

Abstract

A nickel-catalyzed N–N cross-coupling for the synthesis of hydrazides is reported. O-Benzoylated hydroxamates were efficiently coupled with a broad range of aryl and aliphatic amines via nickel catalysis to form hydrazides in an up to 81% yield. Experimental evidence implicates the intermediacy of electrophilic Ni-stabilized acyl nitrenoids and the formation of a Ni(I) catalyst via silane-mediated reduction. This report constitutes the first example of an intermolecular N–N coupling compatible with secondary aliphatic amines.
Nitrogen–nitrogen bonds are prevalent motifs in biologically active small molecules and natural products, featured prominently in pharmaceutical and agricultural compounds (Figure 1A).1−5 Moreover, a variety of druglike heterocyclic scaffolds can be accessed from hydrazines and hydrazides.2,6 The synthesis of N–N containing compounds typically entails a linear, stepwise process of hydrazine protection and derivatization, which hampers rapid access to highly substituted products.2 Improved methods for the convergent cross-coupling of two complex nitrogen-containing compounds would not only simplify the preparation of known hydrazines and hydrazides but also accelerate access to new chemical space. For these reasons, new N–N bond forming reactions are of high value to the synthetic chemistry community.
Figure 1.

Compounds featuring N–N bonds and nitrene-mediated synthetic strategies.
While several impressive examples of N–N cross-coupling have been developed to form hydrazines via oxidative or reductive pathways, the majority of methods furnish fully aromatic products.7,8 Transition-metal-catalyzed nitrene insertion has recently gained attention as a powerful tool for N–N coupling (Figure 1B).9 Both Ag and Rh have been utilized as catalysts for nitrene transfer to tertiary amines, although these methods only lend access to tailored synthetic frameworks.9a,9b In 2021, Chang and Chen disclosed the first N–N cross-coupling method for the formation of hydrazides utilizing Fe or Ir catalysis (Figure 1B).9c They propose the formation of an electrophilic metal-bound nitrenoid that is subject to outersphere attack by an amine to generate the hydrazide product. In a subsequent communication, Chen demonstrated the compatibility of this Ir-catalyzed transformation with O-benzoylated hydroxamates.9d While these methods enable efficient coupling of a wide array of aliphatic electrophiles and secondary anilines, other classes of amine nucleophiles, most notably aliphatic amines, remain incompatible. As part of an industrial-academic collaboration aimed at developing new modular approaches for N–N coupling, we pursued the development of a new nitrene-mediated N–N cross-coupling with the goal of accessing diverse hydrazides from both aryl and aliphatic coupling partners (Figure 1C).
After an unsuccessful survey of Cu and Fe catalysts, we were pleased to discover that reaction with N-(benzoyloxy)benzamide 1a and p-toluidine (2a) in the presence of Zn(0) and catalytic Ni(PPh3)2Cl2 resulted in a 30% yield of the desired hydrazide product 3a (Table 1, entry 1). Iminophosphorane 4 was observed as a side product of this reaction, which is suggestive of the intermediacy of a nitrenoid species.10 Surprisingly, we observed no urea formation, indicating robust stabilization of the nitrene intermediate against Lossen-type rearrangement.11
Table 1. Reaction Optimization of Aryl Aminesa.


Reactions performed on 0.05 mmol scale.
72 h.
Excess hydroxamate 1a (1.5 equiv).
30 °C.
In the absence of Zn, poor conversion was observed, providing a similar 30% yield of 3a only after an extended reaction time with significant unconsumed starting material (entry 2). We postulate that Ni(II) may be effecting the desired transformation via an alternative mechanism, such as Lewis acid activation. Use of Ni(COD)2 yielded no reaction, suggesting that a Ni(0) species is not involved in the catalytic cycle (entry 3). Use of bidentate ligands dppe or bpy resulted in a complete loss of reactivity (entries 4 and 5); however, strongly σ-donating N-heterocyclic carbene (NHC) SIPr afforded a 32% yield of 3a with no observed nitrene transfer to the ligand (entry 6). Use of alternative NHC ligands resulted in diminished yields (Supporting Information). Given the stark difference in reactivity observed between monodentate and bidentate ligands, we hypothesized that the catalyst must be coordinatively unsaturated to enter the catalytic cycle.
