

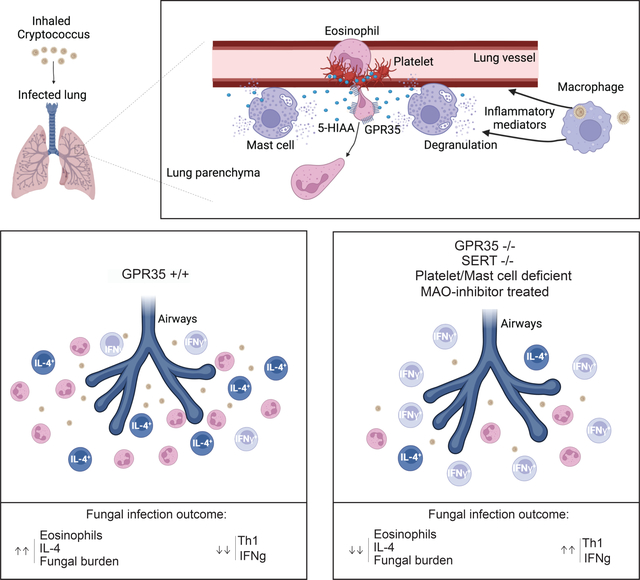
Summary
Cryptococcus neoformans is the leading cause of fungal meningitis and is characterized by pathogenic eosinophil accumulation in the context of type-2 inflammation. The chemoattractant receptor GPR35 is expressed by granulocytes and promotes their migration to the inflammatory mediator 5- hydroxyindoleacetic acid (5-HIAA), a serotonin metabolite. Given the inflammatory nature of Cryptococcal infection, we examined the role of GPR35 in the circuitry underlying cell recruitment to the lung. GPR35-deficiency dampened eosinophil recruitment and fungal growth, whereas overexpression promoted eosinophil homing to airways and fungal replication. Activated platelets and mast cells were the sources of GPR35 ligand activity and pharmacological inhibition of serotonin conversion to 5-HIAA, or genetic deficiency in 5-HIAA production by platelets and mast cells resulted in more efficient clearance of Cryptococcus. Thus, the 5-HIAA-GPR35 axis is an eosinophil chemoattractant-receptor system that modulates the clearance of a lethal fungal pathogen, with implications for the use of serotonin metabolism inhibition in the treatment of fungal infections.
In Brief:
Cryptococcus neoformans infection, a leading cause of fungal meningitis, involves pathogenic eosinophil accumulation. De Giovanni et al. reveal that the chemoattractant receptor GPR35 and its platelet- and mast cell-derived ligand 5-hydroxyindoleacetic acid (5-HIAA), a serotonin metabolite, promotes eosinophil recruitment to the infected lung and exacerbation of disease, Modifiers of serotonin metabolism may be useful in the treatment of fungal infections.
Graphical Abstract:
Introduction
Fungal pathogens have been described as ‘the next deadly plague’. They are responsible for 75,000 hospitalizations in the US each year. An estimated 300 million people are infected with fungal disease worldwide with 2.3 million deaths every year, a number comparable to deaths from Tuberculosis and Malaria (1, 2). The lack of effective diagnostics, therapies and vaccines is largely responsible for the high mortality (3). C. neoformans, a common cause of lethal meningitis in immunocompromised patients, is responsible for ~200,000 deaths annually worldwide, mostly in the AIDS population (1, 4). The infectious process begins when the host inhales desiccated cryptococcal yeast or spores. Some particles find their way into the alveoli where the resident cells of the innate immune system respond to invading pathogens (5).While the majority of infections are asymptomatic and limited to the lungs, in immunocompromised patients C. neoformans may enter the circulation, leading to disseminated disease, including meningoencephalitis which is uniformly lethal in the absence of treatment (4).
Resistance against C. neoformans primarily involves immune effector mechanisms and T helper (Th) cells are key players (6–8). Whereas IL-12–dependent Th1 responses are protective (6, 7, 9), Th2 cells producing IL-4, IL-13, and IL-5 are detrimental to protection (10–14). Pulmonary eosinophilia is a common manifestation of the host response during fungal infection, but it is generally associated with non-protective Th2 immunity (11, 14–17). Indeed, susceptible C57BL/6 mice have many eosinophils in their lungs, moderately resistant BALB/c mice have transient influx of eosinophils, and highly resistant CBA/J mice show only a few eosinophils in their lungs (11). Eosinophils store preformed IL-4 within intracellular granules that are rapidly secreted upon cell activation (18), and eosinophil-derived IL-4 contributes to the development of Th2 cells in allergic disorders (19) and fungal infections (14, 15, 17, 20). In accordance with these observations, one study found that eosinophil-deficient mice are resistant to C. neoformans infection and show reduced Th2 responses (14).
The receptor CCR3 and its eotaxin ligands play a critical role in eosinophil recruitment to the lung in allergy models (21–23), but their role during fungal infections remains to be clarified. Platelets (24) (25) and mast cells (2, 5) are activated upon Cryptococcal infection and communicate with eosinophils to sustain their activation, recruitment and survival through the production of several mediators including eotaxin(s) (26, 27). However, reductions or increases in recruited eosinophils do not necessarily correlate with differences in eotaxin levels (16, 28) suggesting other mechanisms could shape eosinophil recruitment in some contexts. Thus, despite the strong evidence that eosinophils have a detrimental effect on clearance of Cryptococcus from the lungs, the mechanisms responsible for eosinophil recruitment to the infected lung are not well understood.
The G-protein coupled receptor GPR35 is widely expressed by myeloid cells in humans and mice. In recent work, we established that the serotonin metabolite 5-HIAA functions as a physiological GPR35 ligand (29). Expression of Gpr35 is increased in activated neutrophils and the 5-HIAA – GPR35 ligand-receptor system contributes to recruitment of neutrophils to the inflamed peritoneum and skin (29). Platelets and mast cells were identified as important sources of 5-HIAA in these inflamed tissues. Work in vitro and in zebrafish has suggested GPR35 may also function as chemoattractant or adhesion-promoting receptor in monocytes and macrophages (30), but how widely this is the case and whether GPR35 functions as a recruitment receptor in other myeloid cell types is not clear. Moreover, given the multiple and often distinct sets of chemoattractants induced under different inflammatory conditions and in different tissues, it is currently difficult to predict which chemoattractants will be most important in a given disease setting.
Here we examined the impact of GPR35 in the response to pulmonary C. neoformans infection. GPR35 was highly expressed in activated eosinophils and expression in eosinophils contributed to their entry to the infected lung and thereby to type 2-skewing of the effector T cell response and less efficient fungal clearance. 5-HIAA production by platelets and mast cells promoted eosinophil recruitment from blood vessels into the lung parenchyma. Our findings identify 5-HIAA and GPR35 as a chemoattractant receptor system acting on eosinophils to promote their recruitment to the lungs, and demonstrate the contribution of this recruitment system to fungal pathogenesis.
Results
GPR35 expression in bone marrow-derived cells supports eosinophil recruitment and fungal infection
To test whether GPR35 has a role in the immune response to respiratory C. neoformans infection, Gpr35+/− and Gpr35−/− mice were infected intranasally with 1×105 colony forming units (CFU) of the KN99 strain. Measurements of lung and spleen fungal CFU 11 days later revealed a significant protective effect of GPR35-deficiency (Fig. 1A, B). Histological analysis showed that GPR35 −/− mice had reduced lung pathology (Fig. S1A, B). GPR35 is expressed by a range of non-hematopoietic as well as hematopoietic cells (29, 31–34). To determine if the susceptibility-promoting effect of GPR35 reflected action in hematopoietic cells, irradiated WT mice that had been reconstituted with GPR35+/− or GPR35−/− bone marrow (BM) were intranasally infected with C. neoformans. Again, GPR35-deficiency was associated with a lower fungal burden (Fig. 1C, D). These data indicate that GPR35 in a hematopoietic cell type negatively impacts the ability to clear Cryptococci from the lung, resulting in increased systemic spread of the pathogen. Analysis of inflammatory cell accumulation in the inflamed lung revealed few neutrophils were present on day 11 as expected and these were unaffected by GPR35-deficiency (Fig. S1C). Flow cytometric analysis showed GPR35 was minimally expressed in neutrophils from infected mice (Fig. S1D). Monocyte and alveolar macrophage frequencies were similarly unaffected by GPR35-deficiency (Fig. S1E, F). In contrast, eosinophil frequencies and numbers were significantly reduced in the lungs of infected GPR35−/− mice (Fig. 1E–H). Eosinophil frequencies in BM and blood at steady state were unaltered by GPR35-deficiency (Fig. S1G, H). Since it was possible that the reduced lung eosinophil number was secondary to the reduced CFU burden at day 11 we next examined mice at an earlier time point after infection. At day 4 post-infection, lung fungal burden in WT and GPR35−/− mice was similar but eosinophils counts were significantly lower in GPR35−/− mice (Fig. 1I, J). Analysis of neutrophil recruitment at an early time point showed it was not significantly altered by GPR35-deficiency (Fig S1I). Thus, GPR35 deficiency led to less eosinophil accumulation in the lung prior to any effect on lung fungal burden.
Figure 1. GPR35 in BM-derived cells promotes increased Cryptococcus burden and eosinophil recruitment to the infected lung.
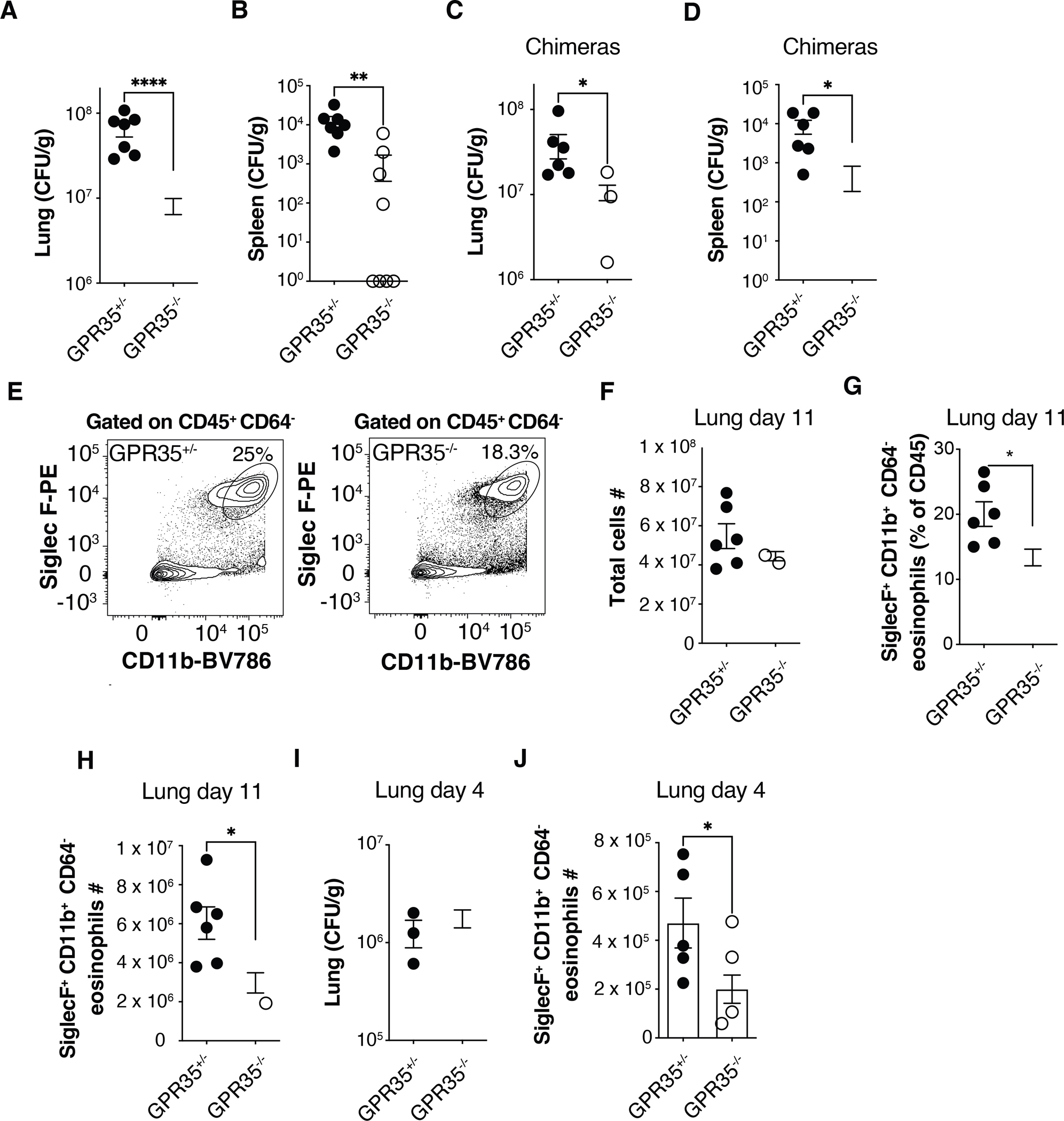
(A-D) Quantification of C. neoformans CFUs in lung and spleen of GPR35+/− and GPR35−/− mice (A, B) or full chimeras (C, D) 11 days after intranasal infection. A, B, n=7–9; C, D, n=6. Data are pooled from 2 independent experiments. (E) Flow cytometry plots showing percentages of SiglecF+ CD11b+ eosinophils in GPR35+/− (left) and GPR35−/− (right) out of CD45+ CD64− cells in the lung. (F-H) Quantification of total cell numbers (F), SiglecF+ CD11b+ CD64− eosinophil percentages (out of CD45+ cells, G) and numbers (H) in the lung of GPR35+/− and GPR35−/− full chimeras 11 days after intranasal infection. n=6. Data are pooled from two independent experiments. (I, J) Quantification of C. neoformans CFUs (I) and SiglecF+ CD11b+ CD64− eosinophil numbers (J) in lungs of GPR35+/− and GPR35−/− mice 4 days after intranasal infection. F-H, n=6; I, n=3–5; J=5–7. Data are pooled from two independent experiments. * p<0.05; ** p<0.005; **** p<0.0001. Data are presented as mean ± SEM. See also Figure S1.
