

Summary
The Maghreb is a key region for understanding the dynamics of cattle dispersal and admixture with local aurochs following their earliest domestication in the Fertile Crescent more than 10,000 years ago. Here, we present data on autosomal genomes and mitogenomes obtained for four archaeological specimens of Iron Age (∼2,800 cal BP–2,000 cal BP) domestic cattle from the Eastern Maghreb, i.e. Althiburos (El Kef, Tunisia). D-loop sequences were obtained for an additional eight cattle specimens from this site. Maternal lineages were assigned to the elusive R and ubiquitous African-T1 haplogroups found in two and ten Althiburos specimens, respectively. Our results can be explained by post-domestication hybridization of Althiburos cattle with local aurochs. However, we cannot rule out an independent domestication in North Africa considering the shared ancestry of Althiburos cattle with the pre-domestic Moroccan aurochs and present-day African taurine cattle.
Subject areas: Biological sciences, Omics, Genomics, Paleobiology, Paleogenetics
Graphical abstract
Highlights
-
•
The first genome-wide study of Iron Age cattle from the North-Eastern Maghreb
-
•
The oldest R-mitogenomes (∼2,800 cal BP–2,000 cal BP) of domestic cattle
-
•
Genomic affinity between Althiburos cattle and a pre-domestic Northwest African aurochs
-
•
Shared ancestry between Iron Age Althiburos cattle and the African taurine N’Dama breed
Biological sciences; Omics; Genomics; Paleobiology; Paleogenetics
Introduction
Cattle are one of the principal domesticated animals in Eurasia and Africa and, as attested by figurines, rock art, and craftwork produced across many cultures, have high symbolic value.1 The wild ancestor of domestic cattle, the aurochs (Bos primigenius), was widespread until it became extinct in AD 1627 in Poland most probably due to overhunting and habitat loss.1 The reconstruction of the shared history of cattle and humans, i.e. of the domestication and dispersion processes, is challenging. Archaeological and genetic data revealed that European taurine (Bos taurus) and humped zebu (Bos indicus) cattle were domesticated independently.2 The former derive from Bos primigenius primigenius and were domesticated in the Fertile Crescent, during the Neolithic ∼10,500 years BP (before present).2 Zebu domestication occurred in the Indus Valley from Bos primigenius nomadicus ∼8,000 years BP.3 The origins of the divergent North African taurine cattle are still debated, and it is not yet clear whether there was a third domestication event in this region. Recently, the analysis of SNP array data using approximate Bayesian computation modeling in worldwide cattle and one aurochs from England suggested there were only two domestication events.4 The distinct genetic variation of African taurine cattle could then result from hybridization with local aurochs (B. primigenius opisthonomous) following the dispersal of cattle domesticated in the Near East,4 but diachronic archaeogenomics data from cattle across North Africa are needed to test alternative demographic scenarios.
The analysis of DNA markers of autosomal, maternal, and paternal inheritance in comprehensive datasets of worldwide cattle disclosed overall patterns of genetic diversity and breed relationships. In cattle, mitochondrial variation is geographically structured. The almost fixation of the T1-matriline in Africa is consistent with a founder effect, associated with the early colonization of this region by Near Eastern cattle, and the subsequent expansion of haplotypes within this haplogroup.2,5,6 However, the possibility that some T1 sub-haplogroups originated locally, from the North African aurochs, could not be ruled out.7 Distinct R-matrilines were detected in a few native breeds from Italy and interpreted as evidence of gene flow from local aurochs.8 In addition, Y chromosome variation allows us to distinguish between taurine and zebu patrilines, i.e. the Y1 and Y2 from Y3 haplogroups, respectively.9,10,11 African cattle carry unique lineages with Y2 and Y3 patrilines almost equally represented.12,13,14 The complementary analysis of autosomal DNA depicts the well-differentiated European taurine, African taurine, and South Asian zebu ancestry clusters, and the patterns of admixture between these groups at the scale of individual genomes, breeds, and geographic regions.12,15,16,17,18,19 For example, native breeds from Italy, such as Chianina, Marchigiana, and Romagnola, have high levels (i.e., slightly over 10%) of indicine introgression, along with African taurine ancestry.20,21 These cattle showed genomic regions under positive selection associated with signatures of indicine introgression, which is compatible with increased fitness.20 Multiple routes of cattle migration originating in the Near East toward the Italian Peninsula support these patterns of adaptive indicine introgression.20 Likewise, African autosomal data depict an East to West decreasing gradient of Indicine cattle introgression associated with adaptive selection for warmer and drier conditions.22,23 The recent finding of some level of incompatibility between proteins encoded by genes of the zebu nuclear genome and of the taurine mitochondria, as well as evidence for positive selection of taurine alleles in admixed populations, could explain the scarcity of indicine maternal haplogroups in African cattle.24 The indicine contribution to African taurine cattle (N’Dama) is not observed in the sex chromosome X compared to the autosomes corroborating an extensive male-mediated gene flow.25 The effect of introgression in the autosomes is larger than in the sex chromosomes due to the low effective size of the latter. In addition, the Y chromosome effective size in some breeds can be extremely low due to artificial breeding, making it more prone to lose variants by drift.
Ancient DNA studies have been crucial to infer the origins and evolutionary trajectories of domestic animals.26 Heterochronous genetic data allow a direct investigation of changes within populations overtime, and of the patterns of admixture between wild and domestic animals.23,26 Genomic analyses of aurochs and early domestic cattle have confirmed mosaic origins and independent events of introgression of wild stock.27 Particularly, genomic data of a pre-domestic aurochs specimen collected in Morocco (∼9,000 years BP; Epipalaeolithic) and carrying the R-matriline suggested some level of proximity with early cattle from the southern Levant (∼6,300–2,800 BP; Neolithic to the Iron Age).27 However, the genetic composition of the prehistoric cattle that ranged across North Africa remains unknown. Here, we present genome-wide high-throughput sequence (HTS) data and mitogenomes for four archaeological specimens of domestic cattle from the Eastern Maghreb, namely from the archaeological site of Althiburos (El Kef, Tunisia), a permanent settlement of local Numidian agropastoralists.28,29 Additionally, we obtained PCR-based sequence data for a fragment of the mitochondrial hypervariable D-loop region for these samples and a further eight cattle specimens also collected in Althiburos. Five specimens were directly radiocarbon dated to ∼2,800–2,000 cal BP (calibrated radiocarbon years before present), i.e. 9th to 1st century cal BCE (BCE). Four of these yielded sufficient autosomal genome coverages (0.01X to 0.10X) for population genomic analyses. The data collected from Althiburos specimens were used to: (i) identify their maternal haplogroups; (ii) determine their biological sex; (iii) infer the proportions of European taurine, African taurine, and Indicine autosomal ancestry; and ultimately, (iv) investigate their genetic affinities with the distinct Moroccan aurochs, past Levantine cattle, and present-day breeds from North Africa.
Results and discussion
The Althiburos archaeological site is located in the Eastern Maghreb, Tunisia, in a fluvial valley of the El Kef province, close to the Algerian border and about 180 km from Carthage (Figure 1). Occupation of the site dates to the 9th century cal BCE and continued into Roman and Medieval times. The archaeobiological data collected thus far provided key information on the autochthonous sedentary people that inhabited this area, the settlement of Numidian states and their trade with the Phoenicians from Carthage.30,31 We processed 12 cattle specimens using established ancient DNA protocols, each corresponding to a single individual (Table 1). These materials comprised five metapodials, two tibiae, one first phalanx, and four molar teeth from the Iron Age—Numidian—phases of occupation (Table S1 and Figure S1). Four specimens yielded genome-wide sequence data and a further eight also yielded D-loop sequences from PCR assays (Table 1). Typical ancient DNA deamination patterns, i.e., an excess of cytosine to thymine misincorporation at 5′ ends, and complementary guanine to adenine misincorporations at 3′ ends, as well as DNA fragmentation (average read lengths ∼80 bp),32 allowed for HTS authentication asserting the ancient age of these samples (Figure S2). Five individuals were directly radiocarbon dated (Beta Analytics, UK), and gave dates spanning from ∼2,800 cal BP–2,000 cal BP (Table 1 and Figure S3). Six specimens were retrieved from stratigraphic levels that correspond to the early phases of the Numidian period, where traces of Phoenician pottery imports were found, whereas the other six specimens originate from the Middle and Late Numidian phases.30 These latter levels show evidence of increased trade with Carthage and other coastal Punic cities, and the presence of pottery imported from Italy.30 While the first contacts with the Phoenicians may have occurred at the end of the 8th century BCE, the substantial influence of Carthaginians in the region dates after the middle 5th century BCE.33 Hence, some of our specimens are representative of the cattle raised by indigenous—Numidian—populations in the El Kef area.