Having identified a more suitable ligand, we re-examined the role of Zn as an additive. Previous reports have demonstrated the ability of Ni(I) dimer 6 to form a bridged Ni-nitrene; we considered the possibility that Zn-mediated reduction may be forming a similar species in situ.12,13 Unfortunately, examination of 6 both with and without Zn resulted in no reactivity (entry 7, 8), leading us to hypothesize a mononuclear Ni(I) complex as the active catalyst.14 While further exploration of reductants revealed that phenylsilane improved reactivity (see Supporting Information), yields were highly variable across batches of starting materials and reagents (entry 9). Despite an extensive effort to assess the purity of our reagents, we were ultimately unable to identify the cause of irreproducibility.
Aiming to achieve more consistent results, we explored well-defined Ni complexes and were intrigued by reports of NHC-ligated Ni(II) half-sandwich catalysts, which have recently been utilized for catalytic oxidative N–N coupling of ammonia to dinitrogen.15−18 These easily synthesized and air-stable complexes have been implicated to undergo hydride-mediated reduction to Ni(I).15,16 Additionally, the substituent cyclopentadienyl (Cp) ligand has demonstrated surprisingly facile equilibration among η5, η3, and η1 binding modes, which we hypothesized could satisfy the previously observed requirement for a coordinatively unsaturated catalyst.15,17 After evaluating a variety of Ni half-sandwich catalysts, we found that the use of 7a afforded a reproducible 61% yield of the desired product (entry 10). Exploration of solvent effects revealed that a 1:4 mixture of CH2Cl2/THF further improved the yield to 80% (entry 11).
Having identified optimal conditions for aryl amine nucleophiles, we sought to expand the scope of the transformation to aliphatic amines. Reaction with unprotected secondary aliphatic amine 8a resulted in significant O-to-N benzoyl transfer from the hydroxamate to the amine, yielding product 10 and minimal formation of the desired hydrazide (Table 2, entry 1). Surmising that silylation of these more challenging nucleophiles might serve as a transient protection strategy until either transmetalation with the catalyst or benzoate-mediated desilylation, we explored reactivity with silylamine 8b.19 To our satisfaction, the reaction of 8b with excess 1a afforded the desired product 9a in 27% yield (entry 2). Use of excess amine was found to improve the product distribution, with 1.5 equiv of 8b resulting in a 57% yield (entries 3, 4). Gratifyingly, the use of IPr- substituted catalyst 7b afforded a 69% yield of the desired hydrazide 9a, minimizing the formation of undesired side-product 10 (entry 5). We posit the O-to-N benzoyl transfer may be accelerated within the coordination sphere of the catalyst and that increasing steric hindrance around the metal center may block this competing pathway. Although the efficiency of these conditions utilizing presilylated amines was satisfactory, a more operationally expedient route was developed via in situ silylation of amine 8a with MSTFA, affording a 59% yield of product in a single synthetic step (entry 6).
Table 2. Reaction Optimization of Aliphatic Aminesa.


Reactions performed on 0.05 mmol scale.
Excess hydroxamate 1a (1.5 equiv).
With optimized conditions in hand, we examined the scope of compatible coupling partners (Scheme 1). Aniline derivatives with electron-donating substituents at the para-position provided the desired products in good yields (3a and 3b), while electron-deficient amines gave slightly diminished yields (3c and 3d). Halide substituents on the arene (3e–3h) were well tolerated under the reaction conditions and showed no signs of protodehalogenation. Functional handles for further derivatization, such as an aryl iodide (3h) and boronic ester (3i), remained intact under the reaction conditions. Sterically hindered ortho-substituted aniline derivatives afforded products 3j and 3k in good yields. Amines bearing unprotected ketone (3l) and hydroxyl (3m and 3n) moieties were also found to be compatible. Moreover, aminoglutethimide, a drug used in the treatment of Cushing’s disease, was efficiently derivatized to the corresponding hydrazide (3o), highlighting the potential of this method for late-stage functionalization of complex molecules.20 Although exploration of a secondary aniline in reaction with 1a yielded only trace hydrazide (3p), we observed an 81% yield of the product when using N-(benzoyloxy)acetamide as an electrophile (3q). We postulate the difference in reactivity between aryl and aliphatic hydroxamates with secondary anilines reveals the stereoelectronic constraints of this transformation—although secondary anilines are more nucleophilic than primary anilines,21 the increased steric bulk may bar their reactivity with more encumbered aryl electrophiles.
