


Abstract
The Sirtuin family of NAD+-dependent enzymes plays an important role in maintaining genome stability upon stress. Several mammalian Sirtuins have been linked directly or indirectly to the regulation of DNA damage during replication through Homologous recombination (HR). The role of one of them, SIRT1, is intriguing as it seems to have a general regulatory role in the DNA damage response (DDR) that has not yet been addressed. SIRT1-deficient cells show impaired DDR reflected in a decrease in repair capacity, increased genome instability and decreased levels of γH2AX. Here we unveil a close functional antagonism between SIRT1 and the PP4 phosphatase multiprotein complex in the regulation of the DDR. Upon DNA damage, SIRT1 interacts specifically with the catalytical subunit PP4c and promotes its inhibition by deacetylating the WH1 domain of the regulatory subunits PP4R3α/β. This in turn regulates γH2AX and RPA2 phosphorylation, two key events in the signaling of DNA damage and repair by HR. We propose a mechanism whereby during stress, SIRT1 signaling ensures a global control of DNA damage signaling through PP4.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
The members of the Sirtuin family of NAD+-dependent enzymes play a key role in the response to metabolic, oxidative and genotoxic stress (1). Sirtuins coordinate an efficient adaptation to these stress conditions at different levels including protection of genome stability, metabolic homeostasis, and increased survival. Among the most relevant roles of Sirtuins is their direct implication in maintaining genome integrity which involves chromatin organization and structure, mitotic progression and DNA repair (2,3). In this functional context, Sirtuins have been implicated in the regulation of both DNA damage response (DDR) signaling and DNA repair itself (4).
The DDR is tightly regulated by a wide range of protein post-translation modifications. Among the most relevant and conserved of them is phosphorylation of histone and non-histone proteins, which participate in all crucial steps of DDR. Phosphorylation of serine 139 in the histone H2A variant H2AX (γH2AX) is a hallmark of double strand break (DSB) signaling in the DDR (5–7). The establishment of γH2AX, catalyzed by the kinases ATM, ATR or DNA-PKcs is an early step in the DDR that promotes interactions and sequential accumulation of other key regulatory factors such as the MRN complex (MRE11–RAD50–NBS1) and MDC1 (8,9). After DNA repair is completed, γH2AX is dephosphorylated, allowing the cell to deactivate the DNA damage checkpoint and to resume the cell cycle (10). Several phosphatases have been described to play this role upon completion of repair including PP2A, PP4 or Wip1 (11). Among them, PP4 was shown to antagonize ATR signaling during replication stress, and its depletion induces γH2AX hyperphosphorylation and a block in DNA damage efficiency (12,13).
PP4 phosphatase is a Ser/Thr PP2A-like multisubunit complex involved in many stress-associated processes beyond the DDR, such as NF-kB signaling, immune response, meiosis, centrosome maturation, splicing, cell cycle regulation and glucose metabolism (14). Reflecting its physiological relevance, PP4 is frequently deregulated in a wide range of cancers (15,16). The PP4 complex is a trimeric/tetrameric complex containing a catalytic subunit PP4c and different combinations of the regulatory subunits PP4R1, PP4R2, PP4R3α and PP4R3β (also known as SMEK1 and 2, respectively), and PP4R4 (17). Given the high degree of identity between PP4c and the PP2 catalytic subunits (65%), it is the regulatory subunits that mediate the specific roles of PP4, including binding to different substrates and other specific factors (18,19). Despite the important role of this complex, little is known about the way this activity is regulated. The most clearly conserved PP4 complex is formed by PP4c–R2–R3 (3α and 3β in mammals), that is responsible for γH2AX dephosphorylation and hypersensitivity to DNA damage-inducing agents, like cisplatin (18). PP4R2 is required to stabilize PP4c structure, and its deficiency decreases the specific activity of PP4c (12,20). In contrast, PP4R3α and β play a more complex role. While they are key to recognize substrates through their N-terminal WH1 domain, they also seem to restrict PP4c activity in vitro, suggesting that they control PP4 enzymatic activity (21). Besides γH2AX, the PP4c–R2–R3 complex also dephosphorylates other key targets such as RPA2, KAP1 or 53BP1 in the context of DDR (22,23). RPA2 is a subunit of the trimeric RPA complex, which binds to ssDNA in the repair of DSBs and base damage. RPA2 phosphorylation by ATR on S33 and T21 during the S and G2 phases (24) participate in HR pathway regulation, replication stress tolerance and mitotic exit, among other functions (25). Thus, RPA2 phosphorylation is associated with DSB pathway choice, as RPA2 dephosphorylation induces foci formation of the HR-specific factor RAD51 (26).
So far, Sirtuins SIRT1,2,6 and 7 have been linked to the DDR. Of them, SIRT1 and SIRT6 seem to have a more prominent role, and in contrast to SIRT2 or SIRT7, their loss impairs the phosphorylation of H2AX, which results in reduced DNA repair efficiency (27–29). SIRT6 has a major role in the initial steps of DSB repair as a pioneering factor that recognizes and binds to DNA damage, initiating the DSB repair process (30). SIRT1 loss inhibits foci formation of NBS1, RAD51, BRCA1 and γH2AX (31,32). However, although several SIRT1-dependent mechanisms have been described involving deacetylation of NBS1, Ku70, WRN, MOF or TIP60, the underlying mechanism and how are they interconnected in the context of a global SIRT1 function, are not well understood (33). The link between SIRT1 and γH2AX is particularly puzzling as the reported functional relationship between SIRT1 and NBS1 or ATM cannot explain the decrease of this modification observed in SIRT1-deficient cells (34–36). Here, we identify and characterize a direct functional antagonism between SIRT1 and PP4 complex through deacetylation of regulatory subunits PP4R3α and PP4R3β. Our evidence helps to explain the decreased γH2AX levels observed in Sirt1−/− cells and suggests a global coordinated control of DDR progression by SIRT1 through regulation of PP4 function.
MATERIALS AND METHODS
Reagents
Camptothecin (CPT) and hydroxyurea(HU) were obtained from Sigma-Aldrich, okadaic acid(OA) from Calbiochem and Puromycin from Invivogen. The following antibodies were used: HA (Sigma-Aldrich, H6908), FLAG (Sigma-Aldrich F1804), c-Myc (Cell Signaling 2276S), actin (Sigma-Aldrich A1978), α-tubulin (Sigma-Aldrich T6199), SIRT1 (Millipore, 07–131; Cell Signaling 8469) SIRT6 (ABCAM AB62739), histone H3 (Cell Signaling, 9715), GFP (MERCK, MAB2510), RPA32 (Cell Signaling 2208), PP4R2 (Bethyl, A300-838A), PP4R3α (Bethyl, A300-840A), PP4R3β (Bethyl, A300-842A), PP4C (Abcam, ab171870), anti-phospho-histone H2A.X (Ser139) (abcam ab 2893 and Merck Millipore, JBW301), phospho-RPA2 (Ser33) (Novus, NB100-544 and Bethyl, A300-246A), phospho-RPA2 (Ser4/8) (Sigma-Aldrich, PLA0071) and Fibrillarin (Santa Cruz Biotechnology, Sc-166001).
Cell culture studies
Wild type and Sirt1−/− Mouse embryonic fibroblasts (MEFs) were cultured in DMEM supplemented with 20% fetal bovine serum, Pen-strep (10 000 IU/ml, 10 mg/ml), non-essential amino acids and sodium pyruvate according to the manufacturer's instructions. HeLa, HEK293T and U2OS Wt and Sirt1−/− cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum. All the cells were grown at 37°C in an atmosphere containing 5% CO2, and 100% humidity. Unless stated otherwise, at 48 h of transfection (if indicated) the cells were treated with the following conditions before harvesting at indicated times: 2 mM of H2O2 (MERCK) for 1 h, 5 mM hydroxyurea (Sigma-Aldrich) for 4 h, 1 μM campthotecin (Sigma-Aldrich) for 1 h and irradiation with X rays at 7.5 and 10 Gy. The cells were exposed to different concentrations of EX527 (as indicated) for 24 h. For cell subfractionation, U2OS cells were treated with 2 mM H2O2 for 1 h. The nucleoplasm fraction was obtained with the Dignam protocol extraction (fraction C) (37), and the remaining chromatin fraction was then solubilized in 1 × Laemmli buffer and sonicated with Branson 250 sonicator.
Endogenous SIRT1 purification
HeLa nuclear extract (2 g) was loaded on a phosphocellulose P11 cation exchange column (Whatman, USA) in a BC buffer (50 mM Tris–HCl pH 7.9, 0.2 mM EDTA, 10% glycerol, 1 mM DTT, 0.2 mM PMSF with a gradient from 100 mM to 1 M KCl). The Flow-Through (FT) of the P11 column was loaded on the anion exchange column DE52 and resolved in a linear gradient of BC buffer from 100 to 600 mM KCl. The SIRT1-containing fractions, monitored by western-blot, were pooled and dialyzed to BC buffer 80 mM KCl and fractionated on the weak anion exchange column DEAE-5PW (Diethylaminoethyl cellulose 52; TosoHaas, Montgomeryville, USA). The DEAE column was resolved by using a linear gradient of 80mM to 725 mM KCl in buffer BC. The fractions containing SIRT1 were pooled and precipitated with saturated Ammonium sulphate 5 M Tris 50 mM pH 7.9 at 40%. The precipitate was then resuspended in BD buffer (BC buffer with 50 mM ammonium sulphate) with 500 mM KCl and fractionated by molecular weight on the gel filtration column Sephacryl S-400 (GE healthcare, USA) with a fractionation range of 2 × 104–8 × 106 Da. Fractions containing SIRT1 were pooled, dialyzed to BD buffer 1 M KCl, applied to hydrophobic interaction chromatography on a phenyl-superose column and resolved in a linear gradient of BD buffer from 1 M to 350 mM KCl. SIRT1 eluted around BD buffer 700 mM KCl and the fractions were pooled and dialyzed to BC buffer with 40 mM KCl. Purified SIRT1 was subjected to a MonoQ anion exchange column (GE healthcare, USA) and resolved in a linear gradient from BC buffer with 40 to 400 mM KCl. The Purified fractions containing SIRT1 were subjected to SDS-PAGE and visualized by silver staining. The fractions 25 and 26 were pooled, loaded on a single lane and subjected to SDS-PAGE followed by staining with colloidal blue (Invitrogen, USA). Bands were cut and containing proteins were identified by mass-spectrometry (see Supplementary information).
