


Abstract
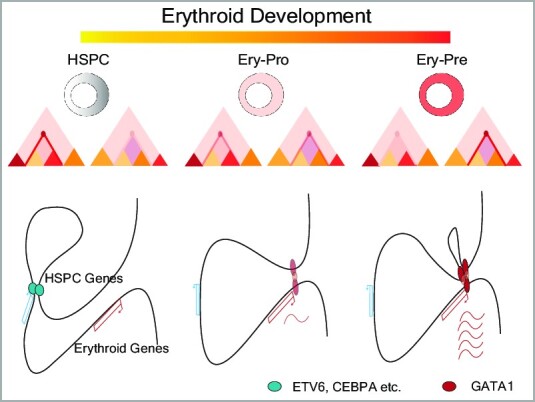
Mammalian erythroid development can be divided into three stages: hematopoietic stem and progenitor cell (HSPC), erythroid progenitor (Ery-Pro), and erythroid precursor (Ery-Pre). However, the mechanisms by which the 3D genome changes to establish the stage-specific transcription programs that are critical for erythropoiesis remain unclear. Here, we analyze the chromatin landscape at multiple levels in defined populations from primary human erythroid culture. While compartments and topologically associating domains remain largely unchanged, ∼50% of H3K27Ac-marked enhancers are dynamic in HSPC versus Ery-Pre. The enhancer anchors of enhancer–promoter loops are enriched for occupancy of respective stage-specific transcription factors (TFs), indicating these TFs orchestrate the enhancer connectome rewiring. The master TF of erythropoiesis, GATA1, is found to occupy most erythroid gene promoters at the Ery-Pro stage, and mediate conspicuous local rewiring through acquiring binding at the distal regions in Ery-Pre, promoting productive erythroid transcription output. Knocking out GATA1 binding sites precisely abrogates local rewiring and corresponding gene expression. Interestingly, knocking down GATA1 can transiently revert the cell state to an earlier stage and prolong the window of progenitor state. This study reveals mechanistic insights underlying chromatin rearrangements during development by integrating multidimensional chromatin landscape analyses to associate with transcription output and cellular states.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
The process of erythropoiesis is characterized by stepwise proliferation and differentiation from hematopoietic stem and progenitor cells (HSPCs) to mature red blood cells (RBCs) through functionally and morphologically distinct stages (1). Normally, approximately 2 million RBCs are produced per second in human bone marrow through this tightly regulated erythroid development process (2). Defects in this process lead to various types of anemia and associate with a wide range of acquired and inherited diseases, such as Diamond-Blackfan anemia and polycythemia vera (3–6).
Previous studies of genetically modified mouse models and human congenital anemia have identified several regulators of erythropoiesis, including the transcription factors GATA1, GATA2 and KLF1 (7–17). Comprehensive analyses of select stages of erythropoiesis using bulk- and single-cell transcriptomics (18–21) and proteomic profiling (22,23), combined with analyses of the epigenetic landscape such as chromatin accessibility (24,25), DNA methylation (24,26) and histone modifications (27–29), provide different point of view to understand this process.
The three-dimensional (3D) organization of chromosomes promotes long-range chromatin interactions, especially between enhancers and promoters, which are of vital importance for maintaining sophisticated gene regulatory networks (30,31). The 3D genome architecture in select stages of erythropoiesis have been studied in key erythroid loci, such as the globin genes (32–35). However, detailed 3D genome characterization throughout the entire process of human erythroid development remains lacking, with particular regard to the genome-wide enhancer–promoter (E–P) dynamic interactions. Therefore, these dynamic erythropoiesis-related interactions require further study to achieve a more comprehensive understanding of chromatin and gene expression regulation and their relationship with red blood cell diseases.
In this study, we used Micro-C and in situ Hi-C followed by chromatin immunoprecipitation (HiChIP) assays to provide comprehensive profiling of chromatin architecture, particularly E–P interactions, in clearly defined stages of human erythropoiesis: HSPC, erythroid progenitor (Ery-Pro) and erythroid precursor (Ery-Pre). We integrated these data with dynamic occupancy of key transcription factors, such as GATA1 and GATA2, and transcriptome profiling at the HSPC, Ery-Pro and Ery-Pre stages to give a systematic understanding of the molecular characteristics of human erythropoiesis. Our data show that stage-specific transcription factors are involved in the rewiring of E–P interactions from the HSPC to Ery-Pro to Ery-Pre stages. Furthermore, the master regulator GATA1 gradually dominates the E–P interactions throughout these transitions. During the transition from Ery-Pro to Ery-Pre, GATA1 exhibits a relatively stable occupancy pattern, but a gain in the distal GATA1 occupancy drives significant chromatin contact changes, promoting productive erythroid gene expression. In addition, our results indicate that the dosage of GATA1 is essential for maintaining the Ery-Pro state, and a transient shortage of GATA1 in the Ery-Pro promotes the conversion of cells to an earlier progenitor state, which has a higher self-renewal capacity.
Overall, our profile offers a valuable resource to advance the understanding of human erythropoiesis, and our integrated analysis provides not only a comprehensive understanding of the 3D gene regulation throughout the differentiation process, but also reveals how stage-specific transcription factors likely participate in rewiring the chromatin connectome, particularly how dynamic GATA1 occupancy and dosage associate with step-wise gene regulation and cellular state. These results may therefore further a wide range of studies related to inherited and acquired diseases of red blood cells.
MATERIAL SAND METHODS
Reagents
All information on reagents and antibodies can be found in Supplementary Table S1.
Biological resources
CD34+ cells (HSPC) were obtained from the Cord Blood Bank of Beijing.
Ex vivo erythroid culture of CD34+ HSPCs and HUDEP-1 cells
Erythroid differentiation was induced using erythroid differentiation medium (EDM): IMDM supplemented with 5% human AB serum, 10% FBS, 300 μg/ml holo-Transferrin, 2 mM l-glutamine, 10 ng/ml heparin, 10 μg/ml, 3 IU/ml erythropoietin (EPO), 50 ng/ml hSCF and 10 ng/ml hIL-3, with cell density kept below 0.5 million/ml. HUDEP-1 cells were maintained in the expansion medium, consisting of StemSpan SFEM supplemented with 50 ng/ml SCF, 3 IU/ml EPO, 1 × 10−6 M dexamethasone, and 1 mg/ml doxycycline, with cell density kept <0.5 million/ml(36). Differentiation of HUDEP-1 cells was achieved by a 5-day culture in EDM supplemented with 1 mg/ml doxycycline. Erythroid differentiation was monitored by flow cytometry analysis of CD71, CD117 and CD235a expression at day3, day5, day7 and day11 for CD34 + erythroid culture and at day 3 and day 5 for HUDEP-1 differentiation.
Isolation of Ery-Pro and Ery-Pre cells
Ery-Pro cells were isolated by a fluorescence-activated cell sorting (FACS) scheme derived from previously described methods with several modifications (20,37–39). Briefly, at day 5 of CD34+ erythroid cell culture, cells were spun down and re-suspended in staining buffer (2% FBS in PBS). Subsequently, the following antibodies were added for 30 min at 4°C per 1 × 106 cells: 5 μl anti-CD235a(APC, HIR2), 5 μl anti-CD45RA(APC, HI100), 5 μl anti-CD123(APC, 7G3), 5 μl anti-CD7(APC, M-T701), 5 μl anti-CD10(APC, HI10a), 5 μl anti-CD90(APC, 5E10), 5 μl anti-CD135(APC, BV10A4H2), 5 μl anti-CD41a(APC, HIP8), 20 μl anti-Hematopoietic Lineage(APC), 5 μl anti-CD71(FITC, OKT9), 5 μl anti-CD34(PE, 4H11), 6 μl anti-CD38(PerCP-Cy5, HIT2), 5 μl anti-CD36(APC-Cy7, 5–271) and 5 μl anti-CD105(PE-CF594, 266). After staining, cells were washed with staining buffer and 1 μg/ml DAPI was added. FACS was performed on the FACSAria-II SORP flow cytometer (BD Biosciences). The DAPI-Lin*− (Lin− CD7− CD10− CD45RA− CD123− CD90− CD135− CD41a−) CD34+ CD38+ CD71+ CD36+ CD105+ cells are Ery-Pro cells. Proerythroblast cells (Erythroblast, Ery-Pre) were sorted on erythroid differentiation day 11 as previously described (40), and defined as CD235a+ CD71+ CD45low CD36+ CD117high CD105high.
Analytical flow cytometry was performed on the LSRFortessa SORP Cell Analyzer (BD Biosciences) by staining with anti-CD71 (1:200), anti-CD34 (1:200), anti-CD38 (1:200), anti-CD36 (1:200), anti-CD117 (1:200), anti-CD235a (1:200) and either DAPI (for differentiation screening) or anti-CD235a (1:200), 2 μg/ml Hoechst33324 and 1 μg/ml propidium iodide (for enucleation screening). CountBright™ Absolute Counting Beads were added to each sample to measure the cell number.
CFU assays and subsequent detection of lineage markers
HSPC, Ery-Pro or Ery-Pre cells were plated in 2.5 ml methylcellulose medium at 200 cells per dish. After 14 days incubation, the colonies were counted, and images of each individual colony were obtained with the DM 1000 microscope (Leica, Wetzler, Germany). Gross images of the colony dish were taken with the iPhone 13 Pro with macro mode (Apple, Cupertino, CA, USA). Identification of colonies is described as follows. Colony-forming unit-erythroid (CFU-E): colonies containing a total of 8–200 erythroblasts. Burst-forming unit-erythroid (BFU-E): colonies containing >200 erythroblasts in single or multiple clusters. Colony-forming unit-granulocyte, macrophage (CFU-GM): colonies containing >40 granulocytes (CFU-G), macrophages (CFU-M), or cells of both lineages (CFU-GM). Colony-forming unit-granulocyte, erythrocyte, macrophage, megakaryocyte (CFU-GEMM): colonies containing erythroblasts and cells of at least two other recognizable lineages.
Erythroid and myeloid marker (CD235a/CD11b) detection was performed on the LSRFortessa SORP Cell Analyzer after washing-out the colonies from the dishes.
CUT&RUN assay
The CUT&RUN experiments were conducted as previously described with modifications (41). Briefly, 10 000 cells for each replicate were captured with BioMagPlus Concanavalin A for 10 min at room temperature in Wash buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM spermidine, 1 × protease inhibitor cocktail). Permeabilization of cells and binding of primary antibodies (1:100) were performed overnight at 4°C in 150 μl antibody buffer (wash buffer with 0.05% digitonin and 2mM EDTA). After washing away unbound antibody twice with 1 ml Dig-wash buffer (wash buffer with 0.05% digitonin), protein A-MNase (homemade, 340ng/ μl) was added at a 1:100 ratio and incubated for 2 h at 4°C in 100 μl Dig-wash buffer. Cells were washed twice again with 1 ml Dig-wash buffer and placed in a 0°C metal block. CaCl2 was then added to a final concentration of 2 mM to activate the protein A-MNase. The reaction was carried out for 30 min and stopped by adding an equal volume of 2× STOP buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.05% digitonin, 100 μg/ml RNAse A and 50 μg/ml glycogen) in 37°C for 30 min. The protein–DNA complex was then released by centrifugation. DNA was extracted using the Monarch PCR & DNA Cleanup Kit, and eluted with 15 μl H2O. Quality control analysis was performed using the Qubit Fluorometer and bioanalyzer (Thermo Fisher Scientific). Protein A-MNase was expressed and purified from E. coli BL21(DE3) carrying pET-pA-MN (derived from Plasmid #86973, Addgene, Watertown, MA, USA).
The CUT&RUN library preparation was performed with the VAHTS Universal DNA Library Prep Kit according to the manufacture's protocol with minor modifications (42). Briefly, 10 μl of DNA was used for library construction. The temperature for dA-tailing was decreased to 50°C to avoid DNA melting, and the reaction time was increased to 1 h. After adaptor ligation, a 2× volume of VAHTS DNA Clean Beads was added to the reaction to ensure high recovery efficiency of short fragments. After 15 cycles of PCR amplification, the reaction was cleaned up with a 1.2× volume of VAHTS DNA Clean Beads. The final libraries were quantified using a VAHTS Library Quantification Kit and a D1000 High Sensitivity DNA chip run on a Bioanalyzer 4250 system (Agilent Technologies, Santa Clara, CA, USA). Two biological replicates per condition were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene, Beijing, China).