Scheme 1. Substrate Scope.
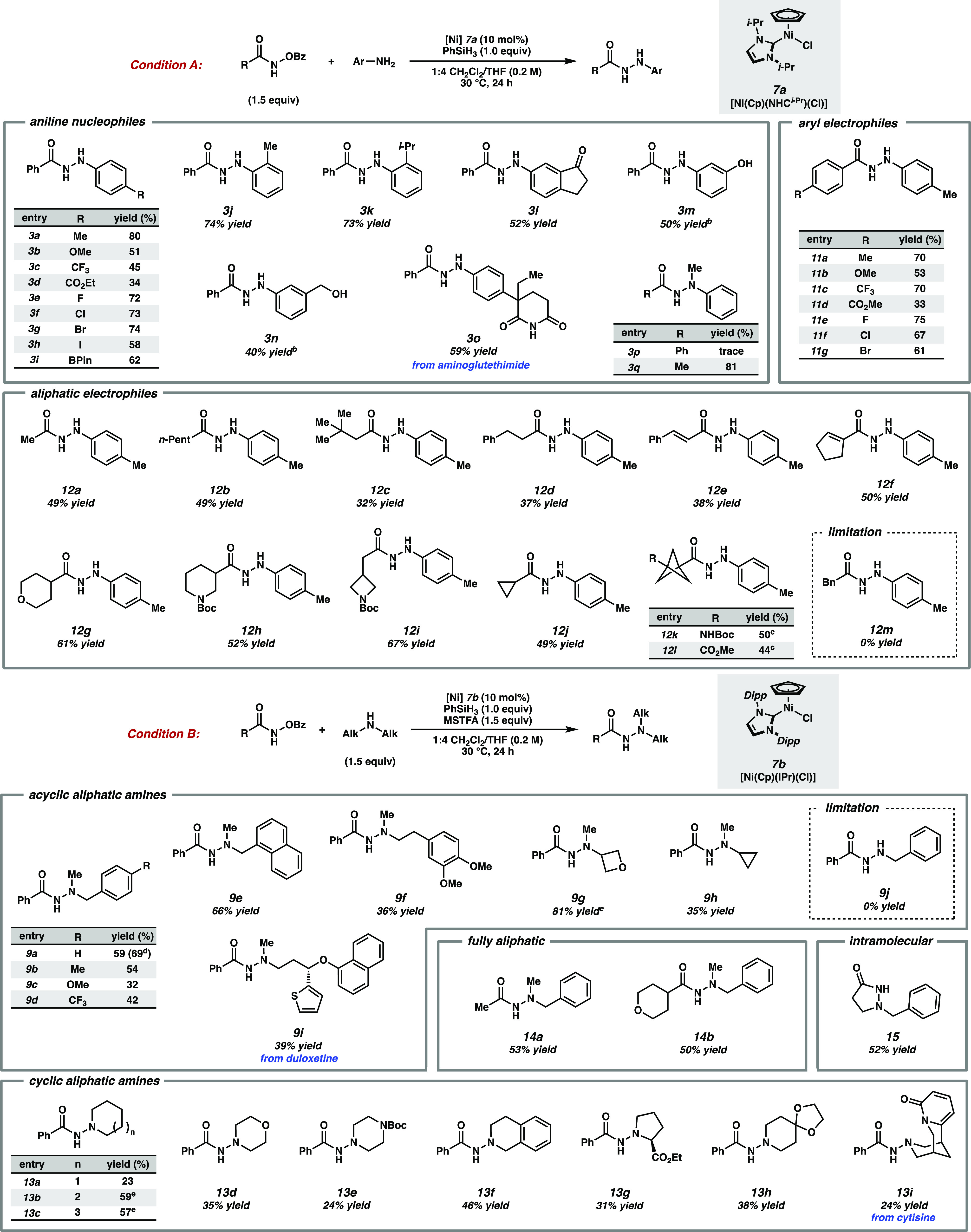
Reactions performed on 0.2 mmol scale.
Protected as TBS ether for isolation.
72 h.
Silylamine (8b) used.
48 h.
