parameters and a proposed model of the slow mobility mode. Slow mobility mode percentage of DNA-binding proteins in DNA droplets (4.1 nM) as a function of molecular weight (A), number of DNA-binding domains (B), and dissociation constant to single short DNA under non-condensed conditions (C). In panel A, the data of GFP are displayed as reference. In panel (C), the data for p53 and its mutants (NTC and TC) were from our previous paper (17). In panels A-C, errors denote the fitting errors, and correlation coefficients (r) for the data of closed symbols except GFP are given. The r values in parentheses in panels A and B represent those for all data including open symbols except GFP. (D) Multivalent interaction model of the slow mobility mode as exemplified by p53. NT (grey), core (light blue), Tet (grey), and CT (pink) show the N-terminal domains, core domains, Tet domains, and C-terminal domains of p53, respectively. Blue lines represent DNA segments. In the slow mobility mode, p53 interacts with multiple DNA segments using different sets of DNA-binding domains (core and CT). The DNA sequence can be scanned using sequence-recognizing core domains, while the protein is tethered to different DNA segments.
Since DNA-binding proteins have different affinities to single DNA segments in non-condensed DNA conditions, we next investigated the relation of the slow mobility mode percentage to the affinity. The dissociation constants of DNA-binding proteins were determined for a 30 bp non-specific DNA fragment, assuming one by one binding, under the same solution condition using fluorescence anisotropy titration measurements (Supplementary Figure S7). The dissociation constant of DNA-binding proteins to the short DNA fragment moderately correlated with the slow mobility mode percentage in DNA droplets (r = −0.72 and P = 0.07 in 4.1 nM DNA; Figure 6C). As the DNA concentration in droplets increased, the correlation with the dissociation constant to the short DNA fragment was smaller (Supplementary Figure S4C). The correlation to molecular size and number of DNA-binding domains in 4.1 nM DNA was larger than to the affinity for single DNA fragment in non-condensed conditions.
To distinguish the importance of molecular size and number of DNA-binding domains, we measured three mutants that maintain the molecular size while reducing the number of DNA-binding domains or that maintain the number of DNA-binding domains while changing the molecular size in 4.1 nM DNA (Figure 6A,B and Supplementary Figure S8). First, dCas9 without MBP, which maintains the number of DNA-binding domains while reducing the molecular size from dCas9-MBP, showed the same slow mobility mode percentage (42 ± 2%) to that of dCas9-MBP (42 ± 1%). Second, the R85A mutation of Fis (number of DNA-binding domains, N = 0), which strongly lowers DNA-binding affinity while maintaining the molecular size (26), lowered the slow mobility mode percentage to 2 ± 1% from 7 ± 1% by wild type Fis (N = 2). Third, the p53-CTD(R/KtoA) mutant (N = 4), which essentially eliminates DNA binding by the C-terminal domain while maintaining the molecular size, lowered the slow mobility mode percentage to 18 ± 2% from 39 ± 1% in wild type (N = 8). The plots of three mutants (open symbols) largely deviated from the data except for the three mutants (closed symbols) in Figure 6A, whereas the plots were within the data except for the three mutants in Figure 6B. As a control, dCas9-MBP fused to the disordered DNA-binding domain of Nhp6A (ST-dCas9-MBP, N = 8) enhanced the slow mobility mode percentage to 47 ± 3% from 42 ± 1% in dCas9-MBP (N = 7). The plot of ST-dCas9-MBP ranged within the other data in Figure 6A and B. Taken together, these results support the importance of the number of DNA-binding domains on the percentage of molecules in the slow mobility mode for different proteins (r = 0.97 and P < 0.001 for all data sets including the four mutants in 4.1 nM DNA), rather than the respective molecular sizes (r = 0.86 and P < 0.001 for the all data sets). Based on these results, we propose that DNA-binding proteins can associate with multiple DNA segments using at least two DNA-binding domains in DNA-condensed droplets to form the long-lived complexes corresponding to the slow mobility mode (Figure 6D).
