


Abstract
Use of synthetic genomics to design and build ‘big’ DNA has revolutionized our ability to answer fundamental biological questions by employing a bottom-up approach. Saccharomyces cerevisiae, or budding yeast, has become the major platform to assemble large synthetic constructs thanks to its powerful homologous recombination machinery and the availability of well-established molecular biology techniques. However, introducing designer variations to episomal assemblies with high efficiency and fidelity remains challenging. Here we describe CRISPR Engineering of EPisomes in Yeast, or CREEPY, a method for rapid engineering of large synthetic episomal DNA constructs. We demonstrate that CRISPR editing of circular episomes presents unique challenges compared to modifying native yeast chromosomes. We optimize CREEPY for efficient and precise multiplex editing of >100 kb yeast episomes, providing an expanded toolkit for synthetic genomics.
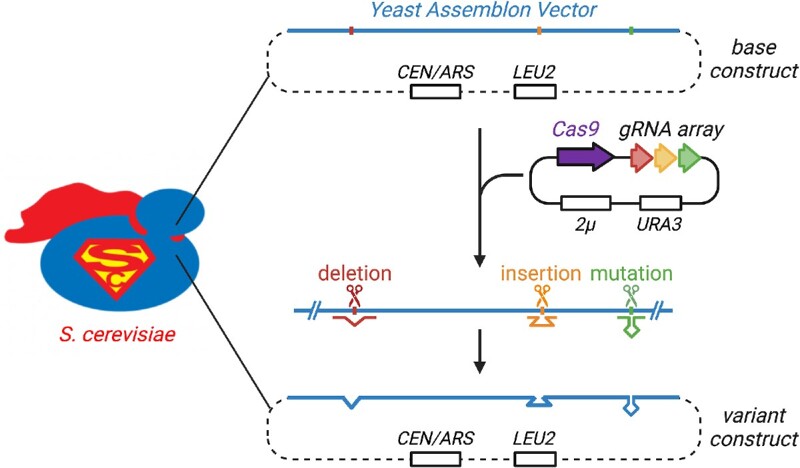
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Synthetic biology approaches have been used to design and assemble ‘big’ DNA to generate chromosomes and even entire genomes, including the de novo assembly of synthetic poliovirus and bacteriophage DNA, as well as the prokaryotic genomes of Mycoplasma and Escherichia coli (1–4). In eukaryotes, the Sc2.0 project aims to build a completely synthetic genome in yeast from the bottom up (5–8). As technology rapidly advances, genome writing in mammalian cells has also become feasible, resulting in new large DNA manipulation and delivery strategies at the locus level (9–15). These studies provide a new lens to understand complex genome architecture, expression regulation, and the genetic basis of human disease.
The human genome, like other mammalian genomes, is complex, with introns and non-coding sequences such as repeats and regulatory elements. Furthermore, most human genome variants implicated in disease map to non-coding, regulatory regions (16). Recently, synthetic genome writing has been used to map the regulatory architecture of the HoxA cluster (12), alpha globin (13) and Sox2 (14), rewrite a ‘cancer-mutation-resistant’ Tp53 gene and a fully-humanized ACE2 receptor gene for SARS-CoV-2 in mice (11).
The assembly of large (>100 kb) DNA constructs in S. cerevisiae is a fundamental step towards synthetic DNA writing of biosynthetic pathways, microbial genomes and mammalian loci (4,17–20). Yeast is tractable as a platform to assemble large DNA constructs because of its efficient homologous recombination (HR) machinery and advanced molecular toolsets available to researchers. Using HR of overlapping segments, linear DNA fragments can be assembled as a large circular episomal construct, also known as ‘YAV’ or Yeast Assemblon Vector, that can be propagated and transferred to bacteria for isolation before delivery to mammalian cells. After the initial assembly, YAV can be edited in yeast to generate panels of designer variants. Compared to building many different constructs from scratch, it is also far more feasible and reliable to introduce designer modifications into a parental base assembly, completely insulating the eventual destination mammalian genome from potential off target effects of CRISPR. Episomal constructs can be delivered to mammalian systems, or further characterized in yeast, as S. cerevisiae is widely used to optimize episomal biosynthetic pathways in metabolic engineering of natural products (20–23).
CRISPR has been widely used for yeast genome editing (24,25). Directed by a sequence-specific single guide RNA (sgRNA), the Cas9 nuclease creates a DNA double strand break (DSB). This DSB can be repaired by homologous recombination with a co-transformed donor DNA containing polymorphisms that prevent further Cas9 binding and cleavage to achieve successful editing. In the absence of such a donor DNA template, the original genomic break may lead to cell cycle arrest or the loss of an essential gene and subsequent death. Compared to mammalian cells, Saccharomyces cerevisiae performs non-homologous end joining (NHEJ) with high fidelity, with the major repair product being simple re-ligation (26,27). As a result, the repaired DNA reforms the original Cas9 cleavage site unless errors in ligation such as lost bases prevent further Cas9 recognition. Typically, when CRISPR/Cas9 targets a chromosomal site that is efficiently cut, the number of surviving colonies in the absence of donor template DNA (designed so as to block ongoing cutting) are 100- to 1000-fold lower than in its presence (24). Previous CRISPR studies have focused mainly on genomic editing, with many well-designed systems established for both simplex and multiplex targets (28–30). However, the efficiency of editing episomal DNA constructs by CRISPR/Cas9 remains unclear and a CRISPR toolbox specifically optimized for episomes, especially those carrying repeat-laden mammalian DNA, is lacking. It is also unknown whether there are any fundamental differences between episomal and chromosomal editing in yeast.
Here, we introduce CRISPR Engineering of EPisomes in Yeast, or CREEPY, a method for episomal YAV engineering. We first compare the efficiency of CRISPR/Cas9 for targeting episomes and chromosomes. With CREEPY optimized for episomal editing, we achieve simplex and multiplex editing, as demonstrated by engineering of a 143-kb mSox2 YAV containing the mouse Sox2 gene and regulatory regions (14). Sox2 is a Yamanaka factor essential for maintaining stem cell pluripotency (31). The mSox2 episome represents a typical mammalian big DNA assembly in length (>100 kb) and complexity. While the majority of episomal edits are successful, unintended modifications can occur. We identified the mechanism underlying these modifications, which occur when internal deletions result from HR or microhomology-mediated end joining (MMEJ) events flanking the initial DSB site rather than from errors introduced by NHEJ. Interestingly, all such events identified in this study were instances of recombination between relatively simple sequence repeats, which are abundantly represented in mammalian DNA. The CREEPY constructs and methods described here can be used to further advance the fields of DNA assembly and metabolic engineering in yeast and mammalian systems.
MATERIALS AND METHODS
Yeast strains and culture
BY4741 was used to test chromosomal editing efficiency, with ADE2 on chromosome XV as the target. To directly compare genomic and episomal editing, we integrated the mSox2 CTCF8 site (40 bp) into chromosome VI (between YFL021W and YFL020C) with a LEU2 marker. This was achieved by assembling the CTCF8 site, which was made up of two annealed oligos, into an entry vector (pAV10, Addgene #63213) together with the LEU2 marker and approximately 500 bp of homologous sequence on each side from chromosome VI, using Golden Gate assembly. The linear fragment for integration was released through NotI digestion and then transformed into BY4741 for integration.
The yeast strain carrying a mSox2 YAV (yLM1371) is a derivative of BY4741 (10). The rad52Δ0 strain was from the yeast knockout library, in which RAD52 was deleted by a KanMX marker in BY4741 background (32). The deletion was confirmed by colony PCR. Yeast strains were grown using YPD as rich medium or defined Synthetic Complete (SC) medium with appropriate nutrients omitted (e.g. SC–Ura lacks uracil) as selective media. All yeast transformations in this study were performed with standard LiAc/SS/PEG method (33).