Hydroxamates with a variety of electron-donating and -withdrawing para-substituents on the aryl ring furnished the N–N products with moderate to high yields (11a–11g). Additionally, primary, secondary, and tertiary aliphatic hydroxamates were compatible with this chemistry (12a–12l). We were pleased to observe styrenyl and alkenyl functional groups did not participate in undesired C–H insertion, hydroamidation, reduction, or aziridination processes, affording moderate yields of products 12e and 12f, respectively.13,22−24 Additionally, several saturated heterocyclic compounds, such as tetrahydropyran-, piperidine-, and azetidine-derived hydroxamates (12g–12i) afforded products in 52–67% yields. Hydroxamates featuring phenyl isostere [1.1.1]-bicyclopentane (BCP) derivatives were also compatible in this transformation (12k and 12l).25 Surprisingly, a benzylic hydroxamate remained inert under the reaction conditions (12m).
N-Methylbenzylamine derivatives with both electron-donating and electron-withdrawing substituents on the aryl ring afforded modest yields of the desired products (9a–9d). Replacement of the benzyl group with a variety of acyclic and cyclic moieties (9e–9i), including a formamide isostere (9g), resulted in efficient product formation.26 Reaction with duloxetine, an antidepressant, yielded the corresponding hydrazide derivative 9i in a synthetically useful yield.27 Although broadly amenable to reaction with secondary aliphatic amines, primary aliphatic amines remain incompatible with this transformation, instead yielding undesired amine acylation arising from O-to-N benzoyl transfer and only trace amount of product (9j).
Cyclic aliphatic amines, including piperidine (13a), azepane (13b), azocane (13c), morpholine (13d), N-Boc piperazine (13e), tetrahydroisoquinoline (13f), (S)-proline ethyl ester (13g), and an azaspirocycle (13h) were competent coupling partners in this chemistry, albeit affording products in diminished yields due to the greater propensity of these nucleophiles to generate O-to-N benzoyl transfer side products. Cytisine, a smoking cessation agent, was able to be derivatized to the corresponding hydrazide in a synthetically useful yield (13i),28 and we were able to access fully aliphatic hydrazides (14a and 14b), including saturated heterocycle 15 resulting from intramolecular bond formation. Lastly, both sets of conditions were demonstrated to be scalable, with hydrazides 3a and 8a obtained in near identical yields on a 2 mmol scale (Scheme 2).
Scheme 2. Large Scale Reactions.

Reactions performed on 2 mmol scale.
To investigate the role of the silane additive, Ni(I) half sandwich 7c was independently synthesized and its catalytic competence was examined (Scheme 3).17 Reaction in the presence and absence of phenylsilane afforded similar product yields with near-identical reaction times, supporting the notion that the active catalyst is a Ni(I) species formed via reduction with silane.15,29
Scheme 3. Ni(I) Catalyst.

Reactions performed on 0.05 mmol scale.
With this evidence in hand, we suggest the following mechanism: phenylsilane can reduce 7a via the formation of Ni(II) hydride A to Ni(I) species B. Reaction with a hydroxamate starting material can form Ni-nitrenoid C with net loss of benzoic acid, which may be enabled by transient slippage of the Cp ligand.15,17 Ni-nitrenoid C can then undergo outer sphere N–N bond formation with an amine, which following proton transfer yields intermediate D. Dissociation of the product regenerates the active catalyst (B) (Figure 2).
Figure 2.

Mechanistic hypothesis.
In summary, we have developed a Ni-catalyzed N–N cross-coupling enabling the convenient formation of complex hydrazides. This transformation is effected by an easily synthesized and air-stable Ni(II) half-sandwich precatalyst. In situ silylation allows unprecedented access to secondary aliphatic amines in N–N coupling methodology. This reaction tolerates an impressive breadth of functionality, including handles for further derivatization. Preliminary mechanistic investigation suggests a mononuclear Ni(I) active catalyst.
Acknowledgments
The authors thank Corteva Agriscience for funding and scientific discussion, as well as NIH-NIGMS (R01GM080269), NIH-NIGMS (R35GM145239), NIH-NIGMS (R35GM118191), Heritage Medical Research Investigators Program, and Caltech for the support of our research program. The authors also thank Dr. Scott Virgil (Caltech) for assistance with instrumentation, Dr. Dave VanderVelde (Caltech) for NMR expertise, Dr. Mona Shahgholi for mass spectrometry, and Dr. Michael Takase (Caltech) for X-ray analysis.