Viral generation and infections
To produce retrovirus, Platinum A cells were seeded into culture dishes a day before transfection in DMEM supplemented with 10% FBS. The cells were co-transfected with 2 μg of pVSVG plasmid and 8 μg of the retroviral vector and supernatant media was collected after 24 and 48 h and stored at –80ºC. For the infection, cells were plated 24 h before infection in 6-well plates. Then, media was replaced by the stocked supernatant containing the retrovirus and incubated for 16 hours, thereafter the media was replaced by fresh media and after 24 h cells were selected with puromycin. For shRNA knockdown with lentiviral infection, Mission shRNAPP4C (pLKO.1-puro, Clon ID #TRCN0000080835) were obtained from Sigma-Aldrich, Inc. HEK293T cells were co-transfected with pMD2.G and psPAX2 (Addgene) together with pLKO-shRNA constructs (PLKO.1 shRNA PP4C) or a non-targeting shRNA (pLKO.1-puro ID # SHC003). Viral supernatants were harvested after 48 h. For infections, SIRT1 MEF cells were incubated with viral supernatants in the presence of 8 μg /ml polybrene. After 48 h, puromycin (5 μg/ml) as selection was added to the medium. Cells were grown in presence of selection for 4–6 days to select for stable knockdown cells.
In vitro enzymatic reactions
For the phosphatase assays, PP4 complex subunits were overexpressed in HeLa cells or SIRT1 MEFs and purified using Flag resin to immunoprecipitate the catalytic subunit PP4C. The bound proteins were eluted by competition with a large excess of free FLAG peptide (0.4 mg/ml). In vitro phosphatase assays were performed in phosphatase reaction buffer (PRB, 50 mM Tris pH 7.2, 0.1 mM CaCl2, 5 mM MnCl2 and 0.2 mg/ml BSA). Purified proteins were first equilibrated with PRB for 10 min at 37°C followed by addition of phospho-H2AX-enriched histone as a substrate and incubation at 37°C for 30min and analyzed by western-blot. For the deacetylation assays HA-SIRT1 and PP4 complex subunits were expressed separately in HeLa cells, immunoprecipitated and purified using HA and Flag peptides in BC100 buffer. Deacetylation assays were performed in deacetylation buffer (50 mM Tris–HCl pH 8, 100 mM NaCl, 2 mM DTT, 5% glycerol) with or without 5 mM of NAD+. SIRT1 was first equilibrated with deacetylation buffer for 10 min at 37°C. Then, substrate were added to the reaction mixture and incubated at 37°C for 1 h and analyzed by western blot.
In the case of PP4R3α/β deacetylation by SIRT1, Biotinylated peptides (Biotin-PNTAYQK(Ac)QQDTLI) previously immobilized on magnetic Streptavidin beads (Dynabeads MyOne Invitrogen™ 65601) were incubated in presence or absence of SIRT1 ± NAD+ (0.5 nM) in a total volume of 100 μl of deacetylase buffer (60 mM Tris–HCl pH 7,8, 40 mM MgCl2, at pH 7.0, 2 mM DTT and protease inhibitor cocktail). The reaction mixtures were incubated at 37°C for 90 min and the beads were washed three times with BC100 and deacetylation of Ac- PP4R3α/β (K64) by SIRT1 was detected by MALDI-TOF (see Supplementary information).
Analysis of metaphase aberrations
Cells were treated with 100 μM H2O2 for 30 min, arrested in metaphase with 100 ng/ml of Colcemid for 1 hour and fixed in 3:1 methanol:acetic acid. Resuspended cells were dropped onto a slide and stained with Giemsa stain (1:12 in 3% methanol). Images of n = 20 metaphases of each condition were acquired with an 100× objective and Metafer software (MetaSystems). Metaphases were analyzed using ImageJ.
HR and NHEJ functional assays
The I-SceI-GFP DR-GFP and EJ-RFP reporters were used to measure HR and NHEJ repair as described elsewhere (38). Briefly, U2OS-EJ-DRs cells were transfected with SCR, SIRT1 and/or PP4C siRNA for 24 h using Dharmafect reagent. To induce I-SceI DSB, cells were incubated with 1 μM Shield-1 and 100 nM triamcinolone acetonide for 24, 48 or 72 h depending on experimental conditions. Cells were then trypsinized and analyzed for GFP and RFP expression in a FACS Canto cytometer. Data was analyzed using Flowjo and results were expressed as the mean percentage of GFP+ and RFP+ cells from three independent experiments.
Cell cycle analysis
Cell cycle analysis was performed using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay kit (C10340, ThermoFisher Scientific). Cells were treated with 10 mM EdU and incubated for 30 min before being trypsinized. After washing with PBS, cells were fixed with 4% PFA for 15 min, washed once with washing buffer (1% BSA, 2 mM EDTA in 1x PBS) and permeabilized (0,5% Triton, 1% BSA in 1× PBS) for 10 min. After washing with washing buffer, click chemistry using Alexa Flour 647 azide was performed for 30 min in the dark, as stated in the manufacturer's instructions. Cells were then washed again in washing buffer and resuspended in analysis buffer (0,02% (wt/vol) sodium azide, 0.5 mg/ml DAPI in 1 mg/ml BSA in 1x PBS). Cell cycle was analysed using a BD FACSCanto™ II System and the FlowJo 7.6 software. Standard gating for cells versus debris and singlet was conducted.
Proliferation and viability
Cell proliferation and viability were measured at the indicated time-points using the trypan blue exclusion assay. Briefly, supernatant and attached cells were collected together, centrifuged 5 min at 1500 rpm, and resuspended in complete media. 20 μl of cells were diluted in 40 μl of trypan blue. 10 μl sample were loaded on to a hemocytometer, and total, alive and dead cells were counted using a Leica DMi1 inverted microscope.
TCGA proteomics, phosphoproteomics and transcriptomics
To evaluate correlations between SIRT1, SIRT6, PP4C protein abundance and pRPA2 (p238), pTRIM28 (pS473) and pATM (pS1981) levels in the Breast Invasive Carcinoma (TCGA) dataset, mass spectrometry levels measured by the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC) were accessed and downloaded through the cBioportal web-based utility (http://www.cbioportal.org). Pearson correlations were calculated using GraphPad Prism version 8.0. A subset of patients with mutations or deep deletions in HDR genes was defined (see Figure 7D) and gene expression and protein levels correlation in this subset was conducted.
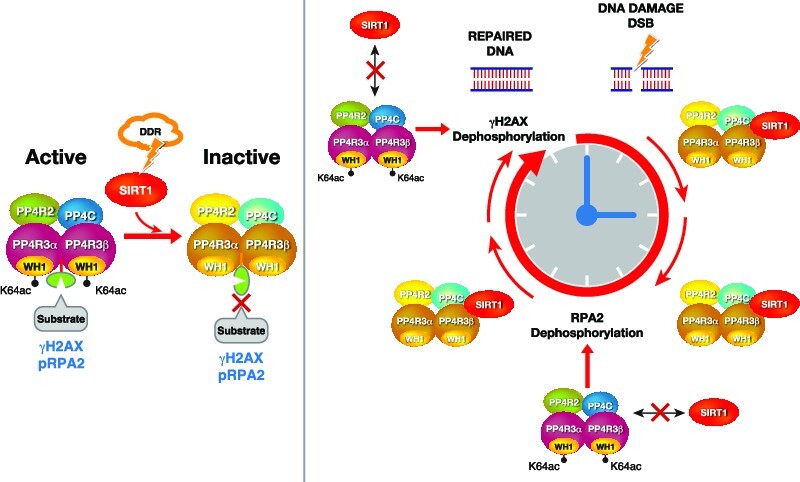
Figure 7.

SIRT1 and PP4 inversely correlate in cancer. (A) Proteomics and phosphoproteomics data from tumor samples was obtained from the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC) analysis of tissues from the Breast Invasive Carcinoma (TCGA) dataset. (B) Heatmap of SIRT1, SIRT6, PPP4C protein abundance and pRPA2 (p238) and pTRIM28 (pS473) levels in samples from the study described in (A). Each column corresponds to one tumor sample. (C) Correlations between the indicated proteins and pRPA2 in each tumor sample were analyzed. Pearson correlation coefficient (r) and P-value (P) for each analysis are shown. (D) List of HDR-associated genes used in the analysis of (E). (E) Samples of the breast cancer cohort containing mutations or deep deletions in the genes in (D) were selected and analyzed as in (C). (F) Proposed model of the antagonistic interplay between SIRT1 and PP4 in HR repair.
Neutral comet assay
To detect DSBs, the neutral comet assay was performed as described previously (39). U20S and SIRT1 MEFs Cells were transfected with either control or PP4c-specific siRNA and treated or not with camptothecin 1 μM for 90 min. The images were acquired using a Leica AF-5000 confocal microscope and analyzed and quantified using the Comet Score program.