CREEPY plasmids and gRNA assembly
Plasmids used in this study are listed in Supplementary Table S1. The original Cas9 and gRNA expression modules were modified from p414-TEF1p-Cas9-CYC1t (Addgene# 43802) and p426-SNR52p-gRNA.CAN1.Y-SUP4t (Addgene# 43803). The LEU2 marker was replaced with a heterologous HIS3MX6 marker (34), referred to here as SpHIS5, by homologous recombination in yeast, generating the plasmid pCTC019. The Cas9 expression module (TEF1 promoter, Cas9 CDS, CYC1 terminator) and gRNA expression module (SNR52 promoter, NotI cutting site, SUP61 terminator) were constructed using Gibson assembly into the pRS416 (CEN-URA3) and pRS426 (2μ-URA3) vectors, generating the single-plasmid systems pYZ462 (Cas9-CEN-URA3) and pYZ463 (Cas9-2μ-URA3), respectively. Plasmids that carry a CEN/ARS element (CEN for short) are reported to propagate in low copy numbers in yeast (around 2 copies per cell), whereas the 2μ element renders plasmids with much higher copy number (around 30 copies per cell) (35).
For the multiplex editing, a tRNA-gRNA array (30) was used to express multiple gRNAs. pYZ463 was modified to introduce a synonymous mutation (G to A, +171 bp from ATG) in Cas9 CDS, eliminating the BsmBI recognition site with Multichange Isothermal (MISO) mutagenesis (36). Then, the tRNA module with a bacterial GFP expression cassette was assembled to replace the original gRNA expression module (SNR52 promoter) (37). The final entry vector with URA3 and 2μ is pYZ960. The same method was used to build another entry vector, pYZ959, with URA3 and CEN/ARS. The gRNAs were assembled as described before (30). Briefly, primers containing the corresponding gRNA sequences, adapters and BsmBI recognition sites were used to amplify a universal PCR template with a tRNAGly and gRNA scaffold (pYZ038). The PCR products were purified using a DNA Clean & Concentrator kit (ZYMO Cat# D4004), and then cloned into an entry vector with Golden Gate assembly, using BsmBI-v2 (NEB Cat# R0739L), T4 DNA ligase (NEB Cat# M0202S) and 10 × T4 DNA ligase reaction buffer (NEB Cat# B0202S), following the NEB Golden Gate Assembly Protocol (38). The reaction was cycled between 42°C and 16°C for 5 min at each temperature, for 30 or 60 cycles for three- or five-gRNA assembly, respectively. This was followed by a 60°C incubation for 5 min, and a final temperature hold at 4°C prior to transformation into E. coli competent cells. GFP-negative colonies, visualized by eye as lacking a green shade, were selected. All plasmids were confirmed by Sanger sequencing.
For instance, to build the Cas9/gRNA.CTCF13, 17, 25 construct (pYZ212) for three edits (Figure 4A), we began with two PCR products amplified from pYZ038 using primers YZ2575/YZ2576 and YZ2577/YZ2578, respectively. The first amplicon contains gRNA.CTCF13 and one half of gRNA.CTCF17, while the second amplicon contains the other half of gRNA.CTCF17 and the entire gRNA.CTCF25. The two amplicons were subsequently purified and cloned into the entry vector pYZ960 via Golden Gate assembly. Similarly, to build the Cas9/gRNA.CTCF3, 8, 13, 17, 25 construct (pYZ214) for five edits, we generated four PCR amplicons with primers of YZ2575/YZ2576, YZ2577/YZ2661, YZ2662/YZ2627 and YZ2628/YZ2631, which were then assembled into the entry vector.
Figure 4.

Multiplex editing of three CTCF sites in mSox2 YAV. (A) Single-plasmid system for multiplex episomal editing. The tRNA-gRNA array was built using Golden Gate assembly with BsmBI. (B) Episomal editing efficiency for at least one or all three designer deletions. Error bars represent mean ± SD (n = 3). (C) Episomal editing efficiency calculated for each designer deletion. Error bars represent mean ± SD (n = 3). (D) Distributions of successful edits of CTCF13Δ, CTCF17Δ and CTCF25Δ. (E) WGS read coverage from colonies obtained in multiplex episomal editing experiments aligned to the mSox2 YAV. (F) Genomic boundaries of unintended deletions from multiple episomal editing experiments mapped to the mouse genome and annotated with relevant repetitive elements mapped at the sites of deletion (see also Supplementary Table S7).
Notably, the entry vector pYZ960 is also compatible with the assembly of one gRNA for single edit, using the sgRNA Small Fragment Assembly protocol described previously (37). All gRNAs used in this study are listed in Supplementary Table S2. The sequences of the universal PCR template and final tRNA-gRNA arrays used for multiplex editing are provided in Supplementary Table S3. The full sequences of CRISPR/gRNA constructs for mSox2 CTCF single edit (pYZ477), three edits (pYZ212), and five edits (pYZ214) are provided as Supplementary Data-S1, -S2 and -S3, respectively, in GenBank format. Primers used to assemble these constructs are listed in Supplementary Table S4.
Yeast transformations for genomic editing
In this study, 250 ng of Cas9/sgRNA plasmids were used in all the experiments to test editing efficiency. The transformations were selected on plates with appropriate SC drop-out media, to select both CRISPR plasmids (with URA3) and the mSox2 YAV (with LEU2). The plates were incubated for three days at 30°C for formation of single colonies, which were then re-streaked to another fresh plate with the same selective media to reduce background in subsequent PCR screening.
For all episomal editing experiments, 1 μg of donor template DNA for each target was transformed. Donor templates were designed as ∼400 bp synthetic gblocks (IDT, Supplementary Table S5) containing ∼200 bp homology at each end. Primers for amplification of donor templates are listed in Supplementary Table S4. Amplified donors were purified with ZYMO DNA Clean & Concentrator kit (ZYMO Cat# D4004). The concentration was measured by Qubit dsDNA HS kits with appropriate dilutions (Invitrogen Q32851), which was also double confirmed visually in an agarose gel where the density of target bands was compared with the DNA ladder (1 Kb Plus, NEB N0469S). All donor DNAs were designed so as to eliminate PAM and associate protospacer sequence within CTCF sites by deletion.
PCR screening for editing efficiency
The PCR screening method for editing efficiency is illustrated in Supplementary Figure S1. The deleted regions for several sites (CTCF3Δ, 8Δ, 17Δ, 25Δ) were short (∼35 bp), rendering it challenging to verify based on the size change of PCR amplicons. Therefore, we utilized primers that target newly formed junctions to generate positive PCR amplifications only when the targeted CTCF sites were successfully deleted. CTCF13, which was deleted in conjunction with CTCF14, produces a ∼2 kb deletion, and was screened based on amplicon size change. We calculated editing efficiency as the ratio of colonies identified as successfully edited relative to the total number of PCR-screened colonies. Colonies that did not pass the PCR screening were further analyzed using whole genome sequencing (WGS) to identify the underlying reason. All primers used in PCR screening are included in Supplementary Table S4.
mSox2 episomal YAV
In this study, one mSox2 YAV that was previously assembled was used as the parent construct, which harbors a 143-kb wild-type mouse Sox2 locus with a backbone containing CEN/ARS and a LEU2 marker. The full sequence is provided in Supplementary Data-S4. Detailed cloning procedures for this construct were described before (10,14).
Whole genome sequencing for episomal constructs
Yeast DNA samples, containing both genomic and episomal DNA, were prepared using a Norgen Biotek fungi/yeast genomic DNA isolation kit (Cat# 27300). Sequencing libraries were prepared using NEBNext Ultra II FS DNA library prep kit (NEB E7805L) with 500 ng DNA as input. Whole genome sequencing was performed using an Illumina NextSeq 500 system and pair-end 36 bp protocol. All raw reads were trimmed to remove adaptor sequence using Trimmomatic (39), and subsequently mapped to original mSox2 YAV and CRISPR constructs as custom references, as well as to the mouse genome (mm10) using Bowtie2 software (40) and Samtools (41). The alignment was visualized using IGV (2.12.2) and the UCSC Genome Browser.