DISCUSSION
In this study, we mimicked LLPS of DNA in cells by reconstituting DNA-condensed droplets in vitro and tracked DNA-binding proteins in the DNA-condensed droplets using single-molecule fluorescence microscopy. Fast and slow mobility modes were observed within DNA-condensed droplets for all four DNA-binding proteins studied. In general, DNA-binding proteins are believed to search for their target sequence of DNA by combining four search mechanisms: 1D sliding along DNA, hopping/jumping along DNA, 3D diffusion, and intersegmental transfer between two DNAs (1–8). We discuss the relation between these proposed mechanisms and the two mobility modes found in this study.
The fast mobility mode in DNA-condensed droplets is interpreted as co-translational diffusion with fast-moving DNA or as 3D diffusion coupled with transient interaction to DNA. For p53, this transient interaction is consistent with sub-millisecond single-molecule tracking studies on isolated DNAs (20,21). This may allow the observed 1D sliding of p53, Nhp6A and Fis (13–15,17,23,26,58) or hopping/jumping of p53 (20,21) along DNA. Similar transient DNA interactions of other DNA-binding proteins have been observed in bacteria (59). Such complex interactions might affect the diffusion manner of molecules in the fast mobility mode in DNA droplets. In fact, analysis with a power-law equation (D = cMα, where c, M, and α are the proportional coefficient, molecular weights of DNA-binding proteins, and the scaling exponent, respectively) demonstrated that the α value in DNA droplets ranged from –0.01 ± 0.01 to –0.110 ± 0.008 (Supplementary Figure S9). The absolute values of α were smaller in DNA droplets than in solution (α = –0.33, Stokes–Einstein relation (60)), under cellular crowding conditions (α = –0.7 ∼ –0.75 (59,60)), and in p53 and FUS droplets (α = –0.33 and –0.22, respectively (39)). In addition to the interaction with DNA, the molecular crowding effect, including a more compact structure of disordered regions for p53 and Nhp6A, might contribute to this small absolute α value. Overall, the fast mobility mode can rapidly transport the DNA-binding proteins, which may extend the search space in DNA droplets.
In the slow mobility mode, DNA-binding proteins within DNA-condensed droplets likely grab multiple DNA segments simultaneously using multiple DNA-binding domains (Figure 6D). This model is supported by the fact that the slow mobility mode percentage was strongly correlated with the number of DNA-binding domains, but only moderately with the affinity to single DNA segment in non-condensed conditions (Figure 6). As the number of DNA-binding domains increases, the probability that a DNA-binding protein interacts with multiple DNA segments increases in DNA-condensed droplets. To explain the linear relation between the slow mobility mode percentage and number of DNA-binding domains (Figure 6B), we built a physical model. In the model, we assumed that DNA-binding domains can associate to DNA segments independently and the DNA segment-association probability as fraction unit, p, is constant for DNA-binding domains, even though there are different physical and chemical interactions for each domain. Based on this assumption, the slow mobility mode percentage, which reflects the percentage of DNA-bound proteins (binding to at least one DNA segment), is estimated to be 100 × (1 − (1 − p)N), where N is the number of DNA-binding proteins. (1 − p)N corresponds to the fraction of DNA-unbound form. If p is much smaller than 1, the slow mode percentage is approximately 100pN. The p value, obtained by the fitting of experimental data with this equation, was 0.06 in 4.1 nM DNA for all data sets. Note that the experimental data in high DNA concentrations deviate from this simple model, likely since the p value in the bound form with multiple DNA segments may be modulated by large electrostatic repulsion between the DNA segments.