Requirement for GPR35 in eosinophils
To test whether GPR35 was required intrinsically in eosinophils for their accumulation in the lung we generated and infected GPR35−/− + WT (~1:1) mixed BM chimeras. Analysis at day 4 and 11 post-infection revealed a selective deficiency in GPR35−/− eosinophil recruitment in the lung (Fig. 2A, B and Fig. S1J, K). Hematopoietic chimerism in the reconstituted mice was determined using B cells since this cell lineage has not been found to express GPR35. As a further approach to test the eosinophil intrinsic requirement of GPR35, we used an in vitro BM culture system to generate WT and GPR35−/− eosinophils (35). One day after transfer into mice previously infected for 5 days, we observed a significant deficit in GPR35−/− eosinophil recruitment to the infected lung (Fig. 2C, D). By in vivo antibody pulse-labeling of blood-exposed cells (36), we found increased percentages of intravascular GPR35-deficient compared to WT eosinophils indicating less efficient extravasation into the lung parenchyma (Fig. 2E, F).
Figure 2. Intrinsic requirement for GPR35 in eosinophils.
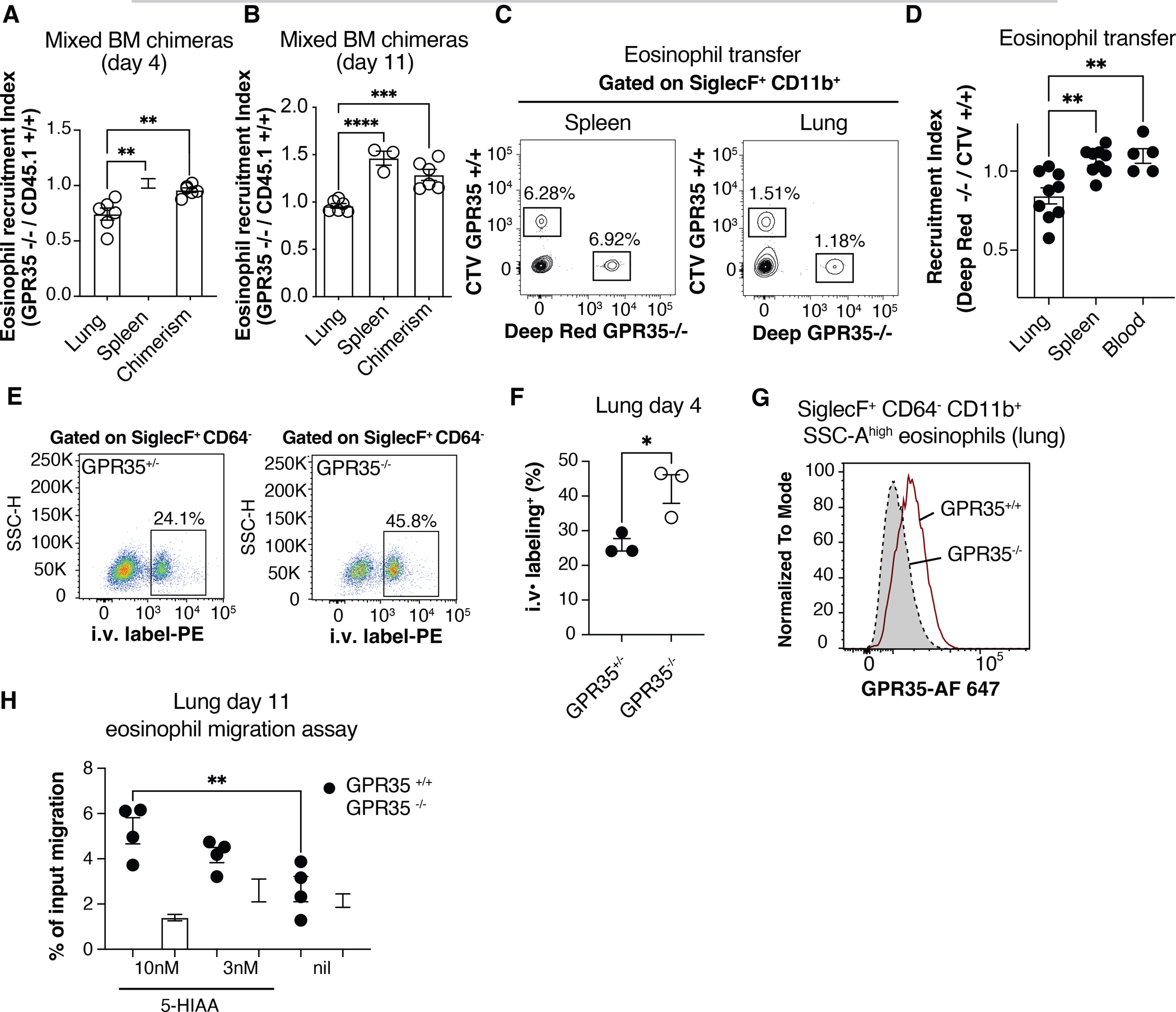
(A, B) Quantification of eosinophil recruitment index in lung and spleen of C. neoformans infected CD45.2+ GPR35−/− + CD45.1+ GPR35+/+ mixed chimeras, 4 (A) or 11(B) days after intranasal infection. The chimerism was calculated as the CD45.2+ / CD45.1+ ratio within blood B220+ cells at the time of tissue isolation. n=3–6. Data are pooled from 2 independent experiments. (C) Flow cytometry plots showing transferred CTV+ GPR35+/+ and Deep Red+ GPR35−/− BM-derived eosinophils in spleen and lung of mice infected 5 days before with C. neoformans, 24 hours after cell injection. (D) Quantification of transferred eosinophils determined as in C, shown as recruitment index in lung, spleen and blood of infected mice, 24 hr after cell transfer. n=5–9. Data are pooled from three independent experiments. (E, F) Flow cytometry plots (E) and quantification (F) of 2 min intravascular labeled (i.v. CD45-PE+) SiglecF+ CD64− eosinophils in lungs of GPR35+/− (left) and GPR35−/− (right) mice 4 days after C. neoformans infection. n=3. Data are representative of two independent experiments. (G) Flow cytometry plot showing GPR35 expression in GPR35+/+ and GPR35−/− SiglecF+ CD11b+ CD64− eosinophils in the lungs 11 days after intranasal infection. Data are representative of at least 2 independent experiments. (H) Transwell migration assay to 5-HIAA (nM) of GPR35+/+ and GPR35−/− eosinophils isolated from C. neoformans infected lung 11 days after infection. Nil indicates no added chemoattractant. n=3–4. Data are representative of 3 independent experiments. * p<0.05; ** p<0.005; *** p<0.0005; **** p<0.0001. Data are presented as mean ± SEM. See also Figure S1.
Using an antibody specific for the GPR35 C-terminus and intracellular staining, GPR35 could be detected in eosinophils from the infected lung (Fig. 2G) but not in eosinophils from the BM of control mice (Fig. S1L). Using a transwell migration assay, we found that GPR35+/+ but not GPR35−/− eosinophils from lungs of C. neoformans infected mice migrated to low nanomolar concentrations of 5-HIAA (Fig. 2H). WT and GPR35−/− cells were similar in their migratory responses to CCL11 (eotaxin-1) and CXCL12 (Fig. S1M). Eosinophil migration to 5-HIAA was inhibited by pretreatment with pertussis toxin, consistent with GPR35 signaling in these cells involving Gαi-containing heterotrimeric G-proteins (Fig. S1N). These findings demonstrate GPR35 acts in activated eosinophils to promote their migration towards 5-HIAA and their accumulation in the infected lung.
GPR35 over-expression augments Cryptococcus accumulation and eosinophil recruitment
The above findings indicated that GPR35 is necessary for augmenting C. neoformans infection and for promoting eosinophil recruitment. To ask whether the receptor is sufficient to have these effects we generated chimeras using BM cells that had been transduced with a GPR35-eGFP encoding or control retroviral construct. The efficiency of reconstitution with transduced (eGFP reporter-expressing) hematopoietic cells was similar for recipients receiving either construct (Fig. S2). Analysis of the BM chimeras 9 and 11 days after C. neoformans infection revealed a higher fungal burden in the GPR35-over-expressing mice (Fig. 3A, B), and a strong enrichment for GPR35 overexpressing eosinophils in the lungs (Fig. 3C–E). Thus, while the retroviral transduction system does not allow us to restrict the increased GPR35-expression to eosinophils, the findings to this point indicate that GPR35 expression is both necessary and limiting for tissue fungal burden and eosinophil accumulation.
Figure 3. GPR35 over-expression augments Cryptococcus infection and eosinophil recruitment.
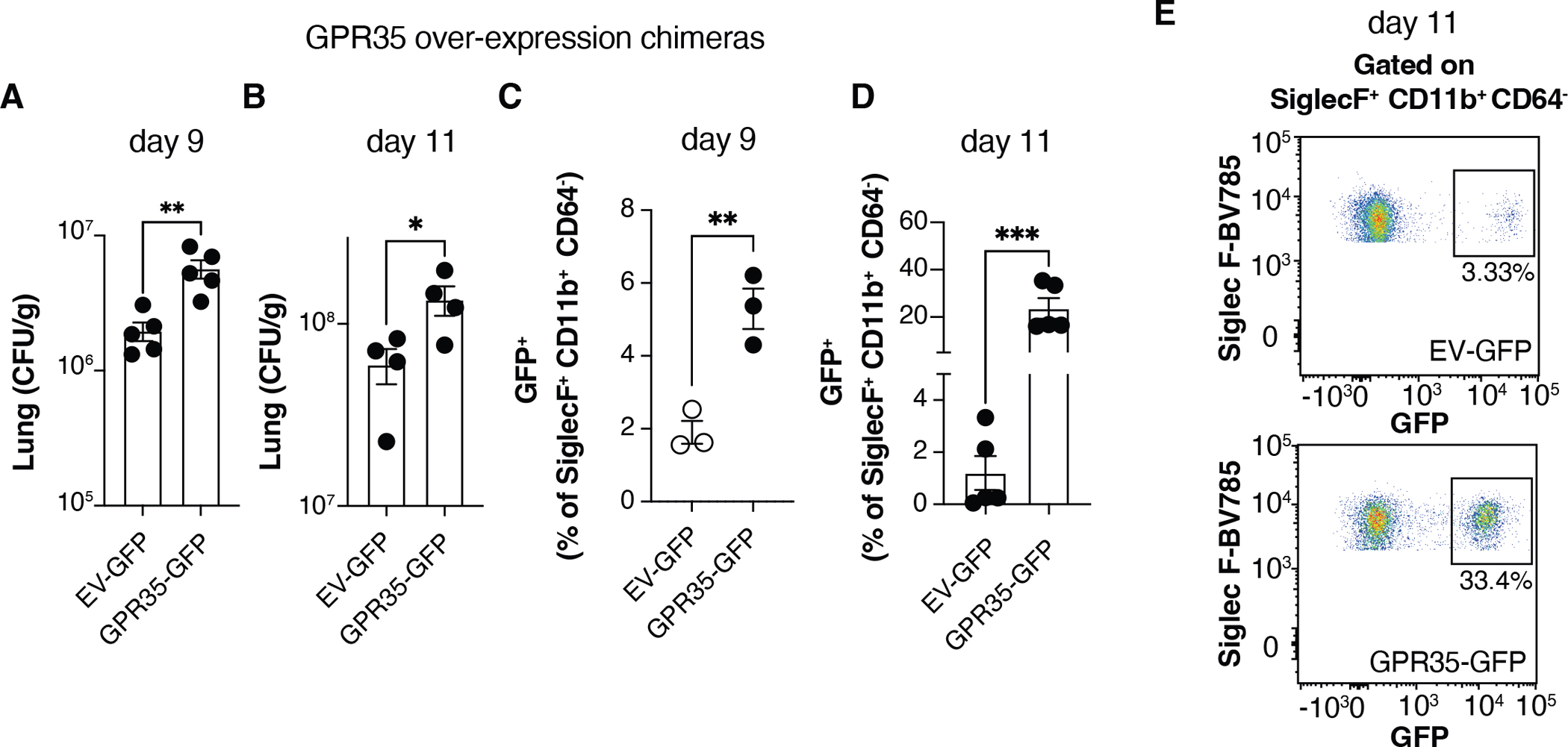
(A-D) Quantification of C. neoformans CFUs (A, B) and GFP+ eosinophil percentages (C, D) in empty vector (EV)-GFP or GPR35-GFP overexpressing chimeras, 9 (A, C) or 11 (B, D) days after intranasal infection. A, B, D, n=4–5; C, n=3. E. Flow cytometry plots showing GFP+ percentages within SiglecF+ CD11b+ CD64− eosinophils quantified in D. Data are pooled (A) or representative (B-D) of two independent experiments. * p<0.05; ** p<0.005; *** p<0.0005. Data are presented as mean ± SEM. See also Figure S2.