RNA-seq and scRNA-seq library preparation
RNA-seq was performed with the Single Cell Full Length mRNA-Amplification Kit, which is based on the SMART-seq2 method (43). Briefly, two hundred cells were lysed and reverse transcribed, followed by 11 cycles of PCR to amplify the transcriptome library. The whole transcriptome library quality was validated using a D5000 DNA Chip run on a Bioanalyzer 4150 system (Agilent Technologies), followed by library preparation using the TruePrep DNA Library Prep Kit and custom index primers according to the manufacturer's instructions. The final libraries were quantified using a VAHTS Library Quantification Kit and a D1000 High Sensitivity DNA chip run on a Bioanalyzer 4250 system (Agilent Technologies). Two biological replicates per condition were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene).
Single cell RNA-seq (scRNA-seq) was performed with the Chromium Next GEM Single Cell 3′ Kit v3.1 targeting 10 000 cells according to the manufacturer's protocol. The final libraries were quantified using a D1000 High Sensitivity DNA chip run on a Bioanalyzer 4250 system (Agilent Technologies). Libraries were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene).
Micro-C assay
The Micro-C assay was carried out as previously described with minor modifications (44), using 100 000 FACS-purified cells for each reaction. In brief, after fixation with 3 mM DSG for 30 min followed by 1% formaldehyde for another 10 min, the crosslinked cells were permeabilized in ice-cold Micro-C Buffer #1 (50 mM NaCl, 10 mM Tris–HCl (pH 7.5), 5 mM MgCl2, 1 mM CaCl2, 0.2% NP-40 and 1× Protease Inhibitor Cocktail) for 20 min. Chromatin from permeabilized cells was digested by 50 U micrococcal nuclease at 37°C for 10 min. After digestion, chromatin fragments were then subjected to dephosphorylation, phosphorylation, and end-chewing processes using T4 PNK and Klenow Fragment in end-repair buffers (50 mM NaCl, 10 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 100 μg/ml BSA, 2 mM ATP, 5 mM DTT, no dNTPs) at 37°C for 30 min. Blunt-end ligation and biotin incorporation were achieved by adding biotin-dATP, biotin-dCTP, dGTP and dTTP and incubating at 25°C for 45 min. Chromatin was ligated by T4 DNA Ligase at room temperature for 2.5 h. Unligated ends containing biotin-dNTPs were then removed by exonuclease III at 37°C for 15 min. After reverse-crosslinking, the DNA was purified and gel size-selected for dinucleosomal DNA at 200–400 bp. DNA fragments containing biotin were immobilized on MyOne Streptavidin C1 Dynabeads, and then library preparation was performed using the VAHTS Universal DNA Library Prep Kit per the manufacturer's protocol. Two biological replicates per condition were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene).
Capture-C assay
The Capture-C assay was carried out using the in situ method as previously described with minor modifications (45), using 1 × 106 cells for each reaction. In brief, after fixation with 1% formaldehyde, cells were lysed in 100 μl HiC buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 0.5% NP-40 and proteinase inhibitor), and the nuclei pellet was solubilized in 20 μl 0.5% SDS and incubated at 62°C for 10 min. The SDS was quenched with 10 μl 10% Triton X-100 at 37°C for 30 min. The nuclei were then digested overnight at 37°C using 50 U of MboI. After MboI inactivation, proximity ligation was carried out overnight at 25°C with 1600 U of T4 DNA Ligase. After reverse-crosslinking, DNA was purified and sheared to 200–600 bp fragments using the M220 sonicator (Covaris, Woburn, MA, USA). Library preparation was then performed using the VAHTS Universal DNA Library Prep Kit per the manufacturer's protocol with 1μg DNA per replicate. Next, 2 μg of indexed and mixed library was used as the input for two rounds of downstream hybridization and biotin capture with biotin-labeled oligos using the GenFisher Hybridization and Wash Kit per the manufacturer's protocol (Supplementary Table S2). Briefly, 2 μg multiplexed DNA from four indexed DNA library (500 ng each) was mixed with 5 μl human repeated DNA and 2 μl universal blockers, and then dried down with vacuum concentrator (Savant™ SpeedVac™ DNA 130). After reconstitution with the hybridization buffer (contain 3 pmol biotin labeled probe) in a PCR tube, the hybridization mix was incubated in a PCR machine for 95°C 30S and 65°C overnight. The hybridization mix was then mixed with an equal volume of pre-washed streptavidin beads and incubated at 65°C for 45 min in a thermomixer at 1200 rpm. After several washes, the DNA-captured beads were reconstituted with 25 μl H2O and 14 cycles of PCR amplification were performed. The second-round capture was performed as the first-round except that eight multiplex sample could be used. Two biological replicates per condition were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene).
HiChIP assay
The HiChIP assay was conducted using the in-situ method as previously described with minor modifications (46). For GATA1 HiChIP, 10 × 106 Day 5 Ery-Pro and Day 12 Ery-Pre cells generated from the ex vivo CD34+ erythroid differentiation culture were used. In brief, after fixation with 1% formaldehyde, the cells were lysed in 1 ml HiC buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 0.5% NP-40 and proteinase inhibitor), and the nuclei pellet was solubilized in 100 μl 0.5% SDS and incubated at 62°C for 5 min. The SDS was quenched with 50 μl 10% Triton X-100 at 37°C for 30 min. Then, the nuclei were digested for 2 h at 37°C using 375 U of MboI. After biotin filling, proximity ligation was carried out with 4000 U of T4 DNA Ligase for 2 h at 25°C. The nuclei pellet was reconstituted with 500 μl nuclear lysis buffer (50 mM Tris–HCl, 10mM EDTA, 1% SDS, 1 × protease inhibitor cocktail). Sonication of re-suspended nuclei was performed using the M220 sonicator (Covaris), then 2 ml HiChIP dilute buffer (0.01% SDS, 1.1% Triton-X 100, 1.2 mM EDTA, 16.7 mM Tris–HCl, 167 mM NaCl, 0.1 × protease inhibitor cocktail) were added to reduce the concentration of SDS. Following an overnight incubation with anti-GATA1 antibody (abcam, ab11852, 15μg/107 cells), 100 μl Dynabeads™ Protein A were added at 4°C and incubated for 2 h. The bound DNA–protein complexes were eluted and reverse-crosslinked after a series of washes.
After reverse-crosslinking, the biotin-containing ligation fragments were immobilized on MyOne Streptavidin C1 Dynabeads, and the library was prepared using the Tn5 Transposome DNA Library Prep Kit per the manufacturer's protocol with 5 ng DNA. After 10 cycles of PCR amplification, the library was then subjected to double-size selection using VAHTS DNA Clean Beads to isolate fragments between 300 and 600 bp. Two biological replicates per condition were sequenced using the Illumina Novaseq 6000 platform (S4, PE150, Novogene).
Reporter gene assay
Reporter gene plasmid transfection was performed with lipofectamine 3000 in K562 cells. In brief, 1 μg of the pGL3-SLC4A1-promoter with either the wild-type (WT) or mutated GATA1 binding sites enhancer and 50 ng of pRL-SV40 were co-transfected into differentiated K562 cells (48 h after 5 U/ml rhEPO treatment) at 200000 cells per well in a 12-well plate. Forty-eight hours post transfection, firefly and renilla luciferase activities were measured consecutively with the Dual-Lumi Luciferase Reporter Gene Assay Kit using a BioTek Cytation 5 luminometer (Agilent Technologies). The reporter gene activity was expressed as the ratio of firefly luciferase relative luminescence units (RLU) to renilla luciferase RLU.
siRNA delivery in Ery-Pro cells
GATA1 knockdown was performed via nucleofecting siRNA (LNA modified, mixture of three different siRNAs on the same target gene) on the purified Ery-Pro cells according to the manufacturer's protocol. In brief, 20000 sorted Ery-Pro cells were spun down and resuspended with 20 μl of solution P3 for primary cells; 15, 30 or 75 pmol of siGATA1 siRNA, or 75 pmol of scrambled siCon siRNA, were then added to the cell suspension; and nucleofection was performed in a 4D-Nucleofector X Unit (Lonza Bioscience) using program EO-100. After nucleofection, 200 cells were plated immediately in 2.5 ml methylcellulose medium for the subsequent 14-day culture. The remaining cells were subsequently cultured in EDM for further growth. The knock-down efficiency was assessed using SMART-seq2 and qRT-PCR after 24, 48 and 72 h of incubation in EDM. The siRNA sequences are provided in Supplementary Table S2.
siRNA delivery and cas9-mediated knock-out in HUDEP-1 cells
In brief, 20 000 or 1000 000 differentiated Day 3 HUDEP-1 cells were spun down and resuspended with 20 μl or 100 μl of solution P3 for primary cells (Lonza Bioscience), respectively, and 75 pmol or 300 pmol of either siGATA1 or Control siRNA was then added to the cell suspension, respectively. Nucleofection was performed in a 4D-Nucleofector X Unit (Lonza Bioscience) using program EO-100. After 48 h of continued culture, gene expressions were confirmed with qRT-PCR.
Cas9–sgRNA RNP complexes were assembled as follows: For each knock-out experiment, 300 pmol of Cas9 protein and 500 pmol of sgRNA (GeneScript) were mixed and incubated for 20 min at room temperature. Next, 1 × 106 differentiated Day 3 HUDEP-1 cells were resuspended with 100 μl of solution P3 for primary cells (Lonza Bioscience), preassembled ribonucleoproteins (RNPs) were then added to the cell suspension and nucleofection was performed in a 4D-Nucleofector X Unit (Lonza Bioscience) using program DZ-100. Cells were centrifuged and transferred to 2 ml of medium for continued growth. To assess genome editing efficiency, cells were lysed to extract genomic DNA (gDNA). Primers spanning the edited sites were used to amplify the genomic region. The guide RNA (gRNA) and genotyping primer sequences are listed in Supplementary Table S2. After two days of differentiation culture, transfected cells were utilized for the downstream analysis. For the precise GATA1 binding site depletion, a single-stranded oligodeoxynucleotide (ssODN) was introduced as the template to co-transfect with the Cas9–RNP the HUDEP-1 cells at maintained stage. After an additional seven days of culture, single clones were picked, and the target genome sequence was confirmed using Sanger sequencing.
Cell proliferation assay
Absolute cell number of Ery-Pro cells was measured using CountBright™ Absolute Counting Beads at Day 3, Day 8, Day 9 and Day 10 post-siRNA delivery while cell differentiation was monitored by flow cytometry. CellTiter-Blue® Cell Viability Assay was used to detect cell proliferation. Briefly, 100 siRNA-delivered Ery-Pro cells were seeded in 200 μl EDM. At the 3-, 6- or 8-day incubation time point, 40 μl CellTiter-Blue reagent was added and incubated for 2 h. The fluorescence signal was measured in triplicate using a BioTek Cytation 5 luminometer (Agilent Technologies).
Micro-C data processing
Micro-C data processing was performed as previously published with minor modifications (47). The raw reads of the Micro-C library were mapped with hg19 and further filtered with HiC-Pro (version 3.10.0, https://github.com/nservant/HiC-Pro) (48). Pairs with multiple hits, MAPQ < 10, singleton, dangling end, self-circle, and PCR duplicates were removed. Paired reads with distances shorter than 100 bp were also disregarded. Correlation of replicates was calculated using the stratum-adjusted correlation coefficient (SCC) method with HiCRep (version 1.11.0, http://github.com/qunhualilab/hicrep) at 10, 20, 50 and 100 kb (49). All biological replicates were processed individually to assess quality and then combined for further analyses and visualization. The valid Micro-C contacts matrices were then normalized using the K-R method, and corresponding .cool files were generated at 200 bp, 400 bp, 600 bp, 800 bp, 1 kb, 2 kb, 4 kb, 10 kb, 20 kb, 100 kb and 200 kb bin size. Visualization of .cool files were conducted on the Higlass 3D genome server (http://higlass.io).
Identification of compartments was conducted by hicCompartmentalization in HiCExplorer (version 3.0, https://github.com/deeptools/HiCExplorer) (50) with matrices at 100 kb resolution and referred to as A or B according to the gene density. The compartments with higher gene density were selected as compartment A, while the compartments with lower gene density were selected as compartment B. Furthermore, the H3K27ac signal was also incorporated to distinguish compartment A or B (51).
TADs and TAD insulation scores were identified and calculated by the hicFindTADs function of the HiCExplorer tool at 20 kb resolution (50). The insulation score is measured using the z-score of the Hi-C matrix and is defined as the mean z-score of all matrix contacts between the left and right regions. The minus value of the insulation score indicates boundary strength (50,52). To calculate the TAD activity (Intra- or Inter-TAD contact ratio), we first merged boundaries observed at ±40 kb across the three stages together, then the TAD activity was express as the Intra- or Inter-TAD contact ratio across the merged TAD regions. The differential TAD was called using the hicDifferentialTAD function of the HiCExplorer tool with threshold P-value <0.05 and FC >1.5.