Figure 1.

The Althiburos archaeological site
Adapted from Sanmartí et al.30
(A) Map of the Mediterranean region and location of Althiburos in Tunisia (red dot).
(B) Photography of Althiburos showing excavation works in the capitol area.
(C) Plan of the Althiburos site showing excavation zones 1 and 2 around the capitol.
(D) Stratigraphy of excavation zone 2 depicting archaeological layers, their corresponding contexts, and cattle specimens: STK233, STK234, and ALT235 (black circles); ALT232 and STK236 (black squares); STK237 and STK239 (black triangles); STK230 and STK231 (black inverted triangles). For more details see Tables 1 and S1.
Table 1.
Althiburos samples information and summary statistics
| Sample name | Molecular sex | Endogenous DNA (%) | Whole Genome coverage (X) | Mitogenome coverage (X) | Mitogenome coverage >3X (%) | Mitochondrial haplogroup | Radiocarbon Date (Cal BP) | Phase/Zone |
|---|---|---|---|---|---|---|---|---|
| STK233 | n.a. | 0.02 | 0.000 | 0.05 | 0 | T1 | n.a. | MN/Z2 |
| STK234 | Female | 13.11 | 0.095 | 7.72 | 96.95 | T1 |
2,544–2,361 2,703–2,630 2,619–2,558 |
MN/Z2 |
| ALT235 | Female | 0.08 | 0.005 | 1.08 | 11.06 | R | 2,765–2,730 | MN/Z2 |
| ALT232 | Female | 0.37 | 0.009 | 8.57 | 96.83 | R |
2,735–2,485 2,475–2,470 |
EN3/Z2 |
| STK236 | Male | 0.91 | 0.007 | 1.21 | 13.53 | T1 | n.a. | EN3/Z2 |
| STK237 | n.a. | 0.32 | 0.002 | 0.56 | 1.51 | T1 | n.a. | EN2/Z2 |
| STK239 | Male | 16.72 | 0.104 | 31.98 | 99.92 | T1 | 2,877–2,759 | EN2/Z2 |
| STK230 | Male | 1.72 | 0.014 | 3.68 | 70.01 | T1 | n.a. | EN1/Z2 |
| STK231 | Male | 0.69 | 0.006 | 1.54 | 19.29 | T1 | n.a. | EN1/Z2 |
| STK241 | n.a. | 0.12 | 0.012 | 0.94 | 7.16 | T1 | n.a. | LN2/Z1 |
| STK243 | Male | 0.93 | 0.013 | 8.98 | 98.76 | T1 |
2,178–2,041 2,302–2,234 2,026–2,003 |
LN2/Z1 |
| STK240 | Male | 0.52 | 0.004 | 1.54 | 20.51 | T1 | n.a. | LN1/Z1 |
C14 radiocarbon dates – two sigma calibration; n.a. not applicable; EN-Early Numidian; MN-Middle Numidian; LN-Late Numidian; Z1-zone 1; Z2-zone 2. See also Table S1 and Figure S3.
The specimens used in subsequent population genomic analysis are highlighted in bold. Mitochondrial haplogroups shown in italic were determined based on 454-sequence data.
Analysis of cattle mitogenomes
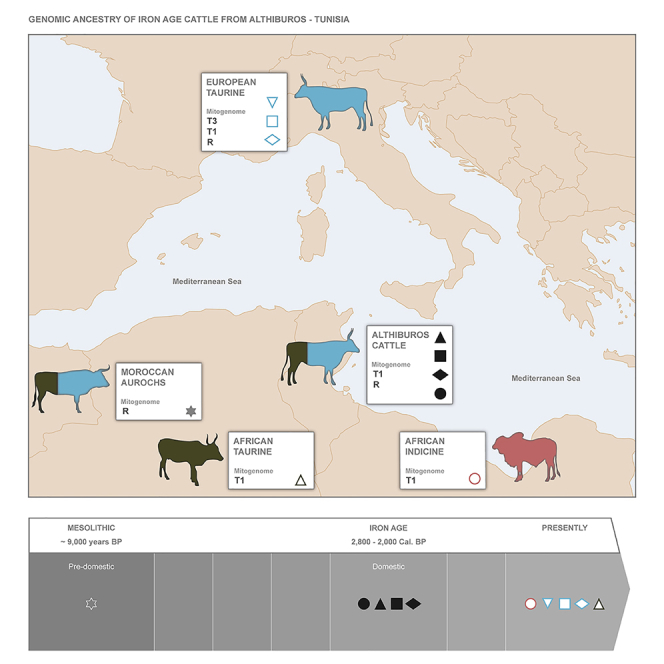
Only four of the 12 specimens for which HTS data were collected complied with the criteria defined for inclusion in a phylogenetic analysis, i.e., over 90% of the mitogenome covered at a minimum 3X, and their mitochondrial genome coverages were between 7.7X and 32X (Table 1). Three of these Althiburos cattle specimens (STK234, STK239, and STK243) belong to the major haplogroup T1, which is almost fixed in present-day African cattle but also frequent in Southern European, Middle Eastern, and Anatolian breeds (Figure 2).5,7,27 One Althiburos specimen (ALT232) carried the distinct R-lineage, which has been detected in a few individuals from four native Italian breeds,34 as well as the pre-domestic aurochs from Northwestern Africa (Morocco) for which whole-genome data were recently retrieved (Figure 2).27 Interestingly, the R-haplogroup was absent in a recent comprehensive genomic study of ancient cattle that included Near Eastern and Anatolian specimens, as well as remains from the Levant which were mostly of the T1-lineage.27 In addition, the haplogroup assignments from our analysis of 12 cattle specimens for a short D-loop sequence (222 bp) using a PCR-based 454-sequencing approach were validated by the HTS data also obtained from four of these specimens. This allowed us to identify the maternal haplogroups of an additional eight specimens collected in Althiburos, including one (ALT235) and seven (STK230, STK231, STK233, STK236, STK237, STK240, and STK241) individuals of the R and T1 lineages, respectively (Table 1 and Figure S4). Both haplogroups were present in the Early and Middle/Late periods of Numidian occupation. These Iron Age Althiburos specimens are the oldest R-mitogenomes described thus far in domesticated cattle and challenge a proposed Italian origin for this maternal lineage.34 Our results also provide support to an early concentration of T1-lineages in Eastern Maghreb cattle, and probably across North Africa and the Levant.
Figure 2.

Bayesian time-scaled phylogeny of cattle mitogenomes
Dates correspond to the node age posterior median and the solid light blue horizontal bars to their 95% highest posterior density interval (HPDI). Reference samples representative of present-day cattle are denoted by the haplogroup followed by GenBank accession numbers (black branches) and the notation of ancient samples corresponds to that of the original publication27 (gray branches, except aurochs that are in green). The four Althiburos specimens are in blue. Only samples with over 90% of the mitogenome covered at 3X were included. For more details see Table S3.
Genetic sex determination
We calculated the number of reads mapping to the Y chromosome as a fraction of the total number of alignments to both sex chromosomes35 to determine the sex of each of the specimens for which HTS data were retrieved (Table 1 and Figure S5; for three out of the 12 samples the poor quality of the data prevented this analysis). Female specimens ALT232, ALT235, and STK234 were assigned statistically with high confidence (Ry ≤ 0.011, 95% CI). In the remaining specimens the confidence interval did not overlap the assignment threshold for females and their Ry ≥ 0.0321 thus suggesting these are males. We also looked at the proportion of reads mapped to chromosomes X and 1 (which have similar sizes, 139.01 and 158.53 Mbp, respectively),36 and the values obtained were ∼1 and ∼0.5 for females and males, respectively, showing an agreement between the two approaches (Table S2). This corroborates previous results obtained for human specimens that show accurate sex determination even for low-coverage resequencing data,35 with male and female samples separated in two clear categories (Figure S5).