FRET
FRET assays were performed as previously described (40). Human SIRT1 and PP4C were amplified by PCR with Sal-I and BamH-I sites added to the 5′ and 3′ ends, respectively. The PCR products were cloned into the mCerulean-C1 and YFP-C1 vector so that both proteins were either N-terminally tagged with the chromophore. All constructs were confirmed by DNA sequencing. HeLa cells were co-transfected with 8 μg YFP-SIRT1 and 1μg CFP-PP4C plasmid DNA per plate and plated on a coverslip. After 48 h, transfected cells were imaged using a Leica TCS SP5 confocal imaging system. To measure FRET signals by acceptor photobleaching, we imaged the fluorescent intensity in a selected region of interest. YFP-SIRT1 fluorescence was then photo-destroyed by repeated scanning with the 568-nm laser line. The fluorescence intensities of images of pre-bleached CFP-PP4C were compared to corresponding regions of post-bleached images. As controls, non-fused chromophores were paired with their corresponding protein-fused chromophores and similarly processed. FRET was measured as an increase in CFP fluorescence intensity following YFP photobleaching, and calculated as 100 × [(CFP post-bleach – CFP pre-bleach)/YFP post-bleach]; taking into account YFP and CFP background noise.
Phospho-H2AX-enriched histone extraction
Hela cells were treated with 2 mM of H2O2 (MERCK) for 1 h, then the cells were scraped down by a cell scraper and washed with PBS and pelleted by centrifugation at 4000 rpm for 1 min, subsequently the cells were resuspended in buffer A (10 mM Tris, pH 7.9, 1,5 mM MgCl2, 10 mM KCl, 0,1 mM PMSF, 0,5 mM DTT, supplemented with protease inhibitor (Sigma-Aldrich)) for 10 min on ice, centrifuged at 12 000 × g for 1 min. The pellet (nuclear fraction) was resuspended with 0.5 M HCl (500 μl/10 cm2 plate). The samples were resuspended by vortex and then incubated on ice for 15 min. After centrifugation at 14 000 × g for 10 min at 4ºC, the supernatant (soluble acid proteins) was collected and precipitated with TCA in a final concentration of 20% and incubated on ice for 1 h. After centrifugation at 14 000 × g for 10 min at 4ºC, the pellet was washed with 100% ice-cold acetone and then centrifuged at 14 000 × g for 10 min at 4ºC. After drying the pellet at RT, it was resuspended with BC100 and stored at –80ºC.
Statistical analysis
The represented values show means of three or more independent experiments (n ≥ 3) with error bars representing standard error of mean (SEM) unless otherwise specified. Statistical analysis of the data was performed, unless stated otherwise, using two-tailed Student's t test (α = 0.05).
Further Materials and Methods information can be found in the supplementary information file.
RESULTS
Purification of endogenous SIRT1 reveals a direct interaction with the PP4 complex
To further understand the role of SIRT1 in genome stability, we purified endogenous SIRT1 to identify any interacting proteins that may shed light on its role in the DDR. Nuclear extracts from HeLaS3 were processed through a sequence of seven chromatography steps (Figure 1A). After each step, fractions containing SIRT1 were pooled before proceeding to the following chromatographic step. Following the final step of an anion exchange monoQ column, proteins were run in SDS-PAGE gel and silver-staining revealed co-elution of the SIRT1 band with three different protein bands of lower molecular weight (Figure 1B). The profile of SIRT1 fractionation was further confirmed by wester-blot analysis (Figure 1C). Analysis of these bands using mass spectrometry identified the members of a previously described PP4 complex, the catalytic subunit PP4c and regulatory proteins PP4R2, PP4R3α and PP4R3β (Supplementary Table S1) (12). Given the well-established role of this PP4–R2–R3α–R3β complex in the DNA damage response, this finding suggested that SIRT1 could participate in the role of PP4c in DNA repair (12).
Figure 1.

Identification of a novel protein complex containing SIRT1 and the PP4 phosphatase complex. (A) Schematic representation of the SIRT1 purification from HeLaS3 nuclear extracts (NE) including the buffers used (BC or BD) and a range of KCl gradient concentrations (from 40 mM to 1 M) depending on each chromatographic step (see Methods and Supplementary information). (B, C) Silver stained SDS-gel (B) and western-blot (C) of the anion-exchange MonoQ column, the final chromatography step of the purification. Fractions 23–25 were pooled and analyzed by mass spectrometry. The analysis showed that PP4 complex components PP4c, PP4R2, PP4R3α and PP4R3β co-fractionate with SIRT1 (Supplementary Table S1).
SIRT1 interacts with the PP4 complex upon genotoxic stress
Given the functional relationship between both factors and the DDR, we tested whether the interaction between SIRT1 and the PP4 complex was specifically boosted by genotoxic stress conditions. Surprisingly, we observed that although the interaction took place under all tested conditions, including replicative stress (HU and CPT) and ionizing irradiation (IR), it was specifically promoted upon oxidative stress (Figure 2A and Supplementary Figure S1A). This oxidative stress-dependent interaction was also confirmed with endogenous proteins (Figure 2B) and with FRET assays between Cerulean (CFP)-PP4c and YFP-SIRT1 (Figure 2C and Supplementary Figure S1B). We further confirmed that stress conditions drive the formation of a stable SIRT1/PP4 complex, as we detected the cofractionation of SIRT1 and PP4 subunits in an approximately 600 kDa complex in gel filtration analysis (Figure 2D). This corresponded to roughly to the sum of the 400 kDa homotetrameric SIRT1 complex (41) and the 240 kDa PP4c complex (42).
Figure 2.

SIRT1-PP4 complex formation is boosted by oxidative stress conditions. (A) SIRT1 and the PP4 complex interaction with SIRT1-FLAG IP experiments in HeLa cells non-stress (NT) or under 5mM H2O2 (2h), 2mM hydroxyurea (4h, HU) and 10 and 20 μM camptothecin (2 h, CPT). (B) Endogenous IP using α-SIRT1 antibodies between the PP4 complex (PP4c, PP4R2, PP4R3α/β) and SIRT1 from whole cell extracts of U2OS cells previously treated or not (C) with 2 mM H2O2 for 1 h. As a negative control the same cell extracts without SIRT1 antibody (no Ab) was used. Inputs (I) and elutions (E) are shown. (C) FRET analysis of SIRT1-YFP (acceptor) and PP4c-CFP (donor) in HeLa cells treated during 1 h with 2 mM H2O2. Cells were imaged with confocal fluorescence microscopy and fluorescence of CFP-PP4c was measured before and after photobleaching in control and oxidative stress conditions. Left, schematic representation of the process. Right, quantification of detected FRET efficiency is shown from n = 3 experiments. Two-tailed t-test analysis (**P< 0.01). (D) SIRT1 and the PP4 complex cofractionate in fractions around 600 kDa (fractions 13–16). Glycerol gradient analysis (12.5–35%) of nuclear extract from HeLa cells expressing FLAG-SIRT1 and treated with H2O2 before harvest (see Methods and Supplementary information). (E) HA and FLAG co-immunoprecipitations of HA-PP4c and FLAG-SIRT1, SIRT1 catalytically inactive point mutant H363Y (SIRT1m) or SIRT6 in whole-cell extracts from HeLa cells expressing the indicated constructs treated or not with 2 mM H2O2 for 1 h before harvest. (F) Time course experiment from 0 to 24 h of SIRT1/PP4 complex assembly upon oxidative stress. PP4c (FLAG) immunoprecipitation of whole-cell extracts from HeLa cells expressing HA-tagged SIRT1 and PP4R2, FLAG-tagged PP4C and Myc-tagged PP4R3α/β treated with H2O2 2 mM for 1h and harvested at the indicated times.
SIRT1 enzymatic activity did not regulate this interaction as both SIRT1 WT and a catalytically-dead point mutant H363Y (SIRT1m) immunoprecipitated PP4 to a similar extent (Figure 2E). The interaction was SIRT1-specific, as we did not detect any interaction between PP4c and SIRT6 under the same conditions (Figure 2E). The SIRT1–PP4 interaction was highly dynamic, as it was detected mainly within 90 min following the treatment with H2O2 (1 h incubation + 30 min after (t30)) with a peak at t0), and to a lesser degree 6–12 h after the treatment (Figure 2F). Together, this indicated that the SIRT1 and PP4 interaction was enhanced by oxidative stress independently of SIRT1 activity.
SIRT1 inhibits PP4c phosphatase activity
As previously reported (31), we detected decreased levels of γH2AX in SIRT1-deficient MEFs, even 48hr after induction of DNA damage (Figure 3A and Supplementary Figure S2A). Considering that γH2AX is also a relevant target of PP4 (12,13), we analyzed the functional implications of the SIRT1–PP4 interaction on γH2AX. For that purpose, we purified the PP4 complex (PP4c–R2–R3α–R3β) from HeLa cells and performed in vitro phosphatase assays in the absence or presence of SIRT1 using γH2AX as a substrate. Incubation of SIRT1 in the presence of NAD+ was able to completely repress the phosphatase activity of the complex, suggesting a direct modulation of PP4 activity through deacetylation (Figure 3B). SIRT1 also had a mild inhibitory effect in the absence of NAD+ (Figure 3B) and the γH2AX phosphatase activity of PP4 purified from SIRT1-deficient MEFs was significantly higher than when purified from Wt MEFs (Figure 3C). Consistently, the activity of the PP4 complex purified from cells treated with the SIRT1 specific inhibitor Ex-527, or with the general Sirtuin inhibitor Nicotinamide (NAM), resulted in a hyperactivation of the phosphatase activity of the complex (Figure 3D). Supporting a role for SIRT1 deacetylase activity in PP4 inhibition, overexpression of active WT SIRT1, but not a catalytically inactive point-mutant (HY), rescued the levels of γH2AX upon oxidative stress conditions (Figure 3E).
Figure 3.