We utilized Mustache (version 1.2.0, https://github.com/ay-lab/mustache) (53) to call loops with balanced contact matrices at resolutions of 400 bp, 600 bp, 800 bp, 1 kb, 2 kb, 4 kb, 10 kb and 20 kb using the calling options –pThreshold 0.1 –sparsityThreshold 0.88 –octaves 2. We then combined all loops at different resolutions. If a loop was detected at different resolutions, we retained the precise coordinates in finer resolutions. We used coolpuppy (version 1.0.0, https://github.com/open2c/coolpuppy) to implement the APA function for the .cool file. To further filter and classify loops, we first defined ±2.5 kb of the gene TSS as the promoter (P) and non-TSS H3K27ac peaks in each stage (HSPC, Ery-Pro, and Ery-Pre) as enhancers (E). GenomicInteractions (version 1.32.0, https://github.com/ComputationalRegulatoryGenomicsICL/GenomicInteractions) (54) was used to designated P–P, E–P and E–E loops in respective stages.
CUT&RUN data processing
CUT&RUN data processing was performed as previously published with minor modifications (55). The raw CUT&RUN sequence data were trimmed to barcode using TrimGalore (version 0.4.4, https://github.com/FelixKrueger/TrimGalore) and reads were aligned to the hg19 version of human genome with bowtie2 (version 2.3.4.1, https://github.com/BenLangmead/bowtie2) (56). Duplicated reads were removed using the Picard tool (version 2.0.1, http://broadinstitute.github.io/picard/) and the low-quality reads (MAPQ < 30) were also removed. Reads with the cutoff of inset size <120 bp were selected for transcription factors occupancy and 150–500 bp for H3K27ac histone modification, as defined in previous studies (41,55,57). Peaks were called using SEACR with default parameters with IgG control (version 1.2, https://github.com/FredHutch/SEACR) (57), and the differentiated binding sites of H3K27ac and GATA1 were calculated using the diffbind R package with the parameter FDR <0.05 (version 1.15.5, https://www.cruk.cam.ac.uk/core-facilities/bioinformatics-core/software/diffbind). Co-binding of transcription factors was performed using the bedtools package with 1 bp overlap (version 2.30.0, https://github.com/arq5x/bedtools2/). The bigwig/bedgraph files were generated using the deepTools bamCoverage tool (version 3.4.0, https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html) (84) to visualize peaks in IGV (https://software.broadinstitute.org/software/igv/) (58) and pyGenomeTracks (version 3.7, https://github.com/deeptools/pyGenomeTracks).
Super enhancer (SE) and typical enhancer (TE) identification was performed as previously described using ROSE (https://bitbucket.org/young_computation/rose/src/master/) with the following parameters: -s 12500 -t 2500 -g hg19 (59).
HiChIP data processing
HiChIP data processing was performed as previously published with minor modifications (60). In brief, the raw reads of the HiChIP library were mapped with hg19 and further filtered with HiC-Pro (version 3.10.0, https://github.com/nservant/HiC-Pro) to remove pairs with multiple hits, MAPQ <10, singleton, dangling end, self-circle, and PCR duplicates (48). All HiChIP GATA1 biological replicates generated from this study were initially processed individually to assess quality and then combined for downstream analyses and visualization. The HiChIP H3K27ac data were obtained from a public dataset without replicates (GSM3769103, GSM5028232) (61,62). Correlation of replicates was calculated using the SCC method with HiCRep (version 1.11.0, http://github.com/qunhualilab/hicrep) at 10, 20 and 50 kb (49). HiChIP datasets were then processed by FitHiChIP (version 10.0, https://github.com/ay-lab/FitHiChIP) after subset to the same valid interaction numbers (60). The following parameters were used to call HiChIP loops for both H3K27ac and GATA1 HiChIP: 5000 bp resolution, 10 000–2000 000 distance threshold, FDR 0.05, coverage specific bias correction, merged nearby peak to all interactions, L model. To simplify the analysis, we designated Day 5 and Day 12 H3K27ac/GATA1 peaks to represent the Ery-Pro and Ery-Pre stage, respectively. GenomicInteractions (https://github.com/ComputationalRegulatoryGenomicsICL/GenomicInteractions) (54) was used to designate the P–P, E–P and E–E loops in each respective stage.
Differential GATA1 HiChIP loops were identified via the ‘DiffAnalysisHiChIP.r’ script of FitHiChIP with the following parameters: FDR <0.01 and FoldChangeThr >2.
Virtual 4C was performed as previously described with minor modifications (63). In brief, the pairs of reads that mapped at the viewpoints region at one side were extracted, and visualization was performed using average CPM signals per condition at 200 bp bin size and 2 kb window.
Capture-C data processing
Capture-C analysis was performed as previously described with capC-MAP (version 1.0, https://github.com/cbrackley/capC-MAP) software (64). In brief, the raw reads were trimmed with cutadapt and mapped to the reduced hg19 reference genome, which is comprised of the reference genome sequences adjacent to the DpnII cut sites using bowtie. The read counts per bin were normalized to the sequencing depth per replicate in counts per million (CPM). Visualization was performed using average CPM signals per condition at a bin size of 200 bp with 2 or 4 kb window.
Locus overlap analysis (LOLA) enrichment analysis
We first set up a customized database using the LOLA package in R with CUT&RUN peaks of GATA1/2, ETV6, RUNX1 and CEBPA for the HSPC, Ery-Pro and Ery-Pre stages individually, as well as the three-stage combined peaks. Next, we mapped the enhancer anchors of the Micro-C- or H3K27ac HiChIP-detected E–P loops back to the original H3K27ac peaks as the input for the LOLA assay. We then showed q-values and odds ratios of the three-stage combined peaks of GATA1/2, ETV6, RUNX1 and CEBPA.
Bulk RNA-seq and scRNA-seq data processing
The bulk RNA-seq data were checked for quality using FastQC (version 0.11.7, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and the data were aligned to the hg19 reference genome using RNA STAR (version 2.7.9a, https://github.com/alexdobin/STAR) (65). The expression level of each gene was calculated using FeatureCounts (version 2.0.3, https://subread.sourceforge.net) and read counts were normalized to transcripts per million (TPM) (66). Genes with TPM >0 were considered to be expressed genes. DESeq2 (version 1.38.0, http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) (67) was then used to calculate the significantly differentially expressed genes, using the thresholds FC > 2 and FDR < 0.05. Gene set enrichment analysis for GO biological processes was performed using the clusterProfiler (version 3.10.1, https://guangchuangyu.github.io/software/clusterProfiler/) package in R (68).
Raw reads of scRNA-seq for each sample were processed using the Cell Ranger (version 6.1.1, 10x Genomics) pipeline software with human reference version GRCh38, then the filtered gene expression matrices were analyzed using the Seurat package (version 4.03, https://github.com/satijalab/seurat) (69). Cells were further filtered with the following criteria: >1000 unique molecular identifiers (UMIs), <25 000 UMIs, <20% UMIs derived from the mitochondrial genome and >500 genes. The gene expression matrices were normalized and 2000 features with high cell-to-cell variation were calculated via the FindVariableFeatures function. Normalized data was subjected to linear transformation and principal component analysis (PCA) based on variable features using the RunPCA function. Graph-based clustering was performed as per gene expression profiles using the FindNeighbors and FindClusters functions with default parameters, and results were visualized using a nonlinear dimensional reduction UMAP technique running RunUMAP and DimPlot functions.
We used the FindIntegrationAnchors function to identify shared sources of variation across the Ery-Pro and 48 h post-knock-down siGATA1 Ery-Pro scRNA datasets, and integrated them via anchors from the IntegrateData function with correlation dimensions of 20. Integrated data were then normalized and subjected to reducing dimension as performed in the individual datasets.
We performed trajectory analysis using the Monocle3 package (https://github.com/cole-trapnell-lab/monocle3) (70) in RStudio on the 48 h post-knock-down siGATA1 Ery-Pro cells and integrated the 48 h post-knock-down siGATA1 Ery-Pro scRNA dataset constructed using Seurat. The Monocle3 UMAP coordinates were then added from the Seurat object. We then fit a principal graph within each cluster using the learn_graph function and visualized the order of cells in pseudotime by plot_cells functions.
Statistical analyses
For Micro-C, HiChIP, CUT&RUN and Capture-C, two biological replicates were conducted. For RNA-seq, three biological replicates were performed, with the exception of siGATA1/ siCon in Ery-Pro, for which two biological replicates were performed. For reporter gene assay, binding sites knock-out by Cas9 and qRT-PCR analysis, four biological replicates were conducted. P values and methods of statistical tests are reported in figure legends. Data analysis and statistical analysis was performed using Python (version 3.7.4), R (version 4.1.2), Excel and Prism 6 software.
Data availability/sequence data resources
The Micro-C, CUT&RUN, HiChIP, Capture-C, RNA-seq and scRNA-seq data was uploaded on GEO: GSE214811.
RESULTS
Dynamic chromatin architecture in human erythropoiesis
To obtain the clearly defined and homogeneous Ery-Pro stage, we established an efficient method to isolate erythroid progenitors derived from primary human cord blood CD34+ cells by modifying previously reported methods (Figure 1A). These Ery-Pro cells are DAPI− Lin*− (CD2, CD3, CD14, CD16, CD19, CD56, CD235a, CD45RA, CD123, CD7, CD10, CD90, CD135, CD41a) CD34+ CD38+ CD71+ CD36+ CD105+ (Supplementary Figure S1A) (37,38,71). The Ery-Pre cells are proerythroblasts, which were separated as previously described: CD71+ CD235a+ CD36+ CD45low CD117(c-kit)high CD105high (40). We next performed the colony forming assay followed by analytical flow cytometry to examine the colony-forming ability and descendant cell markers to validate the Ery-Pro purity (Supplementary Figure S1B). The results showed that Ery-Pro can only generate burst-forming unit-erythroid (BFU-E) and colony-forming unit-erythroid (CFU-E) colonies, with >90% of the cells expressing the erythroid lineage marker CD235a (Supplementary Figure S1C–I). Conversely, CD34 + HSPCs derived from freshly isolated cord blood can give rise to all types of colonies—BFU-E, CFU-E, colony-forming unit-granulocyte/granulocyte-macrophage (CFU-G/GM), colony-forming unit-granulocyte-erythroid-macrophage-megakaryocyte (CFU-GEMM)—with erythroid colonies accounting for <40% (Supplementary Figure S1C–I). Approximately 60% of the cells from the HSPC-derived colonies express the erythroid lineage marker CD235a, most likely due to the large number of erythroid cells in the CFU-GEMM colonies (Supplementary Figure S1H).
Figure 1.

Dynamic chromatin architecture in human erythropoiesis. (A) Schematic description of ex vivo CD34 + erythroid cell differentiation and subsequent separation of HSPC, Ery-Pro, and Ery-Pre cells and illustration of the experimental strategy. (B) Micro-C contact probability as a function of distance in HSPC (blue), Ery-Pro (orange) and Ery-Pre (green) cells. (C) Changes in compartment dynamics during human erythropoiesis. (D) Heatmap showing the boundary strength of HSPC, Ery-Pro and Ery-Pre cells at ±0.5 Mb of boundary regions. (E) Micro-C Hi-C contact matrices and domain scores (top) and H3K27ac signals (middle) of HSPC, Ery-Pro and Ery-Pre stages, and RNA expression (bottom) in HSPC and Ery-Pre stages at chromosome 3 (46.5–51.5 Mb). The pink-shaded area indicates the up-regulated gene LRRC2 in the H3K27ac gained region; the blue-shaded area indicates the down-regulated gene SMARCC1 in the H3K27ac lost region. Abbreviations: HSPC, hematopoietic stem and progenitor cell; Ery-Pro, erythroid progenitor; Ery-Pre, erythroid precursor.