Glossary
Abbreviations
- Cp
cyclopentadiene
- MSTFA
N-trimethylsilyl-N-methyl trifluoroacetamide
- dppe
1,2-bis(diphenylphosphino)ethane
- bpy
bipyridine
- NHC
N-heterocyclic carbene
- IPr
1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium-2-ide
- SIPr
1,3-bis(2,6-diisopropylphenyl)-4,5-dihydro-1H-imidazol-3-ium-2-ide
- COD
1,5-cyclooctadiene
- BCP
bicyclo[1.1.1]pentane
- Boc
tert-butyloxycarbonyl
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c04834.
Experimental procedures, spectroscopic (1H NMR, 13C NMR, IR, HRMS), and crystallographic data (PDF)
Author Contributions
§ V.N.N. and K.R.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- For a general overview of N–N bonds in nature, see:; a Blair L. M.; Sperry J. Natural Products Containing a Nitrogen–Nitrogen Bond. J. Nat. Prod. 2013, 76, 794–812. 10.1021/np400124n. [DOI] [PubMed] [Google Scholar]; b Chen L.; Deng Z.; Zhao C. Nitrogen–Nitrogen Bond Formation Reactions Involved in Natural Product Biosynthesis. ACS Chem. Biol. 2021, 16, 559–570. 10.1021/acschembio.1c00052. [DOI] [PubMed] [Google Scholar]; c He H.-Y.; Niikura H.; Du Y.-L.; Ryan K. S. Synthetic and Biosynthetic Routes to Nitrogen–Nitrogen Bonds. Chem. Soc. Rev. 2022, 51, 2991–3046. 10.1039/C7CS00458C. [DOI] [PubMed] [Google Scholar]; d Narang R.; Narasimhan B.; Sharma S. A Review on Biological Activities and Chemical Synthesis of Hydrazide Derivatives. Curr. Med. Chem. 2012, 19, 569–612. 10.2174/092986712798918789. [DOI] [PubMed] [Google Scholar]
- For a general review of convergent N–N coupling, see:; a Guo Q.; Lu Z. Recent Advances in Nitrogen–Nitrogen Bond Formation. Synthesis 2017, 49, 3835–3847. 10.1055/s-0036-1588512. [DOI] [Google Scholar]; b Tabey A.; Vemuri P. Y.; Patureau F. W. Cross-Dehydrogenative N–N Couplings. Chem. Sci. 2021, 12, 14343–14352. 10.1039/D1SC03851F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira M. M.; Alcântara A. F.; de C.; Piló-Veloso D.; Raslan D. S. NMR Structural Analysis of Braznitidumine: A New Indole Alkaloid with 1,2,9-Triazabicyclo[7.2.1] System, Isolated from Aspidosperma Nitidum (Apocynaceae). J. Braz. Chem. Soc. 2006, 17, 1274–1280. 10.1590/S0103-50532006000700012. [DOI] [Google Scholar]
- Ahmed F. S.; Helmy Y. S.; Helmy W. S. Toxicity and Biochemical Impact of Methoxyfenozide/Spinetoram Mixture on Susceptible and Methoxyfenozide-Selected Strains of Spodoptera Littoralis (Lepidoptera: Noctuidae). Sci. Rep. 2022, 12, 6974–6983. 10.1038/s41598-022-10812-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentué-Ferrer D.; Arvieux C.; Tribut O.; Ruffault A.; Bellissant E. Clinical Pharmacology, Efficacy and Safety of Atazanavir: A Review. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1455–1468. 10.1517/17425250903321514. [DOI] [PubMed] [Google Scholar]
- Majumdar P.; Pati A.; Patra M.; Behera R. K.; Behera A. K. Acid Hydrazides, Potent Reagents for Synthesis of Oxygen-, Nitrogen-, and/or Sulfur-Containing Heterocyclic Rings. Chem. Rev. 2014, 114, 2942–2977. 10.1021/cr300122t. [DOI] [PubMed] [Google Scholar]