GPR35 expressing eosinophils sustain Cryptococcus infection
The transcription factor GATA1 is critical for eosinophil differentiation and mice with a mutation in the GATA1 promoter (referred to as ΔdblGATA) have a selective deficiency in eosinophils (37, 38). To test the importance of eosinophil GPR35 expression in promoting C. neoformans infection of the lung we generated GPR35−/− + ΔdblGATA (1:1) mixed BM chimeras. In these mice, all the eosinophils lack GPR35 whereas all other hematopoietic lineages are ~50% derived from WT BM. Analysis of the BM chimeras at day 11 post-infection showed that selective GPR35-deficiency in eosinophils led to a similar reduction in lung CFU as in mice fully deficient in hematopoietic GPR35 (Fig. 4A). These chimeras were also used to confirm the intrinsic role of GPR35 in eosinophil recruitment to the lung and the block in eosinophil development in full GATA1-deficient BM chimeras (Fig. 4B). Notably, eosinophil deficient ΔdblGATA BM chimeras had a similar reduction in lung CFU as mice lacking GPR35 in eosinophils (Fig. 4A) suggesting GPR35 is critical for the disease-promoting activity of the eosinophils in this infection. We also tested for a possible role of GPR35 in dendritic cells (DC) by generating mixed CD11c-DTR + GPR35−/− mixed BM chimeras and treating with diphtheria toxin (DT) prior to C. neoformans infection. GPR35-deficiency in DCs (and other cells sensitive to CD11c-DTR mediated ablation) did not alter the susceptibility to lung C. neoformans infection (Fig. S3A, B).
Figure 4. GPR35 expressing eosinophils sustain Cryptococcus infection.
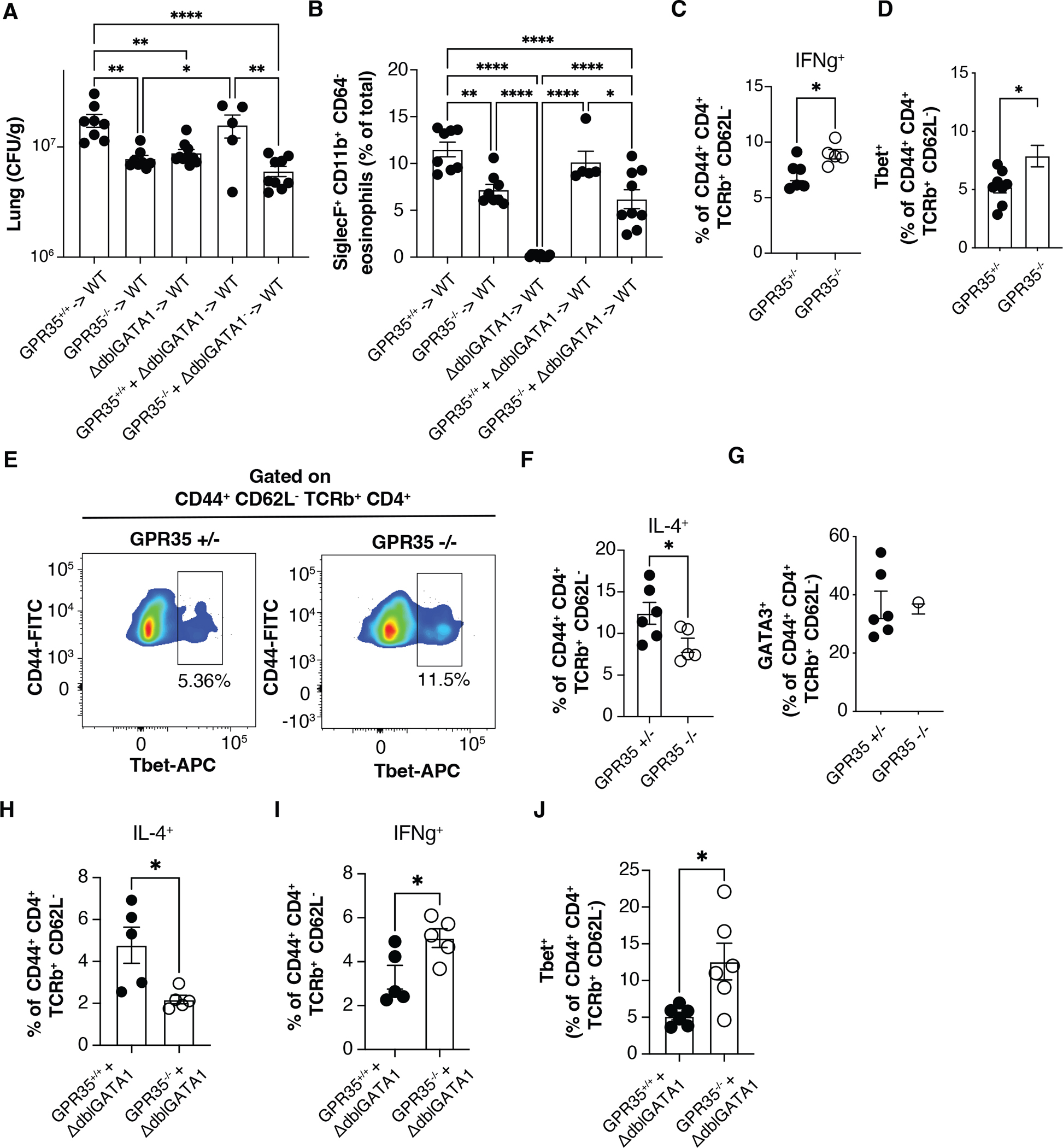
(A, B) Quantification of C. neoformans CFUs (A) and SiglecF+ CD11b+ CD64− eosinophil percentages (B) in the lung of the indicated full and mixed BM chimeras 11 days after intranasal infection. n=5–10. Data are pooled from two independent experiments. (C, D) Quantification of ex-vivo stimulated IFNγ+ cells (C) and Tbet+ Th1 cell percentages (D) out of CD4+ CD44+ TCRβ+ CD62L in the lung of GPR35+/+ and GPR35−/− full chimeras 11 days after intranasal infection. C, n=5–6; d, n=8–9. Data are pooled from 3 (C) or 2 (D) independent experiments. (E) Flow cytometry plot showing Tbet+ Th1 percentages in GPR35+/− (left) and GPR35−/− (right) infected mice quantified in D. (F, G) Quantification of ex-vivo stimulated IL-4+ cell percentages (F) and GATA3+ Th2 cell percentages (G) out of CD4+ CD44+ TCRβ+ CD62L− cells in the lung of GPR35+/+ and GPR35−/− mice 11 days after intranasal infection. n=5–6. (H-J) Quantification of ex-vivo stimulated IL-4+ (H), IFNγ+ (I) and Tbet+ (J) cells out of CD4+ CD44+ TCRβ+ CD62L− cells in the lung of GPR35−/− + ΔdblGATA and control mixed BM chimeras, 11 days after intranasal infection. n=5–6. Data are pooled from two independent experiments. * p<0.05; ** p<0.005; **** p<0.0001. Data are presented as mean ± SEM. See also Figures S3 and S4.
Increased lung Th1 response in the absence of GPR35
Previous studies have established that eosinophils amplify the generation of IL-4 producing T cells in the C. neoformans infected lung, a polarizing activity that is associated with less effective fungal clearance (14, 15, 17, 20). To determine if the reduced eosinophil recruitment caused by GPR35-deficiency led to an altered polarization of the T cell response, we examined the properties of effector CD4 T cells in the lungs at day 11. Infected GPR35-deficient mice had an increase in IFNγ cytokine expression by CD4 T cells and an increased frequency of Tbet+ cells (Fig. 4C–E and Fig. S4A). Importantly, the frequency of IL-4-expressing CD4 T cells was significantly reduced (Fig. 4F and Fig. S4A). The frequency of GATA3+ T cells was unchanged (Fig. 4G), suggesting that GATA3+ Th2 cell generation in lymph nodes was unaffected and that eosinophils promoted upregulation of IL-4 expression in effector CD4 T cells in the lung. To determine if the action of GPR35 in promoting Th2 responses was within eosinophils we generated 1:1 mixed GPR35−/− + ΔdblGATA BM chimeras. At day 11 after infection, lung CD4 T cells in GPR35−/− + ΔdblGATA BM chimeras showed less expression of IL-4 and increased IFNγ (Fig. 4H, I) and there was an increase in the frequency of Tbet+ T cells (Fig. 4J). These findings establish the importance of GPR35-mediated eosinophil recruitment in favoring a Th2 effector response in the lung.
5-HIAA is required for GPR35-mediated eosinophil recruitment to the infected lung
The GPR35 ligand 5-HIAA was strongly upregulated in the lungs at days 4 and 11 after infection (Fig. 5A). To test the importance of 5-HIAA in eosinophil recruitment to the C. neoformans infected lung, mice were treated with phenelzine, a monoamine oxidase (MAO) inhibitor that prevents serotonin metabolism to 5-HIAA (29, 39), starting 3 days before infection. Phenelzine treatment led to a reduction in WT but not GPR35-deficient eosinophil recruitment at day 4 post-infection (Fig. 5B). Phenelzine treatment also reduced eosinophil numbers in the lung at day 11 and lowered lung fungal burden (Fig. 5C, D). ELISA analysis confirmed that lung 5-HIAA abundance was significantly reduced by the treatment (Fig. S4B).
Figure 5. 5-HIAA is required for GPR35-mediated eosinophil recruitment to the infected lung.