With the exception of intersegmental transfer, the multivalent interaction model differs from proposed search mechanisms that consider interactions on only a single DNA segment. Complexes of p53 and Fis with multiple DNA segments is supported by electrophoresis (61,62) and electron microscopy (62), the loop formation of DNA in stretched and relaxed cycles (47,63), and molecular dynamics simulation of p53 with two DNAs (36). Furthermore, this bound form is similar to the intermediate of intersegmental transfer observed in p53 (19,64), Fis (27), and Nhp6A (27). This is also supported by compact collapse of DNA by Cas9 (25). This mode allows these proteins to scan multiple DNA sequences at the same time, which enables efficient target search in droplets. Similar enhanced search of p53 was predicted in condensed nucleosomal DNA compared to less-condensed ones using molecular dynamics simulations (37). DNA-binding proteins often possess one or two disordered tails in addition to structured DNA-binding domains, and these disordered tails are frequently involved in non-specific DNA binding (65), enabling DNA-binding proteins to grab at least two DNAs simultaneously. Considering these facts, the multivalent interaction mode with multiple DNA segments may be general (66). Thus, this slow mobility mode in condensed DNA conditions such as LLPS might function as a recognition mode of target sequences on genomic DNA.
In DNA-condensed droplets, DNA-binding proteins can switch between fast and slow mobility modes inside droplets. This is supported by the fact that the p53 molecules transit from the fast mobility mode to the slow mobility mode and vice versa in several trajectories. This suggests that trapped molecules can be released from locally-condensed DNA regions and that rapidly diffusing molecules can become trapped into locally-condensed DNA regions since the DNA is maintained in a liquid state.
In physiological conditions, the slow mobility mode as well as the fast mobility mode are expected to be used in target searches within DNA-condensed droplets. The concentration of DNA in nuclei is ∼150 mM by base pairs (67). The accessible DNA in vivo is considered to be ∼1%, because the other DNA regions are occupied by histones (68). Accordingly, the concentration of accessible DNA in nuclei is 1.5 mM by base pairs. The maximum concentration of DNA (48.5 kbp) tested in droplets is 33 nM corresponding to 1.6 mM in base pair units, which is comparable to the accessible DNA concentrations in nuclei. In this condition, the slow mobility mode percentage ranges from 43% to 72% for four DNA-binding proteins (Supplementary Figure S4). In addition, the slow mobility mode found in DNA-condensed droplets may explain the gap between in vitro and in vivo data. A long residence time (3.5 s) of p53, corresponding to target-bound complex, was observed within transcription domains of cells (29). In contrast, the residence time of p53 on target sequence of isolated DNAs was 120 ms (16), which was 29-fold shorter than that in transcription domains. Since transcription domains are considered to resemble liquid droplets of condensed DNA segments (69,70), the target binding of p53 in such situation might be stabilized by interacting to multiple DNA segments as proposed for the slow mobility mode in the present study.
DNA-condensed droplets might assist the target search of DNA-binding proteins by confining these proteins within the droplets. 4.3- to 12-fold recruitment of DNA-binding proteins into the droplets suggests that when the proteins encounter the droplet boundary, they may preferentially bounce back into the interior. In fact, we observed several trajectories of p53 confined within a small DNA droplet (Supplementary Figure S10). This implies that the confinement effect of droplets preferentially prevents DNA-binding proteins from escaping from the droplets, giving additional chance to scan DNA sequences located inside the droplets. Furthermore, the DNA droplets can concentrate DNA-binding proteins, which enhances the association with DNA in the droplets. These two properties are unique for LLPS of DNAs.
In future applications, the single-molecule tracking method of DNA-binding proteins in DNA-condensed droplets can be used to measure their actions under complex cell-mimicking conditions including nucleosomal DNA, phase separating factors (proteins and RNAs), and interactions between their disordered regions (69,71,72). We emphasize that this DNA-condensed droplet system is not limited to λ DNA tested in this study, but can also be applied to any long DNA, including purified genomic DNA, since very long DNAs are recruited into dextran-rich droplets.
Supplementary Material
ACKNOWLEDGEMENTS
We thank prof. Reid C. Johnson (UCLA) for contributing proteins and comments on the manuscript.
Contributor Information
Kiyoto Kamagata, Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan; Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan.
Ryo Kusano, Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan; Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan.
Saori Kanbayashi, Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan.
Trishit Banerjee, Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan; Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan.
Hiroto Takahashi, Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan.