- For examples of oxidative N–N coupling, see:; a Rosen B. R.; Werner E. W.; O’Brien A. G.; Baran P. S. Total Synthesis of Dixiamycin B by Electrochemical Oxidation. J. Am. Chem. Soc. 2014, 136, 5571–5574. 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lv S.; Han X.; Wang J.-Y.; Zhou M.; Wu Y.; Ma L.; Niu L.; Gao W.; Zhou J.; Hu W.; Cui Y.; Chen J. Tunable Electrochemical C–N versus N–N Bond Formation of Nitrogen-Centered Radicals Enabled by Dehydrogenative Dearomatization: Biological Applications. Angew. Chem., Int. Ed. 2020, 59, 11583–11590. 10.1002/anie.202001510. [DOI] [PubMed] [Google Scholar]; c Wang F.; Gerken J. B.; Bates D. M.; Kim Y. J.; Stahl S. S. Electrochemical Strategy for Hydrazine Synthesis: Development and Overpotential Analysis of Methods for Oxidative N–N Coupling of an Ammonia Surrogate. J. Am. Chem. Soc. 2020, 142, 12349–12356. 10.1021/jacs.0c04626. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vemuri P. Y.; Patureau F. W. Cross-Dehydrogenative N–N Coupling of Aromatic and Aliphatic Methoxyamides with Benzotriazoles. Org. Lett. 2021, 23, 3902–3907. 10.1021/acs.orglett.1c01034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of reductive N–N coupling, see:; a Li G.; Miller S. P.; Radosevich A. T. PIII/PV=O-Catalyzed Intermolecular N–N Bond Formation: Cross-Selective Reductive Coupling of Nitroarenes and Anilines. J. Am. Chem. Soc. 2021, 143, 14464–14469. 10.1021/jacs.1c07272. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tombari R. J.; Tuck J. R.; Yardeny N.; Gingrich P. W.; Tantillo D. J.; Olson D. E. Calculated Oxidation Potentials Predict Reactivity in Baeyer–Mills Reactions. Org. Biomol. Chem. 2021, 19, 7575–7580. 10.1039/D1OB01450A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of N–N coupling involving a nitrene, see:; a Maestre L.; Dorel R.; Pablo Ó.; Escofet I.; Sameera W. M. C.; Álvarez E.; Maseras F.; Díaz-Requejo M. M.; Echavarren A. M.; Pérez P. J. Functional-Group-Tolerant, Silver-Catalyzed N–N Bond Formation by Nitrene Transfer to Amines. J. Am. Chem. Soc. 2017, 139, 2216–2223. 10.1021/jacs.6b08219. [DOI] [PubMed] [Google Scholar]; b Kono M.; Harada S.; Nemoto T. Chemoselective Intramolecular Formal Insertion Reaction of Rh–Nitrenes into an Amide Bond Over C–H Insertion. Chem.—Eur. J. 2019, 25, 3119–3124. 10.1002/chem.201805878. [DOI] [PubMed] [Google Scholar]; c Wang H.; Jung H.; Song F.; Zhu S.; Bai Z.; Chen D.; He G.; Chang S.; Chen G. Nitrene-Mediated Intermolecular N–N Coupling for Efficient Synthesis of Hydrazides. Nat. Chem. 2021, 13, 378–385. 10.1038/s41557-021-00650-0. [DOI] [PubMed] [Google Scholar]; d Song F.; Zhu S.; Wang H.; Chen G. Iridium-Catalyzed Intermolecular N–N Coupling for Hydrazide Synthesis Using N-Benzoyloxycarbamates as Acyl Nitrene Precursor. Chin. J. Org. Chem. 2021, 41, 4050–4058. 10.6023/cjoc202105044. [DOI] [Google Scholar]
- Zhou Z.; Chen S.; Hong Y.; Winterling E.; Tan Y.; Hemming M.; Harms K.; Houk K. N.; Meggers E. Non-C2-Symmetric Chiral-at-Ruthenium Catalyst for Highly Efficient Enantioselective Intramolecular C(sp3)–H Amidation. J. Am. Chem. Soc. 2019, 141, 19048–19057. 10.1021/jacs.9b09301. [DOI] [PubMed] [Google Scholar]
- a Hong S. Y.; Park Y.; Hwang Y.; Kim Y. B.; Baik M.-H.; Chang S. Selective formation of γ-lactams via C–H amidation enabled by tailored iridium catalysts. Science 2018, 359, 1016–1021. 10.1126/science.aap7503. [DOI] [PubMed] [Google Scholar]; b Franklin E. C. The Hofmann-Beckmann-Curtius-Lossen Rearrangements. Chem. Rev. 1934, 14 (2), 219–250. 10.1021/cr60048a002. [DOI] [Google Scholar]