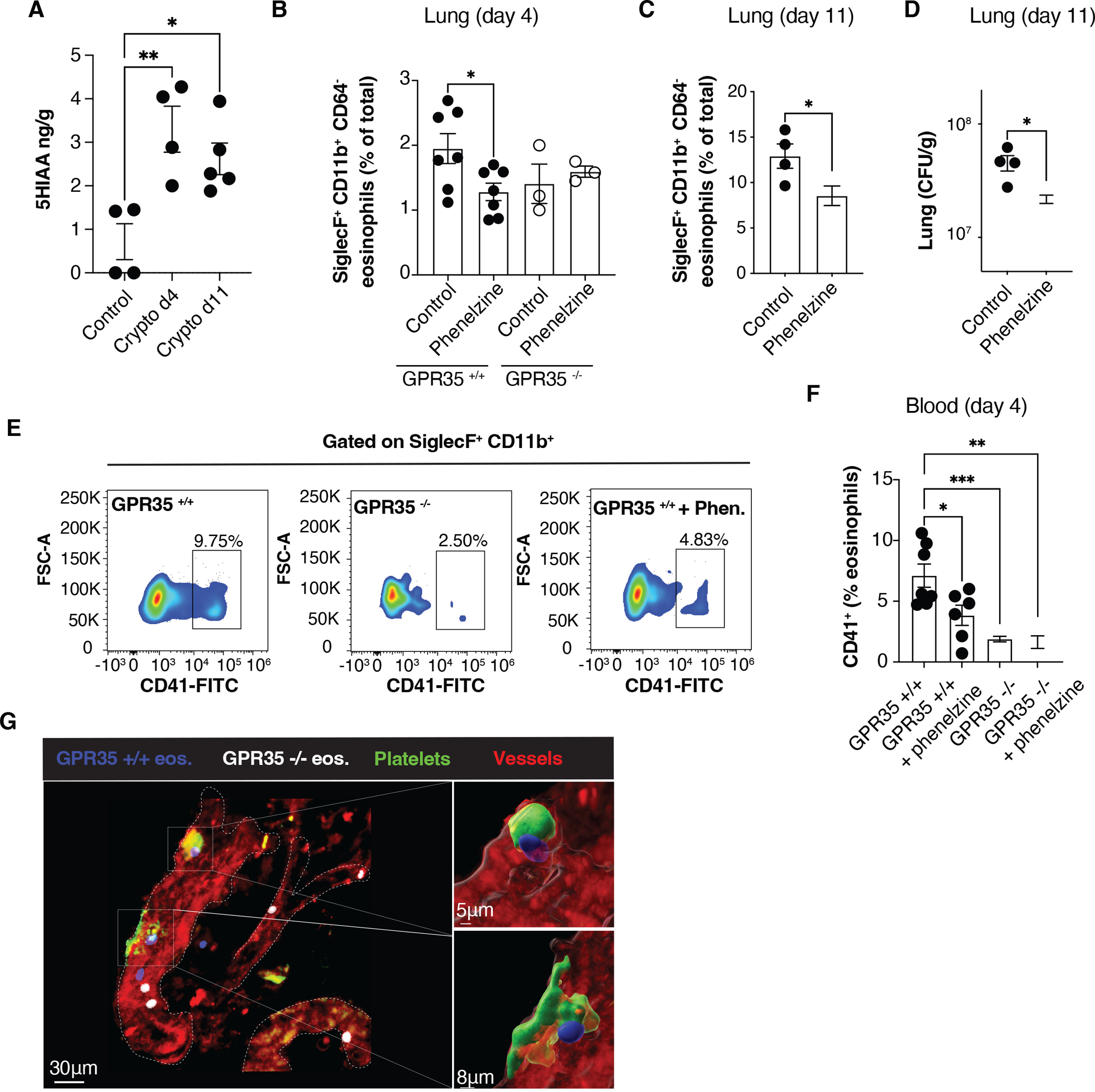
(A) Quantification by ELISA of 5-HIAA concentrations in the lung of uninfected mice or C. neoformans infected mice (4 or 11 days after infection). n=4–5. Data are pooled from two independent experiments. (B-D) Quantification of SiglecF+ CD11b+ CD64− eosinophil percentages (B, C) and C. neoformans CFUs (D) in the lung of GPR35+/+ and GPR35−/− mice not treated or treated with phenelzine, 4 (B) or 11 (C, D) days after intranasal infection. B, n=3–7; C, D, n=4. Data are pooled from two independent experiments. e, f. Flow cytometry plots (E) and quantification (F) of CD41+ SiglecF+ CD64− eosinophils in the blood of not treated or phenelzine-treated GPR35+/+ and GPR35−/− mice, 4 days after intranasal infection. n=3–7. Data are pooled from two independent experiments. (G) Multiphoton micrograph of C. neoformans infected lung from platelet-reporter mice (Pf4-Cre x mTmG mice), 4 days after intranasal infection. Transferred BM-derived GPR35+/+ (CTV+, blue) and GPR35−/− (Deep Red+, white) eosinophils, and endogenous platelets (green) and vessels (red) are shown. Data are representative of at least 2 independent experiments. * p<0.05; ** p<0.005; *** p<0.0005. Data are presented as mean ± SEM.
Activated platelets are a source of 5-HIAA and previous work has shown that platelets can contribute to eosinophil recruitment to sites of inflammation (40, 41). As a measure of the extent of intravascular platelet-eosinophil interaction following infection, we assessed eosinophil acquisition of the platelet marker CD41 (29, 42). Compared to infected WT mice there was an approximately 4-fold reduction in the frequency of CD41+ eosinophils in infected GPR35−/− mice (Fig. 5E, F). Phenelzine treatment also led to a reduction in the CD41+ eosinophil frequency in WT mice (Fig. 5E, F). In accord with phenelzine acting by reducing GPR35 ligand production, the drug had no effect on the frequency of CD41+ eosinophils in GPR35-deficient mice (Fig. 5F).
To examine the impact of GPR35 on tissue eosinophils and platelet interactions, we performed multiphoton imaging of lung from day 4 infected mTmG PF4-Cre mice that: 1) harbor GFP+ platelets and have strong tdTom expression in endothelium, and 2) received a mixture of WT and GPR35−/− BM-derived eosinophils 6 hrs earlier. These studies revealed examples of WT eosinophils (blue) interacting with platelet clusters (Fig. 5G and Movies S1 and S2) while GPR35−/− eosinophils (white) in the same vessels showed less association with the platelet clusters (Fig. 5G and Movies S1 and S2).
To quantitively assess the contribution of platelets and mast cells to eosinophil recruitment, we infected mice lacking either of these cell types. At day 4 post-infection both platelet-deficient mice and mast cell-deficient mice showed reduced eosinophil recruitment in lungs and a higher fraction of eosinophils within the blood, similarly to GPR35−/− mice (Fig. 6A–C). These data are consistent with platelets and mast cells augmenting the post-attachment transendothelial migration step. In addition, reduced eosinophil recruitment correlated with a reduction in CFUs at day 11 in the absence of platelets or mast cells (Fig. 6D). Furthermore, GPR35 influence on eosinophil recruitment was lost in the absence of platelets or mast cells (Fig. 6E, F), consistent with the conclusion that platelet- and mast cell-derived 5-HIAA sustains eosinophil recruitment to infected lungs. ELISA analysis confirmed that lung 5-HIAA abundance was significantly reduced by platelet deficiency and mast cell deficiency (Fig. S4C).
Figure 6. Platelets and mast cells are required for GPR35-mediated eosinophil recruitment to the infected lung.
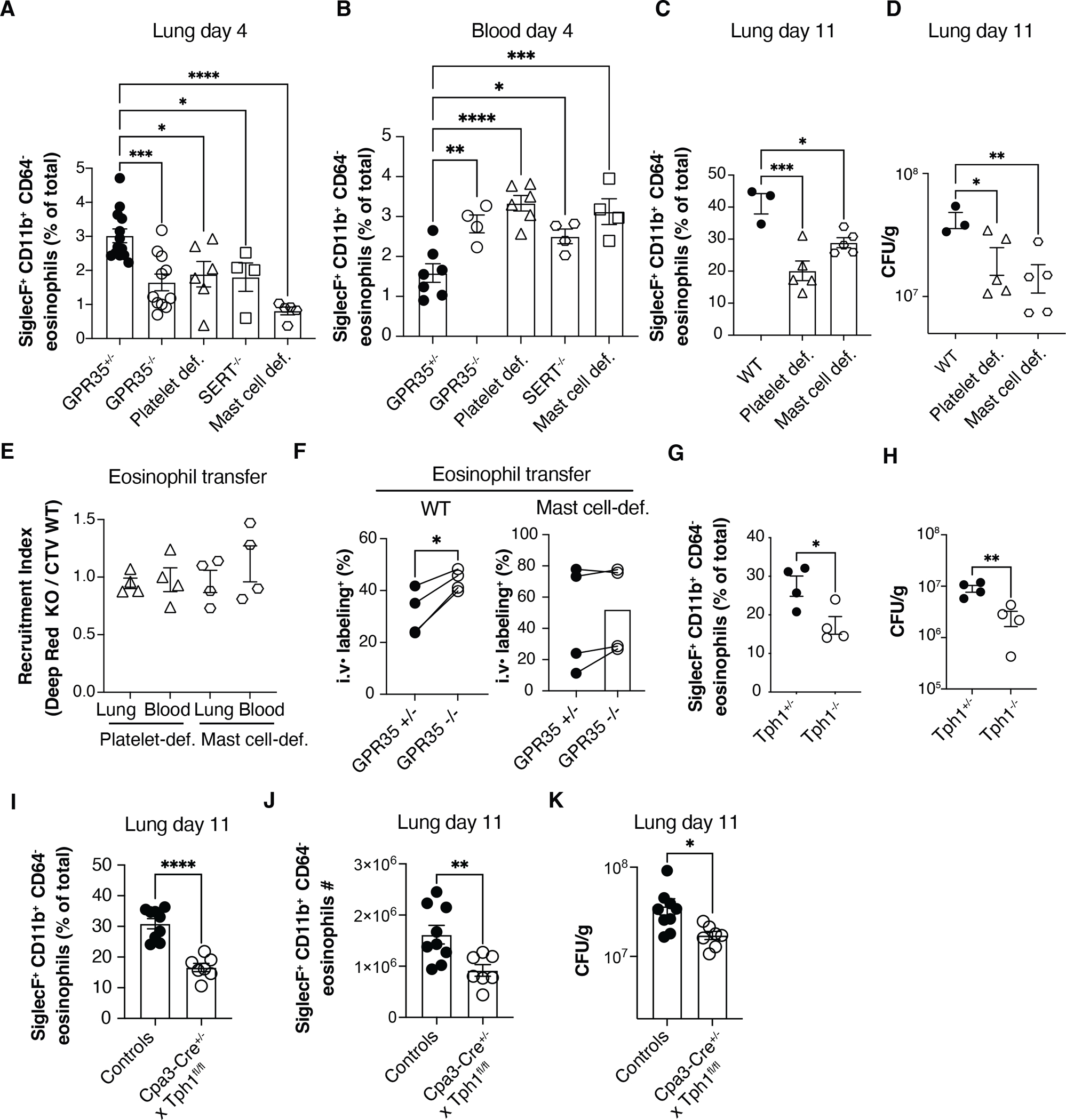
(A, B) Quantification of SiglecF+ CD11b+ CD64− eosinophil percentages in lungs (A) and blood (B) of C. neoformans infected GPR35+/+, GPR35−/−, platelet-deficient (cmlp−/−), SERT−/− and mast cell-deficient (KitW/v) mice, 4 days after intranasal injection. A, n=4–13; B, n=4–7. (C, D) Quantification of SiglecF+ CD11b+ CD64− eosinophil percentages (C) and Cryptococcus CFUs (D) in the lung of WT, platelet-deficient or mast cell-deficient mice, 11 days after intranasal infection. n=3–5. Data are pooled from two independent experiments. (E, F) Quantification of transferred BM-derived CTV+ GPR35+/+ and Deep Red+ GPR35−/− eosinophil recruitment index (E) and intravascular labeling (F), in the lung and blood of C. neoformans infected (day 5) WT, platelet-deficient and/or mast cell-deficient mice, 24 hours after cell transfer. n=4. (G, H) Quantification of SiglecF+ CD11b+ CD64− eosinophil percentages (G) and C. neoformans CFUs (H) in the lung of Tph1+/− and Tph1−/− full BM chimeras, 11 days after intranasal infection. n=4. (I-K) Quantification of SiglecF+ CD11b+ CD64− eosinophil percentages (I), absolute numbers (J) or Cryptococcus CFUs in the lung (K) of Cpa3-Cre+ x Tph1fl/fl mice and littermate controls (Cpa3-Cre+ Tph1fl/wt and Cpa3-Cre− x Tph1fl/fl) 11 days after intranasal infection. n=7–9. * p<0.05; ** p<0.005; *** p<0.0005, **** p<0.0001. Data are pooled from at least 2 independent experiments. Data are presented as mean ± SEM.
The serotonin transporter (SERT) is important for platelets to acquire serotonin, and thus SERT−/− mice lack MAO-generated 5-HIAA in their platelets (43). At day 4 post-infection SERT−/− mice showed a similar reduction in lung eosinophils as GPR35-deficient and platelet deficient mice (Fig. 6A) and they reciprocally showed a similar increase in blood eosinophils (Fig. 6B). At day 11, SERT−/− mice continued to show lower lung eosinophils (Fig. S4D) and CFUs were reduced (Fig. S4E). In contrast to platelets, mast cells express Tryptophan hydroxylase-1 (Tph1) enabling them to synthesize serotonin from tryptophan; they also express MAO (Immgen.org) and convert serotonin to 5-HIAA (29, 44–47). Mast cells are the main Tph1+ hematopoietic cell type (Immgen.org). In WT mice that had been reconstituted with Tph1-deficient BM such that their mast cells lacked the ability to generate 5-HIAA, there was less efficient eosinophil recruitment to the C. neoformans infected lung and reduced fungal CFUs (Fig. 6G, H). To test the importance of 5-HIAA generation in mast cells more definitively, we crossed Tph1 floxed mice with Cpa3-Cre mice that express Cre selectively in mast cells and a fraction of basophils (48). Analysis of Tph1f/f Cpa3-Cre mice at day 11 after C. neoformans infection showed the mice had fewer lung eosinophils (Fig. 6I, J) and a lower lung fungal burden (Fig. 6K). Taken together, these findings are consistent with platelets and mast cells cooperating in promoting GPR35+ eosinophil recruitment to the infected lung through production of 5-HIAA.