RESULTS
CRISPR/cas9 constructs and genomic editing in yeast
To determine whether there are any distinct challenges for episomal YAV editing compared to standard chromosomal editing, we developed CRISPR systems based on previously tested constructs (24). With the two-plasmid system, Cas9 and its cognate sgRNA are expressed by two different plasmids (Figure 1A). Specifically, a human codon-optimized S. pyogenes Cas9 is driven by the yeast TEF1 promoter together with a CYC1 terminator in a CEN/ARS vector (pCTC019, Cas9-CEN-SpHIS5), while the sgRNA is expressed using the RNA polymerase III (pol III) SNR52 promoter and terminated by the poly T sequence in the SUP4 terminator using a separate expression plasmid (pNA306, sgRNA-2μ-URA3). The parent strain is pre-transformed with the Cas9 plasmid, followed by a second transformation of the sgRNA plasmid along with donor DNA. We also subcloned both Cas9 and sgRNA expression modules into a single plasmid, with either a CEN/ARS (pYZ462, Cas9-CEN-URA3) or a 2μ (pYZ463, Cas9-2μ-URA3) backbone, using the same promoters and terminators for Cas9 (Figure 1B). This single-plasmid system enables delivery of all the components required for editing in a single transformation step, thereby increasing the throughput of CRISPR editing.
Figure 1.
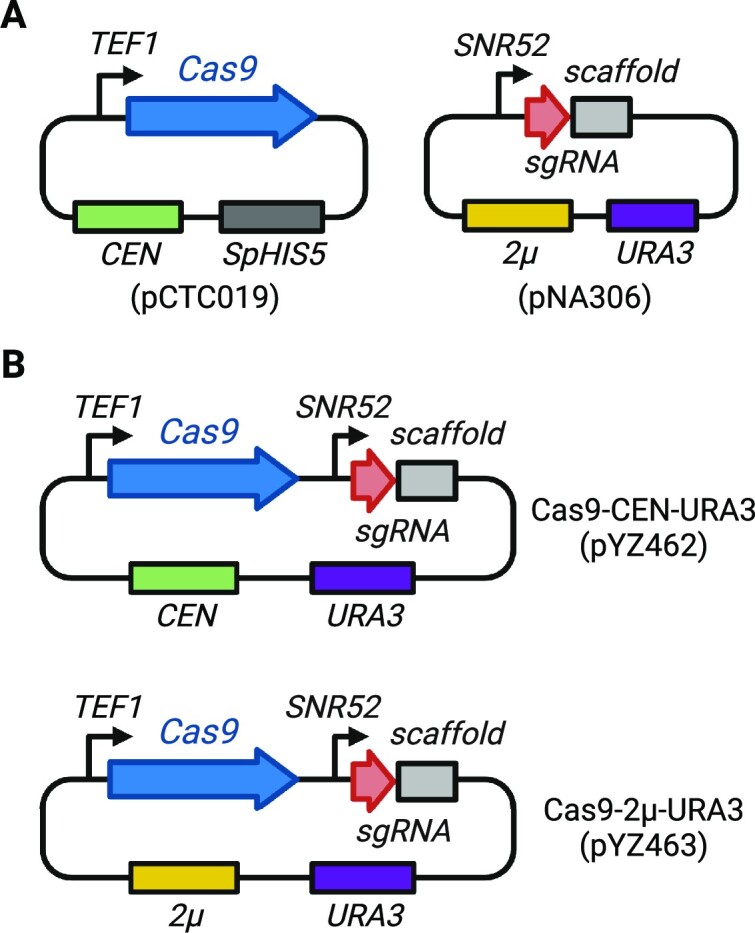
CRISPR/Cas9 constructs used in this study. (A) The two-plasmid system. Cas9 plasmid is pre-transformed into yeast, followed by a second transformation of the sgRNA plasmid. (B) The single-plasmid system. Cas9 and sgRNA are co-expressed from a single plasmid, with a CEN/ARS (top) or a 2μ backbone (bottom).
To compare episomal to genome editing, we first targeted the ADE2 coding sequence (CDS) on yeast chromosome XV using a previously reported sgRNA (30). This sgRNA was assembled into the three construct types described above (two-plasmid, and single-plasmid with either CEN/ARS or 2μ backbone). To delete the ADE2 CDS, the constructs were delivered to BY4741 together with a donor template DNA providing homology arms that span the targeted deletion (Supplementary Table S5). Red colonies on transformation plates, indicating successful deletion of ADE2, were counted to estimate editing efficiencies (42). All three systems achieved efficient editing, with >97% success rate (Figure 2A). Notably, the two-plasmid system in which the Cas9 plasmid was transformed prior to introducing the sgRNA showed the highest efficiency, although the difference was not statistically significant, suggesting that pre-transformed abundant Cas9 in cells may maximize genomic editing efficiency. Despite the slightly reduced efficiency of the single-plasmid systems, the convenience offered by a single transformation makes them a more attractive option. We also calculated the relative survival rate by dividing the colony number from the group transformed with Cas9 and sgRNA by the group transformed only with Cas9. Cas9 and sgRNA, without a donor DNA template, resulted in many fewer colonies (∼1%) (Figure 2B), confirming previous reports that yeast largely rely on HR with donor templates or high-fidelity NHEJ to repair DSBs (24).
Figure 2.

Genomic editing in yeast with one- or two-plasmid systems. (A) Editing efficiency of genomic ADE2. Error bars represent mean ± SD of three replicates. ns, not significant as calculated by an unpaired t-test. (B) Survival rate of yeast transformed with Cas9/sgRNA alone without donor DNA. Error bars represent mean ± SD of three replicates. P< 0.0001 (****), as calculated by an unpaired t-test. (C) Spot assay on SC–Ura plates with BY4741 strains transformed with Cas9 expressed from CEN/ARS or 2μ plasmids. CEN-URA3 and 2μ-URA3 were used as empty plasmid controls. (D) Doubling time of the same yeast strains used in the spot assay, extrapolated from growth liquid rates (Supplementary Figure S2). Error bars represent mean ± SD of three replicates. P< 0.001 (***), as calculated by an unpaired t-test. (E) Method to test the Cas9 toxicity and plasmid stability. Single colonies pre-transformed with either Cas9 or empty plasmids were inoculated in non-selective YPD media, then plated as single colonies and replicated onto SC–Ura selective plates. The proportion of Ura– colonies is calculated. Three single colonies were tested as biological triplicates for each group. (F) Proportion of Ura– colonies. Error bars represent mean ± SD, n = 3 and P< 0.0001 (****), as analyzed with unpaired t test.
While abundant Cas9 protein presumably increases chromosomal editing efficiency, we considered that the Cas9 load may also lead to toxicity in yeast. To assess this directly, we transformed the same Cas9 expression module using either the CEN/ARS or 2μ origin, without any sgRNA, and performed a spot assay to determine strain fitness (Figure 2C). Compared to control empty vectors (CEN-URA3 and 2μ-URA3), transformation of the Cas9-CEN-URA3 construct had a minor effect on cell growth visualized as small colonies, whereas the Cas9-2μ-URA3 construct showed strong toxicity. Growth of these strains in liquid selective medium showed growth curves consistent with the spot assay (Supplementary Figure S2). The doubling time of the strain with the Cas9-2μ plasmid was significantly longer (∼1.5×) compared to the strains with Cas9-CEN or empty vectors (Figure 2D). These results indicate that Cas9 expressed from the high-copy 2μ plasmid leads to obvious toxicity associated with increased Cas9 abundance (43).