SIRT1 inhibits PP4 phosphatase activity. (A) High throughput microscopy (HTM) immunofluorescence analysis of γH2AX in Wt and Sirt1−/− cells treated or not with 7.5 Gy IR for 1 h and analyzed at the indicated times after irradiation. Data are representative of three independent experiments. Two-tailed t-test (****P< 0.001). At least 300 nuclei were analyzed and the mean with SEM is shown for independent cultures. (B) In vitro γH2AX phosphatase activity of a titration of PP4 complexes (1×, 2×, 3×) purified from HeLa cells treated or not with 2 mM H2O2 for 1 h and incubated –/+ SIRT1 in presence or absence of NAD+. (C) In vitro phosphatase activity as in (B) of PP4 complex purified from Wt and Sirt1−/− MEFs under oxidative stress 2 mM H2O2 for 1 h. (D) Assay as in (B) and (C) Purified PP4 complexes in the presence of Ex-527 (1 μM, 10 μM) and 1 mM nicotinamide (NAM). (E) Western-blot of γH2AX, H2AX and the indicated proteins of extracts from HEK293T overexpressing the PP4 complex in presence or absence of FLAG-SIRT1 WT or catalytically-inactive mutant H363Y. (F) Phosphatase assay of whole-cell extracts from Wt and Sirt1−/− cells previously treated with Okadaic Acid (OA) (50 nM) or DMSO (control) for 24 h followed by 1 h incubation of 2 mM H2O2 before harvest. Quantification of the ratio of γH2AX/ H2AX in the phosphatase assay is shown.
Conversely, the inhibition of PP4 activity with Okadaic acid (43) partially recovered the decreased levels of γH2AX detected in SIRT1-deficient cells compared to Wt cells and inhibition of PP4 activity in a SIRT1-deficient background resulted in a milder increase in γH2AX compared to Wt (Figure 3F). SIRT1 loss also resulted in decreased phosphorylation of KAP-1, a well-established target of PP4 (23) (Supplementary Figure S2B), suggesting that the antagonism between SIRT1 and PP4 goes beyond γH2AX regulation. Finally, our results also suggest that the impact of SIRT1 on γH2AX is not related to deacetylation of H2AXK5ac, a mark that was reported to be a SIRT1 target after IR-induced DNA damage (44). We observed that SIRT1 knock-down resulted in decreased levels of both γH2AX and H2AXK5ac under oxidative stress (Supplementary Figure S2B). Altogether, these results suggested that functional interplay between SIRT1 and PP4 regulates γH2AX phosphorylation dynamics.
SIRT1-dependent deacetylation of PP4R3α and PP4R3β regulates the PP4 complex
To determine whether SIRT1-mediated deacetylation of PP4 led to its inhibition, we expressed the PP4 core complex (PP4c-R2) and purified it from Wt and Sirt1−/− MEF cells, resulting in a reconstituted PP4 complex containing all four components (PP4c-R2 and endogenous mouse PP4R3α and 3β). Acetylation analysis of the complex identified two acetylated lysine residues in the PP4R3 subunits in Sirt1−/− cells compared to Wt cells (Figure 4A−C and Supplementary Figures S3A−C); one was common for both PP4R3α and β (K64), and the other ones were specific to each subunit (in K642 for PP4R3α and mouse K777 / human K806 for PP4R3β). K64 is present in the N-terminal WH1 domain, shared by both PP4R3α and β, and is highly conserved in the PP4R3 lineage from Drosophila to humans (Figure 4D). The WH1 domain plays a role in protein-protein interactions and has been found in factors involved in cell signaling, cell cycle, cytoskeleton organization, cell motility, gene expression and chromatin organization (45).
Figure 4.

SIRT1 targets K64 in PP4R3α, β to regulate PP4 activity. (A) PP4 complex was purified from MEFs Wt and Sirt1−/− under oxidative stress (2 mM H2O2 1 h) overexpressing PP4 core complex (PP4c and PP4R2) and analyzed by MS to identify acetylated peptides in the PP4 complex upon loss of SIRT1. Left, Schematic representation of the experiment's pipeline. Right, Levels of the indicated proteins by western blot in the MEFs analyzed. (B) Summary of the acetylated peptides identified in the MS analysis of (A). Differential acetylation was identified in the endogenous PP4R3α and β subunits (see Methods, Supplementary information and Supplementary Table S2). (C) PP4R3α and β protein domains, the acetylation events identified in Sirt1−/− but not in Wt cells, and their position in both subunits. (D) K64 and D67 are two conserved residues in WH1 domain of PP4R3α,β from Drosophila to humans. WH1 domain primary sequence of the homologs of both PP4R3α,β isoforms in Saccaromyces cerevisiae, Drosophila melanogaster, Xenopus laevis, zebrafish, mouse and humans PP4R3 homologs. (E) In vitro γH2AX phosphatase activity assay of human PP4 complex containing the indicated mutations expressed and purified from HeLa cells. A quantification of n = 3 experiments (Supplementary Figure S4D) is shown. Two-tailed t-test (*P< 0.05; **P< 0.01; ***P< 0.005). (F) Quantification of n = 4 experiments (measured by Western blot) as in 3E of the ratio γH2AX/H2AX in HEK293T cells overexpressing the PP4 complex containing PP4R3α/β WT or K64R −/+ SIRT1 treated with 2 mM H2O2 for 1 h. A representative image is shown in Supplementary Figure S3E. Two-tailed t-test assay was performed (*P< 0.005; **P< 0.01; ***P< 0.005). (G) MALDI-MS analysis of an in vitro K64ac deacetylation reaction by SIRT1. The chromatograms for the control reactions without neither SIRT1 nor NAD+ (Ctrl) and with SIRT1 but not NAD+ (SIRT1) are shown together with the complete reaction (SIRT1 + NAD+). The acetylated peptide was detected either alone or in form of a single sodium adduct. Only the complete reaction rendered deacetylation of both species found for the peptide (b and c) in contrast to the acetylated forms of the peptide (a) (see Materials and Methods). (H) Structure of human PP4R3α suggests that K64 and D67 are physically very close and have the potential to interact only if K64 is in its unacetylated form (right upper cartoon). Acetylation of K64 could break the interaction between K64 and D67, allowing D67 to turn to the substrate binding region and interact with the substrate through a lysine residue in the FXXP motif (main and right lower cartoon). (I) Pull-down experiments with HA resin of previously purified PP4 complexes containing PP4R3α WT/K64R/D67N and core histones. Both the PP4 complexes and the core histones were independently purified from HEK293cells previously treated with 2 mM H2O2. (J) γH2AX phosphatase activity of the PP4 complexes in (H).
To test whether the deacetylation of any of these residues explained the SIRT1-dependent inhibition of PP4 complex activity, we mutated these residues to arginine or glutamine to mimic non-acetylated or acetylated lysines, respectively. We purified the PP4 complex containing different combinations of these PP4R3α and β mutants and tested the activity of the complex in an in vitro phosphatase assay towards γH2AX. Of the different combinations we tested, we detected a significant inhibition of the phosphatase activity only when the complex contained both PP4R3α and PP4R3β K64R mutants (Figure 4E and Supplementary Figure S3D). No significant effect was detected when the complex contained at least one K64 residue in either PP4R3 α or β (data not shown). The relevance of K64 acetylation in promoting PP4 activity was further supported by the increased activity observed for PP4 complexes containing both K64Q acetyl-mimicking mutants of PP4R3α and R3β (Figure 4E and Supplementary Figure S3D). Moreover, the decrease in γH2AX levels observed upon expression of the WT PP4c complex in 293 cells was abrogated by SIRT1 overexpression (Figure 4F and Supplementary Figure S3E). In contrast, the expression of PP4c complex containing PP4R3α/β K64R did not decrease the levels of γH2AX, and SIRT1 overexpression did not have any further effect. These observations strongly support a direct interplay between SIRT1 and PP4.
To understand the functional relationship between SIRT1 and K64 acetylation, we first confirmed that K64 was a direct target of SIRT1 in an in vitro deacetylation assay (Figure 4G). The structure of the human PP4R3α EVH1 domain bound to WAPL through a FXXP motif (6R81, 1.52A) (46) and Drosophila Melanogaster’s PP4R3α EVH1 domain bound to CENP-C (4WSF, 1.50A) (46) have been reported. Structural analysis suggests that K64 is not present in the substrate binding site of the domain, but could interact with D67, a conserved residue with the potential to position itself towards the bound substrate, which could result in decreased substrate recognition (Figure 4D and H). In contrast, acetylation of K64 would block this interaction, releasing D67 and allowing its repositioning towards the substrate. Supporting a role for D67 in the catalytic activity of the PP4 complex mutation of this residue to Asn (D67N) in PP4R3α, which abrogates the ability to interact with K64 while keeping a similar physicochemical environment, resulted in a decreased ability of this complex to bind to γH2AX-containing histones in pull-down experiments (Figure 4I). A similar effect was observed with the K64R mutant, indicating that the effect of both mutants on PP4 complex are equivalent (Figure 4I). To analyze the effect of these mutations on PP4 catalytic activity we performed γH2AX phosphatase assays with the same complexes tested in the pull-down experiments. Confirming our hypothesis, the results showed that both K64R and D67N mutations significantly decreased the phosphatase activity of the PP4 complex towards γH2AX (Figure 4J). Taken together, our results demonstrate a functional antagonism between SIRT1 and the PP4 complex upon stress. Further, they suggest that SIRT1 inhibits PP4 complex activity towards γH2AX by blocking PP4 substrate recognition through deacetylation of the WH1 domains of regulatory subunits PP4R3α and β.
SIRT1 antagonizes RPA2 dephosphorylation by PP4
We further analyzed the impact of SIRT1 on another major PP4 target in the DDR, RPA2. Sirt1−/− MEFs showed impaired RPA2-foci formation upon H2O2 or IR treatment without altering overall RPA2 protein levels (Figure 5A–C), suggesting that SIRT1 activity could have an impact on RPA2 function. Both Wt and Sirt1−/− cells showed similar progression through S-phase (Supplementary Figure S4A), indicating that RPA2 foci formation was not caused by impaired fork progression. This effect on RPA2 foci formation correlated with both a global decrease in ph-RPA2 (Figure 5B) and loss of RPA2-S4/8ph foci upon oxidative stress (Supplementary Figure S4B). This was further supported by cell fractionation analysis under the same conditions, as RPA2 levels did not decrease in the chromatin fraction upon loss of SIRT1 despite the loss of RPA2 foci (Figure 5C). RPA2-S33ph also decreased drastically in the same compartment (Figure 5C). The impact of SIRT1 loss on RPA2 phosphorylation, and specifically S33ph, was consistent up to 48 h after DNA damage induced by IR or H2O2 treatment (Figure 5D and Supplementary Figure S4C, D, respectively).