Data Availability
All data generated or analyzed during this study are included in this published article and its Supplementary Data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MEXT/JSPSKAKENHI [JP20K06571, JP21H00379, JP22H05582 to K.K.]; Naito Foundation (to K.K.); Takeda Science Foundation (to K.K.). Funding for open access charge: MEXT/JSPS KAKENHI [JP20K06571, JP21H00379, JP22H05582 to K.K.]; Naito Foundation (to K.K.); Takeda Science Foundation (to K.K.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Von Hippel P.H., Berg O.G.. Facilitated target location in biological systems. J. Biol. Chem. 1989; 264:675–678. [PubMed] [Google Scholar]
- 2. Halford S.E., Marko J.F.. How do site-specific DNA-binding proteins find their targets?. Nucleic Acids Res. 2004; 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tafvizi A., Mirny L.A., van Oijen A.M.. Dancing on DNA: kinetic aspects of search processes on DNA. ChemPhysChem. 2011; 12:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Monico C., Capitanio M., Belcastro G., Vanzi F., Pavone F.S.. Optical methods to study protein-DNA interactions in vitro and in living cells at the single-molecule level. Int. J. Mol. Sci. 2013; 14:3961–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Redding S., Greene E.C.. How do proteins locate specific targets in DNA?. Chem. Phys. Lett. 2013; 570: 10.1016/j.cplett.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kamagata K., Murata A., Itoh Y., Takahashi S.. Characterization of facilitated diffusion of tumor suppressor p53 along DNA using single-molecule fluorescence imaging. J. Photochem. Photobiol. C Photochem. Reviews. 2017; 30:36–50. [Google Scholar]
- 7. Kamagata K., Itoh Y., Subekti D.R.G.. How p53 molecules solve the target DNA search problem: a review. Int. J. Mol. Sci. 2020; 21:1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamagata K. Single-molecule microscopy meets molecular dynamics simulations for characterizing the molecular action of proteins on DNA and in liquid condensates. Front. Mol. Biosci. 2021; 8:795367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheinman M., Kafri Y.. The effects of intersegmental transfers on target location by proteins. Phys. Biol. 2009; 6:016003. [DOI] [PubMed] [Google Scholar]
- 10. Bauer M., Metzler R.. Generalized facilitated diffusion model for DNA-binding proteins with search and recognition states. Biophys. J. 2012; 102:2321–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Veksler A., Kolomeisky A.B.. Speed-selectivity paradox in the protein search for targets on DNA: is it real or not?. J. Phys. Chem. B. 2013; 117:12695–12701. [DOI] [PubMed] [Google Scholar]
- 12. Mahmutovic A., Berg O.G., Elf J.. What matters for lac repressor search in vivo–sliding, hopping, intersegment transfer, crowding on DNA or recognition?. Nucleic Acids Res. 2015; 43:3454–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tafvizi A., Huang F., Leith J.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008; 95:L01–L03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tafvizi A., Huang F., Fersht A.R., Mirny L.A., van Oijen A.M.. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murata A., Ito Y., Kashima R., Kanbayashi S., Nanatani K., Igarashi C., Okumura M., Inaba K., Tokino T., Takahashi S.et al.. One-dimensional sliding of p53 along DNA is accelerated in the presence of Ca2+ or Mg2+ at millimolar concentrations. J. Mol. Biol. 2015; 427:2663–2678. [DOI] [PubMed] [Google Scholar]
- 16. Itoh Y., Murata A., Sakamoto S., Nanatani K., Wada T., Takahashi S., Kamagata K.. Activation of p53 facilitates the target search in DNA by enhancing the target recognition probability. J. Mol. Biol. 2016; 428:2916–2930. [DOI] [PubMed] [Google Scholar]
- 17. Murata A., Itoh Y., Mano E., Kanbayashi S., Igarashi C., Takahashi H., Takahashi S., Kamagata K.. One-dimensional search dynamics of tumor suppressor p53 regulated by a disordered C-terminal domain. Biophys. J. 2017; 112:2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subekti D.R.G., Murata A., Itoh Y., Fukuchi S., Takahashi H., Kanbayashi S., Takahashi S., Kamagata K.. The disordered linker in p53 participates in nonspecific binding to and one-dimensional sliding along DNA revealed by single-molecule fluorescence measurements. Biochemistry. 2017; 56:4134–4144. [DOI] [PubMed] [Google Scholar]
