

Abstract
Ion channels modulate cellular excitability by regulating ionic fluxes across biological membranes. Pathogenic mutations in ion channel genes give rise to epileptic disorders that are among the most frequent neurological diseases affecting millions of individuals worldwide. Epilepsies are triggered by an imbalance between excitatory and inhibitory conductances. However, pathogenic mutations in the same allele can give rise to loss-of-function and/or gain-of-function variants, all able to trigger epilepsy. Furthermore, certain alleles are associated with brain malformations even in the absence of a clear electrical phenotype. This body of evidence argues that the underlying epileptogenic mechanisms of ion channels are more diverse than originally thought. Studies focusing on ion channels in prenatal cortical development have shed light on this apparent paradox. The picture that emerges is that ion channels play crucial roles in landmark neurodevelopmental processes, including neuronal migration, neurite outgrowth, and synapse formation. Thus, pathogenic channel mutants can not only cause epileptic disorders by altering excitability, but further, by inducing morphological and synaptic abnormalities that are initiated during neocortex formation and may persist into the adult brain.
Key Words: developmental and epileptic encephalopathies, epilepsy, K+ channel, KCNB1, Kv2.1, neurodevelopment, potassium channel
Introduction
The formation of the central nervous system is one of the most critical steps in the development of an organism. Particularly crucial is the proper patterning of the cerebral neocortex, the most recently developed structure of the brain, responsible for higher cognitive functions such as intelligence, processing of sensory information, language, and motor function (Kandel et al., 2000). Therefore, correct neuronal positioning in the neocortex is fundamental to guarantee its normal laminarization and, consequently, functionality. To achieve this goal, multiple landmark processes including birth and differentiation of neurons from progenitor stem cells, migration of neurons to their final destinations, outgrowth of axons and dendrites, axon pathfinding, and synapse formation, need to take place in a spatiotemporally controlled manner (Kandel et al., 2000). Hence, the neocortex formation is to be intended as a series of orchestrated events, biological pathways, and cell-to-cell interactions.
Many of the mechanisms underlying cortical development are both hard-wired in the neurons (activity-independent) and/or regulated by the electrical activity of the emergent circuits (activity-dependent) (Gu et al., 1994; Komuro and Rakic, 1996; Hanson et al., 2008). Activity-independent mechanisms in neurons are typically implicated in the processes of differentiation, migration, neurite outgrowth, and axon guidance. Simultaneously, the spontaneous electrical activity of the forming neuronal cells affects neural circuit development and guides the layout of early connectivity maps in many parts of the brain (Corlew et al., 2004; Crepel et al., 2007; Kirkby et al., 2013). The electrical activity of biological cells is generated by the controlled movement of ions across their membranes. Ionic flows are regulated by a fundamental class of integral membrane proteins, the ion channels, that provide the physical pathway for ions (Hille, 2001). This argues that ion channels, which often are embryonically expressed, play important roles in the processes underlying prenatal neurodevelopment. An important corollary to this hypothesis is that aberrant channels, for example resulting from genetic mutations, can give rise to neurodevelopmental pathologies. In fact, gene variants encoding all major families of ion channels can cause encephalopathies in which severe developmental delays (SDDs), intellectual disability (ID), electroencephalogram abnormalities, and brain malformations often co-exist with epileptic seizures (Berg et al., 2010; Guerrini et al., 2023).
Search Strategy
Bibliographic searches were performed in November 2022 in PubMed (National Library of Medicine, National Institutes of Health) using the following keywords: “developmental channelopathy”; “neurodevelopment, sodium channel”; “neurodevelopment, calcium channel”; “neurodevelopment, potassium channel”; “neurodevelopment, integrin”; “neuronal development, channel”.