Figure 5.

SIRT1 regulates RPA2 phosphorylation through PP4. (A) IF analysis of γH2Ax and RPA2 foci formation in Wt or Sirt1−/− MEFs under or not oxidative stress (2 mM H2O2 for 1 h), or ionizing irradiation (IR 30 min 7.5 Gy). (B) Western-blot analysis of the levels of the total pRPA2 in Wt and Sirt1−/− MEFs previously treated with 7.5 Gy IR. Quantification of n = 3 experiments and representative experiment are shown. The shifted phosphorylated form of RPA2 is detected in the longer exposure blots and is marked with *. Two-tail analysis (**P< 0.01). (C) Western blot of SIRT1, RPA2 (total and S33ph) in whole cell extracts (WCE), nuclear soluble fraction (NS) and chromatin insoluble fraction (Chrom) of Wt or Sirt1−/− U2OS cells previously treated with 2 mM H2O2 for 1 h. The levels of fibrillarin and histone H3 were also analyzed as controls of NS and Chrom fractions, respectively. (D) IF Time-course experiment of RPA2-S33P performed as in 3A. A similar experiment is shown by western blot in Figure S4B. (E) Co-IP between FLAG-RPA2 and HA-SIRT1 using FLAG and HA resin under untreated condition or oxidative stress (2 mM H2O2 for 1 h) in HeLa cells. (F) Endogenous IP with α-SIRT1 antibodies of RPA2 and SIRT1 from whole cell extracts of U2OS cells previously treated with 2 mM H2O2 for 1 h. Sirt1−/− extracts were used as a negative control. (G) Levels of the indicated proteins and RPA2 marks upon shRNA-driven downregulation or not of PP4c in Wt and Sirt1−/− MEFs untreated or irradiated with IR (7.5 Gy). Lanes 5 and 10, SIRT1 expression was re-introduced in Sirt1−/− (indicated with *). (H) Similar experiment as in 3D with RPA2-S33P levels.
Oxidative stress boosted SIRT1 specific interaction with overexpressed or endogenous RPA2 (Figure 5E, F). In agreement with a key role of PP4 in SIRT1-dependent regulation of ph-RPA2, RPA2 pS33 and pS4/8 hyperphosphorylation induced by PP4 downregulation in Wt MEFs was significantly reduced in Sirt1−/− MEFs (Figure 5G). This decrease was directly dependent on SIRT1 protein, as re-expression of SIRT1 partially recovered the PTM levels (Figure 5G). Similar to what we observed with γH2AX, the K64R mutation in both PP4R3α/β resulted in increased levels of RPA2 phosphorylation, while K64Q had the converse effect (Figure 5H and Supplementary Figure S4E). Taken together, these results confirmed a direct role for SIRT1-mediated deacetylation in modulating the dynamics of RPA2 phosphorylation through the regulation of PP4 complex activity.
Interplay between SIRT1 and PP4 regulates DNA repair and genome stability
To determine the functional impact of the interplay between SIRT1 and PP4, we analyzed cell cycle progression and DNA repair kinetics. While downregulation of PP4c in Wt U2OS cells resulted in increased G1 phase cells and a mild decrease in S-phase and G2/M, SIRT1 knockdown did not significantly alter the cell cycle. Loss of both SIRT1 and PP4c restored the normal cell cycle, suggesting a functional antagonism between both factors in the control of G1/S (Figure 6A, B and Supplementary Figure S5A). A similar result was observed upon shRNA-mediated downregulation of PP4c in Sirt1−/− MEFs (Supplementary Figure S5B). The increase in G1 induced by PP4c downregulation was accompanied by a mild but significant decrease in cell proliferation that was rescued by SIRT1 depletion (Supplementary Figure S5C). Notably, cell viability was not affected under any condition (Supplementary Figure S5D).
Figure 6.

The interplay between SIRT1 and PP4 regulates DNA repair and genome stability. (A)Downregulation of SIRT1 (siSIRT1) and/or PP4C (siPP4c) expression in U2OS cells using siRNAs (siScr: scramble). (B) Quantification of Propidium iodine (PI)/EdU based cell cycle analysis (n = 3) of the cells used in (A). (C) Cell cycle analysis as in (B) of U2OS cells transfected with an empty (C), wild-type PP4R3α,β (WT) or mutant PP4R3α,β K64Q (Q) expression vectors. (D) Neutral comet assay of U2OS cells Wt or Sirt1−/− expressing siRNA scramble or against PP4c treated with 2 mM of H2O2 for 1 h (upper panel) or 1 μM camptothecin for 90 min (lower panel). A quantification of n = 3 experiments are shown. The % DNA in tail and tail moment are quantified. A similar analysis in absence of stress is included in Supplementary Figure S5E. Two-tailed t-test assay was performed (**P< 0.01; ***P< 0.005; ****P< 0.001). (E) in vivo HR reporter system in U2OS cells upon siRNA-driven downregulation of PP4c and/or SIRT1. Quantification of n = 3 experiments(bottom) are shown. Two-tailed t-test analysis was performed (*P< 0.05; ***P< 0.005; ****P< 0.001). (F, G) HR assays as in (E) upon treatment with either DMSO (control) or 100 μM of SIRT1 inhibitor Ex-527 (F) or upon overexpression of both PP4R3α and β WT or K64R (G). (H) in vivo NHEJ reporter system in U2OS cells upon siRNA-driven downregulation of PP4c and/or SIRT1. Quantification of n = 3 experiments(bottom) are shown. Two-tailed t-test analysis was performed (***P< 0.005; ****P< 0.001). (I–K) Chromosome aberration analysis of Giemsa stained chromosomes in U2OS cells Wt or Sirt1−/− expressing siRNA scramble (siScr) or against PP4 (siPP4), as in (D), treated with 100 μM H2O2 for 30 min. (I) Representative images of each condition. Arrows in zoomed images indicate chromosome aberrations (for Sirt1−/−, denote fusions; and for siPP4c and Sirt1−/−/siPP4c, denote breaks/deletions). (J) Quantification of n = 20 metaphases from each genotype. Two-tailed t-test analysis was performed (*P< 0.05; ****P< 0.001). (K) Table showing frequencies of chromosome aberration types. Frequencies were calculated as the proportion of the number of each chromosome aberration detected in n = 20 metaphases/condition compared to the total number of chromosomes detected in these n = 20 metaphases.
We next tested the effects of PP4R3α/β WT or K64Q overexpression on cell cycle progression. We observed that while both WT and K64Q induced an increase in G2/M cells, we only observed a clear and significant differential effect between PP4R3α/β WT and K64Q in S-phase (Figure 6C). We next analyzed DSB-associated DNA damage by neutral comet assay under oxidative stress and upon induction of S-phase specific replicative damage with the Topoisomerase-1 inhibitor, campothecin (CPT). The results were similar in both cases and confirmed that while downregulation of either SIRT1 or PP4 induced increased levels of DSBs, downregulation of both activities resulted in a significant compensatory effect that was more pronounced in the case of CPT than H2O2 (Figure 6D and Supplementary Figure S5E).
As both PP4 and SIRT1 have been implicated in HR, we measured the impact of their depletion on HR DNA repair using a well-established HR reporter in U2OS cells, DR-GFP (38). Following DSB induction by the nuclease I-SceI, efficient HR results in a reconstitution of intact GFP that can be monitored by flow cytometry (Supplementary Figure S5F). We observed that while siRNA-depletion of SIRT1 resulted in a roughly 33% decrease in the HR capacity of these cells, downregulation of PP4c had a drastic effect on HR efficiency, which dropped to 20% of that observed with the siScramble control (Figure 6E and Supplementary Figure S5G). Supporting the functional antagonism between SIRT1 and PP4, downregulation of both PP4c and SIRT1 resulted in a partial rescue of HR efficiency relative to depletion of only PP4c. The mild recovery of HR efficiency upon loss of both PP4c and SIRT1 may indicate a dominant role for PP4 in the process that includes functions that are not regulated by SIRT1 activity. Chemical inhibition of SIRT1 with EX-527 resulted in a similar partial rescue of the HR defect caused by PP4c depletion (Figure 6F). We confirmed that K64 acetylation regulation was involved in HR regulation, as overexpression of the PP4R3α/β K64R mutants in these cells significantly decreased HR activity in around 35% compared to WT PP4 proteins, strongly supporting a role for PP4R3α/β deacetylation in HR regulation (Figure 6G). In contrast to the reduced HR activity observed following SIRT1 depletion, we observed that SIRT1 downregulation resulted in increased non-homologous end joining (NHEJ) activity, while PP4c depletion drastically decreased NHEJ, in agreement with previous reports (Figure 6H) (47,48). In contrast to HR, we did not observe any compensatory effect when we downregulated both factors, suggesting that the antagonistic interplay between SIRT1 and PP4 does not impact NHEJ and the effects of PP4 loss are dominant to those caused by SIRT1 depletion.
To further study the functional consequences of this interplay, we analyzed the levels of genomic instability following SIRT1 or PP4 downregulation and treatment with oxidative stress. We then measured the number of chromosomal aberrations in metaphase spreads. Surprisingly, while loss of SIRT1 or shRNA-driven downregulation of PP4c resulted in an increased number of chromosomal aberrations, the combined effect of both did not result in any global compensation (Figure 6I, J). However, the analysis of the type of aberrations detected specific effects on some types of aberrations (e.g. bridges, fusions), but not on others (chrom. breaks, dicentric chromosomes) (Figure 6K). This suggested that these aberrations reflect the combined role of both factors in NHEJ, HR and possibly other pathways. Of note, we discarded that our observations were related to cell death, as we did not detect an increase in apoptotic cells in any of these treatments (Supplementary Figure S5H). Altogether, these observations strongly support a functional antagonism between SIRT1 and PP4 in genome stability.