Population genomic analysis of autosomal DNA
A principal component analysis (PCA) of the four Althiburos specimens included in the phylogeny of cattle mitogenomes was done using 87,471 genome-wide autosomal SNPs overlapping HTS data representative of the following: African taurine cattle, i.e. the N’Dama breed;19 African zebu cattle, i.e. the Boran, Kenana and Ogaden breeds;19 commercial taurine cattle, i.e. the Holstein and Angus breeds (Bioproject IDs: PRJNA210521 and PRJNA318087, respectively); more diverse European taurine cattle, i.e. eight individuals each belonging to a primitive Iberian breed;25 and one pre-domestic Northwest African aurochs.27 This PCA revealed a clear separation of the African zebu cattle (PC1; representing 26.3% of the total genomic variation), and a close affinity of the four Althiburos samples and the Moroccan aurochs (Th7), plotting in an intermediate position between the European and African taurine cattle (PC2; representing 6.2% of the total genomic variation) (Figure 3A). Similarly, Verdugo et al.27 observed that Neolithic and Bronze Age cattle specimens from the southern Levant were closely related to the Epipalaeolithic Moroccan aurochs and African taurine cattle, which led the authors to conclude that wild cattle similar to the North African aurochs left their signatures in the genomes of early domesticated cattle from the Levant.
Figure 3.

Population structure analysis of four Althiburos individuals based on autosomal HTS data
(A) Principal component analysis performed on the Althiburos samples which were genetically close to the pre-domestic Northwest African aurochs (gray star).27 Present-day breeds representative of European taurine (blue), African taurine (green), and indicine (pink) cattle were well-differentiated.19,25
(B) Model-based clustering analysis of NGSadmix for K = 3. An European and African taurine ancestry were observed in the Althiburos cattle specimens (from left to right: ALT232, STK243; STK239; STK234) and the pre-domestic Moroccan aurochs (represented by the gray star),27 without zebu influence (except for STK243, from the Late Numidian period, that showed a residual level of African zebu admixture). For more details on the HTS data see Table S4 and Figure S6 for K = 2 to K = 4 clustering results.
The genetic structure inferred by NGSadmix for K = 2 to K = 4 including the same samples is consistent with the PCA (Figures 3B and S6). Althiburos domestic cattle share autosomal genome ancestry with the Moroccan aurochs and, contrasting with what was observed by Verdugo et al.27 of ubiquitous mitochondrial T1-lineages for Levantine specimens, two of these Eastern Maghreb cattle carried the distinctive R-matriline. Moreover, the affinity between Iron Age Althiburos cattle and present-day taurine counterparts is well-depicted by the NGSadmix analysis, including a shared ancestry with the N’Dama breed representing the Northwest African taurine component. The genetic differentiation of the N’Dama probably results from their continued management as a breed, as well as from the effect of genetic drift. Results from D-statistics analyses confirmed a significant excess of shared derived alleles between our Althiburos samples and the African taurine N’Dama, as well as the pre-domestic Moroccan aurochs, when compared to European taurine cattle (Figure S7). Interestingly, a single specimen from the Late Numidian period showed some African zebu admixture (STK243), despite the negligible level of indicine ancestry observed in Althiburos cattle (Figure 3B). This may reflect an intensification of trading between local Numidian herders and other Mediterranean populations in the more recent phases of occupation. Such change in trading practices is supported by archaeological evidence that during the Late Numidian period (subphase 2) up to 36% of the fragments and 33% of the minimum number of vessels were imported from Carthage or other coastal Punic cities.30 Another relevant feature of this subphase is the increased presence of Italic pottery consistent with trade between these regions.30 Recent findings provide support for multiple migration events of cattle into the Italian peninsula associated with evidence for adaptive introgression of indicine-derived ancestry in native breeds.20 A high level of zebu admixture has been observed in cattle across the Mediterranean region.21,37 Thus, it is possible that local cattle raised by Numidian herders could have been sourced in the context of trade and reached the northern Mediterranean coast, then leaving a signature in the genomes of Southern European breeds (e.g. from Italy). Indeed, the finding of a male specimen in Althiburos showing some indicine ancestry is consistent with the early dispersal of paternal lineages of this type, but unfortunately, we lack sufficient coverage to be able to determine the Y-haplogroup for this sample. Additional studies of paternal variation in past and present-day cattle from these regions are needed to depict male gene flow.
Our results corroborate the introgression of aurochs females into the domestic stock of cattle from Althiburos. Present-day European taurine cattle, including Italian breeds, are more closely related to the British (Mesolithic) and Armenian (Neolithic) aurochs than to the well-differentiated pre-domestic Moroccan aurochs (Verdugo et al.27). In contrast, early cattle from the Levant, i.e. from the Neolithic to the Iron Age, already showed significant autosomal allele sharing with the Moroccan aurochs and were differentiated from local Neolithic aurochs.27 Considering the R-matriline is scarce in the archaeological contexts for which genetic data have been retrieved, then the probability that the only African aurochs specimen belongs to this lineage should be extremely low. In addition, bearing in mind that (i) domesticated Iron Age cattle from Althiburos belong to the R and T1 haplogroups, (ii) their autosomal genomes show close affinity with that of the Northwest African aurochs, (iii) and the previously reported absence of R-lineages overtime in the Levant, it is plausible that African taurine cattle may have a local origin in the Maghreb.
Limitations of the study
This study renews the debate on whether there was a third domestication event of taurine cattle in North Africa or if hybridization with local aurochs could explain the patterns of genetic variation observed for cattle from this region. A merit of our study is to provide a first glimpse on the genomic variation of Iron Age cattle from the North-Eastern Maghreb. The inferences we can make are necessarily limited by the nature of ancient DNA, in particular the endogenous DNA content of the Althiburos cattle remains was very little and the average depths of sequencing coverage were extremely low. Thus, we could not use more robust methods to quantify the affinity between the pre-domestic Moroccan aurochs, our Iron Age cattle from Althiburos and present-day taurine cattle from North Africa. Diachronic archaeogenomics data from aurochs and cattle across North Africa and Southern Europe are still lacking. Such information is needed to employ modeling approaches to reconstruct the demographic processes underlying cattle differentiation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| STK232 | Institut National de Patrimoine (INP) | Not Applicable |
| STK235 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 230 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 231 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 233 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 234 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 236 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 237 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 239 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 240 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 241 | Institut National de Patrimoine (INP) | Not Applicable |