- 19. Itoh Y., Murata A., Takahashi S., Kamagata K.. Intrinsically disordered domain of tumor suppressor p53 facilitates target search by ultrafast transfer between different DNA strands. Nucleic Acids Res. 2018; 46:7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subekti D.R.G., Murata A., Itoh Y., Takahashi S., Kamagata K.. Transient binding and jumping dynamics of p53 along DNA revealed by sub-millisecond resolved single-molecule fluorescence tracking. Sci. Rep. 2020; 10:13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Subekti D.R.G., Kamagata K.. The disordered DNA-binding domain of p53 is indispensable for forming an encounter complex to and jumping along DNA. Biochem. Biophys. Res. Commun. 2021; 534:21–26. [DOI] [PubMed] [Google Scholar]
- 22. Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014; 507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamagata K., Mano E., Ouchi K., Kanbayashi S., Johnson R.C.. High free-energy barrier of 1D diffusion along DNA by architectural DNA-binding proteins. J. Mol. Biol. 2018; 430:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamagata K., Ouchi K., Tan C., Mano E., Mandali S., Wu Y., Takada S., Takahashi S., Johnson R.C.. The HMGB chromatin protein Nhp6A can bypass obstacles when traveling on DNA. Nucleic Acids Res. 2020; 48:10820–10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banerjee T., Takahashi H., Subekti D.R.G., Kamagata K.. Engineering of the genome editing protein Cas9 to slide along. DNA. Sci. Rep. 2021; 11:14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kamagata K., Itoh Y., Tan C., Mano E., Wu Y., Mandali S., Takada S., Johnson R.C.. Testing mechanisms of DNA sliding by architectural DNA-binding proteins: dynamics of single wild-type and mutant protein molecules in vitro and in vivo. Nucleic Acids Res. 2021; 49:8642–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giuntoli R.D., Linzer N.B., Banigan E.J., Sing C.E., de la Cruz M.O., Graham J.S., Johnson R.C., Marko J.F.. DNA-segment-facilitated dissociation of fis and NHP6A from DNA detected via single-molecule mechanical response. J. Mol. Biol. 2015; 427:3123–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mazza D., Abernathy A., Golob N., Morisaki T., McNally J.G.. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012; 40:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morisaki T., Muller W.G., Golob N., Mazza D., McNally J.G.. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat. Commun. 2014; 5:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loffreda A., Jacchetti E., Antunes S., Rainone P., Daniele T., Morisaki T., Bianchi M.E., Tacchetti C., Mazza D. Live-cell p53 single-molecule binding is modulated by C-terminal acetylation and correlates with transcriptional activity. Nat. Commun. 2017; 8:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei M.T., Chang Y.C., Shimobayashi S.F., Shin Y., Strom A.R., Brangwynne C.P.. Nucleated transcriptional condensates amplify gene expression. Nat. Cell Biol. 2020; 22:1187–1196. [DOI] [PubMed] [Google Scholar]
- 32. Schneider N., Wieland F.G., Kong D., Fischer A.A.M., Horner M., Timmer J., Ye H., Weber W.. Liquid-liquid phase separation of light-inducible transcription factors increases transcription activation in mammalian cells and mice. Sci. Adv. 2021; 7:eabd3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gamliel A., Meluzzi D., Oh S., Jiang N., Destici E., Rosenfeld M.G., Nair S.J.. Long-distance association of topological boundaries through nuclear condensates. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2206216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M.. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019; 38:e101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander K.A., Coté A., Nguyen S.C., Zhang L., Gholamalamdari O., Agudelo-Garcia P., Lin-Shiao E., Tanim K.M.A., Lim J., Biddle N.et al.. p53 mediates target gene association with nuclear speckles for amplified RNA expression. Mol. Cell. 2021; 81:1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khazanov N., Levy Y.. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011; 408:335–355. [DOI] [PubMed] [Google Scholar]