SIRT1 and PP4 levels inversely correlate in vivo in breast cancer
Given the direct impact of DNA damage and genome stability on human health, we hypothesized that the interplay between SIRT1 and PP4 may play an important role in human pathologies associated with genome instability, such as cancer. To study the impact of this interplay in vivo, we analyzed the degree of correlation between SIRT1 and PP4c levels in a cohort of 94 breast cancer samples from the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium (CPTAC), where a complete proteomic/phosphoproteomic analysis had been performed (Figure 7A). We selected breast cancer in our study as it has a well-established link to genome stability accumulation and DNA repair defects (49). As expected, we observed a general inverse relationship between SIRT1 and PP4c protein abundance (Figure 7B) while SIRT6 showed a direct correlation with PP4c in these tumors. Consistently, the phosphorylation levels of RPA2(p238) and of two other PP4 targets, TRIM28(pS473) and 53BP1(S1030, 1643 and at least other four residues), directly correlated with SIRT1 and inversely correlated with PP4 and SIRT6 (Figure 7C and Supplementary Figure S6A, B). 53BP1 plays a major role in NHEJ, consistent with the opposing roles of SIRT1 and PP4 we identified in this repair pathway. In contrast, neither SIRT1 nor PP4c levels correlated with other phosphorylation events unrelated to PP4, such as pATM-S1981 (Supplementary Figure S6C). We next analyzed whether this correlation was valid in the subset of samples with mutations or deletions in genes associated with HR or PARP inhibitor sensitivity (Figure 7D). Although this filter considerably reduced the number of samples, SIRT1 and PP4C levels showed an anti-correlation at both the protein (Figure 7E) and RNA level (Supplementary Figure S7A). Moreover, we confirmed the opposing correlation between SIRT1 and PP4 at the expression level in another cohort of breast cancer patients (50) (Supplementary Figure S7B). Altogether, this evidence further supports antagonism between SIRT1 and PP4 in tumorigenesis, reflecting the functional interplay between these factors in genome maintenance.
DISCUSSION
Despite the well-established link of Sirtuins with genome stability, we still have a poorly integrated view regarding how these enzymes regulate the DDR. One of the most intriguing observations, the decrease in γH2AX observed in Sirt1−/− mice (31), has remained unaddressed. Our work not only provides a mechanism to explain this early observation, but also suggests a more global perspective about how SIRT1 regulates DDR signaling. We hypothesize that in addition to a specific modulation of HR DNA repair signaling at different stages (NBS1, MOF, TIP60) (51,52), SIRT1 regulates the progression of the whole signaling pathway through a direct control of the PP4 complex function (Figure 7E). In the case of γH2AX, we propose that while stress is active, SIRT1 binding to PP4 ensures that the DDR signaling remains on until completion of repair and the stress signal disappears. This effect is likely unrelated to the described deacetylation of NBS1 by SIRT1 (52), as NBS1 loss does not have a strong impact on the deposition of the mark (7,53).
In contrast to PP2A, loss of PP4 induces increased levels of γH2AX in the absence of external insults, suggesting that PP4 has a key role in basal repair, probably associated with replicative stress. This is further supported by the described antagonism between ATR and PP4 (12,54). The observed decrease of γH2AX in Sirt1−/− MEFs, also in the absence of an external stimuli, further supports the link between SIRT1 and replicative stress. The oxidative stress induced association of SIRT1-PP4 suggests that they play a prominent role in the regulation of genome stability under these conditions. The previously reported phosphorylation of SIRT1 by JNK1 after oxidative stress may be an important regulator of this mechanism, as it promotes its activity and its nuclear localization (55).
Another interesting observation is the striking opposed correlations between SIRT1 or SIRT6 and both PP4 expression and the phosphorylation of its targets in breast cancer samples (15,56,57) (Figure 7A-E and Supplementary Figure S6). This evidence supports a differential role for SIRT1 and SIRT6 in DSB repair and may suggest an unexpected functional interplay between SIRT6 and PP4. SIRT1 has been recently reported to interact with and deacetylate SIRT6 on K33ac to regulate its binding to γH2AX and chromatin (58). However, the fact that SIRT6 was not detected as a component of the SIRT1-PP4 complex (Figure 1) and SIRT6 and PP4 do not seem to interact (Figure 2D) suggests that this correlation is indirect and probably reflects a coordinated regulation of both factors in the DDR process. Further studies will be needed to determine whether SIRT6 has any impact on SIRT1 control of PP4.
Despite the relevance of the interplay between PP4 and SIRT1, our data also suggest that SIRT1 regulates γH2AX through a PP4-independent mechanism, since increased γH2AX observed upon PP4 downregulation or chemical enzymatic inhibition with Okadaic acid, is significantly decreased in a Sirt1−/-background (Figure 3E). This is probably caused by a direct or indirect regulation of ATR or ATM activation by SIRT1, which is not well understood. For instance, while SIRT1 recruitment to DNA damage regions depends on ATM and a direct role of SIRT1 in ATM activation has been shown in post-mitotic neurons, ATM/ATR phosphorylation of the SIRT1 inhibitor DBC1 promotes the inactivation of SIRT1 upon DNA damage (35). Furthermore, SIRT1 deacetylates TOPBP1, an ATR activator under replicative stress (59), an event that inhibits TOBP1 binding to Rad9 and its subsequent activation of the ATR-Chk1 pathway (60). Further studies should clarify the mechanism involved in this observation.
Our observations suggest that the antagonism between SIRT1 and PP4 mainly impacts HR regulation. This is supported by the fact that the compensatory effect of SIRT1 depletion on PP4 downregulation in comet assays is stronger in the case of CPT than H2O2, as the DNA damage induced by the former insult is restricted to S-phase and more dependent on HR for repair. Additionally, we did not observe that SIRT1 knockdown improved the NHEJ defect caused by depletion of PP4c. Although the main roles of both PP4 and SIRT1 in DSB repair appears to be through HR, they have been proposed to target KAP1 (SIRT1 and PP4), Ku70 (SIRT1) and 53BP1 (PP4) to promote NHEJ activity (23,47,48,61–63). While consistent with the strong NHEJ defect we observed with PP4 depletion, loss of SIRT1 actually led to an increase in NHEJ, indicating that impact of SIRT1 in NHEJ may be cell type specific. Interestingly, our results raise the possibility that SIRT1 may regulate the decision to use NHEJ or HR in a context dependent manner. In our analysis of cancer phosphoproteomics, SIRT1 and PP4 expression correlated with 53BP1 phosphorylation in opposing directions (Supplementary Figure S6B), consistent with their antagonistic effects. 53BP1 is a key target in regulating the decision to use NHEJ or HR through the regulation of BRCA1-mediated DSB resection (64). However, as 53BP1 is heavily phosphorylated and the functional importance of many of the individual marks are unknown (65), we cannot make a specific conclusion about the functional relevance of these observations to repair pathway usage without direct analysis of the role of these sites.
In addition to the differential impact of both activities on DNA repair pathways, we also observed differences in the frequency of metaphase aberrations under oxidative stress (Figure 6I, J). In contrast to SIRT1 loss that elevated NHEJ activity and associated aberrations (66,67), such as chromosome fusions and ring chromosomes, siPP4c cells had high levels of chromosome breaks that persisted upon SIRT1 depletion, suggesting potential defects in both NHEJ and HR pathways that are at dependent on the regulation of additional substrates. The specific effects observed for certain types of aberrations suggest that the impact of impairing the functional interplay between SIRT1 and PP4 may depend on specific DSB repair context or type of stress induced.
Another interesting open question is what underlying mechanisms control SIRT1 activation and its subsequent inhibition of PP4 activity. We speculate that SIRT1 has a surveillance role that restrains PP4 activity while DNA is still unrepaired, exemplified by the effects on γH2AX. However, this regulatory mechanism must be highly dynamic to allow the modulation of RPA activity during the repair process. The most likely candidate mechanism for SIRT1 regulation in this case is through phosphorylation. SIRT1 is activated upon stress through phosphorylation by stress-dependent kinases like JNK1, AMPK, DIRK1–3 or CDK1 (55,68–70). The reports that phosphorylated SIRT1 localizes to replication origins to prevent excess origin firing (71), while PP4 dephosphorylation of yeast initiation factors promotes origin firing, may support this hypothesis and also suggest that the SIRT1/PP4 interplay regulates DNA replication. Whether the interaction we describe here is also valid in human origin activation is currently unknown (72), but we hypothesize that the resumption of cell cycle progression after DNA repair may involve SIRT1 inactivation by PP4. Moreover, PP4 may also regulate SIRT1 activity through other factors, such as DBC1 (35).
The functional implications of the SIRT1/PP4 interplay in the regulation of RPA2 phosphorylation remain unclear. While phospho-RPA2 is involved in specific binding to DNA repair factors, its hyper-phosphorylation inhibits RPA complex binding to DNA, delays the formation of RAD51 foci, and blocks DNA replication (26). Interestingly, RPA2 dephosphorylation by PP4 is required to complete DNA repair and resume replication after DNA damage is completed (22). In this sense, the increase of RPA2 phosphorylation by SIRT1-dependent inhibition of PP4 could ensure the activation of the pathway until DNA repair is completed, similar to the case of γH2AX. However, the established role of PP4 in RPA2 regulation and our observation that SIRT1 deficiency also results in a decrease in RPA2 foci formation (Figure 5C), suggests that the dynamic regulation between both factors is key to promote efficient repair of damaged DNA regions.
Another interesting discovery from our work is the identification of PP4R3α and β as the targets of SIRT1 in the regulation of PP4 activity. The identification of K64 in the WH1/EVH1 domain of PP4R3α/β as SIRT1 targets is particularly interesting giving the key role of this domain in the binding to the PP4 substrates (45). The functional crosstalk between SIRT1 and PP4 is likely to impact many other physiological processes, although is not clear whether they will be reliant on the functional antagonism we observed in the DDR. An obvious example is oncogenesis, as PP4 has been described to be overexpressed in several cancer types, including breast cancer (15,16,73,74), while SIRT1 plays a more complex role with both tumor suppressor and oncogenic roles, depending on the context. Our data suggests that both factors could play antagonistic roles in breast cancer and further work will be needed to understand their roles in human malignancies.