In practice, CRISPR plasmids should be removed after editing as soon as possible in order to minimize off-target effects. Plasmid loss also enables repeated use of auxotrophic marker genes for further editing. Cas9 toxicity and plasmid stability may affect this process. To test this, we inoculated single colonies transformed with either the Cas9-CEN-URA3 or Cas9-2μ-URA3 plasmid into non-selective liquid media and then estimated the ratio of cells that lost the plasmid by replica plating (Figure 2E). To provide a baseline measurement, we also determined plasmid loss rates of an empty 2μ plasmid and an empty CEN/ARS plasmid. As expected, Cas9-2μ-URA3 was lost at a much higher rate compared to Cas9-CEN-URA3 and the control plasmids, consistent with its toxicity (Figure 2F) (44). The Cas9-CEN construct showed a similar loss ratio, consistent with minimal toxicity to yeast cells.
CRISPR/cas9 engineering of episomes in yeast: targeting the mSox2 YAV
To test the editing efficiency with CREEPY, we used an episomal YAV containing a 143-kb wild-type mouse Sox2 (mSox2) fragment, which includes the coding sequence and distal regulatory clusters such as DNase I hypersensitive sites (DHSs) and CTCF binding sites (Figure 3A) (10). First, we tested episomal editing by deleting a single CTCF site, CTCF8 (35 bp), in the mSox2 YAV (Figure 3B). The sgRNA was assembled into the same constructs with Cas9 as described above, and co-transformed with the CTCF8Δ donor template DNA (Supplementary Table S5). Following selection of transformants, single colonies were screened using colony PCR for successful designer deletion of CTCF8 using deletion-specific primers (Supplementary Figure S1). We found the single-plasmid Cas9/sgRNA-CEN construct showed the lowest efficiency (∼70%), while the Cas9/sgRNA-2μ construct and the two-plasmid system had high editing efficiency (∼96%) (Figure 3C). This suggests that high Cas9 protein abundance, either from pre-transformed Cas9 plasmid or the high-copy 2μ construct, is important for efficient episomal editing.
Figure 3.

Episomal editing in yeast targeting at mSox2 YAV. (A) Structure of episomal mSox2 YAV. BAC, bacteria artificial chromosome backbone. (B) Episomal editing strategy. Cas9 creates a DSB at CTCF8 which can be repaired with a donor DNA containing CTCF8Δ. (C) Editing efficiency with the single-plasmid (CEN or 2μ backbone) or two-plasmid (CEN/2μ) systems, as determined by genotyping using deletion-specific primers (Supplementary Figure S1). Error bars represent mean ± SD of three replicates. P< 0.05 (*), as calculated by an unpaired t-test. (D) Survival rate of colonies transformed with Cas9/gRNA targeting CTCF8 without donor DNA templates. Error bars represent mean ± SD of three replicates. P< 0.005 (**), as calculated by an unpaired t-test. (E) WGS read coverage of colonies with failed or successful edits aligned to the mSox2 YAV. (F) Unintended deletion boundaries mapped to the indicated mouse genome repetitive elements (see also Supplementary Table S6). (G) Editing strategy of a genome-integrated mSox2 CTCF8 site inserted between YFL021C and YFL021W. (H) Editing efficiency of a genome-integrated mSox2 CTCF8. Error bars represent mean ± SD of three replicates (n = 32 for each group). (I) Survival rate of colonies for genomic and episomal YAV editing without donor DNA templates. Error bars represent mean ± SD of three replicates. P< 0.0001 (****), as calculated by an unpaired t-test. (J) Episomal YAV editing with an SpHIS5 marker adjacent to CTCF8 site. His+ or His–, prototrophic or auxotrophic for histidine, respectively. (K) Ratio of His– colonies surviving episomal YAV editing (shown in J). Error bars represent mean ± SD of three replicates. P< 0.0001 (****), as calculated by an unpaired t-test. Representative images of transformation plates are depicted in Supplementary Figure S5).
Considering all these results, we determined that the best system for genomic editing where the priority is usually to build a yeast strain with designer modifications quickly and efficiently, and avoid non-specific mutations, is the single-plasmid system containing Cas9 on the CEN/ARS backbone, especially given its minor toxicity, minimal effect on cell growth, and efficient loss after culturing in non-selective medium (Figure 2F). In contrast, episomal editing is primarily used for generating DNA variants, and thus the single-plasmid Cas9-2μ system usually provides the best option because of its higher efficiency and rapid loss rate. Its relative toxicity, or potential genomic off-target activity, is of no consequence when the goal is to engineer episomal DNA for transplantation in other systems. In the following episomal engineering experiments, the Cas9-2μ construct was used.
Unintended internal deletions between micro-homologous repeats
For chromosomal editing, very few colonies (∼1%) appeared in the absence of donor DNA (Figure 2B). In contrast, episomal YAV targeting by Cas9 in the absence of donor DNA resulted in a much higher survival rate (∼40%, Figure 3D), suggesting that a different mechanism might be most frequently used to recover from DSBs in episomes than in chromosomes. An alternative explanation is that the repair mechanisms are similar, but the selective pressures involving episomal YAV and chromosomes, the latter contains many essential genes, are different. Moreover, the mammalian genomes are far richer in dispersed repeat sequences compared to the yeast genome. To uncover the mechanism responsible for the background colonies in the absence of donor DNA, from the CTCF8 deletion test, we randomly expanded 8 independent colonies that did not pass PCR screening and subjected them to whole genome sequencing (WGS), together with two colonies that showed positive PCR amplifications as controls. WGS reads were aligned to the mSox2 YAV and the Cas9/gRNA-2μ plasmids references (Figure 3E and Supplementary Figure S3–S4). First, the 35-bp CTCF8 deletion was confirmed in the positive controls, which were otherwise intact (Supplementary Figure S3). However, we found large internal deletions (30–70 kb) that always spanned the original sgRNA target site in all eight colonies that had failed genotyping. These deletions allowed cells to avoid repeated digestion of the episomes by Cas9. This result is dramatically different compared to chromosomal editing, in which survivors tend to present point mutants at the site of cleavage (24). The Cas9/gRNA construct remained intact in all of these samples (Supplementary Figure S4), indicating that the CRISPR system was still active.
Next, we aligned the WGS reads to the mm10 genome assembly and inspected the sequence properties adjacent to the deletion boundaries using RepeatMasker (45). Although the boundaries of unintended deletions were variable, we found that they all mapped to repetitive elements, including microsatellite, SINE and low complexity repeats (Figure 3F and Supplementary Table S6). Considering their length and composition, the deletions were most likely formed by HR or MMEJ, rather than NHEJ. In yeast cells, MMEJ is initiated by DNA resections from the DSB end, and subsequent microhomologous annealing (46). It is an error-prone repair mechanism that is associated with deletions flanking the DSB site (26). In contrast, the products of S. cerevisiae NHEJ are generally precise religations back to the wild-type sequence and HR is generally error free (46). The observed deletions were also much longer than regular indels from non-specific NHEJ. Thus, we speculate that both HR and MMEJ occurred after Cas9 cleavage and led to the survival of cells carrying episomal constructs with internal deletions.
When transformed with the single-plasmid Cas9-2μ system and donor DNA, yeast cells demonstrated high efficiency of simplex episomal editing (∼90%). In the absence of donor DNA, survival rate was relatively high (∼40%), suggesting that yeast cells use HR to repair DSBs when donor DNA is available and that MMEJ is a secondary response when HR with donor DNA is not possible.