To investigate the chromatin architectural changes and enhancer dynamics in human erythropoiesis, we conducted the Micro-C assay, the Cleavage Under Targets & Release Using Nuclease (CUT&RUN) assay detecting H3K27ac, and RNA-seq in primary human HSPC, Ery-Pro and Ery-Pre cells, isolated as shown in Figure 1A. The Micro-C experiment generated over half a billion unique contacts merged from two biological replicates (Supplementary Table S3). Each replicate was generated from 100 000 cells and highly correlated at all resolutions for HSPC, Ery-Pro and Ery-Pre (Supplementary Figure S1J); therefore, biological replicates were then combined for subsequent analysis. Based on the contact probability, we found a higher frequency of long-range intrachromosomal interactions (>1 Mb) in HSPCs but more short-range intrachromosomal interactions (<1 Mb) in Ery-Pro and Ery-Pre stages. HSPC and Ery-Pro had fewer ultra-short range intrachromosomal interactions (<10 kb) than Ery-Pre (Figure 1B). Comparison of compartments A and B among the HSPC, Ery-Pro and Ery-Pre stages based on the principal component 1 (PC1) value showed that 86% of the genome maintained the same compartment state throughout erythropoiesis, and only of 14% regions experienced compartment flip: 2% A to B (active to repressed), 3% B to A (repressed to active) and 9% transient flip (A–B–A or B–A–B, transiently repressed or active) (Figure 1C). We next examined the dynamics of topologically associating domain (TAD) boundaries, and found 5539, 5339 and 5418 boundaries in HSPC, Ery-Pro and Ery-Pre, respectively. Although the boundary strength was enhanced from HSPC to Ery-Pre, we did not observe striking differences among these stages (Figure 1D, E, Supplementary Figure S1K). We next examined the dynamic changes of TAD activity, defined as the ratio of intra-/inter-TAD interactions (72,73). Using the cutoff P < 0.05 and fold-change (FC) > 1.5, we observed only one significantly different TAD region in HSPC relative to Ery-Pro cells, and seven in Ery-Pro relative to Ery-Pre cells (Figure 1E, Supplementary Figure S1L–M). Collectively, these results demonstrate that the TAD boundary and intra-TAD interactions remain relatively stable during human erythroid cell development even with a minor but significant change of compartments. This is different from the previously characterized terminal erythroid nuclear compression process, which shows global disruption of domain boundaries and selective preservation of TADs and boundaries (40).
Enhancer dynamics and enhancer connectome rewiring in erythroid development
We next analyzed the deposition of the active chromatin marker H3K27ac in HSPC, Ery-Pro and Ery-Pre stages (Supplementary Figure S2A). Throughout erythroid progression (HSPC to Ery-Pre), 52% (14628) of the H3K27ac occupancy sites remained stable, >36% (10156) showed a gradual H3K27ac signal decrease, and only 12% (3379) showed an H3K27ac signal increase (Figure 2A). Genomic annotation-based gene ontology (GO) analysis showed that the lost peaks (i.e. HSPC specific), the shared peaks (i.e. present throughout erythroid development), and the gained peaks (i.e. Ery-Pre specific) are all enriched with promoters of genes involved in pathways such as myeloid cell differentiation, histone modification, and erythrocyte differentiation, even though the associated genes are distinct (Figure 2B, Supplementary Table S4). The pathways involved in leukocyte and myeloid development are more highly represented in HSPC specific peaks, while those involved in erythroid development or function have a higher representation in Ery-Pre specific peaks. It was also noted that the chromatin remodeling pathway is significantly enriched in genes associated with the shared peaks, and that certain erythropoiesis related genes are marked in the HSPC stage as described before (Figure 2A, B) (74,75). We further compared the differential H3K27ac sites among the three stages. The lost peaks from the HSPC to the Ery-Pro stage continued to show low signal in the Ery-Pre stage, and the gained peaks from the HSPC to the Ery-Pro stage continued to show high signal in the Ery-Pre stage, whereas only 3% (850) of the sites showed up-regulated H3K27ac deposition from the Ery-Pro to Ery-Pre stage (Supplementary Figure S2B, C). These results indicate that most of the active chromatin regions in the HSPC stage, including some erythroid related active regions, preserve their activity throughout erythropoiesis, while dynamic erythroid related active chromatin regions account for only a small subset of the genome (Supplementary Figure S2B, C). Motif analysis of the HSPC specific H3K27ac regions showed motif enrichment in hematopoietic transcription factors in the ETS family (including MEF), GATA family, RUNX family and IRF family (Supplementary Figure S2D). Similarly, analysis of the Ery-Pro specific H3K27ac regions revealed motif enrichment in the ETS, JUN, RUNX, GATA and MYB families, while Ery-Pre specific H3K27ac regions predominantly showed high GATA family enrichment as well as KLF motifs (Supplementary Figure S2E, F).
Figure 2.

Enhancer dynamics and enhancer connectome rewiring in erythroid progression. (A) H3K27ac CUT&RUN signals at differential H3K27ac occupancy sites of HSPC versus Ery-Pre cells showing lost (n = 10156, top), stable (n = 14 628, middle), and gained (n = 3379, bottom) peaks. (B) GO terms of genes corresponding to the differential H3K27ac peaks of HSPC versus Ery-Pre cells. (C) Connectivity of Micro-C anchors containing SEs, TEs and TSSs in HSPC, Ery-Pro and Ery-Pre cells, respectively. Statistical significance was determined using the Mann–Whiney test. (D) Heatmaps of HSPC, Ery-Pro and Ery-Pre Micro-C data and H3K27ac HiChIP data from Day 0 and Day 12 at HSPC SE and Ery-Pre SE/TE regions: MEIS1 (Chr2: 66 506–68 340 kb), RAB3C (Chr5: 57 646–58 798 kb), α-globin (Chr16: 90–284 kb), and SLC4A1 (Chr17: 42 309–42 410 kb) loci. Circles denote the representative significant Micro-C or H3K27ac HiChIP E–P interactions of the HSPC or Ery-Pre enhancers. Abbreviations: GO, gene ontology; SEs, super enhancers; TEs, typical enhancers; TSSs, transcription start sites.
To elucidate the relationship of fine chromatin architecture changes and enhancer dynamics in human erythroid development, we first used Micro-C to analyze chromatin loops at 400 bp, 600 bp, 800 bp, 1 kb, 2 kb, 4 kb, 10 kb and 20 kb resolution, and then combined the loops with different resolutions together. We identified 24 829, 17 852 and 16 408 loops in the HSPC, Ery-Pro and Ery-Pre cells in the Micro-C data, respectively. Loop connectivity analysis showed that super enhancers (SE) and typical enhancers (TE) display higher loop numbers per anchor than transcription start sites (TSS) (Figure 2C). Furthermore, we compared our data with the published H3K27ac HiChIP datasets ‘Day 0 erythroid culture’ (representing the HSPC stage) (62) and ‘Day 12 erythroid culture’ (representing the Ery-Pre stage), which also showed higher connectivity at SE sites than TE or TSS (Supplementary Figure S2G) (61). The contact heatmap from our Micro-C and H3K27ac HiChIP data of the typical HSPC SE sites MEIS1 and RAB3C (Figure 2D, left) and the Ery-Pre SE/TE sites HBA1/2, SLC4A1 (Figure 2D, right) showed significant enhancer connectome rewiring during human erythropoiesis.
Stage-specific transcription factors are involved in the rewiring of enhancer–promoter contacts
To further our understanding of the relationship between enhancers and gene expression, we next examine connections between the enhancer and promoter. We classified the 24 829 HSPC, 17 852 Ery-Pro and 16 408 Ery-Pre Micro-C loops as enhancer–promoter associated loops (E–P loops) or others (non E–P loops), according to the overlap between the loop anchors with H3K27ac and the promoter (Figure 3A, Supplementary Figure S3A–C, S3F–H). We detected 1737 (7.0%) HSPC, 1308 (7.3%) Ery-Pro and 1194 (7.2%) Ery-Pre E–P loops. To gain a better understanding of the relationship between active transcription and chromatin loops, we analyzed the gene expression profiles and genes associated with E–P loops in the HSPC and Ery-Pre stages. Our results indicate that 1156 genes are only found in HSPC E–P loops, 397 genes are existed in both HSPC and Ery-Pre E–P loops, and 771 genes are only involved in Ery-Pre E–P loops; furthermore, the expression level of these genes is significantly correlated with the E–P loop identity, e.g. genes with promoters located at the HSPC E–P loop anchors have higher expression in the HSPC stage (HSPC E–P loops genes versus constant E–P loops genes: P < 0.0001; constant E–P loops genes versus Ery-Pre E–P loops genes: P < 0.001; Figure 3A). GO analysis of Micro-C loops showed that the HSPC E–P loops are enriched with gene promoters related to the regulation of hematopoietic differentiation, while the Ery-Pre E–P loops are enriched with gene promoters related to erythroid development and cell homeostasis process, etc. (Supplementary Figure S3K, Supplementary Table S5). Analytical results from the erythroid Day 0 and Day 12 H3K27ac HiChIP datasets are similar to our Micro-C results, and the expression level of Day 0 and Day 12 HiChIP E–P associated genes is significantly correlated with the loop identity (Day 0 E–P loops genes versus constant E–P loops genes: P < 0.0001; constant E–P loops genes versus Day 12 E–P loops genes: P < 0.001; Figure 3B, Supplementary Figure S3D–E, S3I–J). GO analysis showed that the HSPC H3K27ac-HiChIP E–P loops are enriched with promoters of genes involved in cell proliferation and adhesion, while the Ery-Pre H3K27ac-HiChIP E–P loops are enriched with promoters of genes involved in chromatin remodeling and the heme biosynthetic process (Supplementary Figure S3L, Supplementary Table S5).
Figure 3.

Stage-specific transcription factors are involved in the rewiring of enhancer–promoter contacts. (A) The number of all loops and E–P loops detected by Micro-C (left) in HSPC, Ery-Pro and Ery-Pre; the gene expression (log2FC HSPC versus Ery-Pre) of corresponding promoter anchors (right) in HSPC specific, constant and Ery-Pre specific loops. Statistical significance was determined using the Mann–Whitney test. (B) The number of all loops and E–P loops detected by H3K27ac-HiChIP (left) in Ery-Day0 and Ery-Day12; the gene expression (log2FC HSPC versus Ery-Pre) of corresponding promoter anchors (right) in HSPC specific, constant and Ery-Pre specific loops. Statistical significance was determined using the Mann–Whitney test. (C) LOLA enrichment analysis for the enhancer anchors of the Micro-C E–P loops in HSPC, Ery-Pro and Ery-Pre stages using our CUT&RUN datasets of transcription factors. LOLA enrichment for each transcription factor was based on combined peaks from all stages of the respective transcription factor. (D) LOLA enrichment analysis for the enhancer anchors of the H3K27ac-HiChIP E–P loops in erythroid culture day 0 (Ery-Day0) and day 12 (Ery-Day12) using our CUT&RUN datasets of transcription factors. LOLA enrichment for each transcription factor was based on combined peaks from all stages of the respective transcription factor. (E) Virtual 4C profile of normalized H3K27ac HiChIP signals around HOXA9 VPs (Chr 7: 26.1–27.3 Mb, top), Micro-C detected E–P loops (middle), and CUT&RUN signals of H3K27ac, GATA1, GATA2, ETV6, CEBPA and RUNX1 (bottom) at HSPC, Ery-Pro and Ery-Pre stages. (F) Virtual 4C profile of normalized H3K27ac HiChIP signals around LCR VPs (Chr 11: 5.20–5.35 Mb, top), Micro-C detected E–P loops (middle), and CUT&RUN signals of H3K27ac, GATA1, GATA2, ETV6, CEBPA and RUNX1 (bottom) at HSPC, Ery-Pro and Ery-Pre stages. Abbreviations: LOLA, locus overlap analysis; CPM, counts per million; VP, viewpoint.
To gain insight into the transcription factors involved in the rewiring of E–P loops during erythropoiesis, we identified predicted transcription factors from our motif analysis of stage-specific H3K27ac peaks and then performed CUT&RUN analysis on those select transcription factors (Supplementary Figure S2D–F, Supplementary Figure S3M–N). The enhancer anchors of HSPC E–P loops were highly enriched with HSPC behavior-regulating transcription factors CEBPA and ETV6 (76–79), moderately enriched with RUNX1, and only slightly enriched with erythroid transcription factors GATA2 and GATA1 (Figure 3C). Conversely, the enhancer anchors of Ery-Pro E–P loops were only minimally enriched with CEBPA and ETV6, moderately enriched with RUNX1 (similar to HSPC), and more enriched with GATA1 and GATA2 than HSPC E–P loops (Figure 3C). However, in the Ery-Pre stage, only GATA1 showed significant enrichment (Figure 3C), which was further validated in the erythroid Day 0 and Day 12 H3K27ac HiChIP dataset (Figure 3D). For example, the H3K27ac signal at the HOXA locus (Chr 7 26.1–27.3 Mb) was highly enriched in the HSPC stage and diminished gradually with the Ery-Pro and Ery-Pre stages. Virtual 4C of the H3K27ac HiChIP and Micro-C E–P loops displayed high connectivity of the HOXA cluster with the target genes SKAP2, SNX10 and CBX3, and CUT&RUN analysis showed that ETV6 and CEBPA were highly enriched in these anchor regions (Figure 3E). The erythroid associated β-globin locus control region (LCR) (Chr 11 5.20–5.35 Mb), showed an accumulation of H3K27ac signals with erythroid development. Virtual 4C of the H3K27ac HiChIP signal and Micro-C E–P loops revealed that connectivity of the LCR and globin genes, which is inactive in HSPCs, became activated in both the Ery-Pro and Ery-Pre stages (Figure 3F). As expected from the results shown in Figure 3C–D, the GATA1 signal increased during erythroid development, but GATA2 and RUNX1 were highly enriched in the Ery-Pro stage only. While H3K27ac signal is commonly used to indicate enhancers, it may not be sufficient to accurately define them. Therefore, we integrated the stage-specific H3K27ac signals with the ATAC-seq data from Day0 and Day12 erythroid cells to better represent the enhancers in HSPC, Ery-Pro and Ery-Pre stages. The analysis revealed results similar to those obtained using only H3K27ac signal to define enhancers (Supplementary Figure S3O–R) (61). These findings indicate that throughout erythropoiesis, GATA1 governs E–P loops and regulates most erythroid genes; however, in the Ery-Pro stage, GATA1 may cooperate with additional transcription factors such as GATA2, RUNX1, and SPI1, thereby participating in the sophisticated gene regulation of erythroid development and maintenance of hematopoietic cell function (16,80–87).