| STK 243 | Institut National de Patrimoine (INP) | Not Applicable |
| Chemicals, peptides, and recombinant proteins | ||
| Proteinase K | VWR Sweden | Cat#1.24568.0100 |
| EDTA buffer solution pH 8.0 (0.5 mol/l) for biotechnology, sterile | VWR Sweden | Car#E522-100ML |
| Sodium acetate | Sigma Aldrich Sweden | Cat#S2889 |
| Guanidine hydrochloride | Sigma Aldrich Sweden | Cat#50933 |
| Isopropanol | Sigma Aldrich Sweden | Cat#67-63-0 |
| Tween-20 | Sigma Aldrich Sweden | Cat#9005-64-5 |
| Sodium phosphate PH 6.0 | VWR Sweden | Cat#101447-426 |
| ATP | Fermentas/Thermo Scientific | Cat#R0441 |
| T4 Polynucleotide Kinase | Thermo Scientific | Cat#EK0032 |
| T4 DNA Polymerase | Fermentas/Thermo Scientific | Cat#EP0062 |
| Bst polymerase (supplied with 10X ThermoPol reaction buffer) | NEB/BioNordika | Cat#M0275S |
| AmplitTaq Gold | Invitrogen/life technologies | Cat#4311816 |
| ATP | Fermentas/Thermo Scientific | Cat#R0441 |
| 10X Tango Buffer | Fermentas/Thermo Scientific | Cat#BY5 |
| High Pure Viral Nucleic Acid Large Volume Kit | Roche | Cat#5114403001 |
| T4 DNA Ligase | Fermentas/Thermo Scientific | Cat#EL0011 |
| Min Elute PCR Purification Kit | QIAGEN | Cat#28006 |
| PEG-4000 | Sigma | Cat#1546569 |
| Agencourt AMPure XP beads (60ml) | Beckman Coulter | Cat#A63881 |
| dntp's | Thermo Scientific | Cat#R1121 |
| Critical commercial assays | ||
| High Sensitivity DNA (chips + reagents) (Bioanalyzer 2100) | Agilent Technologies | Cat#5067-4626 |
| SYBR Green qPCR master mix (2X) | Life Technologies | Cat#K0221 |
| Deposited data | ||
| European Nucleotide Archive | National Center for Biotechnology Information (nih.gov) | PRJNA803479 |
| 454-sequence data | DRYAD | https://doi.org/10.5061/dryad.v9s4mw71n |
| Oligonucleotides | ||
| IS1_adapter.P5: 5'-A∗C∗A∗C∗TCTTTCC CTACACGACGCTCTTCCG∗A∗T∗C∗T-3' (∗ indicates a PTO bond) |
Meyer & Kircher (2010)38 | Biomers |
| IS2_adapter.P7: 5'-G∗T∗G∗A∗CTGGAGT TCAGACGTGTGCTCTTCCG∗A∗T∗C∗T-3' (∗ indicates a PTO bond) |
Meyer & Kircher (2010)38 | Biomers |
| IS3_adapter.P5+P7: 5'-A∗G∗A∗T∗CGG AA∗G∗A∗G∗C-3'(∗ indicates a PTO bond) |
Meyer & Kircher (2010)38 | Biomers |
| IS4: (5'-AATGATACGGCGACCACCGAGATC TACACTCTTTCCCTACACGACGCTCTT 3' ) |
Meyer & Kircher (2010)38 | Biomers |
| P7 indexing: (50-CAAGCAGAAGACGGCA TACGAGATxxxxxxxGTGACTGGAGTTC AGACGTGT 30 ) where x is one of 228 different 7 bp indexes provided in McHugo et al.26 |
Meyer & Kircher (2010)38 | Biomers |
| IS7 qPCR primer: 5' - ACACTCTTTC CCTACACGAC - 3' |
Meyer & Kircher (2010)38 | Biomers |
| IS8 Qpcr PRIMER: 5' - GTGACTGGAGTTC AGACGTGT - 3' |
Meyer & Kircher (2010)38 | Biomers |
| Software and algorithms | ||
| AdapterRemoval version 2 | Schubert, Lindgreen & Orlando (2016)39 | https://github.com/MikkelSchubert/adapterremoval |
| Burrows-Wheeler Aligner BWA aln version 0.7.17 | Li & Durbin (2009)40 | https://github.com/lh3/bwa |
| Samtools version 1.8 | Li (2011)41 | http://www.htslib.org/ |
| bcftools from the SAMtools package | Kallala et al.29 | http://www.htslib.org/ |
| Picard toolkit version 2.18.5 | Broad Institute, GitHub Repository | https://broadinstitute.github.io/picard |
| GATK version 3.7 | McKenna et al. (2010)42 | https://software.broadinstitute.org/gatk |
| MapDamage version 2.0 | Jónsson et al. (2013)43 | https://github.com/ginolhac/mapDamage |
| Qualimap | Okonechnikov, Conesa & García-Alcalde (2016)44 | http://qualimap.bioinfo.cipf.es/ |
| Geneious prime version 2019.0.4 | Biomatters, Inc. | https://www.geneious.com/ |
| ANGSD version 0.931 | Korneliussen, Albrechtsen & Nielsen (2014)45 | http://www.popgen.dk/angsd |
| Muscle version 3.8.425 | Edgar (2004)46 | http://www.drive5.com/muscle/ |
| BEAST version 1.10.4 | Suchard et al. (2018)47 | https://beast.community/ |
| ModelTest-NG version 0.1.7 | Darriba et al. (2020)48 | https://github.com/ddarriba/modeltest/releases |
| Tracer version 1.7.1 | Rambaut et al. (2018)49 | https://beast.community/tracer |
| FigTree version 1.4.4 | Institute of Evolutionary Biology, University of Edinburgh, UK | http://tree.bio.ed.ac.uk/software/figtree/ |
| PCAngsd version 0.1 | Meisner & Albrechtsen (2018)50 | http://www.popgen.dk/software/index.php/PCAngsd |
| NGSadmix version 32 | Skotte, Korneliussen & Albrechtsen (2013)51 | http://www.popgen.dk/software/index.php/NgsAdmix |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Catarina Ginja (catarinaginja@cibio.up.pt).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Archaeological information and context
The sampling of the archaeological materials involved archaeologists, zooarchaeologists and local authorities in the framework of a collaborative project established between the Institut National de Patrimoine (INP) - Tunisia, the Catalan Institute for Classical Archaeology (ICAC) and the University of Barcelona (UB) - Spain. This research involved the sub-sampling of zooarchaeological specimens for DNA extraction and subsequent genomic analyses, as well as radiocarbon dating to confirm their historic context. Through collaborations with archaeologists and zooarchaeologists, we used well-documented specimens, and sub-sampling was carried out based on agreements and partnership protocols for multidisciplinary research in which the results are of interest to all the institutions involved (INP, ICAC and UB). Sub-sampling for ancient DNA analyses was carried out at the Centre for Palaeogenetics (CPG), Stockholm University, Sweden, following international standards and routine procedures optimized to minimisethe damage in the bone/tooth specimens, despite abundance of livestock materials in most archaeological contexts of human occupation. There was no risk to the individuals taking part in this research, and we do not think this project raises potential ethical issues.
Method details
DNA extraction
Analyses of ancient DNA were conducted in the dedicated ancient DNA (aDNA) facilities at the Centre for Palaeogenetics (CPG), Stockholm University, Sweden. The bone/tooth specimens were submitted to UV sterilisation and removal of ∼1 mm from the outermost surfaces with a clean razor blade to eliminate potential surface contaminants. We obtained approximately 90 mg of bone powder from each specimen using a multitool drill (DremelTM) at low rpm to avoid local heating. One negative control was included in every batch of 6 samples. The bone/tooth powder was incubated overnight in 1 mL of digestion buffer (0.45 M EDTA pH 8.0 and 0.25 mg/ml of proteinase K) at 38°C under constant rotation. DNA was extracted and purified using a silica-binding method optimised for short DNA fragments.52,53 After incubation, the samples were centrifuged and 1 ml of supernatant was added to a 13 ml of binding buffer (5 M guanidine hydrochloride, 40% v/v isopropanol, 0.05% Tween-20 and 90 mM sodium acetate pH 5.2) in a 50 ml Falcon tube. This solution containing the binding buffer and the extraction supernatant was transferred into a 50 ml silica column (Roche, High Pure Viral Nucleic Acid Large Volume Kit). Following centrifugation for 6 min at 4,000 rpm, the Roche falcon tube was removed and the silica column containing each sample was placed into a new 2 mL collection tube. The silica column tube was then centrifuged for 1 min at 6,000 rpm, and the DNA purified by adding 750 μl PE buffer (QIAGEN) to the silica column followed by centrifugation for 1 min at 13,200 rpm. The flow-through was discarded and this washing step was repeated. The flow-through was discarded and the silica column was dried by centrifugation for 1 min at maximum speed (16,000 rpm) and placed in a new 1.5 mL collection tube. We added 22μl of TET buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.05% Tween-20) to the centre of the silica membrane for elution and, after 5 min incubation at 37°C, the DNA was collected by centrifugation for 1 min at maximum speed (16,000 rpm). This step was repeated to obtain a final volume of 44μl of DNA extract (stored at -20°C).