Genomic CRISPR/cas9 editing of an integrated CTCF8 site
To directly compare genomic and episomal YAV editing utilizing the same gRNA target, we integrated the CTCF8 site into chromosome VI along with a LEU2 marker cassette (Figure 3G and Materials and Methods). We then employed the same Cas9/gRNA.CTCF8 construct to evaluate genomic editing efficiency. Consistently, with a donor template, we observed a high editing efficiency (∼98%) (Figure 3H), similar to ADE2 editing. We also compared the relative survival rate when editing an episomal vs genomic CTCF8 site, in the absence of the donor template. Similar to ADE2 editing, ‘donorless’ genomic CTCF8 editing produced very few colonies (∼0.1% vs the no-gRNA control), and significantly lower than episomal CTCF8 editing (∼30%) (Figure 3I).
NHEJ in episomal YAV editing
In S. cerevisiae, the vast majority of NHEJ events are precise religations that restore the wild-type sequence (27), leading to continuous cleavage by Cas9. For genomic editing without a donor template, survivors could present either point mutants or indels at the site of cleavage, which prevent subsequent recognition and re-cutting (23). In contrast, for episomal editing of the mSox2 YAV, our WGS results indicated that most yeast cells survived thanks to large deletions surrounding the gRNA targeting site.
To further assess the impact of NHEJ-induced variants on episomal YAV editing, we first integrated an SpHIS5 marker adjacent to CTCF8 in the mSox2 YAV, and subsequently transformed the Cas9/gRNA.CTCF8 plasmid without any donor template (Figure 3J). Colonies with small, localized NHEJ-induced variants should remain His+ (i.e. prototrophic for histidine), since mutations or small indels are unlikely to disrupt the his5 open reading frame. Conversely, large deletions, such as those described above (Figure 3F), are likely to extend into this marker, resulting in colonies that are His– (i.e. auxotrophic for histidine). Interestingly, we found that almost all donor-free survivors were His–, with only ∼0.2% His+ colonies (Figure 3K and Supplementary Figure S5), indicating that local NHEJ-mediated mutations represent a very infrequent mechanism for resolving episomal dsDNA breaks.
Multiplex episomal editing using a tRNA-gRNA array
Multiplex editing can significantly accelerate introduction of multiple designer variations in parallel. With this goal in mind, we evaluated the efficiency of multiplex editing of episomes. With abundant Cas9 enzyme in cells, a key challenge is whether more than one sgRNA can be expressed at once. In yeast, several genomic editing strategies have been reported in which sgRNA maturation is assisted by viral ribozymes, Csy4 cleavage or tRNA processing (25,29,30). In this study, we used a tRNA-gRNA array to express multiple sgRNAs simultaneously (Figure 4A). The sgRNAs are co-transcribed with a tRNA by pol III, and are then released from the primary transcript by endogenous RNase P and RNase Z (30,47). This design ensures the co-expression of multiple gRNAs within a single cell and avoids complicated strain engineering such as integration of Csy4 (29). Recently, it was demonstrated by RNA-seq that this strategy results in even gRNA expression from all positions in the array (48).
To test multiplex editing of mSox2 YAV, we attempted to delete three different CTCF sites in parallel: CTCF13, CTCF17 and CTCF25 (Figure 3A). We first built an entry vector (pYZ960) with the same Cas9 expression module and 2μ plasmid backbone with URA3, and containing BsmBI sites for integration of the sgRNAs. Three mSox2-targeting sgRNAs were assembled with Golden Gate cloning (see Methods) and the resulting Cas9/sgRNA-array construct was transformed with all three corresponding donor DNAs mediating CTCF site deletions. Using PCR genotyping, we found that most colonies had at least one edit (∼80%), consistent with the high editing efficiency of CREEPY (Figure 4B). More importantly, ∼25% of colonies presented all three edits, demonstrating successful multiplex episomal editing. We found that deletion of CTCF13 and CTCF25 showed similarly high efficiencies, whereas the CTCF17 deletion was less efficient, and was mostly responsible for reducing overall multiplex editing efficiency (Figure 4C).
To understand why some colonies had one or two, but not three modifications, we investigated the distribution of these edits as shown in a Venn diagram (Figure 4D). Most of the colonies with a successful CTCF17 deletion also contained the other two deletions (27 out of 30). In other words, as long as the CTCF17 designer deletion occurred, the other two edits were likely to also have occurred. In contrast, for episomes with CTCF13Δ, the majority also contained an additional edit of CTCF25Δ (61 out of 70), but all three edits occurred in only 27 of 70 colonies. A similar ratio was observed for CTCF25Δ. Consistent with its low editing efficiency, CTCF17Δ seems to represent the bottleneck limiting multiplex editing.
To understand the reasons behind this phenomenon, we performed WGS and aligned reads to the mSox2 YAV and CRISPR plasmid from three independent colonies of each of the following edit types: all three edits, CTCF13Δ only, CTCF25Δ only, CTCF13Δ and CTCF25Δ, and no detected edits, as determined by PCR genotyping (Figure 4E). For colonies with all three edits, the designer deletions were confirmed and the mSox2 episomes were otherwise intact (Supplementary Figure S6). Interestingly, for colonies with CTCF13Δ only, in addition to validating the deletion of CTCF13, internal deletions in the downstream region flanking CTCF17 and CTCF25 were observed, completely eliminating those Cas9/sgRNA recognition sites. Similar events were observed in colonies with CTCF25Δ only, where upstream internal deletions removed the CTCF13 and CTCF17 sites. Consistently, for colonies with both CTCF13Δ and CTCF25Δ but not CTCF17Δ, unintended deletions were only detected for the CTCF17 site. Notably, the CRISPR plasmids from all these groups remained intact with full-length tRNA-sgRNA arrays (Supplementary Figure S7). These results indicate that all three sgRNAs, including gRNA.CTCF17, were functional and expressed well, as cleavage happened in all CTCF sites. CTCF17 is located centrally in the mSox2 YAV while CTCF13 and CTCF25 sites are more terminal. Thus, there is a larger number of repetitive elements situated on both sides of CTCF17. Also, it is notable that the CTCF17 site is flanked on both sides by mouse SINE B1 and B2 retrotransposons, most of which range in length from 150–180 bp. Due to their length, these repeats provide rather extensive opportunities for both HR- and MMEJ-mediated repair and this may also contribute significantly to the relatively-high frequency of recovering this class of deletions. Rather than sgRNA performance, this ‘position effect’ is probably the major cause of lower efficiency of CTCF17 deletion. Finally, for colonies with zero edits, wild-type mSox2 was observed without any modifications. Instead, in those cases, deletions occurred in the Cas9/sgRNA-array plasmid, disrupting the Cas9 CDS (Supplementary Figure S7).
We also aligned the sequencing reads to the mouse genome (mm10). Consistent with the simplex editing experiment, we mapped all the deletion boundaries to micro-homologous or homologous repeats, such as micro-satellites and SINEs, respectively (Figure 4F and Supplementary Table S7). As speculated previously, this finding directly demonstrated that yeast cells can resolve Cas9 cleavage in YAV through formation of extensive internal deletions.
Internal deletions mainly result from HR and MMEJ, not NHEJ
To further investigate the mechanism behind unintended internal deletions, we first used Sanger sequencing to precisely map the junctions from multiplex editing experiments (Supplementary Figure S8). All unintended internal deletions were confirmed and mapped (Supplementary Figure S9). Several deletions, such as in colony 13, had homologous sequences substantially longer than 40 bp of sequence identity (Figure 5A). Other deletions, such as those identified in colony 17 and 18, most likely resulted from MMEJ as their homologous arms were shorter (Supplementary Figure S9). None of these deletions resulted from error-free NHEJ.
Figure 5.