Discussion
Precise, selective control of immune cell recruitment is critical for determining the outcome of inflammation. Here we present evidence that 5-HIAA is a novel driver of eosinophil recruitment to the lung during the initial pulmonary phase of Cryptococcal infection. We propose a model whereby fungal infection causes lung platelet and mast cell activation and release of 5-HIAA. GPR35hi eosinophils that have attached to lung blood vessels encounter the 5-HIAA and it cooperates with other locally produced factors to augment eosinophil adhesion in association with platelet clusters. 5-HIAA emanating from perivascular mast cells then helps promote eosinophil extravasation into the tissue. We present findings with GPR35-deficient and over-expressing eosinophils, platelet and mast cell deficient mice, and mice unable to generate 5-HIAA in platelets or mast cells, that support this model. Furthermore, we show that GPR35-deficiency causes a shift towards a type I immune response and improved fungal clearance. Tissue eosinophils produce mediators, including IL-4, that contribute to type II skewing of the immune response. It has been speculated (49) that C. neoformans evolved to promote a type II immune response as a mechanism of evading the type I response that is more effective at killing intracellular organisms. We suggest that by augmenting eosinophil recruitment to the lung, 5-HIAA and GPR35 favor a type II immune response and thus contribute to the C. neoformans immune evasion mechanism. An implication of these findings is that agents that reduce 5-HIAA or antagonize GPR35 might have therapeutic benefit in C. neoformans infections.
Previous work established a role for GPR35 and 5-HIAA in neutrophil recruitment to the inflamed peritoneum and inflamed skin and lymph nodes (29). The lack of an effect of GPR35-deficiency on neutrophil recruitment to the C. neoformans infected lung may reflect a lack of GPR35-induction on neutrophils during Cryptococcal infection and possible redundancy with other inflammation-induced neutrophil chemoattractants. Multiple chemoattractants have also been defined for eosinophils, including eotaxins (CCL11, CCL24 and CCL26), CCL5, CXCL12, prostaglandins and oxysterols (27, 50). It is thus striking that GPR35 makes a non-redundant contribution to eosinophil recruitment to the Cryptococcus infected lung. It will be important in future work to define the signals promoting GPR35 upregulation in activated eosinophils and to examine the contribution of 5-HIAA to their recruitment during different inflammatory insults in the lung and other tissues.
Platelets have a well-established role in augmenting neutrophil attachment to and transmigration across inflamed endothelium (51). Several studies have also shown platelets promoting eosinophil accumulation in inflamed tissues, including the lung (40, 41). Multiple platelet-derived factors are likely to be involved in engaging eosinophils, including P-selectin, integrin aIIbB3, platelet factor-4 (PF4), CCL5 and platelet activating factor (PAF) (27, 40, 41). We suggest that 5-HIAA co-operates with other platelet-derived factors to promote eosinophil adhesive interactions with endothelium-attached activated platelet clusters. This attachment is expected to augment subsequent transendothelial migration. In accord with our imaging and functional data, activated platelets in the lung vasculature have been observed in several inflammatory conditions (40) including in the lungs of COVID19 patients (52). Our finding of a new platelet-derived mediator contributing to eosinophil recruitment to the lung highlights the diverse roles of platelets in shaping the lung inflammatory cell recruitment process during infection.
Peri-vascular mast cells are numerous in the lung and skin (53–57). The stimuli promoting mast cell activation during fungal infection are not well studied but may include β-glucans and ligands for TLR4 and CCR1 (3, 49). Like platelets, mast cells release chemoattractive mediators, including PAF and CCL5, that can act on eosinophils (27, 57). However, in contrast to their production of neutrophil-attracting chemokines, they have not yet been shown to produce the best-defined eosinophil chemoattractants, the eotaxins CCL11, CCL24 and CCL26. Mast cell mediators, including histamine and serotonin, cause changes in the endothelium that facilitate platelet and eosinophil attachment and cell transmigration. 5-HIAA is likely to cooperate with other mast cell-derived factors in promoting eosinophil recruitment into the infected lung.
Our work was all performed in the mouse and future studies will be needed to test the function of GPR35 in human eosinophils. Possibly in accord with GPR35 contributing to Cryptococcal pathogenesis in humans, a GPR35 nonsense mutation predicted to cause C-terminus truncation and hypothetically increased receptor activity has been identified in a patient with non-HIV cryptococcal meningoencephalitis (58). While GPR35 and 5-HIAA action in mouse eosinophils has an adverse effect on the response to C. neoformans, we anticipate that this chemoattractant-receptor system will play critical roles in protection from other lung pathogens, especially in cases where eosinophils or neutrophils are required for protection. An important question that emerges from this study is whether clinically approved drugs that block serotonin uptake and thereby deplete platelets of serotonin and 5-HIAA, or that block serotonin metabolism to 5-HIAA, would have efficacy in reducing eosinophil recruitment, type II effector T cell induction and Cryptococcal disease. In this regard, it is notable that a clinical study provided preliminary evidence that treatment of patients with a SERT inhibitor may be protective from C. neoformans disease (59). While the mechanism has been thought to involve direct anti-fungal actions of this agent (60, 61), we suggest that it may be acting by reducing 5-HIAA availability in the lung and decreasing eosinophil recruitment.
Limitations of the study
Although we used multiple approaches to modulate 5-HIAA production (phenelzine treatment, SERT−/−, Tph1−/− BM chimeras, Tph1f/f Cpa3-Cre mice), each of these approaches also impacts on serotonin production. While not a ligand for GPR35, serotonin can engage any of 14 receptors and can have pleiotropic actions in tissues (43). We cannot exclude that some of the effects observed under conditions of altered 5-HIAA generation also reflect effects of altered serotonin abundance or the abundance of other serotonin metabolites or related amines. Use of a 5-HIAA catabolic enzyme to selectively deplete 5-HIAA from tissues would be a valuable approach but to our knowledge no such enzyme has been identified. In addition, while our data indicate that GPR35-expressing eosinophils modulate Th1/Th2 balance within infected airways, whether alterations of this balance are required for GPR35-mediated fungal outgrowth remains to be established. Finally, we note that performing our experiments with the widely studied C. neoformans KN99 strain allows our work to be placed in context with a large body of existing literature. However, we acknowledge that this is a laboratory adapted strain and it remains important in the future to perform studies with human C. neoformans isolates.
Material and Methods
Resource availability
Lead contact
Further information and requests for reagents will be fulfilled by Dr. Jason Cyster (jason.cyster@ucsf.edu).
Materials availability
A list of critical reagents (key resources) is included in the key resources table. Some material may require requests to collaborators and/or agreements with various entities. Material that can be shared will be released via a Material Transfer Agreement.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PerCP/Cy5.5 rat anti-mouse CD45.1 (A20) | BioLegend | #110728 |
| FITC-conjugated rat anti-mouse CD45.2 (104) | BioLegend | #109806 |
| BV785-conjugated rat anti-mouse SiglecF (E50-2440) | BD | #740956 |
| PE-conjugated rat anti-mouse SiglecF (E50-2440) | BioLegend | #552126 |
| BV785-conjugated rat anti-mouse/human CD11b (M1/70) | BioLegend | #101243 |
| PE-conjugated rat anti-mouse/human CD11b (M1/70) | BioLegend | # 101208 |
| APC-conjugated rat anti-mouse CD64 (X54-5/7.1) | BioLegend | #139306 |
| BV605-conjugated rat anti-mouse CD45.2 (104) | BioLegend | #109841 |
| PB-conjugated rat anti-mouse CD45.2 (104) | BioLegend | #09820 |
| BV605-conjugated rat anti-mouse CD45.1 (A20) | BioLegend | #110738 |
| AF700-conjugated rat anti-mouse CD45.1 (A20) | BioLegend | #110724 |
| PB-conjugated rat anti-mouse CD45.1 (A20) | BioLegend | #110722 |
| APC-Cy7-conjugated rat anti-mouse Ly6G (1A8) | TONBO | #25-1276 |
| FITC-conjugated rat anti-mouse Ly6C (AL-21) | BD | #553104 |
| BV605-conjugeted rat anti-mouse Ly6C (HK1.4) | BioLegend | #128036 |
| BV650-conjugeted rat anti-mouse Ly6C (HK1.4) | BioLegend | #128049 |
| BV570-conjugated rat anti-mouse/human CD11b (M1/70) | BioLegend | #101233 |
| FITC- conjugated rat anti-mouse CD41 (MWReg30) | BioLegend | #133903 |
| PE- conjugated anti-mouse CD45.2 (104) | BioLegend | #109808 |
| PerCP/Cy5.5-conjugated rat anti-mouse CD4 (GK1.5) | BioLegend | #100434 |
| FITC-conjugated rat anti-mouse CD44 (IM7) | BioLegend | #103006 |
| BV421-conjugated rat anti-mouse TCR β chain (H57-597) | BioLegend | #109230 |
| APC-Cy7-conjugated rat anti-mouse CD62L (MEL-14). | BioLegend | # 104428 |
| PE-conjugated rat anti mouse/human GATA3 (REA174) | Miltenyi | #130-100-664 |
| Alexa647-conjugated rat anti mouse anti-T-bet (4B10) | BioLegend | #644803 |
| APC-conjugated anti-mouse IL-4 (11B11) | eBioscience | #17-7041-82 |
| PE/Cy7-conjugated rat anti-mouse IFNg (XMG1.2) | BioLegend | #505826 |
| APC anti- mouse IFNg (XMG1.2) | BioLegend | #505810 |
| Rabbit polyclonal anti-GPR35 | Biomatik | None |
| AF647-Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Ab | Fisher Scientific | # A21245 |
| Bacterial and virus strains | ||
| Cryptococcus neoformans (strain KN99) | Madhani lab, UCSF | None |
| Biological samples | ||
| None | ||
| Chemicals, peptides, and recombinant proteins | ||
| 5-HIAA | Sigma | #H8876 |
| CXCL12 (SDF1a human) | Peprotech | #300-28A |
| Diphtheria Toxin (DT) | Millipore/Sigma | #322326 |
| FMS-like tyrosine kinase 3 ligand Peprotech) | Peprotech | #250-31 |
| IL-5 | Peprotech | #215-15 |
| IL-3 | Peprotech | #213-13 |
| IL-6 | Peprotech | #216-16 |
| Stem cell factor | Peprotech | #250-03 |
| Cell trace Violet | Fisher | #C34557 |
| Deep red | Life Tech | #C34565 |
| Critical commercial assays | ||
| Mouse 5-Hydroxyindoleacetic acid (5HIAA 5-HIAA) ELISA Kit | AssayGenie | # MOEB2528 |
| Deposited data | ||
| None | ||
| Experimental models: Cell lines | ||
| Plat-e cells | Susan Schwab lab, NYU | |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson | B6 background |
| BoyJ (CD45.1) | Jackson | B6 background |
| Gpr35−/− | EMMA (EM09677; Gpr35tm1b(EUCOMM)Hmgu) | B6 background |
| Pf4-Cre x mTmG (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) | Looney lab, UCSF (63) | B6 background |
| Kit/v x Kit/W | Jackson | B6 background |
| TPH1 het and ko (Bone Marrow) | Huaquing Wang and Waliul Khan lab, McMaster Univ. | B6 background |
| GATA1-deficient ΔdblGATA (C.129S1(B6)-Gata1tm6Sho/J) | Jackson | B6 background |
| Tph1 floxed mice | Gerard Karsenty, Columbia Univ. (66) | B6 background |
| Cpa3-Cre mice | Paul Bryce, Northwestern Univ. (48) | B6 background |
| SERT-deficient mice (B6.129(Cg)-Slc6a4tm1Kpl/J) | Jackson (64) | B6 background |
| Oligonucleotides | ||
| ACGCGGGGACAGAGGTATG-Fwd (Gpr35 qPCR) | ||
| TGAGGGTGCTGTTACAGGTTG-Rev (Gpr35 qPCR) | ||
| Recombinant DNA | ||
| None | ||
| Software and algorithms | ||
| IMARIS (v.9.6.0) | ||
| Flowjo (v.10.6.2) | ||
| Prism (GraphPad 9.0.1) | ||
| Seurat R package (version 2.2) | ||
| R studio (3.5) | ||
Data and code availability
All data reported in this paper are available in the main text or supplementary information.