- 37. Kanada R., Terakawa T., Kenzaki H., Takada S.. Nucleosome crowding in chromatin slows the diffusion but can promote target search of proteins. Biophys. J. 2019; 116:2285–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kamagata K., Iwaki N., Hazra M.K., Kanbayashi S., Banerjee T., Chiba R., Sakomoto S., Gaudon V., Castaing B., Takahashi H.et al.. Molecular principles of recruitment and dynamics of guest proteins in liquid droplets. Sci. Rep. 2021; 11:19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kamagata K., Iwaki N., Kanbayashi S., Banerjee T., Chiba R., Gaudon V., Castaing B., Sakomoto S.. Structure-dependent recruitment and diffusion of guest proteins in liquid droplets of FUS. Sci. Rep. 2022; 12:7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsumoto K., Arai M., Nakatani N., Watanabe S.N., Yoshikawa K.. Does DNA exert an active role in generating cell-sized spheres in an aqueous solution with a crowding binary polymer?. Life. 2015; 5:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakatani N., Sakuta H., Hayashi M., Tanaka S., Takiguchi K., Tsumoto K., Yoshikawa K.. Specific spatial localization of actin and DNA in a water/water microdroplet: self-emergence of a cell-like structure. ChemBioChem. 2018; 19:1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamagata K., Kanbayashi S., Honda M., Itoh Y., Takahashi H., Kameda T., Nagatsugi F., Takahashi S.. Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains. Sci. Rep. 2020; 10:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.Y., Liu D.R.. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015; 33:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Terry B.R., Matthews E.K., Haseloff J.. Molecular characterisation of recombinant green fluorescent protein by fluorescence correlation microscopy. Biochem. Biophys. Res. Commun. 1995; 217:21–27. [DOI] [PubMed] [Google Scholar]
- 45. Wang H., Kelley F.M., Milovanovic D., Schuster B.S., Shi Z.. Surface tension and viscosity of protein condensates quantified by micropipette aspiration. Biophys. Rep. 2021; 1:100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kamagata K., Ariefai M., Takahashi H., Hando A., Subekti D.R.G., Ikeda K., Hirano A., Kameda T.. Rational peptide design for regulating liquid-liquid phase separation on the basis of residue-residue contact energy. Sci. Rep. 2022; 12:13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Igarashi C., Murata A., Itoh Y., Subekti D.R.G., Takahashi S., Kamagata K.. DNA garden: a simple method for producing arrays of stretchable DNA for single-molecule fluorescence imaging of DNA-binding proteins. Bull. Chem. Soc. Jpn. 2017; 90:34–43. [Google Scholar]
- 48. Stracy M., Lesterlin C., Garza de Leon F., Uphoff S., Zawadzki P., Kapanidis A.N.. Live-cell superresolution microscopy reveals the organization of RNA polymerase in the bacterial nucleoid. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4390–E4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stracy M., Kapanidis A.N.. Single-molecule and super-resolution imaging of transcription in living bacteria. Methods. 2017; 120:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hansen A.S., Woringer M., Grimm J.B., Lavis L.D., Tjian R., Darzacq X.. Robust model-based analysis of single-particle tracking experiments with Spot-On. Elife. 2018; 7:e33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thompson J.F., Moitoso de Vargas L., Koch C., Kahmann R., Landy A.. Cellular factors couple recombination with growth phase: characterization of a new component in the lambda site-specific recombination pathway. Cell. 1987; 50:901–908. [DOI] [PubMed] [Google Scholar]
- 52. Pan C.Q., Johnson R.C., Sigman D.S.. Identification of new Fis binding sites by DNA scission with fis-1,10-phenanthroline-copper(I) chimeras. Biochemistry. 1996; 35:4326–4333. [DOI] [PubMed] [Google Scholar]