Preparation of genomic libraries
Double stranded genomic libraries were prepared from 20 μl of DNA extract using the blunt end ligation protocol described by Meyer and Kircher38 with modifications as in Günther et al.54 Due to ancient DNA damage the initial fragmentation step was not done. Indexing PCRs were set up in a total volume of 25 μl with a final concentration of 1x Gold Buffer (Invitrogen/life technologies), 2.5 mM Magnesium Chloride (Invitrogen/life technologies), 250 μM dNTP (each), 3 μl of DNA library, 0.2 μM IS4 PCR primer (5’- AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGTACACTCTTTCCCTACACGACGCTCTT - 3’) and 0.2 μM indexing primer (5’- CAAGCAGAAGACGGCATACGAGATxxxxxxxGTGACTGGAGTTCAGACGTGT - 3’) (where x is one of the 228 different 7 bp indexes provided by Korlević55 and 0.1 U/μl of AmpliTaq Gold (Invitrogen/ life technologies). Cycling conditions were the following: 12 min activation step at 94°C; 8-15 cycles of 30 s at 94°C, 30 s at 60°C, 45 s at 72°C; a final extension step of 10 min at 72°C. The optimal number of PCR cycles for amplification of the genomic libraries was determined using qPCR and varied between 7 and 13.38 Each library was amplified in four replicates along with two PCR blanks using an index in the P7 primer. PCR products from the same library were pooled and purified with Agencourt AMPure XP beads (Beckman Coulter). The concentration and size profiles of the purified libraries were identified on a Bioanalyzer 2100 using the High Sensitivity DNA chip (Agilent). Purified libraries were pooled in equimolar concentration and sequenced (2 x 100 bp) on an Illumina HiSeq X instrument at the National Genomics Infrastructure (NGI, SciLifeLab) in Stockholm, Sweden. The DNA was not subjected to USER treatment prior to library preparation.
Quantification and statistical analysis
Sequence pre-processing and mapping
Adapter sequences were trimmed from our Illumina shotgun resequencing data and overlapping paired-end reads were merged requiring an overlap of 11 bp using the AdapterRemoval software version 2.39 Reads shorter than 30 bp length and with quality score below 20 were discarded. The merged reads were then mapped as single-end reads using BWA aln version 0.7.1740 to: the Bos taurus reference genome version UMD 3.1.1 (bosTau8; GenBank accession number: GCA_000003055.5); Btau_4.6.1 (bosTau7; GenBank accession number: GCA_000003205.4), which contains an assembled Y-chromosome; and the reference mitogenome (GenBank accession number: NC_006853.1). The mapping was done with modified settings for deactivated seeding (-l 1000), more substitutions (-n 0.01) and up to two gaps (-o 2). Only the reads with mapping quality higher than 25 were selected for the downstream process using SAM tools version 1.8.56 For each sample, the BAM files of independent resequencing runs were merged and PCR duplicates were removed with the Picard tools software version 2.18.5 (MarkDuplicates REMOVE_DUPLICATES=true option) and the generated reads were realigned with GATK version 3.7.42 We used Qualimap version 2.2.144 to generate summary reports from the BAM files obtained for each sample, to assess the quality of the samples based on their endogenous DNA content, i.e., the percentage of reads mapping to the cattle genome, and calculate the mean depths of coverage.
Damage patterns
We used the MapDamage software version 2.043 to verify the authenticity of the ancient DNA resequencing data. All genomic libraries presented post-mortem damage patterns, as expected due to the biological processes following cell death and which are characteristic of ancient DNA sequences, i.e., typical C-to-T and G-to-A deaminations at the 5’- and 3’-termini, and high levels of fragmentation. We then apply the --rescale option to the filtered bam files to downscale the quality scores at positions likely affected by deamination according to their initial quality values, position in reads and damage patterns. The new rescaled bam files generated were used for downstream analyses.
Phylogenetic bayesian analysis of mitogenomes
The phylogenetic relationships among our Althiburos cattle specimens and other published archaeological and extant samples (N=94, see Table S3) were studied using complete mitogenomes together with the Bayesian inference implemented in BEAST version 1.10.4.47 Consensus sequences were obtained using the software ANGSD version 0.931,45 considering only the reads with base quality above 20, mapping quality above 30 and with more than 3X coverage to call confident bases. The ambiguous low-coverage positions were defined as undetermined (N), using −doFasta 2. Consensus sequences were also visually inspected using Geneious Prime version 2019.0.4 (https://www.geneious.com). The consensus sequences were aligned with mitogenomes available from public repositories, representative of each maternal haplogroup7,8,57 and including extant Iberian breeds,25 using MUSCLE version 3.8.425.46 The analysis was restricted to sequences with at least 90% of the mitogenome covered at 3X, with a total length of 16,348 bp. We assessed the best-fit model of nucleotide evolution on ModelTest-NG version 0.1.748 and selected the HKY+I+G458 substitution model under the Akaike Information Criteria. In order to account for cattle population size changes through time59 and serial sampling, a GMRF Bayesian skyride model was assumed60,61 to describe the coalescent process over the phylogenetic tree. Mean calibrated radiocarbon ages (see Table S3) were used as priors for all ancient samples. Bovid mitochondrial mutation rate was taken from a broad exponential distribution with mean 1.0E-7 per bp per year and a strict molecular clock was assumed. The overall mean mutation rate was estimated to be 5.45E-8 (95% HPD interval 4.34E-8 - 6.58E-8), which agrees with previous studies.8,62 Three independent Markov Chain Monte Carlo (MCMC) chains, each with 20 million iterations, sampled at every 1,000 iterations were run. The results of each MCMC chain were analysed using Tracer version 1.7.149 to check for their stability, convergence and if for all parameters sufficient sampling was reached by examining the effective sample size values (i.e. equal or higher than 200) . The trees of the individual runs were combined using Logcombiner version 1.10.4 (10% burn-in) and summarized using TreeAnnotator version 1.10.4 (both packages within BEAST47). The resulting Maximum Clade Credibility tree, where median heights were used as node heights (or ages), was then visualized in FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
PCR-based 454-sequence data
The sub-sampling was done as above. Following, we used the Yang et al.52 guanidinium/silica-based method for DNA extraction with modifications as in Svensson et al.63 In brief, after overnight proteinase K digestion (EDTA 0.5 M pH 8.0, Urea 1 M, 0.20 mg/ml proteinase K), the lysates were concentrated to 100 μl (Millipore-Amicon→ Ultra-4 30k Da, Merck KGaA, Darmstadt, Germany) and commercial MinElute spin columns (Qiagen Nordic, Sweden) used for ancient DNA recovery following the manufacturer’s protocol. Two negative extraction controls were included in every batch of 6 samples. We analysed two overlapping mitochondrial D-loop fragments of 157 bp and 139 bp with the oligonucleotide primer pairs AN2_F: 5’ - GCCCCATGCATATAAGCAAG - 3’/AN1_R: 5’ - CACGCGGCATGGTAATTAAG -3’64 and KO1: 5’ - ACCATTAGATCACGAGCTTAA - 3’/KO2: 5’ - GAAGAAAGAACCAGATGCC - 3’,65 respectively. These primers are specific for cattle and allow us to recover the majority of the haplotypic variability comprised within the maternal haplogroups observed in worldwide breeds. They are also useful to infer matrilines in ancient samples of domestic cattle and aurochs. Amplification reactions contained 2 μL of genomic DNA, 1x Taq polymerase buffer (Naxo Ltd, Tartu, Estonia), 2.5 mM of MgCl2 (Applied Biosystems), 0.20 mM of each deoxynucleoside triphosphate (dNTP) (Applied Biosystems), 0.4 μM of each PCR primer, 2.5 U of Hot start Taq DNA polymerase (Naxo Ltd, Tartu, Estonia) and PCR water (Qiagen) in a final volume of 25 μL. Amplification conditions were as follows: an initial denaturation/activation step of 95°C for 15 min; 60 cycles of denaturation at 94°C (15 s), annealing at 55°C (30 s for AN2_F/AN1_R) or 57°C for (30 s for KO1/KO2), and extension at 72°C (30 s); a final extension step at 72°C for 10 min. Negative controls were used and all extraction blanks were subject to PCR amplification. Amplifications were verified by the electrophoresis of 5 μL of each PCR product in 2% ethidium bromide agarose gels in Tris/Borate/EDTA (TBE) (0.5x) buffer. Electrophoresis ran at 10V/cm for 30 min. Samples that yielded PCR products of the expected sizes were sequenced following purification with the QIAquick Gel Extraction Kit (Qiagen) according to the manufacturer's recommendations. Tagged primers66 were used for multiplexing in emulsion PCR and individual identification subsequently to 454-sequencing as detailed in Malmström et al.67 We used the Quant-iTTM dsDNA High-Sensitivity Assay Kit (Invitrogen) to quantify the purified PCR products, which were then pooled in equimolar mixtures and sequenced on the Genome Sequencer FLX System (Roche). The GALAXY platform (http://galaxy.psu.edu/), a web-based genome analysis package,68 was used to obtain sequence files for each sample. Sequence reads were identified through unique combinations of primers and tags for each sample and aligned against reference NCBI sequences. Stringent criteria for ancient DNA analysis and sequence authentication were followed.68,69 For multiple alignments, the Geneious Prime version 2019.0.4 (https://www.geneious.com) software was used, and consensus sequences were generated for each individual from independent amplifications. A neighbor-joining phylogeny of genetic distances was obtained using the same software with the HKY model of sequence evolution58 for determination of the maternal haplogroups of the Althiburos specimens.