Dynamic GATA1 connectome is associated with stage specific GATA1 occupancy and coordinates gene expression in erythroid progression
GATA1 is involved in the E–P interaction remodeling during erythropoiesis. To dissect the GATA1-engaged chromatin interactions, we next performed GATA1 HiChIP in the Ery-Pro abundant stage (erythroid culture Day 5) and the Ery-Pre stage (erythroid culture Day 12) using two biological replicates, which were highly correlated at all resolutions of Day 5 and Day 12 (Supplementary Figure S4A, Supplementary Table S3). Differential looping analysis indicated three clusters of GATA1-centered interactions: gained loops in the Ery-Pre stage (n = 9626), stable (n = 10 963), and lost loops (n = 7798) (Figure 4A, Supplementary Figure S4B, Supplementary Table S6). Although previous studies have shown that GATA1 is involved in both gene activation and repression (84,88), we observed that genes within the gained and lost GATA1 loops were significantly up- and down-regulated, respectively, in comparison to the expression of all genes (Figure 4B). These observations are further supported by the overlap of GATA1 and H3K27ac occupancy: Nearly 90% of GATA1 at TSS regions co-localized with H3K27ac, while approximately 60% of GATA1 occupancy sites at non-TSS regions correlated with H3K27ac in both the Ery-Pro and Ery-Pre stages (Supplementary Figure S4C). GO analysis indicated that the lost GATA1 loops at Day 5, which corresponds to the Ery-Pro stage, were enriched with promoters of genes involved in mononuclear cell differentiation and phosphate metabolism (Figure 4C, Supplementary Table S7). Both the stable and gained GATA1 loops were enriched with promoters of genes associated with similar GO terms such as chromatin remodeling, nucleosome/telomere organization, and erythrocyte differentiation, which showed similar P-values but higher gene ratios in genes associated with gained loops (Figure 4C). To further understand how GATA1 is involved in dynamic looping, we categorized the dynamic GATA1 loops into two groups: 3D differential loops, which exhibit changes solely in the 3D structure, and GATA1 differential occupancy loops, which exhibit changes in GATA1 occupancy in at least one anchor of the loop (60). We found that a large fraction of the dynamic GATA1 loops were GATA1 differential occupancy loops (79.7%, n = 13 891) (Figure 4D).
Figure 4.

Dynamic GATA1 connectome is associated with stage specific GATA1 occupancy and coordinates gene expression in erythroid progression. (A) Heatmap of gained (n = 9626, top), stable (n = 10 963, middle) and lost (n = 7798, bottom) GATA1 loops detected by GATA1 HiChIP in Ery-Pro (Day5) and Ery-Pre (Day12) cells. (B) Change in gene expression between Ery-Pre and Ery-Pro cells whose promoters were involved in lost (n = 2741 genes), stable (n = 4123 genes), or gained (n = 1847 genes) GATA1 loops (Mann–Whiney test). (C) GO terms of genes corresponding to lost, stable, or gained GATA1 loops. (D) Classification of dynamic GATA1 loops: 3D differential loops (n = 3533, 20.3%) indicate no GATA1 occupancy changes in both anchors; GATA1 differential binding loops (n = 13 891, 79.7%) denote GATA1 occupancy changes in at least one anchor. (E) Heatmap showing the stable (unchanged from Ery-Pro to Ery-Pre, n = 8041), lost (Ery-Pro specific, n = 702), and gained (Ery-Pre specific, n = 479) GATA1 occupancy sites in Ery-Pro and Ery-Pre cells using CUT&RUN. (F) Classification of stable and dynamic GATA1 (Ery-Pro versus Ery-Pre) occupancy sites based on genomic positions (TSS or non-TSS). (G) Aggregate Peak Analysis for genome-wide averaged contact signals of promoter loops associated with gained non-TSS GATA1 peaks (upper, n = 835) and lost non-TSS GATA1 peaks (bottom, n = 376). GATA1 HiChIP loops in Day5 and Day12 cells were used in this analysis (resolution: 2 kb). (H–K) Virtual 4C profile of normalized GATA1 HiChIP signals (top) and CUT&RUN signals of H3K27ac, GATA1 and GATA2 (middle) and RNA-seq (bottom) at Ery-Pro and Ery-Pre stages of the representative gained GATA1 loops regions: SLC4A1 locus, Chr17: 42 310–42 380 kb (G), METTL7A locus, Chr12: 51 150–51 430 kb (H); and the representative lost GATA1 loops regions: ENO locus, Chr1: 8895–8960 kb (I), and DPH5 locus, Chr1: 101 450–101 610 kb (J). Arrowheads denote the dynamic GATA1 occupancy sites. CUT&RUN signals are expressed as RPKM, and RNA-seq signals are expressed as CPM. (L) RNA-seq-based gene expression of gained GATA1 occupancy site-associated genes (non-TSS peaks determined by GATA1 HiChIP to corresponding genes, n = 696) in HSPC, Ery-Pro and Ery-Pre cells, respectively (Wilcoxon test). (M) Gene expression change of select gained GATA1 occupancy site-associated genes from panel L in Ery-Pro and Ery-Pre cells using qRT-PCR (n = 4). Data are presented as mean ± SEM. Abbreviations: TSS, transcription start site; CPM, counts per million; RPKM, reads per kilobase million; VP, viewpoint.
Unlike MyoD or YY1, which promote chromatin contacting via homodimer formation, previous studies have demonstrated that GATA1 forms loops by interacting with the looping factors such as LDB1, FOG1 or BRG1 (89–95). To exclude complicating factors that may affect GATA1 loops, we focused on the dominant population - GATA1 differential occupancy loops. We first examined the GATA1 differential occupancy in Ery-Pro versus Ery-Pre and found that most peaks exhibited a stable occupancy pattern (n = 8072, 87.2%), whereas 7.6% of Ery-Pro GATA1 peaks (n = 702) showed decreased signal and only 5.2% of Ery-Pre GATA1 peaks (n = 479) showed increased signal during the erythroid progenitor-precursor transition (Figure 4E). We also observed a positive correlation between H3K27ac signal and changes in GATA1 occupancy. Specifically, sites with increased GATA1 occupancy also showed an increase in H3K27Ac signal, which supports the idea that GATA1 primarily has a positive regulatory effect on gene expression (Supplementary Figure S4D).
Interestingly, during the development from erythroid progenitors to precursors, most of the dynamic GATA1 occupancy sites were distributed in non-TSS regions: 93.9% of gained GATA1 occupancy sites and 94.3% of lost GATA1 occupancy sites—whereas only 68.2% of the stable GATA1 occupancy sites were located within non-TSS regions (Figure 4F). The majority of the GATA1 TSS occupancy sites were stable (97.3%, 2554 sites), which reveals that GATA1 already occupies most TSS regions that are necessary for erythroid development in the progenitor stage. We next examined the loop intensity of gained and lost non-TSS GATA1 binding sites with their target promoters. The results indicate that the occupancy of GATA1 on non-TSS sites is positively correlated with their magnitude of interaction with target promoters (Figure 4G). Representative examples of gained GATA1 occupancy regions, such as the SLC4A1 and METTL7A loci, showed an increased interaction signal between the gained GATA1 occupancy site and the local gene promoter, as well as an augmented H3K27ac signal on the gained GATA1 occupancy site (arrowhead, Figure 4H–I, Supplementary Figure S4E–G). On the contrary, ENO1 and DPH5 loci showed decreased interaction signals between the lost GATA1 occupancy site and the local gene promoter, as well as a decline in the H3K27ac signal on the lost GATA1 occupancy site (Figure 4J–K, Supplementary Figure S4H). Similarly, in mouse fetal liver erythroid progenitor cells (Cd71-Ter119- or Cd71medTer119-) and erythroblasts (Cd71+ Ter119+), around 85% of GATA1 occupancy sites were stable, and the dynamic GATA1 occupancy sites were primarily located at non-TSS sites (Supplementary Figure S4I, GSE171383). RNA-seq analysis showed that the gained GATA1 occupancy sites coordinated genes that were up-regulated in the progenitor-precursor transition but stable during the differentiation from HSPC to Ery-Pro, and the lost GATA1 occupancy sites coordinated genes that were down-regulated in the progenitor-precursor transition but stable during the differentiation from HSPC to Ery-Pro (Figure 4L–M, Supplementary Figure S4J).
Collectively, our integrated GATA1 HiChIP, CUT&RUN, and RNA-seq analyses showed that GATA1 associated loops are highly dynamic during the erythroid progenitor-precursor transition, that gene expression correlated with the formation/deformation of GATA1 loops, and that the majority of GATA1 occupancy is stable, especially at TSSs. Limited GATA1 occupancy changes (12.8%), which are primarily located within non-TSS regions and are highly correlated with H3K27ac signals, engage more than 80% of the dynamic GATA1 loops.
Ery-Pre cells acquired distal GATA1 occupancy sites that promote gene expression and local chromatin rewiring
We found that the gained GATA1 occupancy sites correlated with productive erythroid gene expression and were accompanied by GATA1-associated chromatin contact changes during the development from progenitors to precursors. To further investigate the role of gained GATA1 occupancy sites on gene expression and local chromatin architecture, we first examined effects of GATA1 knock-down in differentiated HUDEP-1 cells. Our results showed that abrogation of GATA1 expression repressed the expression of GATA1 target genes bearing the gained GATA1 occupancy site at their distal regions, which were annotated as enhancer, namely, SLC4A1, TLCD4, METTL7A, ATF1, SLC11A2, CHID1 and KLF13 (Supplementary Figure S5A-B, Figure 4, and Supplementary Figure S4) (96–98). To exclude the global effects of GATA1 knock-down on gene expression, we disrupted the gained GATA1 occupancy sites in differentiated HUDEP-1 cells with Cas9 and detected the nearby gene expression. We found that knocking out the gained GATA1 peaks at the SLC4A1 distal enhancer, the TLCD4 intron, and the CHID1 distal enhancer down-regulated the corresponding gene expression (Figure 5A, Supplementary Figure S5C). Disruption of the gained GATA1 peaks at the METTL7A distal enhancer reduced the expression of METTL7A and SLC11A2 but had no notable effects on ATF1 expression. Knocking out the gained GATA1 peaks at the KLF13 distal enhancer did not significantly alter KLF13 expression, suggesting GATA1 may not play a major role in regulating these genes (Figure 5A, Supplementary Figure S5C). These functional assays show an overall correlation between the degree of gene expression abrogation after GATA1 knockdown and that after knocking out distal GATA1 binding sites. Knocking out of distal GATA1 binding sites results in more minor but precise control of GATA1-mediated transcription activity.
Figure 5.