- 53. Izeddin I., Récamier V., Bosanac L., Cissé I.I., Boudarene L., Dugast-Darzacq C., Proux F., Bénichou O., Voituriez R., Bensaude Oet al.. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife. 2014; 3:e02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hansen A.S., Amitai A., Cattoglio C., Tjian R., Darzacq X.. Guided nuclear exploration increases CTCF target search efficiency. Nat. Chem. Biol. 2020; 16:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mazzocca M., Fillot T., Loffreda A., Gnani D., Mazza D. The needle and the haystack: single molecule tracking to probe the transcription factor search in eukaryotes. Biochem. Soc. Trans. 2021; 49:1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iida S., Shinkai S., Itoh Y., Tamura S., Kanemaki M.T., Onami S., Maeshima K.. Single-nucleosome imaging reveals steady-state motion of interphase chromatin in living human cells. Sci. Adv. 2022; 8:eabn5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Weinberg R.L., Veprintsev D.B., Fersht A.R.. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004; 341:1145–1159. [DOI] [PubMed] [Google Scholar]
- 58. Leith J.S., Tafvizi A., Huang F., Uspal W.E., Doyle P.S., Fersht A.R., Mirny L.A., van Oijen A.M.. Sequence-dependent sliding kinetics of p53. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16552–16557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stracy M., Schweizer J., Sherratt D.J., Kapanidis A.N., Uphoff S., Lesterlin C.. Transient non-specific DNA binding dominates the target search of bacterial DNA-binding proteins. Mol. Cell. 2021; 81:1499–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mika J.T., Poolman B.. Macromolecule diffusion and confinement in prokaryotic cells. Curr. Opin. Biotechnol. 2011; 22:117–126. [DOI] [PubMed] [Google Scholar]
- 61. Skoko D., Yoo D., Bai H., Schnurr B., Yan J., McLeod S.M., Marko J.F., Johnson R.C.. Mechanism of chromosome compaction and looping by the Escherichia coli nucleoid protein Fis. J. Mol. Biol. 2006; 364:777–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kearns S., Lurz R., Orlova E.V., Okorokov A.L.. Two p53 tetramers bind one consensus DNA response element. Nucleic Acids Res. 2016; 44:6185–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Skoko D., Yan J., Johnson R.C., Marko J.F.. Low-force DNA condensation and discontinuous high-force decondensation reveal a loop-stabilizing function of the protein Fis. Phys. Rev. Lett. 2005; 95:208101. [DOI] [PubMed] [Google Scholar]
- 64. Takada S., Kanada R., Tan C., Terakawa T., Li W., Kenzaki H.. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015; 48:3026–3035. [DOI] [PubMed] [Google Scholar]
- 65. Vuzman D., Azia A., Levy Y.. Searching DNA via a “Monkey Bar” mechanism: the significance of disordered tails. J. Mol. Biol. 2010; 396:674–684. [DOI] [PubMed] [Google Scholar]
- 66. Zhou H., Song Z., Zhong S., Zuo L., Qi Z., Qu L.J., Lai L.. Mechanism of DNA-induced phase separation for transcriptional repressor VRN1. Angew. Chem. Int. Ed. Engl. 2019; 58:4858–4862. [DOI] [PubMed] [Google Scholar]
- 67. Iwahara J., Clore G.M.. Direct observation of enhanced translocation of a homeodomain between DNA cognate sites by NMR exchange spectroscopy. J. Am. Chem. Soc. 2006; 128:404–405. [DOI] [PubMed] [Google Scholar]
- 68. Hesselberth J.R., Chen X., Zhang Z., Sabo P.J., Sandstrom R., Reynolds A.P., Thurman R.E., Neph S., Kuehn M.S., Noble W.S.et al.. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009; 6:283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhat P., Honson D., Guttman M.. Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol. 2021; 22:653–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li W., Jiang H.. Nuclear protein condensates and their properties in regulation of gene expression. J. Mol. Biol. 2022; 434:167151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brodsky S., Jana T., Mittelman K., Chapal M., Kumar D.K., Carmi M., Barkai N.. Intrinsically disordered regions direct transcription factor in vivo binding specificity. Mol. Cell. 2020; 79:459–471. [DOI] [PubMed] [Google Scholar]
- 72. Nozawa R.S., Yamamoto T., Takahashi M., Tachiwana H., Maruyama R., Hirota T., Saitoh N.. Nuclear microenvironment in cancer: control through liquid-liquid phase separation. Cancer Sci. 2020; 111:3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Supplementary Data.