Sex determination
For sex assignment of the Althiburos specimens we used the resequencing data mapped to the Bos taurus reference genome version Btau_4.6.1. (bosTau7; GenBank accession number: GCA_000003205.4) to obtain Y chromosome reads. We then used the method described by Skoglund and colleagues,35 which is adequate for low-coverage ancient DNA resequencing data, to determine the biological sex of the Althiburos specimens. The number of alignments to the Y-chromosome (nY) was calculated as a fraction of the total number of alignments to both sex chromosomes (nX+nY), where RY=nY/(nX+nY), with 95% confidence intervals. The proportion of reads mapped to the X-chromosome and chromosome 1 (which have similar sizes) was also estimated.
Population structure
Population structure was assessed using whole-genome data obtained for four Althiburos specimens and 60 samples from public repositories, as follows: 10 Holstein (Bioproject ID: PRJNA210521); 3 Angus (PRJNA318087); 10 N’Dama, 9 Kenana, 9 Ogaden and 10 Boran (PRJNA312138); 1 Brava de Lide (fighting cattle), 1 Barrosã, 1 Maronesa, 1 Arouquesa, 1 Mertolenga, 1 Preta, 1 Alentejana and 1 Mirandesa (PRJNA526824); and 1 pre-domestic Northwest African aurochs (PRJEB31621). For more details on these data see Table S4. These analyses were carried out using methods that take the statistical uncertainty associated with genotype calling in low coverage sequencing data into account. SNPs were called with ANGSD version 0.93145 using the filtered BAM files with genotype likelihoods estimated with the samtools model,41 considering: only bases with quality above 20, reads with mapping quality above 30, autosomal sites, a minimum minor allele frequency of 0.05, a maximum of 10% missing data (58 out of a total of 64 individuals) and a p-value <1e-6 (angsd -bam bam.list -doGlf 2 -GL 1 -minQ 20 -minMapQ 30 -rf autosomes.list -doMajorMinor 1 -doMaf 2 -SNP_pval 1e-6 -doCounts 1 -minInd 58 -minMaf 0.05). We selected variants that were present in at least one of the ancient samples to avoid bias from having missing data mainly in the Althiburos specimens. The resulting beagle file with a total of 87,471 SNPs was used to calculate a covariance matrix in PCAngsd version 0.1.50 This matrix was then loaded into R 3.4.4 (https://www.R-project.org) where the function eigen was used to perform a principal component analysis (PCA). The same beagle file was used in NGSadmix software version 3251 to define the ancestry clusters and admixture proportions. NGSadmix was run with K ranging from 2 to 4 ancestral populations, which was ideal for determining the patterns of Indicine, African and European taurine cattle ancestry, for 500 seed values, i.e. enough to reach convergence in each run. We also estimated D-statistics to explicitly test for introgression by determining if there is an excess of shared derived alleles between Althiburos cattle (H1 group) and the Moroccan aurochs (H3) or N’Dama cattle (H3) in comparison with an European taurine breed (H2), i.e. Holstein-Friesian, using the Yak (Bos mutus; Bioproject ID: PRJNA74739)70 as an outgroup. We applied an extended version of the D-statistic71 which can use multiple individuals per population sequenced at low coverage and is implemented in ANGSD45 (keeping only autosomal sites with minimum base quality of 20 and mapping quality of 30, discarding transitions and triallelic sites). It takes observed allele frequencies for each individual in a population which are then combined linearly to find an unbiased estimator of population frequency while minimizing the variance.71
Acknowledgments
The authors gratefully acknowledge the following for funding their research: Fundação Nacional para a Ciência e a Tecnologia (FCT), Portugal, contract grants 2020.02754.CEECIND (C.G.) and DL57/2016/CP1440/CT0029 (A.E. Pires), Project grant PTDC/CVTLIV/2827/2014 co-funded by COMPETE 2020 POCI-01-0145-FEDER-016647 and LISBOA-01-0145-FEDER-016647 (C.G. and A.E.P); R.D.F. acknowledges the support of the Villum Fonden for the Center for Global Mountain Biodiversity (grant no 25925) and ERC-StG ZooMWest (ERC-StG 716298). This study was also co-funded by the project NORTE-01-0246-FEDER-000063, supported by Norte Portugal Regional Operational Programme (NORTE2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). We acknowledge the use of computational resources from UPPMAX—the Uppsala Multidisciplinary Centre for Advanced Computational Science under the projects b2014175 and SNIC 2018/8-54. We are grateful for the excellent service and support provided by the National Genomics Infrastructure (NGI, SciLifeLab) in Stockholm, Sweden. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Vendela Kempe Lagerholm, Maja Krzewinska, Petter Larsson, and Nicolas Dussex for the cameradage and technical support during our work at CPG, Stockholm, Sweden. We thank Emma Svensson for fruitful discussions on the 454-sequence data and the broad subject of cattle dispersal and evolution. We thank Dan Bradley for his comments and careful reading of the manuscript. Tribute: Joan Sanmartí was an Archaeologist specialized in Mediterranean protohistory and Professor of Archaeology at the University of Barcelona. Member of the Institut d'Estudis Catalans, in 2009 he received the ICREA – Catalan Institution for Research and Advanced Studies Academy Award. He joined numerous research and excavation projects, including Althiburos archaeological site in El Kef, Tunisia. Without his precious collaboration, this study could not have been accomplished.
Author contributions
C.G., S.V.L., and A.E.P. designed and supervised the study with input from A.G., C.G., S.G., and L.S. carried out the sub-sampling, DNA extraction, and the NGS laboratory work. R.D.F., R.R., S.G., and C.S. performed the bioinformatics and statistical analyses of the genetic data with contributions from C.G. and A.G. M.C.B., N.K., J.R.T., J.S., A.M.A., C.D., S.D., and J.M. provided samples and/or input about the genomic, archaeological, zooarchaeological, and anthropological contexts. C.G., S.G., and S.V.L. wrote the manuscript with contributions from all authors. All authors have read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107196.
Supplemental information
Data and code availability
-
•
Data: The raw reads comprising newly generated genome data were deposited in the Sequence Read Archive (SAMN25653738-SAMN25653746) with the corresponding Bioproject PRJNA803479. The 454-sequence data of the mitochondrial D-loop fragments is available at DRYAD (https://doi.org/10.5061/dryad.v9s4mw71n).
-
•
Code: This paper does not report original code.