Ery-Pre cells acquired GATA1 occupancy sites that promote gene expression and local chromatin rewiring. (A) Gene expression of SLC4A1, TLCD4, METTL7A, ATF1, SLC11A2, CHID1 and KLF13 after knock-out of the corresponding gained GATA1 binding sites in Ery-Pre cells (see panel 5B–D and Figure 4) in differentiated HUDEP-1 cells. Data are shown as mean ± SEM (n = 4). Statistical significance was determined using the Student's t-test; *P < 0.05, **P < 0.01, ***P < 0.001. (B–D) Top, Capture-C contact profiles plotted as overlays, comparing contacts of the control (siCon, blue) and GATA1 knock-down (siGATA1, red) in differentiated HUDEP-1 cells at the SLC4A1 locus (Chr17: 42310–42380 kb) (B), TLCD4 locus (Chr1: 95 543–95 653 kb) (C) and METTL7A locus (Chr12: 51 150–51 430 kb) (D). The y-axis shows the Capture-C sequencing coverage expressed as mean RPM with 200 bp bin and 2000 bp window (B, C) or 200 bp bin and 4000 bp window (D) (n = 2). VP represents the viewpoint of the SLC4A1 promoter (B), TLCD4 intronic GATA1 binding site (C) and METTL7A distal GATA1 binding site (D). Bottom, GATA1 CUT&RUN tracks of siCon and siGATA1, respectively. Arrowheads indicate the gained GATA1 peaks in the erythroid progenitor-precursor transition. CTCF CUT&RUN signal, CTCF-site orientations, and H3K27ac peaks are indicated at the bottom. Dash lines below the Capture-C contact profiles indicate statistically significant regions (EDGER, P < 0.05, log2FC > 1.5). (E) Top, Capture-C contact profiles plot, comparing contacts of the WT, single GATA1 binding site mutant (ΔGATA1 BS1), and dual GATA1 binding site mutant (ΔGATA1 BS1&2) in differentiated HUDEP-1 clones at the SLC4A1 locus (Chr17: 42 310–42 380 kb). The y-axis shows Capture-C sequencing coverage expressed as mean RPM with 200 bp bin and 2000 bp window (n = 2). VP represents the viewpoint of the SLC4A1 promoter. Bottom, GATA1 CUT&RUN tracks of WT, single GATA1 binding site mutant (ΔGATA1 BS1), and dual GATA1 binding site mutant (ΔGATA1 BS1&2) HUDEP-1 clones in differentiated and maintained stages, respectively. Arrowheads show the degenerated peaks. Enh indicates the enhancer sequence used for the reporter gene assay in panel F (Chr17: 42 358 715–42 361 352 bp). Dash lines below the Capture-C contact profiles indicate statistically significant regions (EDGER, P < 0.05, log2FC > 1.5). (F) Enhancer reporter gene assay of WT, ΔGATA1 BS1, and ΔGATA1 BS1&2 enhancer with the SLC4A1 promoter (mean ± SEM, n = 4). Statistical significance was determined using the Student's t-test, ** P < 0.01, *** P < 0.001. (G) Gene expression of WT, ΔGATA1 BS1, and ΔGATA1 BS1&2 HUDEP-1 clones in undifferentiated (maintained) and differentiated condition (mean ± SEM, n = 4, normalized to maintained WT). Statistical significance was determined using the Student's t-test; ** P < 0.01, *** P < 0.001. GPATCH8 is the reference gene located distal to SLC4A1 but not controlled by GATA1. Abbreviations: WT, wild-type; RPM, reads per million; VP, viewpoint
We next performed Capture-C assay in SLC4A1, TLCD4 and METTL7A loci to dissect the local chromatin interaction changes semi-quantitatively. Our results indicated that GATA1 knock-down reduced the contacts between the SLC4A1 promoter and the distal gained GATA1 enhancer region (Figure 5B), as well as the contacts between the TLCD4 intronic gained GATA1 occupancy enhancer region and the TLCD4 promoter (Figure 5C). Interactions between the distal gained GATA1 peak at the METTL7A locus also exhibited decreased signal with the nearby gene promoter regions for METTL7A and SLC11A2 (Figure 5D). Furthermore, we generated knock-out HUDEP-1 cell lines of GATA1 binding sites located at the distal enhancer of SLC4A1 to further validate the role of gained GATA1 binding sites on local chromatin rewiring. Knocking out both the single gained GATA1 binding site (ΔGATA1 BS1) and the double gained GATA1 binding site (ΔGATA1 BS1&2) reduced GATA1 occupancy as well as the contact signals between the enhancer region and the SLC4A1 promoter (Figure 5E, Supplementary Figure S5D). The E–P reporter gene assay showed that both single- and double-mutated gained GATA1 binding sites repressed the reporter gene activity (Figure 5F). Additionally, SLC4A1 gene expression in both maintained and differentiated HUDEP-1 stages was repressed by mutation of the GATA1 binding sites (Figure 5G). The ΔGATA1 BS1 cell line, with one GATA1 binding site disrupted without affecting the nearby KLF1 motif, exhibited significant repression of SLC4A1 gene expression, whereas the ΔGATA1 BS1&2 cell line, with two GATA1 and a nearby KLF1 binding sites interrupted, resulted in further SLC4A1 repression (Supplementary Figure S5D).
Here, we found that the gained GATA1 occupancy sites at non-promoter regions in the Ery-Pre stage furthered the enhancer contact with nearby promoters and increased the transcriptional activity in several erythroid genes. These results demonstrate that during the erythroid progenitor-precursor transition, certain erythroid genes require additional GATA1 binding in the distal region to guaranty exquisite gene regulation for stage-specific erythroid function during development.
GATA1 dosage controls erythroid progression and progenitor amplification
We found that most GATA1 occupancy sites remain stable throughout the erythroid progenitor-precursor transition, especially the sites at the TSS region, but that the dynamic GATA1 occupancy sites, primarily located at non-promoter regions, regulate the productive erythroid gene expression and local chromatin rewiring. Previous studies have shown that GATA1 is indispensable in erythropoiesis and megakaryopoiesis in both humans and mice (7–10). Moreover, insufficient GATA1 levels lead to erythroid maturation arrest but are still adequate for the proliferation of progenitors, even resulting in leukemogenesis (99,100). To elucidate the role of GATA1 in managing the erythroid progenitor-precursor transition and progenitor amplification in ex vivo human erythropoiesis, we performed a transient GATA1 inhibition assay using siRNA (Supplementary Figure S6A). Our results showed that delivery of GATA1 siRNA blocked GATA1 mRNA expression within 48 hours (Supplementary Figure S6B). Transient knock-down of GATA1 promoted an approximately 3-fold increase in erythroid cell production but with a similar colony composition as the control, which indicates that inhibition of GATA1 in Ery-Pro results in increased production of terminally differentiated erythroid cells without disturbing the lineage fate (Figure 6A–B, Supplementary Figure S6C–F). However, at early stages post-siRNA delivery (Day 6), GATA1 knock-down actually caused cell growth arrest instead of proliferation promotion (Figure 6C, Supplementary Figure S6F). Flow cytometry showed that GATA1 knock-down 48 h post-siRNA delivery in Ery-Pro cells predominantly blocked the expression of early erythroid markers CD71 and CD36 but retained the expression of progenitor marker CD38 (Figure 6D–E). Bulk RNA-seq at 24 h, 48 h, and 72 h post-siRNA delivery showed that transcription factors that are highly expressed in Ery-Pro cells (e.g. ETV6 and RUNX1) were up-regulated, but that terminal erythroid development (e.g. KLF1) and proliferation associated transcription factors (e.g. MYC) were down-regulated (Figure 6F, Supplementary Figure S6G–H, Supplementary Table S8). We next performed single cell RNA-seq in GATA1 knock-down Ery-Pro cells 48 h post-siRNA delivery. Dimensionality reduction and unsupervised clustering of the integrated GATA1 knock-down and control Ery-Pro datasets yielded five major clusters: EP1, EP2, Earlier EP1, Earlier EP2, and Myelo cells (Supplementary Figure S6I). GATA1 knock-down cells contributed to all five cell clusters, but the original Ery-Pro cells primarily contributed to clusters EP1 and EP2 (Supplementary Figure S6J); furthermore, GATA1 expression was lower in GATA1 knock-down cells (Supplementary Figure S6K). Erythroid lineage-associated genes TFRC, HBA1 and HBB were highly expressed in the EP1 and EP2 clusters only. Progenitor genes CEBPA, SPI1, RUNX1, and GATA2 were enriched in the Earlier EP1 and Earlier EP2 clusters (Supplementary Figure S6L). In analysis including only the GATA1 knock-down cells, dimensionality reduction and unsupervised clustering gave rise to four clusters: Ery1–4 (Figure 6G). Erythroid lineage-associated genes TFRC and HBB were highly expressed in the Ery3 and Ery4 clusters only. Progenitor genes CEBPA, ETV6, SPI1 and RUNX1 were enriched in the Ery1 cluster, and GATA2 was highly expressed in the Ery1 and Ery2 clusters (Figure 6H). Furthermore, the pseudotime value increased from Ery1 to Ery4 (Supplementary Figure S6M). These results indicate that transient abrogation of GATA1 in Ery-Pro promotes the output of erythroid cells by restricting progenitor differentiation and redirecting them to an earlier stage. Furthermore, these results indicate the presence of earlier erythroid progenitors. We next examined the top 10 marker genes of each cluster, which showed that the earlier Ery1 erythroid cells highly expressed hematopoietic stem cell or myeloid associated genes such as SRGN, PRG2, and LYZ, and that the Ery2 cells highly expressed TIMP1, ID2, CD52 and CCL2. Erythroid genes such as TFRC, KLF1 and HBB were enriched in the Ery3 and Ery4 clusters; additionally, Ery3 cells also expressed HIST1 genes, which indicates a distinct cell cycle between Ery3 and Ery4 (Supplementary Figure S6N, Supplementary Table S9). GO analysis showed that Ery1 is significantly enriched in ER stress-associated pathways and Ery2 is enriched with cytoskeleton remodeling and immunomodulating-associated pathways (Supplementary Figure S6O–P, Supplementary Table S10). Erythroid functional pathways such as gas transport were highly enriched in the Ery3 and Ery4 clusters (Supplementary Figure S6Q–R, Supplementary Table S10).
Figure 6.

GATA1 dosage controls erythroid progression and progenitor amplification. (A) Colony morphology of Ery-Pro cells under microscope after delivery of scramble (top, siCon) or anti-GATA1 siRNA (bottom, siGATA1). The colonies were analyzed on day 14. (B) Colony quantification of different types of colonies after delivery of siCon or siGATA1 in Ery-Pro cells plated at 200 cells/well. Data are shown as mean ± SEM (n = 3). The colonies were analysis at day 14. (C) Cell proliferation quantification using CellTiter-Blue after delivery of siCon or siGATA1 in Ery-Pro cells. Data are presented as mean ± SEM (n = 4). Statistical significance was determined using the Student's t-test; * P < 0.05, ** P < 0.01, *** P < 0.001. (D, E) Expression of CD235a and CD71 (D) and CD38 and CD36 (E) 48 h after delivery of siCon or siGATA1 in Ery-Pro cells (n = 2). (F) Differentially expressed transcription factors 24 h after delivery of siCon or siGATA1 in Ery-Pro cells with various siRNA concentrations (top bar). (G) The UMAP projection of 12 200 cells from Ery-Pro cells 48 h after delivery of siGATA1, resulting in four clusters shown with their respective labels (Ery1, Ery2, Ery3, Ery4). Each dot corresponds to a single cell, colored according to cluster. (H) Dot plot showing the expression level and percentage of erythroid differentiation relevant-genes across the four clusters from panel G. Abbreviations: Ery-Pro, erythroid progenitor; BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid; CFU-G/GM, colony-forming unit-granulocyte/granulocyte-macrophage; CFU-GEMM, colony-forming unit-granulocyte-erythroid-macrophage-megakaryocyte; EP, erythroid progenitors; Myelo, myeloid progenitors.
To explore the putative surface markers of earlier erythroid progenitors, we examined the cluster-specific cell surface antigens. The earlier erythroid progenitor, Ery1, exhibited highly expressed genes such as CD38, IL3RA and CXCR4, whereas Ery2 was enriched with CD52, SLC44A1 and KIT. The erythroid genes such as TFRC, CD36, GYPC and RHAG were enriched in the Ery3 and Ery4 clusters (Supplementary Figure S6S). These findings are consistent with previous studies showing that TFRC and CD36 begin to express in the middle of the erythroid progenitor stage. These results may facilitate the discovery of new markers for identifying and separating earlier erythroid progenitors and erythropoiesis screening (20,38).
Collectively, our findings suggest that transiently reducing the level of GATA1 during the erythroid progenitor stage can delay differentiation, extend the proliferation window of progenitor cells, and ultimately increase erythroid cell production.
DISCUSSION
Here, we performed genome-wide profiling of the chromatin connectome, enhancers, vital transcription factor occupancy, and transcriptome dynamics in defined human erythroid developmental stages: HSPC, Ery-Pro and Ery-Pre. By integrating these datasets, we provide a comprehensive analysis of stage-specific multidimensional chromatin architecture and address the functional association in human erythropoiesis, which not only fills the knowledge gap of 3D chromatin landscape in erythropoiesis, but also may lead to further mechanistic insights into hematopoiesis or other developmental processes.