This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Experimental model and subject details
Mice
C57BL/6J and BoyJ (CD45.1) mice were bred in an internal colony and 7–12-week-old mice of both sexes were used. Gpr35−/− mice were obtained from EMMA (EM09677; Gpr35tm1b(EUCOMM)Hmgu) and maintained on a B6 background. Platelet-deficient mice (c-mpl−/−), Pf4-Cre x mTmG (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) platelet reporter mice (62, 63) and CD11c-DTR-GFP mice were all on a B6 background. Mast cell-deficient Kit/v x Kit/W mice and SERT-deficient mice (64) were obtained from Jackson Laboratories and maintained on a B6 background. Tph1+/− and Tph1−/− (65) BM were provided by Huaquing Wang and Waliul Khan (McMaster Univ.). GATA1-deficient ΔdblGATA (C.129S1(B6)-Gata1tm6Sho/J) mice were obtained from Jackson Laboratories. Tph1 floxed mice (66) were kindly provided by Gerard Karsenty (Columbia Univ.). Cpa3-Cre mice (48) were kindly provided by Paul Bryce (Northwestern Univ). Co-caged littermate controls (+/+, +/− and −/−) were used for experiments, mice were allocated to control and experimental groups randomly, sample sizes were chosen based on previous experience to obtain reproducible results and the investigators were not blinded.
Methods details
Generation and transfer of bone marrow-derived eosinophils
To produce BM-derived eosinophil, total BM cells were isolated from GPR35+/+ and GPR35−/− and cultured in Iscove’s modified Dulbecco’s media (IMDM) (#12440053; Fisher), containing 10% heat-inactivated FBS (#FB-02; Omega Scientific), 1% Pen Strep (#MT-30–001-CI; Fisher), 2 mM glutamine (#MT 25–005-CI; Fisher), and 55 μM 2-mercaptoethanol (#21985023; Fisher). During the 14-day culture, medium was supplemented with FMS-like tyrosine kinase 3 ligand (# 250–31, Peprotech) and stem cell factor (#250–03, Peprotech) at 100 ng/mL, for the first 4 days. At day 4, medium was removed and fresh IMDM supplemented with IL-5 (10 ng/mL, #215–15, Peprotech) was added for the following 10 days, similarly to what was previously described (35). Eosinophil purity (>95%) was checked before cell transfer by flow cytometry (using SiglecF, CD64 and CD11b staining). For co-transfer experiments, BM-derived GPR35+/+ and GPR35−/− eosinophils were labeled for 30 min at 37°C with Cell trace Violet (#C34557, Fisher) or Deep red (#C34565, Life Tech) similarly to what was previously described (29)and mixed 50:50 before injection (3 × 107 eosinophil / mouse in 200μl saline) in mice previously infected with C. neoformans (day 5).
Infection, treatments, and bone marrow chimeras
Cryptococcus neoformans (strain KN99) was from the Madhani lab. Individual colonies from C. neoformans plates were cultured overnight in 10 ml YPAD at 30 °C. The next day, yeast were counted on a hemocytometer and diluted to 2 × 106 cells/ml in saline. Mice were anaesthetized with a mixture of oxygen and Isoflurane and then hung by their front teeth using surgical thread. 50 μl yeast (1 × 105 CFU) were then pipetted onto the nasal flares and taken up by aspiration, as previously described (49).To deplete total generation of 5-HIAA, mice were treated i.p. with Phenelzine (30mg/kg, #P6777, Millipore Sigma) starting 3 days before infection. The treatment was repeated every other day (15mg/kg) until the end of the experiment. To produce mixed chimeras, CD45.1 congenic Boy/J mice were lethally irradiated with 1300 rad in split doses and reconstituted with 5 × 106 BM cells (~50:50) as indicated. Mice were analyzed 7–8 weeks later. Since the hematopoietic stem cell content in BM from different donor mice may not be identical, reconstitution of the B220+ B cell compartment was used to assess chimerism after reconstitution. B cells lack GPR35 expression and are not expected to be affected by GPR35-deficiency. Animals were housed in a pathogen-free environment in the Laboratory Animal Resource Center at the University of California, San Francisco, and all experiments conformed to ethical principles and guidelines that were approved by the Institutional Animal Care and Use Committee. To generate mice with full GPR35-deficiency restricted to dendritic cells, CD11c-DTR-GFP BM was mixed 1:1 with GPR35+/+ or GPR35−/− BM and used to reconstitute WT mice. After 7 weeks reconstitution the chimeras were treated with 20ng/g diptheria toxin i.v. (DT, #322326, Millipore) starting 3 days before infection and every other day with 80ng DT (i.v.). To generate mice with full GPR35-deficiency restricted to eosinophils, GATA1−/− BM was mixed 1:1 with GPR35+/+ or GPR35−/− BM and used to reconstitute WT mice. Chimeric mice were infected 7–8 weeks after reconstitution. For detection of cells within the vascular system versus parenchyma of the lung, mice were injected with 2ug of anti-CD45.2-PE 5 min before sacrifice and tissue isolation. During this short period of in vivo antibody exposure, cells in blood vessels and blood exposed spaces become labeled while cells in the parenchyma remain unlabeled (36).
Generation of over-expressing bone marrow chimeras
Mice to be used as BM donors (C57BL/6J) were injected intravenously with 3 mg 5-fluorouracil (Sigma). BM was collected after 4 days and cultured in DMEM containing 15% (v/v) FBS, antibiotics (penicillin (50 IU/ml) and streptomycin (50mg/ml); Fisher) and 10 mM HEPES, pH 7.2 (Cellgro), supplemented with IL-3 (#213–13, Peprotech), IL-6 (#216–16, Peprotech and stem cell factor (#250–03, Peprotech) at concentrations of 20, 50 or 100 ng/ml, respectively). Cells were spininfected twice at days 1 and 2 with viral supernatant (MSCV-EV-GFP and MSCV-GPR35-GFP) and transferred into irradiated CD45.1 B6 recipient mice on day 3, similarly to what was previously described (67).
Transwell migration assay
GPR35+/+(CD45.1+) and GPR35−/− (CD45.2+) lungs were mashed in 70 μm strainers and resuspended in 5ml of migration medium (RPMI containing 0.5% fatty acid-free BSA, 10 mM HEPES and 50 IU/L penicillin/streptomycin RPMI) in 15ml conical tubes. Samples were centrifuged at 20g (300 rpm) for 3 minutes to remove large clumps and cell-containing supernatants were transferred into clean 15ml conical tubes. Samples were than centrifuged at 526g (1500 rpm) for 5 minutes and pellets were ACK lysed. Cells were washed twice in pre-warmed migration medium and mixed 50:50 (CD45.1:CD45.2). The cells were resuspended in migration medium at 2–5 × 106 cells / ml and resensitized for 30 min in a 37 °C water bath in migration plus medium. Transwell filters (6 mm insert, 5 μm pore size, Corning) were placed on top of each well, and 100 μl containing 2–5 × 105 cells was added to the transwell insert. In some cases cells were pretreated with pertussis toxin (100ng/ml) for 2 hr prior to the assay. 600ul of chemoattractant (5-HIAA at indicated concentrations, CXCL12 100ng/ml or CCL11 1ug/ml) or no chemoattractant (Nil) was added to the lower well. The cells were allowed to migrate for 3 hr, after which the cells in the bottom well were counted by flow cytometry. Representative experiments for each migration assay are plotted as a percentage of input migration.
5-HIAA ELISA
5-HIAA (#H8876) was purchased from Sigma and diluted in DMSO. To quantify 5-HIAA concentrations, lungs were weighed, carefully minced into small pieces and mashed through 100μM strainer (Fisher Scientific) in 1ml PBS per g of tissue (1ml/g). Samples were than centrifuged at 10000 rpm for 30min at 4°C to obtain lung supernatants. To quantify 5-HIAA, Mouse 5-Hydroxyindoleacetic acid (5-HIAA) ELISA Kit (AssayGenie) was used following manufacturer instructions.
Flow cytometry
Eosinophils were identified as CD45+ SiglecF+ CD64− CD11b+ or CD45+ SiglecF+ CD64− cells as indicated. Cells were stained with the following antibodies: PerCP/Cy5.5 rat anti-mouse CD45.1 (A20, #110728); FITC-conjugated rat anti-mouse CD45.2 (104, #109806, BioLegend); BV785-conjugated rat anti-mouse SiglecF (E50–2440, #740956; BD); PE-conjugated rat anti-mouse SiglecF (E50–2440, #552126, BioLegend); BV785-conjugated rat anti-mouse/human CD11b (M1/70, #101243, BioLegend); PE-conjugated rat anti-mouse/human CD11b (M1/70, # 101208, BioLegend); APC-conjugated rat anti-mouse CD64 (X54–5/7.1, #139306, BioLegend). For mixed chimeras experiments the following additional antibodies were used to discriminate between GPR35+/+ and GPR35−/− cells: BV605-conjugated rat anti-mouse CD45.2 (104, #109841, BioLegend); PB-conjugated rat anti-mouse CD45.2 (104, #09820, BioLegend); BV605-conjugated rat anti-mouse CD45.1 (A20, #110738, BioLegend); AF700-conjugated rat anti-mouse CD45.1 (A20, 110724, BioLegend); PB-conjugated rat anti-mouse CD45.1 (A20, #110722, BioLegend). To identify neutrophils, monocytes and alveolar macrophages, the following antibodies (in addition to previous ones) were used: APC-Cy7-conjugated rat anti-mouse Ly6G (1A8, # 25–1276, TONBO); FITC-conjugated rat anti-mouse Ly6C (AL-21, #553104, BD); BV605-conjugeted rat anti-mouse Ly6C (HK1.4, #128036, BioLegend); BV650-conjugeted rat anti-mouse Ly6C (HK1.4, #128049, BioLegend); BV570-conjugated rat anti-mouse/human CD11b (M1/70, #101233, BioLegend). For flow cytometry on lung samples, mice were infected or challenged as indicated and then lungs were dissected and minced using scissors. The minced lung tissue was then incubated for 30 min shacking (1000rpm) at 37 °C in 1ml of digestion medium (RPMI, 1% NBCS, 0.25 mg ml–1, Liberase TM Research Grade (Sigma), 0.025 mg ml–1 DNaseI (Sigma)). To stop the digestion, 100μl of quenching solution (RPMI, 0.1M EDTA, 50% NBCS) were added to each sample. The digested lung tissue was then mashed through a 100-μm strainer (Fisher Scientific) and washed with in RPMI (2% FCS, 1 mM EDTA). Spleens were mashed through a 100-μm strainer 70-μm in RPMI, 1%NBCS, 0.1m EDTA. Blood was collected by retro-orbital bleeding using heparinized micro-hematocrit capillary tubes (Fisher). Red blood cells were then lysed (2x) for 5 min on ice using ACK Lysis Buffer. Cells were resuspended in FACS buffer (1× PBS, 2% FCS, 1 mM EDTA) for staining. To identify intravascular cells, 2 μg / 200μl saline of PE- conjugated anti-mouse CD45.2 (104, #109808, Biolegend) were injected 5 min before sacrifice as indicated. To identify Th1 and Th2 cells in infected lungs, lung cell surface markers were stained with the following antibodies: BV605-conjugated rat anti-mouse CD45.2 (104, #109841, BioLegend); PerCP/Cy5.5-conjugated rat anti-mouse CD4 (GK1.5, #100434, BioLegend); FITC-conjugated rat anti-mouse CD44 (IM7, # 103006, BioLegend); BV421-conjugated rat anti-mouse TCR β chain (H57–597, # 109230, BioLegend); APC-Cy7-conjugated rat anti-mouse CD62L (MEL-14, # 104428, BioLegend). Cells were then washed, fixed/permeabilized (FoxP3 fixation/permeabilization concentrate and diluent, eBioscience) and stained with the following antibodies: PE-conjugated rat anti mouse/human GATA3 (REA174, #130–100-664, Miltenyi); Alexa647-conjugated rat anti mouse anti-T-bet (4B10, #644803, BioLegend). For ex vivo re-stimulation, processed lung cells from Cryptococcus infected GPR35+/− and GPR35−/− full BM chimeras (day 11) were incubated with resting medium (RPMI containing 10% FBS, 10 mM HEPES, 2 mM glutamine and 50 IU/L penicillin/streptomycin plus GolgiPlug Protein Transport Inhibitor (#555029, BD)) or stimulation medium (RPMI containing 10% FBS, 10 mM HEPES, 2 mM glutamine and 50 IU/L penicillin/streptomycin plus GolgiPlug Protein Transport Inhibitor and PMA/Ionomycin (Millipore Sigma)) for 3.5 hours at 37°C. Cells were than washed and stained for the surface markers with the following antibodies: BV605-conjugated rat anti-mouse CD45.2 (104, #109841, BioLegend); PerCP/Cy5.5-conjugated rat anti-mouse CD4 (GK1.5, #100434, BioLegend); FITC-conjugated rat anti-mouse CD44 (IM7, #103006, BioLegend); BV421-conjugated rat anti-mouse TCR β chain (H57–597, # 109230, BioLegend); APC-Cy7-conjugated rat anti-mouse CD62L (MEL-14, # 104428, BioLegend). Cells were than washed, fixed/permeabilized (Cytofix/Cytoperm Fixation and Permeabilization Solution, BD) and stained with the following antibodies APC-conjugated anti-mouse IL-4 (11B11, #17–7041-82, eBioscience); PE/Cy7-conjugated rat anti-mouse IFNg (XMG1.2, #505826); APC anti- mouse IFNg (XMG1.2, #505810). Rabbit polyclonal anti-GPR35 was produced by Biomatik (using as immunogen the C terminus peptide: MAREFQEASKPATSSNTPHKSQDSQILSLT) and affinity-purified. To reduce non-specific background, anti-GPR35 polyclonal antibody was pre-absorbed against GPR35−/− lung cells overnight at 4°C. Cells were surface-stained, fixed and permeabilized (eBioscience™, #00552100) before intracellular staining. AF647-Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Ab (A21245, Fisher Scientific) was used as secondary antibody. To quantify eosinophil-platelet interaction, 100 μl of blood were obtained by retro-orbital bleeding (heparinized capillaries) and drawn into tubes containing 1ml of Lyse/Fix Buffer 5X (Ox-7, #558049, BioLegend) and incubated for 15 min. Cells were than washed and resuspended in staining buffer. FITC- conjugated anti-mouse CD41 (MWReg30, # 133903, BioLegend) was used to stain eosinophils that acquired platelet membrane material. Data were acquired using a BD LSR II flow cytometer or a Cytek Aurora. Flow cytometry data were analyzed using Flowjo (v.10.6.2).