Internal deletion mechanism and multiplex editing with five targets. (A) Junctions from the internal deletions in colony 13. More junction sequences are shown in Supplementary Figure S9. (B) Survival rate with Cas9 and gRNA.CTCF17, in wild-type or rad52Δ0 background. (C) WGS read coverage from three colonies obtained in CTCF17 cleavage experiments in rad52Δ0 strain background, aligned to the original mSox2 YAV. (D) CREEPY construct used for multiplex editing with five targets. (E) Editing efficiency with five targets. Colonies (n = 96) were first screened for CTCF17Δ (left). Positive colonies (n = 10, green) were subsequently selected for further screening of all other targets. The raw PCR screening gel image is shown in Supplementary Figure S12.
To find more direct evidence for MMEJ, we transformed the full-length mSox2 YAV into a yeast strain lacking RAD52 (rad52Δ0), which is required for HR, but not for MMEJ (49). We then transformed a Cas9 plasmid with or without the sgRNA targeting CTCF17 into wild-type or rad52Δ0 yeast strains (Supplementary Figure S10). Interestingly, while 45% of transformants survived in the wild-type yeast strain, in the rad52Δ strain, the survival rate was reduced to only ∼10%, presumably due to unavailable HR repair pathways (Figure 5B). This result suggests that MMEJ occurs after DNA double-strand break formation while HR may still be the major mechanism to repair the cleavage. Both scenarios could result in unintended internal deletions that eliminate Cas9/sgRNA recognition sites.
We then selected three colonies from the rad52Δ background and analyzed the Sox2 episomes by WGS. Similar internal deletions were detected spanning the CTCF17 site (Figure 5C). The boundaries were also mapped to repetitive elements (Supplementary Table S8). The junction sequences were further confirmed by Sanger sequencing, showing similar MMEJ patterns (Supplementary Figure S11).
Multiplex editing with up to five edits
Finally, we challenged the practical upper limit of multiplex episomal editing with CREEPY. We assembled a tRNA–gRNA array consisting of five gRNAs targeting CTCF13, CTCF17, CTCF25, CTCF3 and CTCF8 (Figure 5D). This plasmid was co-transformed with corresponding donor DNAs to introduce five simultaneous deletions. Learning from our previous experiments, we performed a first-round screening for the successful CTCF17Δ edit, which was the most difficult one due to its ‘position effect’. We found that 10/96 (10.4%) of colonies were positive for the precise designer CTCF17Δ deletion (Figure 5E and Supplementary Figure S12). Among these positive colonies, a second-round screening for the remaining deletions identified 4/10 colonies with all five planned deletions. We submitted three for WGS and confirmed that they all have expected designer deletions of five CTCF sites (Supplementary Figure S13).
DISCUSSION
In this study, we used CREEPY to perform CRISPR editing of episomes. CRISPR has been widely used for strain construction and genome engineering. Both the Cas9 and Cas12a systems have been implemented for genomic editing in yeast for simplex and multiplex targets, and optimized for a variety of functions (50–54). Here, we used Cas9 with human-optimized codons, driven by the constitutively active TEF1 promoter, as described in its first implementation in S. cerevisiae (24). Other groups have tested the same Cas9 enzyme with codons optimized for yeast or using the original coding sequence from S. pyogenes (25,28,55). All versions of the Cas9 enzyme showed similarly high genomic editing efficiency, indicating that the effects of codon-optimization are minimal. However, more direct comparisons of efficiency and toxicity are lacking and represent one area of further study.
We demonstrated that a single-plasmid system with Cas9 in a 2μ backbone for yeast genomic editing led to high toxicity and reduced growth rate. In other experiments unrelated to this study, we found that genome editing in haploid strains with the Cas9-2μ construct might in some instances lead to whole genome endoreduplication, generating a diploid cell with successful editing but two copies of each chromosome (data not shown). Thus, we recommend using the Cas9-CEN construct to reduce off-target effects while maintaining optimal host strain fitness, i.e. for yeast genome engineering. However, for big DNA applications focused on mammalian genome rewriting, yeast is used as a platform for DNA assembly only. The final constructs are extracted and transplanted into a target system (mammalian cells in this case). The accompanying yeast genome is no longer of any consequence. Thus, we recommend using the Cas9-2μ construct for CREEPY due to its higher editing efficiency in this context.
To test episomal editing, we used CREEPY to delete CTCF sites in the mSox2 YAV. We also found the background colonies with failed designer editing contained internal deletions that eliminated the CRISPR/sgRNA recognition sites. These deletions were due to HR and MMEJ between repetitive sequences within Sox2 as in mammalian genomes, instead of NHEJ. All these repeats were mapped and annotated with RepeatMasker (Supplementary Figure S14). Beyond this work, using CREEPY, over 60 constructs with deletions, inversions and surgical alterations of DHSs and CTCF sites in mSox2, were built in yeast and delivered to mESCs in order to study its regulatory architecture (14). In practice, we also tried another single-plasmid system, pYTK-Cas9, built with the yeast tool kit (Supplementary Figure S15A) (56). It contained a yeast-codon optimized Cas9, driven by the constitutive yeast PGK1 promoter in a CEN/ARS backbone. Different auxotrophic markers for the Cas9 plasmid backbone were also used, including URA3 (pNA304, pYZ462 and pYZ463), ScHIS3 (pNA519) and SpHIS5 (pCTC019) (Supplementary Figure S15B). All of these constructs worked efficiently. For sgRNA expression, the SNR52 promoter and endogenous tRNA were used in this study. Other pol III promoters like RPR1 can also be used to express sgRNAs in yeast (57,58). All these pol III promoters worked with high efficiency.
We conducted further analysis to evaluate the NHEJ-induced variants for mSox2 episome editing and discovered that in the absence of donor templates, nearly all colonies displayed deletions following Cas9 cleavage. It is noteworthy that while point mutations or indels are infrequent, this does not imply that NHEJ repair itself is rare. Rather, S. cerevisiae is proficient in NHEJ repair, but exhibiting much higher fidelity than mammalian systems, producing nearly 100% precise relegation to the wild-type sequence (27). Precise NHEJ repair preserves DNA integrity, rendering it susceptible to ongoing rounds of Cas9 cleavage. For genomic editing, the lack of flanking repetitive regions likely leads to the observed lethality. Consequently, only a small fraction of colonies survived, and most of them are presumed to carry ‘local’ NHEJ mutations. Extensive internal deletions are significantly more frequent for episomal YAV editing, as the wild-type mammalian genomic fragment is richer in repetitive sequences, allowing for facile recombination-based repair, whereas the native yeast genomic DNA is not. This is also consistent with our observations that internal deletion boundaries were all mapped to mammalian repetitive elements.
We achieved both simplex and multiplex episomal editing in our study, with up to five targets within one single step. We tried multiplex editing for six targets with one extra gRNA targeting CTCF4, but unfortunately, we have yet to succeed obtaining a colony with all six deletions. Facilitated by a tRNA–gRNA array, the simultaneous expression of multiple gRNAs is quite feasible. We speculate that the limiting step is the available copy number of each of the multiple donor DNA fragments required. The successful multiplex editing requires the presence of all donor DNAs in one cell. Missing one or some of these templates may lead to survival but inefficient editing as demonstrated here. This process may become increasingly challenging with additional targets and represents an avenue for further improvements to multiplex editing in yeast.
In this study, we utilized the common wild-type yeast strain BY4741 as a platform to implement our CREEPY system. However, the editing efficiency might be further enhanced by using genetically engineered strains. Mutations in various non-essential genes have been suggested as potential candidates to improve the efficiency of the system, such as the MRE complex, particularly MRE11 (49), and the nucleotide excision repair complex, including RAD1, RAD10, RAD14 (59), to inhibit MMEJ, or overexpression of RAD18 to promote HR (60). Nevertheless, systematic screening and quantitative analysis will be necessary to compare the efficiency of HR, NHEJ and MMEJ in these strains. It is crucial to ensure that HR efficiency is not compromised, as it is vital for accurate CRISPR editing through recombination with donor templates.