-
•
Other items: Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Wright E. In: Cattle and People. Wright E., Ginja C., editors. Lockwood Press; 2022. The Aurochs in the European Pleistocene and Early Holocene Origins, Evidence and Body Size; pp. 3–27. [DOI] [Google Scholar]
- 2.Loftus R.T., MacHugh D.E., Bradley D.G., Sharp P.M., Cunningham P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA. 1994;91:2757–2761. doi: 10.1073/pnas.91.7.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S., Lin B.Z., Baig M., Mitra B., Lopes R.J., Santos A.M., Magee D.A., Azevedo M., Tarroso P., Sasazaki S., et al. Zebu cattle are an exclusive legacy of the South Asia neolithic. Mol. Biol. Evol. 2010;27:1–6. doi: 10.1093/molbev/msp213. [DOI] [PubMed] [Google Scholar]
- 4.Pitt D., Sevane N., Nicolazzi E.L., MacHugh D.E., Park S.D.E., Colli L., Martinez R., Bruford M.W., Orozco-terWengel P. Domestication of cattle: Two or three events? Evol. Appl. 2019;12:123–136. doi: 10.1111/eva.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenstra J., Ajmone-Marsan P., Beja-Pereira A., Bollongino R., Bradley D., Colli L., De Gaetano A., Edwards C., Felius M., Ferretti L., et al. Meta-Analysis of mitochondrial DNA reveals several population bottlenecks during worldwide migrations of cattle. Diversity. 2014;6:178–187. doi: 10.3390/d6010178. [DOI] [Google Scholar]
- 6.Bradley D.G., MacHugh D.E., Cunningham P., Loftus R.T. Mitochondrial diversity and the origins of African and European cattle. Proc. Natl. Acad. Sci. USA. 1996;93:5131–5135. doi: 10.1073/pnas.93.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonfiglio S., Ginja C., de Gaetano A., Achilli A., Olivieri A., Colli L., Tesfaye K., Agha S.H., Gama L.T., Cattonaro F., et al. Origin and spread of Bos taurus: New clues from mitochondrial genomes belonging to haplogroup T1. PLoS One. 2012;7:e38601. doi: 10.1371/journal.pone.0038601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Achilli A., Olivieri A., Pellecchia M., Uboldi C., Colli L., Al-Zahery N., Accetturo M., Pala M., Hooshiar Kashani B., Perego U.A., et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr. Biol. 2008;18:157–158. doi: 10.1016/j.cub.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 9.Edwards C.J., Ginja C., Kantanen J., Pérez-Pardal L., Tresset A., Stock F., European Cattle Genetic Diversity Consortium. Gama L.T., Penedo M.C.T., Bradley D.G., et al. Dual origins of dairy cattle farming - Evidence from a comprehensive survey of european Y-chromosomal variation. PLoS One. 2011;6:e15922. doi: 10.1371/journal.pone.0015922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Pardal L., Royo L.J., Beja-Pereira A., Chen S., Cantet R.J., Traore A., Curik I., Solkner J., Bozzi R., Fernandez I., et al. Multiple paternal origins of domestic cattle revealed by Y-specific interspersed multilocus microsatellites. Heredity. 2010;105:511–519. doi: 10.1038/hdy.2010.30. [DOI] [PubMed] [Google Scholar]
- 11.Ginja C., Telo Da Gama L., Penedo M.C.T. Y chromosome haplotype analysis in Portuguese cattle breeds using SNPs and STRs. J. Hered. 2009;100:148–157. doi: 10.1093/jhered/esn080. [DOI] [PubMed] [Google Scholar]
- 12.Ginja C., Gama L.T., Cortés O., Burriel I.M., Vega-Pla J.L., Penedo C., Sponenberg P., Cañón J., Sanz A., do Egito A.A., et al. The genetic ancestry of American Creole cattle inferred from uniparental and autosomal genetic markers. Sci. Rep. 2019;9:11486. doi: 10.1038/s41598-019-47636-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanotte O., Tawah C.L., Bradley D.G., Okomo M., Verjee Y., Ochieng J., Rege J.E. Geographic distribution and frequency of a taurine Bos taurus and an indicine Bos indicus Y specific allele amongst sub-saharan African cattle breeds. Mol. Ecol. 2000;9:387–396. doi: 10.1046/j.1365-294x.2000.00858.x. [DOI] [PubMed] [Google Scholar]
- 14.Pérez-Pardal L., Sánchez-Gracia A., Álvarez I., Traoré A., Ferraz J.B.S., Fernández I., Costa V., Chen S., Tapio M., Cantet R.J.C., et al. Legacies of domestication, trade and herder mobility shape extant male zebu cattle diversity in South Asia and Africa. Sci. Rep. 2018;8:18027. doi: 10.1038/s41598-018-36444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen N., Cai Y., Chen Q., Li R., Wang K., Huang Y., Hu S., Huang S., Zhang H., Zheng Z., et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018;9:2337. doi: 10.1038/s41467-018-04737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bovine HapMap Consortium. Gibbs R.A., Taylor J.F., Van Tassell C.P., Barendse W., Eversole K.A., Gill C.A., Green R.D., Hamernik D.L., Kappes S.M., et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324:528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decker J.E., McKay S.D., Rolf M.M., Kim J., Molina Alcalá A., Sonstegard T.S., Hanotte O., Götherström A., Seabury C.M., Praharani L., et al. Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. PLoS Genet. 2014;10:e1004254. doi: 10.1371/journal.pgen.1004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanotte O., Bradley D.G., Ochieng J.W., Verjee Y., Hill E.W., Rege J.E.O. African Pastoralism: Genetic Imprints of Origins and Migrations. Science. 2002;296:336–339. doi: 10.1126/science.1069878. [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Hanotte O., Mwai O.A., Dessie T., Bashir S., Diallo B., Agaba M., Kim K., Kwak W., Sung S., et al. The genome landscape of indigenous African cattle. Genome Biol. 2017;18:34. doi: 10.1186/s13059-017-1153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbato M., Hailer F., Upadhyay M., Del Corvo M., Colli L., Negrini R., Kim E.S., Crooijmans R.P.M.A., Sonstegard T., Ajmone-Marsan P. Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci. Rep. 2020;10:1279. doi: 10.1038/s41598-020-57880-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Upadhyay M., Bortoluzzi C., Barbato M., Ajmone-Marsan P., Colli L., Ginja C., Sonstegard T.S., Bosse M., Lenstra J.A., Groenen M.A.M., Crooijmans R.P.M.A. Deciphering the patterns of genetic admixture and diversity in southern European cattle using Genome-wide SNPs. Evol. Appl. 2019;12:951–963. doi: 10.1111/eva.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman A.R., Bradley D.G., Nagda S., Gibson J.P., Hanotte O. Combination of multiple microsatellite data sets to investigate genetic diversity and admixture of domestic cattle. Anim. Genet. 2006;37:1–9. doi: 10.1111/j.1365-2052.2005.01363.x. [DOI] [PubMed] [Google Scholar]
- 23.Frantz L.A.F., Bradley D.G., Larson G., Orlando L. Animal domestication in the era of ancient genomics. Nat. Rev. Genet. 2020;21:449–460. doi: 10.1038/s41576-020-0225-0. [DOI] [PubMed] [Google Scholar]
- 24.Ward J.A., McHugo G.P., Dover M.J., Hall T.J., Ng’ang’a S.I., Sonstegard T.S., Bradley D.G., Frantz L.A.F., Salter-Townshend M., MacHugh D.E. Genome-wide local ancestry and evidence for mitonuclear coadaptation in African hybrid cattle populations. iScience. 2022;25:104672. doi: 10.1016/j.isci.2022.104672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Da Fonseca R.R., Ureña I., Afonso S., Pires A.E., Jørsboe E., Chikhi L., Ginja C. Consequences of breed formation on patterns of genomic diversity and differentiation: The case of highly diverse peripheral Iberian cattle. BMC Genom. 2019;20:334. doi: 10.1186/s12864-019-5685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McHugo G.P., Dover M.J., MacHugh D.E. Unlocking the origins and biology of domestic animals using ancient DNA and paleogenomics. BMC Biol. 2019;17:98. doi: 10.1186/s12915-019-0724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verdugo M.P., Mullin V.E., Scheu A., Mattiangeli V., Daly K.G., Maisano Delser P., Hare A.J., Burger J., Collins M.J., Kehati R., et al. Ancient cattle genomics, origins, and rapid turnover in the Fertile Crescent. Science. 2019;365:173–176. doi: 10.1126/science.aav1002. [DOI] [PubMed] [Google Scholar]
- 28.Kallala N., Sanmartí J., Althiburos I. Documenta. Vol. 18. Universitat de Barcelona; Institut Català d’Arqueologia Clàssica; Institut National du Patrimoine Tunisia; Tarragona: 2011. La fouille dans l’aire du capitole et dans la nécropole méridionale. [Google Scholar]
- 29.Kallala N., Sanmartí J., Belarte M.C., editors. II. La fouille dans l’aire du capitole et dans la nécropole méridionale: études. Vol. 28. Universitat de Barcelona; Institut Català d’Arqueologia Clàssica; Institut National du Patrimoine Tunisia; Tarragona: 2016. (Documenta). [Google Scholar]
- 30.Sanmartí J., Kallala N., Belarte M.C., Ramon J., Telmini B.M., Jornet R., Miniaoui S., Fadrique T., López D., Morell N., et al. Filling gaps in the protohistory of the Eastern Maghreb: The Althiburos archaeological project (El Kef, Tunisia) J. African Arch. 2012;10:21–44. doi: 10.3213/2191-5784-10213. [DOI] [Google Scholar]