During human erythropoiesis, we observed stable TAD and intra-TAD interactions, but highly dynamic compartment and E–P interactions, which correlated with erythroid gene activation. H3K27ac profiling revealed that 52% of the active chromatin regions in the HSPC stage, which contain a certain degree of erythroid related active regions, maintain their activity throughout erythroid progression, while only 12% of the genome regions became activated during development. Furthermore, only 3% of the H3K27ac regions were gained and 12% were lost during the transition from Ery-Pro to Ery-Pre. These findings are consistent with previous studies which indicate that the vast majority of H3K27ac regions are pre-established within lineage progression (29,101). Our analysis of E–P interactions suggests that GATA1 is involved in most E–P interactions, particularly during the Ery-Pre stage, and exhibits a gradually enriched pattern at enhancer anchor sites. Other hematopoietic transcription factors such as GATA2, RUNX1, and SPI1, participate in E–P interactions in cooperation with GATA1 by controlling the complicated gene regulation during the Ery-Pro stage.
We found that GATA1 occupancy was generally stable from the progenitor to precursor stage, while GATA1 HiChIP revealed a quite dynamic 3D GATA1 interactome. Surprisingly, over 80% of the dynamic GATA1 loops resulted from only 12.8% of the dynamic GATA1 occupancy events. More interestingly, over 90% of the dynamic GATA1 occupancy sites were located in non-TSS regions, whereas GATA1 occupancy at TSSs was relatively stable. The gain of GATA1 occupancy sites at non-promoter regions in the Ery-Pre stage rewired the enhancer contacts with nearby promoters, thereby promoting transcription. These findings indicate that GATA1 binding remains stable at the TSSs of erythroid genes, while additional GATA1 binding occurs at distal regions to ensure exquisite transcriptional regulation during the erythroid progenitor-precursor transition for certain GATA1 regulated genes. Additionally, we found that the transient abrogation of GATA1 in the Ery-Pro stage could delay erythroid progenitor differentiation, revert cells to an earlier cell state, and ultimately increase the production of erythroid cells.
However, although we have provided a substantial amount of data and detailed integrative analysis to support these notions, some key factors remain to be addressed: Firstly, KLF1 cooperated with GATA1 via adjacent motifs to regulate terminal erythropoiesis and acted as a pioneer factor to facilitate GATA1 occupancy on the chromatin. Although in the single ΔGATA1 BS1 SLC4A1 mutant we did not disrupt the KLF1 motif—which excluded the possibility that the KLF1 motif could affect SLC4A1 expression in this clone—the double ΔGATA1 BS1&2 SLC4A1 mutant did disrupt the KLF1 motif, which may have affected its gene expression (Supplementary Figure S5D) (14,102). Therefore, for the global gained GATA1 occupancy-regulated genes, we could not rule out the effect of KLF1 on their expression. Secondly, dynamic CTCF occupancy is also involved in the chromatin architecture and erythropoiesis, but we did not test whether they work together in the erythroid progenitor-precursor transition (61).
In summary, our study provides the first comprehensive analysis of chromatin architecture, especially E–P interactions and specific transcription factor occupancy, in clearly defined stages of human erythropoiesis. During this process, TAD and intra-TAD interactions remained relatively stable, whereas E–P interactions were highly variable and correlated with erythroid gene activation. This study sheds light on a previously uncharacterized function of GATA1 by showing that it promotes the productive expression of erythroid genes during the progenitor–precursor switch through gained occupancy at non-promoter sites involved in enhancer and local promoter contact rewiring. Meanwhile, the majority of GATA1 occupancy at promoter sites is stable. The findings provide insights into how the master erythroid regulator GATA1 gradually contributes to establishing stage-specific chromatin connectome and heightens our understanding of its three-dimensional roles in the developmental progression of erythroid cells. Our study also implies that temporary inhibition of GATA1 in the Ery-Pro stage hinders subsequent progenitor differentiation and reverts the cells to an earlier state, eventually enhancing the output of erythroid cells. Our profiling and integrated analysis provides a valuable resource for advanced understanding of 3D genome associated transcriptional regulation during erythroid cell differentiation and a comprehensive reference of erythroid gene regulation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Ryo Kurita and Yukio Nakamura (Cell Engineering Division, RIKEN BioResource Center, Tsukuba, Japan) for providing the HUDEP-1 cells. We thank the High-Performance Computing Platform of the Center for Life Sciences (Peking University) for supporting data analysis. We thank the Flow Cytometry Core of the National Center for Protein Sciences at Peking University (PKU). We are grateful to Dr Hongxia Lyu, Fei Wang, Yinghua Guo, Xuefang Zhang and Huan Yang for technical support. We thank Dr Zheng Wang at NIBS for the creative discussion and proofreading of the manuscript. We thank Drs Xiong Ji, Yongpeng Jiang and Rui Wang at PKU for assistance with the HiChIP method.
Authors’ contributions: D.L. and H.Y.L. conceptualized this project and designed the experiments. D.L., X.Y.Z., S.Z. and Q.H. performed the experiments. D.L. conducted the bioinformatic analyses. D.L. and H.Y.L. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Contributor Information
Dong Li, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China.
Xin-Ying Zhao, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China.
Shuo Zhou, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China.
Qi Hu, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China.
Fan Wu, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China.
Hsiang-Ying Lee, Ministry of Education Key Laboratory of Cell Proliferation and Differentiation, School of Life Sciences, Peking University, Beijing 100871, China; Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China; Peking University People's Hospital, Peking University Institute of Hematology, National Clinical Research Center for Hematologic Disease, Beijing 100871, China.
Data Availability
The Micro-C, CUT&RUN, HiChIP, Capture-C, RNA-seq and scRNA-seq data can be downloaded at GEO: GSE214811. Original code is not reported in this paper; however, all relevant data and custom scripts are available upon reasonable request.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2022YFA1103300]; National Natural Science Foundation of China [81970110]; Peking-Tsinghua Center for Life Sciences and School of Life Sciences, Peking University (to H.Y.L.). Funding for open access charge: Ministry of Science and Technology of the People's Republic of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Dzierzak E., Philipsen S.. Erythropoiesis: development and differentiation. Cold Spring Harb. Perspect. Med. 2013; 3:a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. 2014; 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sankaran V.G., Weiss M.J.. Anemia: progress in molecular mechanisms and therapies. Nat. Med. 2015; 21:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caulier A.L., Sankaran V.G.. Molecular and cellular mechanisms that regulate human erythropoiesis. Blood. 2022; 139:2450–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flygare J., Karlsson S.. Diamond-blackfan anemia: erythropoiesis lost in translation. Blood. 2007; 109:3152–3154. [DOI] [PubMed] [Google Scholar]
- 6. Tefferi A., Vannucchi A.M., Barbui T.. Polycythemia vera: historical oversights, diagnostic details, and therapeutic views. Leukemia. 2021; 35:3339–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsai S.F., Martin D.I., Zon L.I., D’Andrea A.D., Wong G.G., Orkin S.H.. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 1989; 339:446–451. [DOI] [PubMed] [Google Scholar]
- 8. Evans T., Felsenfeld G.. The erythroid-specific transcription factor Eryf1: a new finger protein. Cell. 1989; 58:877–885. [DOI] [PubMed] [Google Scholar]
- 9. Fujiwara Y., Browne C.P., Cunniff K., Goff S.C., Orkin S.H.. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Nat. Acad. Sci. U.S.A. 1996; 93:12355–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiss M.J., Orkin S.H.. Transcription factor GATA-1 permits survival and maturation of erythroid precursors by preventing apoptosis. Proc. Nat. Acad. Sci. U.S.A. 1995; 92:9623–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gnanapragasam M.N., McGrath K.E., Catherman S., Xue L., Palis J., Bieker J.J.. EKLF/KLF1-regulated cell cycle exit is essential for erythroblast enucleation. Blood. 2016; 128:1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gnanapragasam M.N., Bieker J.J.. Orchestration of late events in erythropoiesis by KLF1/EKLF. Curr. Opin. Hematol. 2017; 24:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tallack M.R., Perkins A.C.. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. IUBMB Life. 2010; 62:886–890. [DOI] [PubMed] [Google Scholar]
- 14. Tallack M.R., Whitington T., Yuen W.S., Wainwright E.N., Keys J.R., Gardiner B.B., Nourbakhsh E., Cloonan N., Grimmond S.M., Bailey T.L.et al.. A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 2010; 20:1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Im H., Grass J.A., Johnson K.D., Kim S.I., Boyer M.E., Imbalzano A.N., Bieker J.J., Bresnick E.H.. Chromatin domain activation via GATA-1 utilization of a small subset of dispersed GATA motifs within a broad chromosomal region. Proc. Nat. Acad. Sci. U.S.A. 2005; 102:17065–17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson K.D., Conn D.J., Shishkova E., Katsumura K.R., Liu P., Shen S., Ranheim E.A., Kraus S.G., Wang W., Calvo K.R.et al.. Constructing and deconstructing GATA2-regulated cell fate programs to establish developmental trajectories. J. Exp. Med. 2020; 217:e20191526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ludwig L.S., Lareau C.A., Bao E.L., Liu N., Utsugisawa T., Tseng A.M., Myers S.A., Verboon J.M., Ulirsch J.C., Luo W.et al.. Congenital anemia reveals distinct targeting mechanisms for master transcription factor GATA1. Blood. 2022; 139:2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barile M., Imaz-Rosshandler I., Inzani I., Ghazanfar S., Nichols J., Marioni J.C., Guibentif C., Gottgens B.. Coordinated changes in gene expression kinetics underlie both mouse and human erythroid maturation. Genome Biol. 2021; 22:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tusi B.K., Wolock S.L., Weinreb C., Hwang Y., Hidalgo D., Zilionis R., Waisman A., Huh J.R., Klein A.M., Socolovsky M.. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018; 555:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J., Hale J., Bhagia P., Xue F., Chen L., Jaffray J., Yan H., Lane J., Gallagher P.G., Mohandas N.et al.. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood. 2014; 124:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. An X., Schulz V.P., Li J., Wu K., Liu J., Xue F., Hu J., Mohandas N., Gallagher P.G.. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014; 123:3466–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gillespie M.A., Palii C.G., Sanchez-Taltavull D., Shannon P., Longabaugh W.J.R., Downes D.J., Sivaraman K., Espinoza H.M., Hughes J.R., Price N.D.et al.. Absolute quantification of transcription factors reveals principles of gene regulation in erythropoiesis. Mol. Cell. 2020; 78:960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palii C.G., Cheng Q., Gillespie M.A., Shannon P., Mazurczyk M., Napolitani G., Price N.D., Ranish J.A., Morrissey E., Higgs D.R.et al.. Single-cell proteomics reveal that quantitative changes in Co-expressed lineage-specific transcription factors determine cell fate. Cell Stem Cell. 2019; 24:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schulz V.P., Yan H., Lezon-Geyda K., An X., Hale J., Hillyer C.D., Mohandas N., Gallagher P.G.. A unique epigenomic landscape defines Human erythropoiesis. Cell Rep. 2019; 28:2996–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ludwig L.S., Lareau C.A., Bao E.L., Nandakumar S.K., Muus C., Ulirsch J.C., Chowdhary K., Buenrostro J.D., Mohandas N., An X.et al.. Transcriptional states and chromatin accessibility underlying Human erythropoiesis. Cell Rep. 2019; 27:3228–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shearstone J.R., Pop R., Bock C., Boyle P., Meissner A., Socolovsky M.. Global DNA demethylation during mouse erythropoiesis in vivo. Science. 2011; 334:799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murphy Z.C., Murphy K., Myers J., Getman M., Couch T., Schulz V.P., Lezon-Geyda K., Palumbo C., Yan H., Mohandas N.et al.. Regulation of RNA polymerase II activity is essential for terminal erythroid maturation. Blood. 2021; 138:1740–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong P., Hattangadi S.M., Cheng A.W., Frampton G.M., Young R.A., Lodish H.F.. Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood. 2011; 118:e128–e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang J., Liu X., Li D., Shao Z., Cao H., Zhang Y., Trompouki E., Bowman T.V., Zon L.I., Yuan G.C.et al.. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell. 2016; 36:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dekker J., Mirny L.. The 3D genome as moderator of chromosomal communication. Cell. 2016; 164:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu M., Ren B.. The Three-Dimensional Organization of Mammalian Genomes. Rev. Cell Dev. Biol. 2017; 33:265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oudelaar A.M., Beagrie R.A., Gosden M., de Ornellas S., Georgiades E., Kerry J., Hidalgo D., Carrelha J., Shivalingam A., El-Sagheer A.H.et al.. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nat. Commun. 2020; 11:2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hua P., Badat M., Hanssen L.L.P., Hentges L.D., Crump N., Downes D.J., Jeziorska D.M., Oudelaar A.M., Schwessinger R., Taylor S.et al.. Defining genome architecture at base-pair resolution. Nature. 2021; 595:125–129. [DOI] [PubMed] [Google Scholar]