As big DNA design and writing technologies are advancing rapidly, the ability to engineer designer variations quickly and efficiently is essential. By manipulating large synthetic constructs of >100 kb we can study the relationship between genomic architecture and functional elements, and their association with developmental regulation, human disease and evolution. CREEPY with its simplex and multiplex editing capabilities, will directly benefit these studies and accelerate such engineering.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the center for Synthetic Regulatory Genomics (SyRGe) at NYU Langone Health for general help and discussions. We thank Hannah Ashe, Gwen Ellis and Matthew Maurano for assistance with sequencing. We also thank Nicolette Somogyi and Megan Hogan for helpful discussions about mSox2 design and constructs. This work was supported by NIH/NHGRI grant 1RM1HG009491. Figures were created with BioRender.com.
Author contributions: Y.Z. and J.D.B. conceived the project. Y.Z., M.M. and C.C. performed the experiments and collected data. Y.Z., S.L. and J.D.B. analyzed the results. J.M.L. built the pYTK plasmid. R.B. provided the wild-type mSox2 YAV and designed most primers and gblocks used in this study. Y.Z., S.L., R.B. and J.D.B. prepared the manuscript with comments from other authors. This project was supervised by R.B. and J.D.B.
Notes
Present address: Stephanie Lauer, School of STEM, St. Thomas Aquinas College, Sparkill, NY 10976, USA.
Contributor Information
Yu Zhao, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA.
Camila Coelho, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA.
Stephanie Lauer, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA.
Miłosz Majewski, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA; Maastricht Science Programme, Maastricht University, Maastricht, 6200, MD, The Netherlands.
Jon M Laurent, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA.
Ran Brosh, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA.
Jef D Boeke, Institute for Systems Genetics, NYU Langone Health, New York, NY, 10016, USA; Department of Biochemistry and Molecular Pharmacology, NYU Langone Health, New York, NY, 10016, USA; Department of Biomedical Engineering, NYU Tandon School of Engineering, Brooklyn, NY11201, USA.
Data Availability
All data and materials used in this study are available upon request. The CREEPY vectors (pYZ038, pYZ462, pYZ463, pYZ959 and pYZ960) have been deposited to Addgene (Supplementary Table S1). Their sequence files and maps are also available at Addgene. The full sequence of episomal wild-type mSox2 (143 kb) YAV is included as Supplementary Data-S4. The raw sequencing data were uploaded to the Short Read Archive (SRA) database under BioProject PRJNA944213.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH/NHGRI [1RM1HG009491]. Funding for open access charge: NIH/NHGRI [1RM1HG009491].
Conflict of interest statement. J.D.B. is a Founder and Director of CDI Labs, Inc., a Founder of and consultant to Neochromosome, Inc., a Founder, SAB member of and consultant to ReOpen Diagnostics, LLC and serves or served on the Scientific Advisory Board of the following: Sangamo, Inc., Modern Meadow, Inc., Rome Therapeutics, Inc., Sample6, Inc., Tessera Therapeutics, Inc. and the Wyss Institute. The other authors declare no competing interests.
REFERENCES
- 1. Cello J., Paul A.V., Wimmer E.. Chemical synthesis of poliovirus cDNA: generation of infectious virus in the absence of natural template. Science. 2002; 297:1016–1018. [DOI] [PubMed] [Google Scholar]
- 2. Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A.et al.. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008; 319:1215–1220. [DOI] [PubMed] [Google Scholar]
- 3. Smith H.O., Hutchison C.A. 3rd, Pfannkoch C., Venter J.C.. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fredens J., Wang K., de la Torre D., Funke L.F.H., Robertson W.E., Christova Y., Chia T., Schmied W.H., Dunkelmann D.L., Beranek V.et al.. Total synthesis of Escherichia coli with a recoded genome. Nature. 2019; 569:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Annaluru N., Muller H., Mitchell L.A., Ramalingam S., Stracquadanio G., Richardson S.M., Dymond J.S., Kuang Z., Scheifele L.Z., Cooper E.M.et al.. Total synthesis of a functional designer eukaryotic chromosome. Science. 2014; 344:55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dymond J.S., Richardson S.M., Coombes C.E., Babatz T., Muller H., Annaluru N., Blake W.J., Schwerzmann J.W., Dai J., Lindstrom D.L.et al.. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature. 2011; 477:471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchell L.A., Wang A., Stracquadanio G., Kuang Z., Wang X., Yang K., Richardson S., Martin J.A., Zhao Y., Walker R.et al.. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science. 2017; 355:eaaf4831. [DOI] [PubMed] [Google Scholar]
- 8. Richardson S.M., Mitchell L.A., Stracquadanio G., Yang K., Dymond J.S., DiCarlo J.E., Lee D., Huang C.L., Chandrasegaran S., Cai Y.et al.. Design of a synthetic yeast genome. Science. 2017; 355:1040–1044. [DOI] [PubMed] [Google Scholar]
- 9. Mitchell L.A., McCulloch L.H., Pinglay S., Berger H., Bosco N., Brosh R., Bulajic M., Huang E., Hogan M.S., Martin J.A.et al.. De novo assembly and delivery to mouse cells of a 101 kb functional human gene. Genetics. 2021; 218:iyab038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brosh R., Laurent J.M., Ordonez R., Huang E., Hogan M.S., Hitchcock A.M., Mitchell L.A., Pinglay S., Cadley J.A., Luther R.D.et al.. A versatile platform for locus-scale genome rewriting and verification. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2023952118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang W., Golynker I, Brosh R., Wudzinska A.M., Zhu Y., Carrau L., Damani-Yokota P., Khairallah C., Chalhoub N., Huang E.et al.. Mouse genomic rewriting and tailoring: synthetic Trp53 and humanized ACE2. 2022; bioRxiv doi:23 June 2022, preprint: not peer reviewed 10.1101/2022.06.22.495814. [DOI]
- 12. Pinglay S., Bulajic M., Rahe D.P., Huang E., Brosh R., Mamrak N.E., King B.R., German S., Cadley J.A., Rieber L.et al.. Synthetic regulatory reconstitution reveals principles of mammalian Hox cluster regulation. Science. 2022; 377:eabk2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blayney J.W., Francis H., Camellato B.R., Mitchell L., Stolper R., Boeke J., Higgs D.R., Kassouf M.. Super-enhancers require a combination of classical enhancers and novel facilitator elements to drive high levels of gene expression. 2022; bioRxiv doi:21 June 2022, preprint: not peer reviewed 10.1101/2022.06.20.496856. [DOI]
- 14. Brosh R., Coelho C., Ribeiro-Dos-Santos A.M., Ellis G., Hogan M.S., Ashe H.J., Somogyi N., Ordonez R., Luther R.D., Huang E.et al.. Synthetic regulatory genomics uncovers enhancer context dependence at the Sox2 locus. Mol. Cell. 2023; 83:1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camellato B.R., Brosh R., Maurano M.T., Boeke J.D.. Genomic analysis of a synthetic reversed sequence reveals default chromatin states in yeast and mammalian cells. 2022; bioRxiv doi:22 June 2022, preprint: not peer reviewed 10.1101/2022.06.22.496726. [DOI]
- 16. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J.et al.. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lartigue C., Vashee S., Algire M.A., Chuang R.Y., Benders G.A., Ma L., Noskov V.N., Denisova E.A., Gibson D.G., Assad-Garcia N.et al.. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science. 2009; 325:1693–1696. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y., Coelho C., Hughes A.L., Lazar-Stefanita L., Yang S., Brooks A.N., Walker R.S., Zhang W., Lauer S., Hernandez C.. Debugging and consolidating multiple synthetic chromosomes reveals combinatorial genetic interactions. 2022; bioRxiv doi:11 April 2022, preprint: not peer reviewed 10.1101/2022.04.11.486913. [DOI] [PubMed]