Altogether, our work identifies a novel regulatory interplay between SIRT1 and PP4 and suggests that broader Sirtuin-dependent control of the DDR may modulate the cascade of phosphorylation events involved. Future work will be needed to determine if this mechanism has further biological implications in development and cancer.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Anne-Claude Gingras (LTRI, Toronto) for providing PP4c expression vector and Dr Ranjit S Bindra (Yale Univ, New Haven) and Dr Simon N. Powell (MSKCC, NYC) for providing the DR-HR U2OS cell line. Thanks to the members of the Vaquero laboratory for stimulating discussions.
Contributor Information
George Rasti, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Maximilian Becker, Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Berta N Vazquez, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Maria Espinosa-Alcantud, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Irene Fernández-Duran, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain.
Andrés Gámez-García, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain.
Alessandro Ianni, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Department of Cardiac Development and Remodeling, Max-Planck-Institute for Heart and Lung Research, Ludwigstrasse 43, 61231, Bad Nauheim, Germany.
Jessica Gonzalez, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Laia Bosch-Presegué, Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain; Tissue Repair and Regeneration Laboratory (TR2Lab), Institut de Recerca i Innovació en Ciències de la Vida i de la Salut a la Catalunya Central (IrisCC). Experimental Sciences and Methodology Department. Faculty of Health Sciences and Welfare (FCSB), University of Vic - Central University of Catalonia (UVic-UCC), Vic, Spain.
Anna Marazuela-Duque, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Anna Guitart-Solanes, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain.
Sandra Segura-Bayona, Institute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Barcelona, Spain; Current affiliation: The Francis Crick Institute, 1 Midland Road, London NW1 1AT, UK.
Joan-Josep Bech-Serra, Proteomic Unit, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916, Badalona, Barcelona, Spain.
Michael Scher, Howard Hughes Medical Institute, Division of Nucleic Acids Enzymology, Department of Biochemistry, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, NJ, 08854, USA.
Lourdes Serrano, Department of Science, BMCC, The City University of New York (CUNY), 199 Chambers Street N699P, New Yirk, NY, 10007, USA.
Uma Shankavaram, Radiation Oncology Branch, National Cancer Institute, Bethesda, MD, 20892, USA.
Hediye Erdjument-Bromage, Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, New York, NY, 10065, USA; Department of Cell Biology, New York University School of Medicine, New York, NY, 10016, USA.
Paul Tempst, Molecular Biology Program, Memorial Sloan-Kettering Cancer Center, New York, NY, 10065, USA.
Danny Reinberg, Howard Hughes Medical Institute, Division of Nucleic Acids Enzymology, Department of Biochemistry, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, NJ, 08854, USA; Howard Hughes Medical Institute, Department of Biochemistry, New York University School of Medicine, New York, NY, 10016, USA.
Mireia Olivella, Bioinfomatics and Medical Statistics Group, Faculty of Science, Technology and Engineering. University of Vic-Central University of Catalonia, Vic, Spain.
Travis H Stracker, Institute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Barcelona, Spain; Radiation Oncology Branch, National Cancer Institute, Bethesda, MD, 20892, USA.
Carolina de la Torre, Proteomic Unit, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916, Badalona, Barcelona, Spain.
Alejandro Vaquero, Chromatin Biology Laboratory, Josep Carreras Leukaemia Research Institute (IJC), Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; Chromatin Biology Laboratory, Cancer Epigenetics and Biology Program (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), Av. Gran Via de l’Hospitalet, 199-203, 08908 L’Hospitalet de Llobregat, Barcelona, Spain.
Data Availability
The mass spectrometry data generated in this work is included in the main figures and Supplementary Table S1 and S2. Mass spectrometry analysis of PP4 complex acetylation has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository database (http://www.ebi.ac.uk/pride) with the data set identifier PXD036905.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Economy and Competitiveness (MINECO) [SAF2014-55964R, SAF2017-88975R, PID2020-117284RB-I00 to A.V., PGC2018-095616-B-100/GINDATA to T.H.S.]; FEDER funds/European Regional Development Fund (ERDF)—A Way to Build Europe; The European Commission's Horizon 2020 research and innovation programme—Marie Skłodowska-Curie [895979 to B.N.V.]; Catalan Government Agency AGAUR [2014-SGR-400, 2017-SGR-148 to A.V.]; Centres of Excellence Severo Ochoa award (to T.H.S.); La Marató de TV3 Foundation (to A.V.); NIH Intramural Research Program (to T.H.S., U.S.); CERCA Programme/Generalitat de Catalunya for institutional support; Beatriu de Pinos fellowship AGAUR (to B.N.V.); MINECO [FPI BES-2015-071251 to M.E.A., FI-AGAUR 2021-FI-B1-00228 to A.G.-G.]; ‘la Caixa’ Foundation [LCF/PR/GN14/10270002 to S.S.B.]. Funding for open access charge: Internal funding from Josep Carreras Leukaemia Research Institute; Spanish Ministry of Science and Innovation [PID2020-117284RB-I00].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kupis W., Palyga J., Tomal E., Niewiadomska E.. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016; 72:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guarente L. Franklin H. Epstein Lecture: sirtuins, aging, and medicine. N. Engl. J. Med. 2011; 364:2235–2244. [DOI] [PubMed] [Google Scholar]
- 3. Haigis M.C., Sinclair D.A.. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010; 5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bosch-Presegue L., Vaquero A.. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015; 282:1745–1767. [DOI] [PubMed] [Google Scholar]
- 5. Sedelnikova O.A., Rogakou E.P., Panyutin I.G., Bonner W.M.. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat. Res. 2002; 158:486–492. [DOI] [PubMed] [Google Scholar]
- 6. Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M.. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998; 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 7. Celeste A., Fernandez-Capetillo O., Kruhlak M.J., Pilch D.R., Staudt D.W., Lee A., Bonner R.F., Bonner W.M., Nussenzweig A.. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003; 5:675–679. [DOI] [PubMed] [Google Scholar]
- 8. Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J.. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001; 276:42462–42467. [DOI] [PubMed] [Google Scholar]
- 9. Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017; 66:801–817. [DOI] [PubMed] [Google Scholar]
- 10. Campos A., Clemente-Blanco A.. Cell cycle and DNA repair regulation in the damage response: protein phosphatases take over the reins. Int. J. Mol. Sci. 2020; 21:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freeman A.K., Monteiro A.N.. Phosphatases in the cellular response to DNA damage. Cell Commun Signal. 2010; 8:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chowdhury D., Xu X., Zhong X., Ahmed F., Zhong J., Liao J., Dykxhoorn D.M., Weinstock D.M., Pfeifer G.P., Lieberman J.. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol. Cell. 2008; 31:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakada S., Chen G.I., Gingras A.C., Durocher D.. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008; 9:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cohen P.T., Philp A., Vazquez-Martin C.. Protein phosphatase 4–from obscurity to vital functions. FEBS Lett. 2005; 579:3278–3286. [DOI] [PubMed] [Google Scholar]
- 15. Wang B., Zhao A., Sun L., Zhong X., Zhong J., Wang H., Cai M., Li J., Xu Y., Liao J.et al.. Protein phosphatase PP4 is overexpressed in human breast and lung tumors. Cell Res. 2008; 18:974–977. [DOI] [PubMed] [Google Scholar]
- 16. Weng S., Wang H., Chen W., Katz M.H., Chatterjee D., Lee J.E., Pisters P.W., Gomez H.F., Abbruzzese J.L., Fleming J.B.et al.. Overexpression of protein phosphatase 4 correlates with poor prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 2012; 21:1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hastie C.J., Carnegie G.K., Morrice N., Cohen P.T.. A novel 50 kDa protein forms complexes with protein phosphatase 4 and is located at centrosomal microtubule organizing centres. Biochem. J. 2000; 347:845–855. [PMC free article] [PubMed] [Google Scholar]
- 18. Gingras A.C., Caballero M., Zarske M., Sanchez A., Hazbun T.R., Fields S., Sonenberg N., Hafen E., Raught B., Aebersold R.. A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol. Cell. Proteomics. 2005; 4:1725–1740. [DOI] [PubMed] [Google Scholar]
- 19. Kloeker S., Reed R., McConnell J.L., Chang D., Tran K., Westphal R.S., Law B.K., Colbran R.J., Kamoun M., Campbell K.S.et al.. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr Purif. 2003; 31:19–33. [DOI] [PubMed] [Google Scholar]
- 20. Park J., Lee D.H.. Functional roles of protein phosphatase 4 in multiple aspects of cellular physiology: a friend and a foe. BMB Rep. 2020; 53:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karman Z., Rethi-Nagy Z., Abraham E., Fabri-Ordogh L., Csonka A., Vilmos P., Debski J., Dadlez M., Glover D.M., Lipinszki Z.. Novel perspectives of target-binding by the evolutionarily conserved PP4 phosphatase. Open Biol. 2020; 10:200343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee D.H., Pan Y., Kanner S., Sung P., Borowiec J.A., Chowdhury D.. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat. Struct. Mol. Biol. 2010; 17:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee D.H., Goodarzi A.A., Adelmant G.O., Pan Y., Jeggo P.A., Marto J.A., Chowdhury D.. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012; 31:2403–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olson E., Nievera C.J., Klimovich V., Fanning E., Wu X.. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem. 2006; 281:39517–39533. [DOI] [PubMed] [Google Scholar]