- 34. Oudelaar A.M., Harrold C.L., Hanssen L.L.P., Telenius J.M., Higgs D.R., Hughes J.R.. A revised model for promoter competition based on multi-way chromatin interactions at the alpha-globin locus. Nat. Commun. 2019; 10:5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang P., Keller C.A., Giardine B., Grevet J.D., Davies J.O.J., Hughes J.R., Kurita R., Nakamura Y., Hardison R.C., Blobel G.A.. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 2017; 31:1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurita R., Suda N., Sudo K., Miharada K., Hiroyama T., Miyoshi H., Tani K., Nakamura Y.. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One. 2013; 8:e59890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao X., Lee H.Y., da Rocha E.L., Zhang C., Lu Y.F., Li D., Feng Y., Ezike J., Elmes R.R., Barrasa M.I.et al.. TGF-beta inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood. 2016; 128:2637–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yan H., Ali A., Blanc L., Narla A., Lane J.M., Gao E., Papoin J., Hale J., Hillyer C.D., Taylor N.et al.. Comprehensive phenotyping of erythropoiesis in human bone marrow: evaluation of normal and ineffective erythropoiesis. Am. J. Hematol. 2021; 96:1064–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iskander D., Psaila B., Gerrard G., Chaidos A., En Foong H., Harrington Y., Karnik L.C., Roberts I., de la Fuente J., Karadimitris A.. Elucidation of the EP defect in Diamond-Blackfan anemia by characterization and prospective isolation of human eps. Blood. 2015; 125:2553–2557. [DOI] [PubMed] [Google Scholar]
- 40. Li D., Wu F., Zhou S., Huang X.J., Lee H.Y.. Heterochromatin rewiring and domain disruption-mediated chromatin compaction during erythropoiesis. Nat. Struct. Mol. Biol. 2023; 30:463–474. [DOI] [PubMed] [Google Scholar]
- 41. Skene P.J., Henikoff J.G., Henikoff S.. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018; 13:1006–1019. [DOI] [PubMed] [Google Scholar]
- 42. Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R.et al.. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018; 173:430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Picelli S., Bjorklund A.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R.. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013; 10:1096–1098. [DOI] [PubMed] [Google Scholar]
- 44. Hansen A.S., Hsieh T.S., Cattoglio C., Pustova I., Saldana-Meyer R., Reinberg D., Darzacq X., Tjian R.. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Mol. Cell. 2019; 76:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davies J.O., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R.. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016; 13:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mumbach M.R., Rubin A.J., Flynn R.A., Dai C., Khavari P.A., Greenleaf W.J., Chang H.Y.. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods. 2016; 13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krietenstein N., Abraham S., Venev S.V., Abdennur N., Gibcus J., Hsieh T.S., Parsi K.M., Yang L., Maehr R., Mirny L.A.et al.. Ultrastructural details of mammalian chromosome architecture. Mol. Cell. 2020; 78:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang T., Zhang F., Yardimci G.G., Song F., Hardison R.C., Noble W.S., Yue F., Li Q.. HiCRep: assessing the reproducibility of hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017; 27:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wolff J., Rabbani L., Gilsbach R., Richard G., Manke T., Backofen R., Gruning B.A.. Galaxy HiCExplorer 3: a web server for reproducible hi-C, capture hi-C and single-cell hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2020; 48:W177–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng H., Xie W.. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019; 20:535–550. [DOI] [PubMed] [Google Scholar]
- 52. Ramirez F., Bhardwaj V., Arrigoni L., Lam K.C., Gruning B.A., Villaveces J., Habermann B., Akhtar A., Manke T.. High-resolution tads reveal DNA sequences underlying genome organization in flies. Nat. Commun. 2018; 9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roayaei Ardakany A., Gezer H.T., Lonardi S., Ay F.. Mustache: multi-scale detection of chromatin loops from hi-C and Micro-C maps using scale-space representation. Genome Biol. 2020; 21:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harmston N., Ing-Simmons E., Perry M., Baresic A., Lenhard B.. GenomicInteractions: an R/bioconductor package for manipulating and investigating chromatin interaction data. BMC Genomics [Electronic Resource]. 2015; 16:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hainer S.J., Boskovic A., McCannell K.N., Rando O.J., Fazzio T.G.. Profiling of pluripotency factors in single cells and early embryos. Cell. 2019; 177:1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meers M.P., Tenenbaum D., Henikoff S.. Peak calling by Sparse Enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin. 2019; 12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A.. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bhattacharyya S., Chandra V., Vijayanand P., Ay F.. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019; 10:4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qi Q., Cheng L., Tang X., He Y., Li Y., Yee T., Shrestha D., Feng R., Xu P., Zhou X.et al.. Dynamic CTCF binding directly mediates interactions among cis-regulatory elements essential for hematopoiesis. Blood. 2021; 137:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Montefiori L.E., Bendig S., Gu Z., Chen X., Polonen P., Ma X., Murison A., Zeng A., Garcia-Prat L., Dickerson K.et al.. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 2021; 11:2846–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Di Giammartino D.C., Kloetgen A., Polyzos A., Liu Y., Kim D., Murphy D., Abuhashem A., Cavaliere P., Aronson B., Shah V.et al.. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat. Cell Biol. 2019; 21:1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Buckle A., Gilbert N., Marenduzzo D., Brackley C.A.. capC-MAP: software for analysis of Capture-C data. Bioinformatics. 2019; 35:4773–4775. [DOI] [PubMed] [Google Scholar]
- 65. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wagner G.P., Kin K., Lynch V.J.. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012; 131:281–285. [DOI] [PubMed] [Google Scholar]
- 67. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hao Y., Hao S., Andersen-Nissen E., Mauck W.M. 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M.et al.. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qiu X., Mao Q., Tang Y., Wang L., Chawla R., Pliner H.A., Trapnell C.. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 2017; 14:979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mori Y., Chen J.Y., Pluvinage J.V., Seita J., Weissman I.L.. Prospective isolation of human erythroid lineage-committed progenitors. Proc. Nat. Acad. Sci. U.S.A. 2015; 112:9638–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ortabozkoyun H., Huang P.Y., Cho H., Narendra V., LeRoy G., Gonzalez-Buendia E., Skok J.A., Tsirigos A., Mazzoni E.O., Reinberg D. CRISPR and biochemical screens identify MAZ as a cofactor in CTCF-mediated insulation at Hox clusters. Nat. Genet. 2022; 54:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stadhouders R., Vidal E., Serra F., Di Stefano B., Le Dily F., Quilez J., Gomez A., Collombet S., Berenguer C., Cuartero Y.et al.. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 2018; 50:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jimenez G., Griffiths S.D., Ford A.M., Greaves M.F., Enver T.. Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc. Nat. Acad. Sci. U.S.A. 1992; 89:10618–10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ranzoni A.M., Tangherloni A., Berest I., Riva S.G., Myers B., Strzelecka P.M., Xu J., Panada E., Mohorianu I., Zaugg J.B.et al.. Integrative single-cell RNA-seq and ATAC-seq analysis of Human developmental hematopoiesis. Cell Stem Cell. 2021; 28:472–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hock H., Meade E., Medeiros S., Schindler J.W., Valk P.J., Fujiwara Y., Orkin S.H.. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004; 18:2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ye M., Zhang H., Amabile G., Yang H., Staber P.B., Zhang P., Levantini E., Alberich-Jorda M., Zhang J., Kawasaki A.et al.. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat. Cell Biol. 2013; 15:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hasemann M.S., Lauridsen F.K., Waage J., Jakobsen J.S., Frank A.K., Schuster M.B., Rapin N., Bagger F.O., Hoppe P.S., Schroeder T.et al.. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014; 10:e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hock H., Shimamura A.. ETV6 in hematopoiesis and leukemia predisposition. Semin. Hematol. 2017; 54:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Suzuki M., Kobayashi-Osaki M., Tsutsumi S., Pan X., Ohmori S., Takai J., Moriguchi T., Ohneda O., Ohneda K., Shimizu R.et al.. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells. 2013; 18:921–933. [DOI] [PubMed] [Google Scholar]
- 81. Wu W., Cheng Y., Keller C.A., Ernst J., Kumar S.A., Mishra T., Morrissey C., Dorman C.M., Chen K.B., Drautz D.et al.. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011; 21:1659–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Monteiro R., Pouget C., Patient R.. The gata1/pu.1 lineage fate paradigm varies between blood populations and is modulated by tif1gamma. EMBO J. 2011; 30:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cheng Y., Wu W., Kumar S.A., Yu D., Deng W., Tripic T., King D.C., Chen K.B., Zhang Y., Drautz D.et al.. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 2009; 19:2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fujiwara T., O’Geen H., Keles S., Blahnik K., Linnemann A.K., Kang Y.A., Choi K., Farnham P.J., Bresnick E.H.. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell. 2009; 36:667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Willcockson M.A., Taylor S.J., Ghosh S., Healton S.E., Wheat J.C., Wilson T.J., Steidl U., Skoultchi A.I.. Runx1 promotes murine erythroid progenitor proliferation and inhibits differentiation by preventing Pu.1 downregulation. Proc. Nat. Acad. Sci. U.S.A. 2019; 116:17841–17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang P., Zhang X., Iwama A., Yu C., Smith K.A., Mueller B.U., Narravula S., Torbett B.E., Orkin S.H., Tenen D.G.. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000; 96:2641–2648. [PubMed] [Google Scholar]
- 87. Nerlov C., Querfurth E., Kulessa H., Graf T.. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000; 95:2543–2551. [PubMed] [Google Scholar]
- 88. Yu M., Riva L., Xie H., Schindler Y., Moran T.B., Cheng Y., Yu D., Hardison R., Weiss M.J., Orkin S.H.et al.. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell. 2009; 36:682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Soler E., Andrieu-Soler C., de Boer E., Bryne J.C., Thongjuea S., Stadhouders R., Palstra R.J., Stevens M., Kockx C., van Ijcken W.et al.. The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev. 2010; 24:277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Deng W., Rupon J.W., Krivega I., Breda L., Motta I., Jahn K.S., Reik A., Gregory P.D., Rivella S., Dean A.et al.. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014; 158:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A., Blobel G.A.. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012; 149:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Weintraub A.S., Li C.H., Zamudio A.V., Sigova A.A., Hannett N.M., Day D.S., Abraham B.J., Cohen M.A., Nabet B., Buckley D.L.et al.. YY1 Is a structural regulator of enhancer–promoter loops. Cell. 2017; 171:1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang R., Chen F., Chen Q., Wan X., Shi M., Chen A.K., Ma Z., Li G., Wang M., Ying Y.et al.. MyoD is a 3D genome structure organizer for muscle cell identity. Nat. Commun. 2022; 13:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim S.I., Bultman S.J., Kiefer C.M., Dean A., Bresnick E.H.. BRG1 requirement for long-range interaction of a locus control region with a downstream promoter. Proc. Nat. Acad. Sci. U.S.A. 2009; 106:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Letting D.L., Chen Y.Y., Rakowski C., Reedy S., Blobel G.A.. Context-dependent regulation of GATA-1 by friend of GATA-1. Proc. Nat. Acad. Sci. U.S.A. 2004; 101:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M.et al.. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017; 2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Alvarez-Dominguez J.R., Knoll M., Gromatzky A.A., Lodish H.F.. The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell Rep. 2017; 19:2503–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gao T., Qian J.. EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic. Acids. Res. 2020; 48:D58–D64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shimizu R., Kuroha T., Ohneda O., Pan X., Ohneda K., Takahashi S., Philipsen S., Yamamoto M.. Leukemogenesis caused by incapacitated GATA-1 function. Mol. Cell. Biol. 2004; 24:10814–10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. McDevitt M.A., Shivdasani R.A., Fujiwara Y., Yang H., Orkin S.H.. A “knockdown" mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc. Natl Acad. Sci. U.S.A. 1997; 94:6781–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rubin A.J., Barajas B.C., Furlan-Magaril M., Lopez-Pajares V., Mumbach M.R., Howard I., Kim D.S., Boxer L.D., Cairns J., Spivakov M.et al.. Lineage-specific dynamic and pre-established enhancer–promoter contacts cooperate in terminal differentiation. Nat. Genet. 2017; 49:1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gillinder K.R., Magor G., Bell C., Ilsley M.D., Huang S., Perkins A.. KLF1 Acts As a pioneer transcription factor to open chromatin and facilitate recruitment of GATA1. Blood. 2018; 132:501–501.29739754 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Micro-C, CUT&RUN, HiChIP, Capture-C, RNA-seq and scRNA-seq data was uploaded on GEO: GSE214811.
The Micro-C, CUT&RUN, HiChIP, Capture-C, RNA-seq and scRNA-seq data can be downloaded at GEO: GSE214811. Original code is not reported in this paper; however, all relevant data and custom scripts are available upon reasonable request.