- 19. Boeke J.D., Church G., Hessel A., Kelley N.J., Arkin A., Cai Y., Carlson R., Chakravarti A., Cornish V.W., Holt L.et al.. GENOME ENGINEERING. The Genome Project-Write. Science. 2016; 353:126–127. [DOI] [PubMed] [Google Scholar]
- 20. Zhao M., Zhao Y., Yao M., Iqbal H., Hu Q., Liu H., Qiao B., Li C., Skovbjerg C.A.S., Nielsen J.C.et al.. Pathway engineering in yeast for synthesizing the complex polyketide bikaverin. Nat. Commun. 2020; 11:6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo X., Reiter M.A., d’Espaux L., Wong J., Denby C.M., Lechner A., Zhang Y., Grzybowski A.T., Harth S., Lin W.et al.. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature. 2019; 567:123–126. [DOI] [PubMed] [Google Scholar]
- 22. Galanie S., Thodey K., Trenchard I.J., Filsinger Interrante M., Smolke C.D.. Complete biosynthesis of opioids in yeast. Science. 2015; 349:1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Awan A.R., Blount B.A., Bell D.J., Shaw W.M., Ho J.C.H., McKiernan R.M., Ellis T.. Biosynthesis of the antibiotic nonribosomal peptide penicillin in baker's yeast. Nat. Commun. 2017; 8:15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DiCarlo J.E., Norville J.E., Mali P., Rios X., Aach J., Church G.M.. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013; 41:4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ryan O.W., Skerker J.M., Maurer M.J., Li X., Tsai J.C., Poddar S., Lee M.E., DeLoache W., Dueber J.E., Arkin A.P.et al.. Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife. 2014; 3:e03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sfeir A., Symington L.S. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway?. Trends Biochem. Sci. 2015; 40:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shrivastav M., De Haro L.P., Nickoloff J.A.. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008; 18:134–147. [DOI] [PubMed] [Google Scholar]
- 28. Bao Z., Xiao H., Liang J., Zhang L., Xiong X., Sun N., Si T., Zhao H.. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 2015; 4:585–594. [DOI] [PubMed] [Google Scholar]
- 29. Ferreira R., Skrekas C., Nielsen J., David F. Multiplexed CRISPR/Cas9 genome editing and gene regulation using Csy4 in Saccharomyces cerevisiae. ACS Synth. Biol. 2018; 7:10–15. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y., Wang J., Wang Z., Zhang Y., Shi S., Nielsen J., Liu Z.. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 2019; 10:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–676. [DOI] [PubMed] [Google Scholar]
- 32. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H.et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 33. Gietz R.D., Woods R.A.. Yeast Protocol. 2006; Springer; 107–120. [Google Scholar]
- 34. Wach A., Brachat A., Alberti-Segui C., Rebischung C., Philippsen P.. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast. 1997; 13:1065–1075. [DOI] [PubMed] [Google Scholar]
- 35. Karim A.S., Curran K.A., Alper H.S.. Characterization of plasmid burden and copy number in Saccharomyces cerevisiae for optimization of metabolic engineering applications. FEMS Yeast Res. 2013; 13:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitchell L.A., Cai Y., Taylor M., Noronha A.M., Chuang J., Dai L., Boeke J.D.. Multichange isothermal mutagenesis: a new strategy for multiple site-directed mutations in plasmid DNA. ACS Synth. Biol. 2013; 2:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horwitz A.A., Walter J.M., Schubert M.G., Kung S.H., Hawkins K., Platt D.M., Hernday A.D., Mahatdejkul-Meadows T., Szeto W., Chandran S.Set al.. Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst. 2015; 1:88–96. [DOI] [PubMed] [Google Scholar]
- 38. Pryor J.M., Potapov V., Bilotti K., Pokhrel N., Lohman G.J.S. Rapid 40 kb genome construction from 52 parts through data-optimized assembly design. ACS Synth. Biol. 2022; 11:2036–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones E.W Regulation of amino acid and nucleotide biosynthesis in yeast. Mol. Biol. Yeast Saccharomyces. 1982; 2:181–299. [Google Scholar]
- 43. Christianson T.W., Sikorski R.S., Dante M., Shero J.H., Hieter P.. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992; 110:119–122. [DOI] [PubMed] [Google Scholar]
- 44. Jayaram M., Li Y.Y., Broach J.R.. The yeast plasmid 2mu circle encodes components required for its high copy propagation. Cell. 1983; 34:95–104. [DOI] [PubMed] [Google Scholar]
- 45. Chen N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004; Chapter 4:Unit 4.10. [DOI] [PubMed] [Google Scholar]
- 46. Wang H., Xu X.. Microhomology-mediated end joining: new players join the team. Cell Biosci. 2017; 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie K., Minkenberg B., Yang Y.. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. NAtl. Acad. Sci. U.S.A. 2015; 112:3570–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yuan Q., Gao X.. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 2022; 13:2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Truong L.N., Li Y., Shi L.Z., Hwang P.Y., He J., Wang H., Razavian N., Berns M.W., Wu X.. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. NAtl. Acad. Sci. U.S.A. 2013; 110:7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ciurkot K., Gorochowski T.E., Roubos J.A., Verwaal R.. Efficient multiplexed gene regulation in Saccharomyces cerevisiae using dCas12a. Nucleic Acids Res. 2021; 49:7775–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Verwaal R., Buiting-Wiessenhaan N., Dalhuijsen S., Roubos J.A.. CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae. Yeast. 2018; 35:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swiat M.A., Dashko S., den Ridder M., Wijsman M., van der Oost J., Daran J.M., Daran-Lapujade P.. FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017; 45:12585–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Y., Boeke J.D.. Construction of designer selectable marker deletions with a CRISPR-Cas9 toolbox in Schizosaccharomyces pombe and new design of common entry vectors. G3 (Bethesda). 2018; 8:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao Y., Boeke J.D.. CRISPR-Cas12a system in fission yeast for multiplex genomic editing and CRISPR interference. Nucleic Acids Res. 2020; 48:5788–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Utomo J.C., Hodgins C.L., Ro D.K.. Multiplex genome editing in yeast by CRISPR/Cas9-A potent and agile tool to reconstruct complex metabolic pathways. Front. Plant Sci. 2021; 12:1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee M.E., DeLoache W.C., Cervantes B., Dueber J.E.. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth. Biol. 2015; 4:975–986. [DOI] [PubMed] [Google Scholar]
- 57. Tran V.G., Cao M., Fatma Z., Song X., Zhao H.. Development of a CRISPR/Cas9-based tool for gene deletion in Issatchenkia orientalis. Msphere. 2019; 4:e00345-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shan L., Dai Z., Wang Q.. Advances and opportunities of CRISPR/Cas technology in bioengineering non-conventional yeasts. Front. Bioeng. Biotechnol. 2021; 9:765396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee K., Ji J.H., Yoon K., Che J., Seol J.H., Lee S.E., Shim E.Y.. Microhomology selection for microhomology mediated end joining in Saccharomyces cerevisiae. Genes (Basel). 2019; 10:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nambiar T.S., Billon P., Diedenhofen G., Hayward S.B., Taglialatela A., Cai K., Huang J.W., Leuzzi G., Cuella-Martin R., Palacios A.et al.. Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat. Commun. 2019; 10:3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials used in this study are available upon request. The CREEPY vectors (pYZ038, pYZ462, pYZ463, pYZ959 and pYZ960) have been deposited to Addgene (Supplementary Table S1). Their sequence files and maps are also available at Addgene. The full sequence of episomal wild-type mSox2 (143 kb) YAV is included as Supplementary Data-S4. The raw sequencing data were uploaded to the Short Read Archive (SRA) database under BioProject PRJNA944213.