- 31.Valenzuela-Lamas S. In: Althiburos II: L’aire du capitole et la nécropole méridionale. Kallala N., Sanmartí J., Belarte M.C., editors. Institut Català d’Arqueologia Clàssica; 2016. Alimentation et élevage à partir des restes fauniques; pp. 421–448. [Google Scholar]
- 32.Sawyer S., Krause J., Guschanski K., Savolainen V., Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One. 2012;7:e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ameling W. In: A Companion to the Punic Wars. Hoyos D., editor. Wiley & Sons Ltd; 2011. The Rise of Carthage to 264 BC; pp. 39–57. [DOI] [Google Scholar]
- 34.Bonfiglio S., Achilli A., Olivieri A., Negrini R., Colli L., Liotta L., Ajmone-Marsan P., Torroni A., Ferretti L. The enigmatic origin of bovine mtDNA haplogroup R: Sporadic interbreeding or an independent event of Bos primigenius domestication in Italy? PLoS One. 2010;5:e15760. doi: 10.1371/journal.pone.0015760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skoglund P., Storå J., Götherström A., Jakobsson M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 2013;40:4477–4482. doi: 10.1016/j.jas.2013.07.004. [DOI] [Google Scholar]
- 36.Bro-Jørgensen M.H., Keighley X., Ahlgren H., Scharff-Olsen C.H., Rosing-Asvid A., Dietz R., Ferguson S.H., Gotfredsen A.B., Jordan P., Glykou A., et al. Genomic sex identification of ancient pinnipeds using the dog genome. J. Archaeol. Sci. 2021;127:105321. doi: 10.1016/j.jas.2020.105321. [DOI] [Google Scholar]
- 37.Papachristou D., Koutsouli P., Laliotis G.P., Kunz E., Upadhyay M., Seichter D., Russ I., Gjoko B., Kostaras N., Bizelis I., Medugorac I. Genomic diversity and population structure of the indigenous Greek and Cypriot cattle populations. Genet. Sel. Evol. 2020;52:43. doi: 10.1186/s12711-020-00560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 39.Schubert M., Lindgreen S., Orlando L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes. 2016;9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jónsson H., Ginolhac A., Schubert M., Johnson P.L.F., Orlando L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okonechnikov K., Conesa A., García-Alcalde F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32:292–294. doi: 10.1093/bioinformatics/btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korneliussen T.S., Albrechtsen A., Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinf. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suchard M.A., Lemey P., Baele G., Ayres D.L., Drummond A.J., Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darriba D., Posada D., Kozlov A.M., Stamatakis A., Morel B., Flouri T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020;37:291–294. doi: 10.1093/molbev/msz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meisner J., Albrechtsen A. Inferring population structure and admixture proportions in low-depth NGS data. Genetics. 2018;210:719–731. doi: 10.1534/genetics.118.301336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skotte L., Korneliussen T.S., Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang D.Y., Eng B., Waye J.S., Dudar J.C., Saunders S.R. Technical note: Improved DNA extraction from ancient bones using silica- based spin columns. Am. J. Phys. Anthropol. 1998;105 doi: 10.1002/(SICI)1096-8644(199804)105:4<539::AID-AJPA10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 53.Dabney J., Knapp M., Glocke I., Gansauge M.T., Weihmann A., Nickel B., Valdiosera C., García N., Pääbo S., Arsuaga J.L., Meyer M. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA. 2013;110:15758–15763. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Günther T., Valdiosera C., Malmström H., Ureña I., Rodriguez-Varela R., Sverrisdóttir Ó.O., Daskalaki E.A., Skoglund P., Naidoo T., Svensson E.M., et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc. Natl. Acad. Sci. USA. 2015;112:11917–11922. doi: 10.1073/pnas.1509851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korlević P., Gerber T., Gansauge M.T., Hajdinjak M., Nagel S., Aximu-Petri A., Meyer M. Reducing microbial and human contamination in dna extractions from ancient bones and teeth. Biotechniques. 2015;59:87–93. doi: 10.2144/000114320. [DOI] [PubMed] [Google Scholar]
- 56.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olivieri A., Gandini F., Achilli A., Fichera A., Rizzi E., Bonfiglio S., Battaglia V., Brandini S., De Gaetano A., El-Beltagi A., et al. Mitogenomes from Egyptian cattle breeds: New clues on the origin of haplogroup Q and the early spread of Bos taurus from the Near East. PLoS One. 2015;10:e0141170. doi: 10.1371/journal.pone.0141170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hasegawa M., Kishino H., Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 59.Boitard S., Rodríguez W., Jay F., Mona S., Austerlitz F. Inferring Population Size History from Large Samples of Genome-Wide Molecular Data - An Approximate Bayesian Computation Approach. PLoS Genet. 2016;12:e1005877. doi: 10.1371/journal.pgen.1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minin V.N., Bloomquist E.W., Suchard M.A. Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol. Biol. Evol. 2008;25:1459–1471. doi: 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drummond A.J., Nicholls G.K., Rodrigo A.G., Solomon W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161:1307–1320. doi: 10.1093/genetics/161.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bibi F. A multi-calibrated mitochondrial phylogeny of extant Bovidae (Artiodactyla, Ruminantia) and the importance of the fossil record to systematics. BMC Evol. Biol. 2013;13:166. doi: 10.1186/1471-2148-13-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svensson E.M., Anderung C., Baubliene J., Persson P., Malmström H., Smith C., Vretemark M., Daugnora L., Götherström A. Tracing genetic change over time using nuclear SNPs in ancient and modern cattle. Anim. Genet. 2007;38:378–383. doi: 10.1111/j.1365-2052.2007.01620.x. [DOI] [PubMed] [Google Scholar]
- 64.Edwards C.J., MacHugh D.E., Dobney K.M., Martin L., Russell N., Horwitz L.K., McIntosh S.K., MacDonald K.C., Helmer D., Tresset A., et al. Ancient DNA analysis of 101 cattle remains: Limits and prospects. J. Archaeol. Sci. 2004;31:695–710. doi: 10.1016/j.jas.2003.11.001. [DOI] [Google Scholar]
- 65.Anderung C., Bouwman A., Persson P., Carretero J.M., Ortega A.I., Elburg R., Smith C., Arsuaga J.L., Ellegren H., Götherström A. Prehistoric contacts over the Straits of Gibraltar indicated by genetic analysis of Iberian Bronze Age cattle. Proc. Natl. Acad. Sci. USA. 2005;102:8431–8435. doi: 10.1073/pnas.0503396102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Binladen J., Gilbert M.T.P., Bollback J.P., Panitz F., Bendixen C., Nielsen R., Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malmström H., Gilbert M.T.P., Thomas M.G., Brandström M., Storå J., Molnar P., Andersen P.K., Bendixen C., Holmlund G., Götherström A., Willerslev E. Ancient DNA reveals lack of continuity between neolithic hunter-gatherers and contemporary Scandinavians. Curr. Biol. 2009;19:1758–1762. doi: 10.1016/j.cub.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 68.Afgan E., Baker D., Batut B., Van Den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malmström H., Linderholm A., Skoglund P., Storå J., Sjödin P., Gilbert M.T.P., Holmlund G., Willerslev E., Jakobsson M., Lidén K., Götherström A. Ancient mitochondrial DNA from the northern fringe of the Neolithic farming expansion in Europe sheds light on the dispersion process. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20130373. doi: 10.1098/rstb.2013.0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu Q., Zhang G., Ma T., Qian W., Wang J., Ye Z., Cao C., Hu Q., Kim J., Larkin D.M., et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012;44:946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 71.Soraggi S., Wiuf C., Albrechtsen A. Powerful inference with the D-statistic on low-coverage whole-genome data. G3. 2018;8:551–566. doi: 10.1534/g3.117.300192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: The raw reads comprising newly generated genome data were deposited in the Sequence Read Archive (SAMN25653738-SAMN25653746) with the corresponding Bioproject PRJNA803479. The 454-sequence data of the mitochondrial D-loop fragments is available at DRYAD (https://doi.org/10.5061/dryad.v9s4mw71n).
-
•
Code: This paper does not report original code.
-
•
Other items: Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.