Multiphoton ex vivo imaging, image analysis and PAS staining
To perform multiphoton ex vivo imaging of eosinophil and platelet interactions, we injected 1.5 × 107 (50:50, in 200μl saline) of Cell Trace Violet-labeled (CTV, # C34557, Life Tech) GPR35+/+ and Deep Red-labeled (#C34565, Life Tech) GPR35−/− BM-derived eosinophils in Pf4-Cre x mTmG previously i.n. infected with C. neoformans (day 5). Pf4Cre mTmG mice expressed GFP selectively in platelets; these mice broadly expressed tdTom in other cell types including neutrophils but the intensity of red fluorescence in these cells was weak as reported (68). Accordingly, platelet and vessels were identified based on GFP signal and tdTomato signal, respectively. To further define the vessel lumen, tetramethylrhodamine isothiocyanate (TRITC) Dextran (150kDa, # T1287, Sigma) was injected 20 min before mouse euthanasia. Mice were euthanized 6 hrs after cell transfer and infected lungs were dissected, washed with PBS and cut into pieces (~0.1–0.3cm3) that were glued (VetBond) on Superfrost Plus Microscope Slides (Fisher). Samples were immerged in PBS and imaged with a Zeiss LSM 7MP equipped with a Chameleon laser (Coherent) and a x20 objective. Samples were excited at 820–850nm. For image acquisition, a series of planes of 3μm z-spacing spanning a depth of 30–60 μm were collected. Multiphoton images were imported into IMARIS software (v.9.6.0). Vessel surface was obtained using IMARIS built-in surface function based on dextran-TRITC plus TdTomato (mTmG) signal as indicated. IMARIS built-in functions ‘Background subtraction’ and ‘Gaussian Filter’ were applied. To track single cells and platelets, surface seed points were created and tracked with IMARIS spot built-in function based on signal intensity.
To perform PAS staining, Cryptococcus infected lungs were cut into 3mm pieces and fixed overnight in 4% PFA at 4°C. After 12–18 hrs, the samples were washed and incubated in 70% ethanol for 72 hrs at 4°C. Lung slices (4 μm) were prepared and stained by the BTBTC Core (UCSF).
Quantification and statistical analysis
Prism software (GraphPad 9.0.1) was used for all statistical analyses. The statistical tests used are specified in the figure legends. Two-tailed unpaired t-tests were performed when comparing only two groups, Paired- t-tests were used to compare internally controlled replicates, and ordinary one-way ANOVA using Turkey’s multiple comparisons test was performed when comparing one variable across multiple groups. P < 0.05 was considered significant. In summary graphs, points indicate individual samples and horizontal lines are means or medians as indicated. In bar graphs, bars show means and error bars indicate standard error mean (SEM).
Supplementary Material
Movie S1. GPR35 sustains eosinophil interaction with platelets, related to Figure 5. Multiphoton micrograph showing a projection view of Cryptococcus infected lung (day 5) from Pf4-Cre x mTmG mice injected with BM-derived transferred GPR35+/+ (CTV, blue) and GPR35−/− (Deep Red, white) eosinophils, 6 hours after cell transfer. Platelet (green) and vessels (red) are depicted. Iso-surfaces representing of GPR35+/+ and GPR35−/− eosinophils, platelets (green) and vessels were obtained using IMARIS built-in surface function based on dextran TRITC plus TdTomato (mTmG) signal as indicated. Data are representative of at least 2 independent experiments.
Movie S2. Additional example of GPR35-sufficient eosinophil interaction with platelets, related to Figure 5. Multiphoton micrograph of Cryptococcus infected lung (day 5) from Pf4-Cre x mTmG mice injected with BM-derived GPR35+/+ (CTV, blue) and GPR35−/− (Deep Red, white) eosinophils, 6 hours after cell transfer. Single z-stack image (left) and movie showing steps through the individual z-planes (3μm, right). Platelets are depicted in green. White dashed line indicates vessel limits (based on mTmG/dextran signal, not shown). White arrows indicate intravascular GPR35-sufficient eosinophils (CTV+, blue) in contact with platelet cluster (Pf4-Cre x mTmG, green). The blue arrow points to one GPR35-sufficient eosinophil that extends most of its cellular body out of vessel while still in contact with platelet cluster.
Highlights.
GPR35 functions as a chemoattractant receptor in eosinophils
5-HIAA from platelets and mast cells helps recruit eosinophils to the inflamed lung
GPR35+ eosinophil recruitment alters Th cell activity and reduces fungal clearance
MAOIs reduce eosinophil recruitment and protect from Cryptococcus infection
Acknowledgements
We thank Serena Ranucci and Antonia Gallman for technical help and discussions. We thank Waliul Khan and Huaquing Wang for Tph1-deficient BM, Rebecca Krier and Paul Bryce for Cpa3-Cre mice, and Hong Liu and Gerard Karsenty for Tph1 floxed mice. M.D.G. was supported by a was supported by an EMBO long-term fellowship and is supported by a CRI Irvington Postdoctoral Fellowship. J.G.C. is an investigator of the Howard Hughes Medical Institute. This work was supported part by NIH grants R21AI163036, R01 AI40098, and R01 AI45073.
Footnotes
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci Transl Med 4, 165rv113. 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Saluja R, Metz M, and Maurer M (2012). Role and relevance of mast cells in fungal infections. Front Immunol 3, 146. 10.3389/fimmu.2012.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mok AC, Mody CH, and Li SS (2021). Immune Cell Degranulation in Fungal Host Defence. J Fungi (Basel) 7. 10.3390/jof7060484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyer KR, Revie NM, Fu C, Robbins N, and Cowen LE (2021). Treatment strategies for cryptococcal infection: challenges, advances and future outlook. Nat Rev Microbiol 19, 454–466. 10.1038/s41579-021-00511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopes JP, Stylianou M, Backman E, Holmberg S, Ekoff M, Nilsson G, and Urban CF (2019). Cryptococcus neoformans Induces MCP-1 Release and Delays the Death of Human Mast Cells. Front Cell Infect Microbiol 9, 289. 10.3389/fcimb.2019.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decken K, Kohler G, Palmer-Lehmann K, Wunderlin A, Mattner F, Magram J, Gately MK, and Alber G (1998). Interleukin-12 is essential for a protective Th1 response in mice infected with Cryptococcus neoformans. Infect Immun 66, 4994–5000. 10.1128/IAI.66.10.4994-5000.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoag KA, Lipscomb MF, Izzo AA, and Street NE (1997). IL-12 and IFN-gamma are required for initiating the protective Th1 response to pulmonary cryptococcosis in resistant C.B-17 mice. Am J Respir Cell Mol Biol 17, 733–739. 10.1165/ajrcmb.17.6.2879. [DOI] [PubMed] [Google Scholar]
- 8.Voelz K, Lammas DA, and May RC (2009). Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect Immun 77, 3450–3457. 10.1128/IAI.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leopold Wager CM, Hole CR, Wozniak KL, Olszewski MA, and Wormley FL Jr. (2014). STAT1 signaling is essential for protection against Cryptococcus neoformans infection in mice. J Immunol 193, 4060–4071. 10.4049/jimmunol.1400318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackstock R and Murphy JW (2004). Role of interleukin-4 in resistance to Cryptococcus neoformans infection. Am J Respir Cell Mol Biol 30, 109–117. 10.1165/rcmb.2003-0156OC. [DOI] [PubMed] [Google Scholar]
- 11.Huffnagle GB, Boyd MB, Street NE, and Lipscomb MF (1998). IL-5 is required for eosinophil recruitment, crystal deposition, and mononuclear cell recruitment during a pulmonary Cryptococcus neoformans infection in genetically susceptible mice (C57BL/6). J Immunol 160, 2393–2400. [PubMed] [Google Scholar]
- 12.Muller U, Stenzel W, Kohler G, Werner C, Polte T, Hansen G, Schutze N, Straubinger RK, Blessing M, McKenzie AN, et al. (2007). IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol 179, 53675377. 10.4049/jimmunol.179.8.5367. [DOI] [PubMed] [Google Scholar]
- 13.Osterholzer JJ, Surana R, Milam JE, Montano GT, Chen GH, Sonstein J, Curtis JL, Huffnagle GB, Toews GB, and Olszewski MA (2009). Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol 174, 932–943. 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piehler D, Stenzel W, Grahnert A, Held J, Richter L, Kohler G, Richter T, Eschke M, Alber G, and Muller U (2011). Eosinophils contribute to IL-4 production and shape the T-helper cytokine profile and inflammatory response in pulmonary cryptococcosis. Am J Pathol 179, 733–744. 10.1016/j.ajpath.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez Y, Arora S, Erb-Downward JR, McDonald RA, Toews GB, and Huffnagle GB (2005). Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J Immunol 174, 1027–1036. 10.4049/jimmunol.174.2.1027. [DOI] [PubMed] [Google Scholar]
- 16.Szymczak WA, Sellers RS, and Pirofski LA (2012). IL-23 dampens the allergic response to Cryptococcus neoformans through IL-17-independent and -dependent mechanisms. Am J Pathol 180, 1547–1559. 10.1016/j.ajpath.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma AH, Bueter CL, Rothenberg ME, and Deepe GS (2017). Eosinophils subvert host resistance to an intracellular pathogen by instigating non-protective IL-4 in CCR2(−/−) mice. Mucosal Immunol 10, 194–204. 10.1038/mi.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muniz VS, Weller PF, and Neves JS (2012). Eosinophil crystalloid granules: structure, function, and beyond. J Leukoc Biol 92, 281–288. 10.1189/jlb.0212067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogan SP, Rosenberg HF, Moqbel R, Phipps S, Foster PS, Lacy P, Kay AB, and Rothenberg ME (2008). Eosinophils: biological properties and role in health and disease. Clin Exp Allergy 38, 709–750. 10.1111/j.1365-2222.2008.02958.x. [DOI] [PubMed] [Google Scholar]
- 20.Barrett NA and Austen KF (2009). Innate cells and T helper 2 cell immunity in airway inflammation. Immunity 31, 425–437. 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Xu X, Teng J, Cheng S, Bunjhoo H, Cao Y, Liu J, Xie J, Wang C, Xu Y, et al. (2015). CXCR4 inhibitor attenuates allergen-induced lung inflammation by downregulating MMP-9 and ERK1/2. Int J Clin Exp Pathol 8, 6700–6707. [PMC free article] [PubMed] [Google Scholar]
- 22.Lukacs NW, Berlin A, Schols D, Skerlj RT, and Bridger GJ (2002). AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 160, 1353–1360. 10.1016/S0002-9440(10)62562-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, Zimmermann N, Molkentin JD, and Rothenberg ME (2005). Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem 280, 13952–13961. 10.1074/jbc.M406037200. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Li Y, Sun T, Du W, Zhang Z, Li D, and Ding C (2020). Integrative Proteome and Acetylome Analyses of Murine Responses to Cryptococcus neoformans Infection. Front Microbiol 11, 575. 10.3389/fmicb.2020.00575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nail S, Robert R, Dromer F, Marot-Leblond A, and Senet JM (2001). Susceptibilities of Cryptococcus neoformans strains to platelet binding in vivo and to the fungicidal activity of thrombin-induced platelet microbicidal proteins in vitro. Infect Immun 69, 1221–1225. 10.1128/IAI.69.2.1221-1225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitchford SC, Momi S, Giannini S, Casali L, Spina D, Page CP, and Gresele P (2005). Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 105, 2074–2081. 10.1182/blood-2004-06-2282. [DOI] [PubMed] [Google Scholar]