- 25. Shi W., Feng Z., Zhang J., Gonzalez-Suarez I., Vanderwaal R.P., Wu X., Powell S.N., Roti Roti J.L., Gonzalo S., Zhang J.. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis. 2010; 31:994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee A., Moon B.I., Kim T.H.. BRCA1/BRCA2 pathogenic variant breast cancer: treatment and prevention strategies. Ann Lab Med. 2020; 40:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dobbin M.M., Madabhushi R., Pan L., Chen Y., Kim D., Gao J., Ahanonu B., Pao P.C., Qiu Y., Zhao Y.et al.. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 2013; 16:1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toiber D., Erdel F., Bouazoune K., Silberman D.M., Zhong L., Mulligan P., Sebastian C., Cosentino C., Martinez-Pastor B., Giacosa S.et al.. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell. 2013; 51:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vazquez B.N., Thackray J.K., Simonet N.G., Kane-Goldsmith N., Martinez-Redondo P., Nguyen T., Bunting S., Vaquero A., Tischfield J.A., Serrano L.. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016; 35:1488–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Onn L., Portillo M., Ilic S., Cleitman G., Stein D., Kaluski S., Shirat I., Slobodnik Z., Einav M., Erdel F.et al.. SIRT6 is a DNA double-strand break sensor. Elife. 2020; 9:e51636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang R.H., Sengupta K., Li C., Kim H.S., Cao L., Xiao C., Kim S., Xu X., Zheng Y., Chilton B.et al.. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008; 14:312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deng C.X. SIRT1, is it a tumor promoter or tumor suppressor?. Int. J. Biol. Sci. 2009; 5:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mei Z., Zhang X., Yi J., Huang J., He J., Tao Y.. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016; 35:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guarente L. Overcoming ATM deficiency by activating the NAD(+)/SIRT1 Axis. Cell Metab. 2016; 24:526–528. [DOI] [PubMed] [Google Scholar]
- 35. Zannini L., Buscemi G., Kim J.E., Fontanella E., Delia D.. DBC1 phosphorylation by ATM/ATR inhibits SIRT1 deacetylase in response to DNA damage. J. Mol. Cell Biol. 2012; 4:294–303. [DOI] [PubMed] [Google Scholar]
- 36. Yuan Z., Seto E.. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell Cycle. 2007; 6:2869–2871. [DOI] [PubMed] [Google Scholar]
- 37. Bosch-Presegue L., Raurell-Vila H., Marazuela-Duque A., Kane-Goldsmith N., Valle A., Oliver J., Serrano L., Vaquero A.. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell. 2011; 42:210–223. [DOI] [PubMed] [Google Scholar]
- 38. Bindra R.S., Goglia A.G., Jasin M., Powell S.N.. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013; 41:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martinez-Redondo P., Vaquero A.. Methods to study the role of sirtuins in genome stability. Methods Mol. Biol. 2013; 1077:273–283. [DOI] [PubMed] [Google Scholar]
- 40. Bosch-Presegue L., Raurell-Vila H., Thackray J.K., Gonzalez J., Casal C., Kane-Goldsmith N., Vizoso M., Brown J.P., Gomez A., Ausio J.et al.. Mammalian HP1 isoforms have specific roles in heterochromatin structure and organization. Cell Rep. 2017; 21:2048–2057. [DOI] [PubMed] [Google Scholar]
- 41. Vaquero A., Scher M., Erdjument-Bromage H., Tempst P., Serrano L., Reinberg D.. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007; 450:440–444. [DOI] [PubMed] [Google Scholar]
- 42. Kloeker S., Wadzinski B.E.. Purification and identification of a novel subunit of protein serine/threonine phosphatase 4. J. Biol. Chem. 1999; 274:5339–5347. [DOI] [PubMed] [Google Scholar]
- 43. Swingle M., Ni L., Honkanen R.E.. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol. Biol. 2007; 365:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamagata K., Kitabayashi I.. Sirt1 physically interacts with Tip60 and negatively regulates Tip60-mediated acetylation of H2AX. Biochem. Biophys. Res. Commun. 2009; 390:1355–1360. [DOI] [PubMed] [Google Scholar]
- 45. Ball L.J., Jarchau T., Oschkinat H., Walter U.. EVH1 domains: structure, function and interactions. FEBS Lett. 2002; 513:45–52. [DOI] [PubMed] [Google Scholar]
- 46. Ueki Y., Kruse T., Weisser M.B., Sundell G.N., Larsen M.S.Y., Mendez B.L., Jenkins N.P., Garvanska D.H., Cressey L., Zhang G.et al.. A consensus binding motif for the PP4 protein phosphatase. Mol. Cell. 2019; 76:953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu J., Xu L., Zhong J., Liao J., Li J., Xu X.. Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle. 2012; 11:2643–2649. [DOI] [PubMed] [Google Scholar]
- 48. Zheng X.F., Acharya S.S., Choe K.N., Nikhil K., Adelmant G., Satapathy S.R., Sharma S., Viccaro K., Rana S., Natarajan A.et al.. A mitotic CDK5-PP4 phospho-signaling cascade primes 53BP1 for DNA repair in G1. Nat. Commun. 2019; 10:4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li A., Geyer F.C., Blecua P., Lee J.Y., Selenica P., Brown D.N., Pareja F., Lee S.S.K., Kumar R., Rivera B.et al.. Homologous recombination DNA repair defects in PALB2-associated breast cancers. NPJ Breast Cancer. 2019; 5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krug K., Jaehnig E.J., Satpathy S., Blumenberg L., Karpova A., Anurag M., Miles G., Mertins P., Geffen Y., Tang L.C.et al.. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell. 2020; 183:1436–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li K., Casta A., Wang R., Lozada E., Fan W., Kane S., Ge Q., Gu W., Orren D., Luo J.. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 2008; 283:7590–7598. [DOI] [PubMed] [Google Scholar]
- 52. Yuan Z., Zhang X., Sengupta N., Lane W.S., Seto E.. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell. 2007; 27:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kobayashi J., Tauchi H., Sakamoto S., Nakamura A., Morishima K., Matsuura S., Kobayashi T., Tamai K., Tanimoto K., Komatsu K.. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 2002; 12:1846–1851. [DOI] [PubMed] [Google Scholar]
- 54. Katsube T., Mori M., Tsuji H., Shiomi T., Wang B., Liu Q., Nenoi M., Onoda M.. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014; 156:85–95. [DOI] [PubMed] [Google Scholar]
- 55. Nasrin N., Kaushik V.K., Fortier E., Wall D., Pearson K.J., de Cabo R., Bordone L.. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One. 2009; 4:e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rifai K., Idrissou M., Penault-Llorca F., Bignon Y.J., Bernard-Gallon D. Breaking down the contradictory roles of histone deacetylase SIRT1 in human breast cancer. Cancers (Basel). 2018; 10:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thirumurthi U., Shen J., Xia W., LaBaff A.M., Wei Y., Li C.W., Chang W.C., Chen C.H., Lin H.K., Yu D.et al.. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci. Signal. 2014; 7:ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meng F., Qian M., Peng B., Peng L., Wang X., Zheng K., Liu Z., Tang X., Zhang S., Sun S.et al.. Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. Elife. 2020; 9:e55828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang R.H., Lahusen T.J., Chen Q., Xu X., Jenkins L.M., Leo E., Fu H., Aladjem M., Pommier Y., Appella E.et al.. SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int. J. Biol. Sci. 2014; 10:1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu T., Lin Y.H., Leng W., Jung S.Y., Zhang H., Deng M., Evans D., Li Y., Luo K., Qin B.et al.. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol. Cell. 2014; 56:681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pfeifer G.P. Protein phosphatase PP4: role in dephosphorylation of KAP1 and DNA strand break repair. Cell Cycle. 2012; 11:2590–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin Y.H., Yuan J., Pei H., Liu T., Ann D.K., Lou Z.. KAP1 deacetylation by SIRT1 promotes non-homologous end-joining repair. PLoS One. 2015; 10:e0123935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jeong J., Juhn K., Lee H., Kim S.H., Min B.H., Lee K.M., Cho M.H., Park G.H., Lee K.H.. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007; 39:8–13. [DOI] [PubMed] [Google Scholar]
- 64. Chapman J.R., Taylor M.R., Boulton S.J.. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012; 47:497–510. [DOI] [PubMed] [Google Scholar]
- 65. Mirman Z., de Lange T.. 53BP1: a DSB escort. Genes Dev. 2020; 34:7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burssed B., Zamariolli M., Bellucco F.T., Melaragno M.I.. Mechanisms of structural chromosomal rearrangement formation. Mol Cytogenet. 2022; 15:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zimmermann M., de Lange T.. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014; 24:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nin V., Escande C., Chini C.C., Giri S., Camacho-Pereira J., Matalonga J., Lou Z., Chini E.N.. Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J. Biol. Chem. 2012; 287:23489–23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sasaki T., Maier B., Koclega K.D., Chruszcz M., Gluba W., Stukenberg P.T., Minor W., Scrable H.. Phosphorylation regulates SIRT1 function. PLoS One. 2008; 3:e4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guo X., Williams J.G., Schug T.T., Li X.. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010; 285:13223–13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Utani K., Fu H., Jang S.M., Marks A.B., Smith O.K., Zhang Y., Redon C.E., Shimizu N., Aladjem M.I.. Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res. 2017; 45:7807–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jenkinson F., Tan K.W., Schöpf B., Santos M.M., Phanada I., Zegerman P.. Dephosphorylation of the pre-initiation complex during S-phase is critical for origin firing. Mol. Cell. 2021; 83:12–25. [DOI] [PubMed] [Google Scholar]
- 73. Li M., Li X., Xu S., Xue P., Li Q., Lu Q., Jia Q., Zhang L., Li X., Li X.. Protein phosphatase 4 catalytic subunit is overexpressed in glioma and promotes glioma cell proliferation and invasion. Tumour Biol. 2016; 37:11893–11901. [DOI] [PubMed] [Google Scholar]
- 74. Li X., Liang L., Huang L., Ma X., Li D., Cai S.. High expression of protein phosphatase 4 is associated with the aggressive malignant behavior of colorectal carcinoma. Mol. Cancer. 2015; 14:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data generated in this work is included in the main figures and Supplementary Table S1 and S2. Mass spectrometry analysis of PP4 complex acetylation has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository database (http://www.ebi.ac.uk/pride) with the data set identifier PXD036905.