- Dible B. R.; Sigman; Matthew S.; Arif; Atta M. Oxygen-Induced Ligand Dehydrogenation of a Planar Bis-μ-Chloronickel(I) Dimer Featuring an NHC Ligand. Inorg. Chem. 2005, 44, 3774–3776. 10.1021/ic050445u. [DOI] [PubMed] [Google Scholar]
- Laskowski C. A.; Hillhouse G. L. Group-Transfer Reactions of Ni(II)–Ni(II) Bridging Imido Complexes. Catalytic Formation of Carbodiimides and Isocyanates via Nitrene Transfer from Organoazides. Organometallics 2009, 28, 6114–6120. 10.1021/om900783u. [DOI] [Google Scholar]
- Laskowski C. A.; Miller A. J. M.; Hillhouse G. L.; Cundari T. R. A Two-Coordinate Nickel Imido Complex That Effects C–H Amination. J. Am. Chem. Soc. 2011, 133, 771–773. 10.1021/ja1101213. [DOI] [PubMed] [Google Scholar]
- Banach Ł.; Guńka P. A.; Zachara J.; Buchowicz W. Half-Sandwich Ni(II) Complexes [Ni(Cp)(X)(NHC)]: From an Underestimated Discovery to a New Chapter in Organonickel Chemistry. Coord. Chem. Rev. 2019, 389, 19–58. 10.1016/j.ccr.2019.03.006. [DOI] [Google Scholar]
- Ulm F.; Cornaton Y.; Djukic J.; Chetcuti M. J.; Ritleng V. Hydroboration of Alkenes Catalysed by a Nickel N-Heterocyclic Carbene Complex: Reaction and Mechanistic Aspects. Chem. – Eur. J. 2020, 26, 8916–8925. 10.1002/chem.202000289. [DOI] [PubMed] [Google Scholar]
- a Wu J.; Nova A.; Balcells D.; Brudvig G. W.; Dai W.; Guard L. M.; Hazari N.; Lin P.-H.; Pokhrel R.; Takase M. K. Nickel(I) Monomers and Dimers with Cyclopentadienyl and Indenyl Ligands. Chem.—Eur. J. 2014, 20, 5327–5337. 10.1002/chem.201305021. [DOI] [PubMed] [Google Scholar]; b Abernethy C. D.; Clyburne J. A. C.; Cowley A. H.; Jones R. A. Reactions of Transition-Metal Metallocenes with Stable Carbenes. J. Am. Chem. Soc. 1999, 121, 2329–2330. 10.1021/ja983772s. [DOI] [Google Scholar]
- Stephens D. N.; Szilagyi R. K.; Roehling P. N.; Arulsamy N.; Mock M. T. Catalytic Ammonia Oxidation to Dinitrogen by a Nickel Complex. Angew. Chem., Int. Ed. 2023, 62, e202213462. 10.1002/anie.202213462. [DOI] [PubMed] [Google Scholar]
- a Malapit C. A.; Borrell M.; Milbauer M. W.; Brigham C. E.; Sanford M. S. Nickel-Catalyzed Decarbonylative Amination of Carboxylic Acid Esters. J. Am. Chem. Soc. 2020, 142, 5918–5923. 10.1021/jacs.9b13531. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith C. J.; Early T. R.; Holmes A. B.; Shute R. E. Palladium Catalysed Cross-Coupling Reactions of Silylamines. Chem. Commun. 2004, 17, 1976–1977. 10.1039/B406868H. [DOI] [PubMed] [Google Scholar]; c Verma V.; Koperniku A.; Edwards P. M.; Schafer L. L. N -Silylamines in Catalysis: Synthesis and Reactivity. Chem. Commun. 2022, 58, 9174–9189. 10.1039/D2CC02915D. [DOI] [PubMed] [Google Scholar]
- Thorén M.; Adamson U.; Sjöberg H. E. Aminoglutethimide and Metyrapone in the Management of Cushing’s Syndrome. Acta Endocrinol. (Copenh.) 1985, 109, 451–457. 10.1530/acta.0.1090451. [DOI] [PubMed] [Google Scholar]
- Brotzel F.; Chu Y. C.; Mayr H. Nucleophilicities of primary and secondary amines in water. J. Org. Chem. 2007, 72, 3679–3688. 10.1021/jo062586z. [DOI] [PubMed] [Google Scholar]