- 27.Ramirez GA, Yacoub MR, Ripa M, Mannina D, Cariddi A, Saporiti N, Ciceri F, Castagna A, Colombo G, and Dagna L (2018). Eosinophils from Physiology to Disease: A Comprehensive Review. Biomed Res Int 2018, 9095275. 10.1155/2018/9095275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiesner DL, Smith KD, Kashem SW, Bohjanen PR, and Nielsen K (2017). Different Lymphocyte Populations Direct Dichotomous Eosinophil or Neutrophil Responses to Pulmonary Cryptococcus Infection. J Immunol 198, 1627–1637. 10.4049/jimmunol.1600821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Giovanni M, Tam H, Valet C, Xu Y, Looney MR, and Cyster JG (2022). GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell 185, 815830 e819. 10.1016/j.cell.2022.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaya B, Donas C, Wuggenig P, Diaz OE, Morales RA, Melhem H, Swiss IBDCI, Hernandez PP, Kaymak T, Das S, et al. (2020). Lysophosphatidic Acid-Mediated GPR35 Signaling in CX3CR1(+) Macrophages Regulates Intestinal Homeostasis. Cell Rep 32, 107979. 10.1016/j.celrep.2020.107979. [DOI] [PubMed] [Google Scholar]
- 31.Boleij A, Fathi P, Dalton W, Park B, Wu X, Huso D, Allen J, Besharati S, Anders RA, Housseau F, et al. (2021). G-protein coupled receptor 35 (GPR35) regulates the colonic epithelial cell response to enterotoxigenic Bacteroides fragilis. Commun Biol 4, 585. 10.1038/s42003-021-02014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin LC, Quon T, Engberg S, Mackenzie AE, Tobin AB, and Milligan G (2021). G Protein-Coupled Receptor GPR35 Suppresses Lipid Accumulation in Hepatocytes. ACS Pharmacol Transl Sci 4, 1835–1848. 10.1021/acsptsci.1c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneditz G, Elias JE, Pagano E, Zaeem Cader M, Saveljeva S, Long K, Mukhopadhyay S, Arasteh M, Lawley TD, Dougan G, et al. (2019). GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci Signal 12. 10.1126/scisignal.aau9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyant GA, Yu W, Doulamis IP, Nomoto RS, Saeed MY, Duignan T, McCully JD, and Kaelin WG Jr. (2022). Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 377, 621–629. 10.1126/science.abm1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wen T, Besse JA, Mingler MK, Fulkerson PC, and Rothenberg ME (2013). Eosinophil adoptive transfer system to directly evaluate pulmonary eosinophil trafficking in vivo. Proc Natl Acad Sci U S A 110, 6067–6072. 10.1073/pnas.1220572110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira JP, An J, Xu Y, Huang Y, and Cyster JG (2009). Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol 10, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, Kato Y, Onodera M, Minegishi N, Yamamoto M, Fukao K, et al. (2002). Essential and instructive roles of GATA factors in eosinophil development. J Exp Med 195, 1379–1386. 10.1084/jem.20020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, and Orkin SH (2002). Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med 195, 1387–1395. 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, Cifuni SM, Mauler M, Cicko S, Bader M, et al. (2013). Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 121, 1008–1015. 10.1182/blood-2012-06-437392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah SA, Page CP, and Pitchford SC (2017). Platelet-Eosinophil Interactions As a Potential Therapeutic Target in Allergic Inflammation and Asthma. Front Med (Lausanne) 4, 129. 10.3389/fmed.2017.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yue M, Hu M, Fu F, Ruan H, and Wu C (2022). Emerging Roles of Platelets in Allergic Asthma. Front Immunol 13, 846055. 10.3389/fimmu.2022.846055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Page C and Pitchford S (2013). Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int Immunopharmacol 17, 1176–1184. 10.1016/j.intimp.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 43.Berger M, Gray JA, and Roth BL (2009). The expanded biology of serotonin. Annu Rev Med 60, 355–366. 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freitag A, Wessler I, and Racke K (1995). Characterization of 5-hydroxytryptamine release from isolated rabbit and rat trachea: the role of neuroendocrine epithelia cells and mast cells. Naunyn Schmiedebergs Arch Pharmacol 353, 55–63. 10.1007/BF00168916. [DOI] [PubMed] [Google Scholar]
- 45.Gershon RK, Askenase PW, and Gershon MD (1975). Requirement for vasoactive amines for production of delayed-type hypersensitvity skin reactions. J Exp Med 142, 732–747. 10.1084/jem.142.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehtosalo JI, Uusitalo H, Laakso J, Palkama A, and Harkonen M (1984). Biochemical and immunohistochemical determination of 5-hydroxytryptamine located in mast cells in the trigeminal ganglion of the rat and guinea pig. Histochemistry 80, 219–223. 10.1007/BF00495769. [DOI] [PubMed] [Google Scholar]
- 47.Sjoerdsma A, Waalkes TP, and Weissbach H (1957). Serotonin and histamine in mast cells. Science 125, 1202–1203. 10.1126/science.125.3259.1202. [DOI] [PubMed] [Google Scholar]
- 48.Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J, Yu M, Tsai M, Piliponsky AM, and Galli SJ (2011). Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 118, 6930–6938. 10.1182/blood-2011-03-343962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dang EV, Lei S, Radkov A, Volk RF, Zaro BW, and Madhani HD (2022). Secreted fungal virulence effector triggers allergic inflammation via TLR4. Nature 608, 161–167. 10.1038/s41586-022-05005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohrer AC, Castro E, Tocheny CE, Assmann M, Schwarz B, Bohrnsen E, Makiya MA, Legrand F, Hilligan KL, Baker PJ, et al. (2022). Rapid GPR183-mediated recruitment of eosinophils to the lung after Mycobacterium tuberculosis infection. Cell Rep 40, 111144. 10.1016/j.celrep.2022.111144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deppermann C and Kubes P (2018). Start a fire, kill the bug: The role of platelets in inflammation and infection. Innate Immun 24, 335–348. 10.1177/1753425918789255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, Thomas S, Adler NM, Charytan DM, Gasmi B, et al. (2020). Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 24, 100434. 10.1016/j.eclinm.2020.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng LE, Hartmann K, Roers A, Krummel MF, and Locksley RM (2013). Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity 38, 166–175. 10.1016/j.immuni.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dudeck J, Kotrba J, Immler R, Hoffmann A, Voss M, Alexaki VI, Morton L, Jahn SR, Katsoulis-Dimitriou K, Winzer S, et al. (2021). Directional mast cell degranulation of tumor necrosis factor into blood vessels primes neutrophil extravasation. Immunity 54, 468–483 e465. 10.1016/j.immuni.2020.12.017. [DOI] [PubMed] [Google Scholar]
- 55.Garcia-Rodriguez KM, Bini EI, Gamboa-Dominguez A, Espitia-Pinzon CI, Huerta-Yepez S, Bulfone-Paus S, and Hernandez-Pando R (2021). Differential mast cell numbers and characteristics in human tuberculosis pulmonary lesions. Sci Rep 11, 10687. 10.1038/s41598-021-89659-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karhausen J, Choi HW, Maddipati KR, Mathew JP, Ma Q, Boulaftali Y, Lee RH, Bergmeier W, and Abraham SN (2020). Platelets trigger perivascular mast cell degranulation to cause inflammatory responses and tissue injury. Sci Adv 6, eaay6314. 10.1126/sciadv.aay6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kunder CA, St John AL, and Abraham SN (2011). Mast cell modulation of the vascular and lymphatic endothelium. Blood 118, 5383–5393. 10.1182/blood-2011-07-358432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hargarten J (2021). Characterization of a GPR35 nonsense mutation in a previously healthy patient with non-HIV cryptococcal meningoencephalitis. NIH FARE2021 Abstract. https://www.training.nih.gov/assets/FARE2021_Winners_Sorted_by_Institute_Center.pdf.
- 59.Rhein J, Morawski BM, Hullsiek KH, Nabeta HW, Kiggundu R, Tugume L, Musubire A, Akampurira A, Smith KD, Alhadab A, et al. (2016). Efficacy of adjunctive sertraline for the treatment of HIV-associated cryptococcal meningitis: an open-label dose-ranging study. Lancet Infect Dis 16, 809–818. 10.1016/S1473-3099(16)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loulergue P, Mir O, Rocheteau P, Chretien F, and Gaillard R (2016). Sertraline-induced increase in VEGF brain levels and its activity in cryptococcal meningitis. Lancet Infect Dis 16, 891. 10.1016/S1473-3099(16)30147-5. [DOI] [PubMed] [Google Scholar]
- 61.Zhai B, Wu C, Wang L, Sachs MS, and Lin X (2012). The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infections. Antimicrob Agents Chemother 56, 3758–3766. 10.1128/AAC.00212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lefrancais E, Mallavia B, Zhuo H, Calfee CS, and Looney MR (2018). Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 3. 10.1172/jci.insight.98178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. (2017). The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109. 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengel D, Murphy DL, Andrews AM, Wichems CH, Feltner D, Heils A, Mossner R, Westphal H, and Lesch KP (1998). Altered brain serotonin homeostasis and locomotor insensitivity to 3, 4-methylenedioxymethamphetamine (“Ecstasy”) in serotonin transporter-deficient mice. Mol Pharmacol 53, 649–655. 10.1124/mol.53.4.649. [DOI] [PubMed] [Google Scholar]
- 65.Cote F, Thevenot E, Fligny C, Fromes Y, Darmon M, Ripoche MA, Bayard E, Hanoun N, Saurini F, Lechat P, et al. (2003). Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U S A 100, 13525–13530. 10.1073/pnas.2233056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, et al. (2008). Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135, 825–837. 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu D, Duan L, Rodda LB, Lu E, Xu Y, An J, Qiu L, Liu F, Looney MR, Yang Z, et al. (2022). CD97 promotes spleen dendritic cell positioning and homeostasis through sensing of red blood cells. Science 375, eabi5965. 10.1126/science.abi5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. GPR35 sustains eosinophil interaction with platelets, related to Figure 5. Multiphoton micrograph showing a projection view of Cryptococcus infected lung (day 5) from Pf4-Cre x mTmG mice injected with BM-derived transferred GPR35+/+ (CTV, blue) and GPR35−/− (Deep Red, white) eosinophils, 6 hours after cell transfer. Platelet (green) and vessels (red) are depicted. Iso-surfaces representing of GPR35+/+ and GPR35−/− eosinophils, platelets (green) and vessels were obtained using IMARIS built-in surface function based on dextran TRITC plus TdTomato (mTmG) signal as indicated. Data are representative of at least 2 independent experiments.
Movie S2. Additional example of GPR35-sufficient eosinophil interaction with platelets, related to Figure 5. Multiphoton micrograph of Cryptococcus infected lung (day 5) from Pf4-Cre x mTmG mice injected with BM-derived GPR35+/+ (CTV, blue) and GPR35−/− (Deep Red, white) eosinophils, 6 hours after cell transfer. Single z-stack image (left) and movie showing steps through the individual z-planes (3μm, right). Platelets are depicted in green. White dashed line indicates vessel limits (based on mTmG/dextran signal, not shown). White arrows indicate intravascular GPR35-sufficient eosinophils (CTV+, blue) in contact with platelet cluster (Pf4-Cre x mTmG, green). The blue arrow points to one GPR35-sufficient eosinophil that extends most of its cellular body out of vessel while still in contact with platelet cluster.
Data Availability Statement
All data reported in this paper are available in the main text or supplementary information.
This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.