- a Lyu X.; Zhang J.; Kim D.; Seo S.; Chang S. Merging NiH Catalysis and Inner-Sphere Metal-Nitrenoid Transfer for Hydroamidation of Alkynes. J. Am. Chem. Soc. 2021, 143, 5867–5877. 10.1021/jacs.1c01138. [DOI] [PubMed] [Google Scholar]; b Choi H.; Lyu X.; Kim D.; Seo S.; Chang S. Endo -Selective Intramolecular Alkyne Hydroamidation Enabled by NiH Catalysis Incorporating Alkenylnickel Isomerization. J. Am. Chem. Soc. 2022, 144, 10064–10074. 10.1021/jacs.2c03777. [DOI] [PubMed] [Google Scholar]
- a Hossain I.; Schmidt J. A. R. Cationic Nickel(II)-Catalyzed Hydrosilylation of Alkenes: Role of P, N-Type Ligand Scaffold on Selectivity and Reactivity. Organometallics 2020, 39, 3441–3451. 10.1021/acs.organomet.0c00551. [DOI] [Google Scholar]; b Ulm F.; Poblador-Bahamonde A. I.; Choppin S.; Bellemin-Laponnaz S.; Chetcuti M. J.; Achard T.; Ritleng V. Synthesis, Characterization, and Catalytic Application in Aldehyde Hydrosilylation of Half-Sandwich Nickel Complexes Bearing (κ 1 - C)- and Hemilabile (κ 2 - C, S)-Thioether-Functionalised NHC Ligands. Dalton Trans. 2018, 47, 17134–17145. 10.1039/C8DT03882A. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Zhou Z.; Kürti L. Direct and Stereospecific Synthesis of N-H and N-Alkyl Aziridines from Unactivated Olefins Using Hydroxylamine-O-Sulfonic Acids. Angew. Chem., Int. Ed. 2017, 56, 9886–9890. 10.1002/anie.201705530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse E. G.; Houston S. D.; Williams C. M.; Savage G. P.; Rendina L. M.; Hallyburton I.; Anderson M.; Sharma R.; Walker G. S.; Obach R. S.; Todd M. H. Nonclassical Phenyl Bioisosteres as Effective Replacements in a Series of Novel Open-Source Antimalarials. J. Med. Chem. 2020, 63, 11585–11601. 10.1021/acs.jmedchem.0c00746. [DOI] [PubMed] [Google Scholar]
- Rojas J. J.; Croft R. A.; Sterling A. J.; Briggs E. L.; Antermite D.; Schmitt D. C.; Blagojevic L.; Haycock P.; White A. J. P.; Duarte F.; Choi C.; Mousseau J. J.; Bull J. A. Amino-Oxetanes as Amide Isosteres by an Alternative Defluorosulfonylative Coupling of Sulfonyl Fluorides. Nat. Chem. 2022, 14, 160–169. 10.1038/s41557-021-00856-2. [DOI] [PubMed] [Google Scholar]
- Goldstein D. J. Duloxetine in the Treatment of Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2007, 3, 193–209. 10.2147/nedt.2007.3.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker N.; Howe C.; Glover M.; McRobbie H.; Barnes J.; Nosa V.; Parag V.; Bassett B.; Bullen C. Cytisine versus Nicotine for Smoking Cessation. N. Engl. J. Med. 2014, 371, 2353–2362. 10.1056/NEJMoa1407764. [DOI] [PubMed] [Google Scholar]
- For examples of bimetallic H2 reductive elimination, see:; a Jeon J.; Lee C.; Seo H.; Hong S. NiH-Catalyzed Proximal-Selective Hydroamination of Unactivated Alkenes. J. Am. Chem. Soc. 2020, 142, 20470–20480. 10.1021/jacs.0c10333. [DOI] [PubMed] [Google Scholar]; b Schaefer B. A.; Margulieux G. W.; Small B. L.; Chirik P. J. Evaluation of Cobalt Complexes Bearing Tridentate Pincer Ligands for Catalytic C–H Borylation. Organometallics 2015, 34, 1307–1320. 10.1021/acs.organomet.5b00044. [DOI] [Google Scholar]; c Gu N. X.; Oyala P. H.; Peters J. C. An S = 1/2 Iron Complex Featuring N2, Thiolate, and Hydride Ligands: Reductive Elimination of H2 and Relevant Thermochemical Fe–H Parameters. J. Am. Chem. Soc. 2018, 140, 6374–6382. 10.1021/jacs.8b02603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.