Loss-of-Function and Gain-of-Function Mutant Channels Cause Epileptic Disorders
The notion that ion channels are involved in epileptic disorders began to emerge at the end of the 20th century, when genetic screening of patients affected by idiopathic epilepsies mapped mutations in genes encoding the neuronal nicotinic acetylcholine receptor, the voltage-gated sodium (Na+), potassium (K+), and calcium (Ca2+) channel (Steinlein et al., 1995; Ophoff et al., 1996; Biervert et al., 1998; Charlier et al., 1998; Sing et al., 1998; Wallace et al., 1998; Escayg et al., 2000; Sugawara et al., 2001). As the list of pathogenic variants increased, correlations began to emerge between the functional effects of a mutation and its underlying pathology. In general, mutations in ion channel genes can give rise to two types of proteins: loss-of-function (LOF) or gain-of-function (GOF) variants. Roughly, a LOF mutant is a channel that, under the same experimental conditions (voltage, concentration of ligand, etc.), conducts a smaller current compared to the wild type (WT) channel, and vice versa, a GOF mutant is a channel that conducts a larger current than WT. Thus, LOF and GOF mutants should produce distinct, if not opposite, effects on excitability. For example, a LOF mutation in a voltage-gated K+ channel should make the cell hyperexcitable and thus, is expected to be pro-epileptic since K+ channels mediate the repolarization phase of the action potential (AP). In contrast, a GOF mutation should render the cell hypoexcitable and consequently, be anti-epileptic. The opposite should be true for the channels that depolarize the cell, such as sodium (Na+), calcium (Ca2+), and non-selective cation channels. Nonetheless, the distinction between LOF and GOF is blurry, and for any channel gene, both types can be pathogenic.
Gain-of-Function and Loss-of-Function Na+ Channel Variants Cause Encephalopathies
Gain-of-function and loss-of-function mutations in the genes encoding for the alpha subunits of voltage-gated Na+ channels family 1 (Nav1.x) are known to cause autism spectrum disorders, ID, and developmental and epileptic encephalopathies (DEEs), a group of severe conditions characterized by developmental delays, that often present with seizures and epileptiform abnormalities (Scheffer et al., 2017; Bar et al., 2020; Reynolds et al., 2020; Zaman et al., 2020; Miao et al., 2021). Intriguingly, a significant percentage of Nav1.x LOF variants cause seizures, a fact apparently not consistent with their expected anti-epileptic action. Studies in mice showed that the pyramidal neurons of the prefrontal cortex and striatum that express Nav1.2 are intrinsically hyperexcitable (Spratt et al., 2019, 2021; Zhang et al., 2021). In those neurons, the diminution of Na+ influx is compensated by a decrease in K+ conductances. This results in incomplete neuronal repolarization between consecutive APs along with reduced AP backpropagation into dendrites that cause increased firing frequency and impaired synaptic functionality. The Nav1.x channels provide one of the best examples of the functional heterogeneity through which GOF and LOF variants dysregulate excitability. However, adding to the complexity, ictogenic Na+ channel alleles are often coupled with brain abnormalities known as malformations of cortical development (MCDs) (Kessi et al., 2020). For example, Barba et al. reported the clinical cases of six patients diagnosed with Dravet syndrome, an autosomal dominant genetic epileptic encephalopathy characterized by prolonged febrile seizures (Barba et al., 2014). The patients carried mutations in the SCN1A gene that encodes Nav1.1 – that are consistent with both LOF and GOF products (missense, truncated, spliced) – and presented multiple MCDs, including bilateral periventricular nodular heterotopia in two patients, focal cortical dysplasia in three, and multifocal micronodular dysplasia in the left temporal lobe in one. Pluripotent stem cell derived neural progenitor cells and GABAergic inter-neuronal cells from Dravet syndrome patients exhibited transcriptional changes in Nav1.1 haploinsufficient GABAergic cells, likely giving rise to immature neural differentiation and maturation (Schuster et al., 2019). In mice heterozygous in Scnaq1 Knockout--a model of Dravet syndrome--the axon caliber within the corpus callosum was reduced compared to control littermates, even in the absence of seizures (Richards et al., 2021).
Pathogenic Ion Channel Alleles Give Rise to Electrical and Anatomical Disturbances
The ability to induce severe encephalopathy via a variety of mechanisms, from dysregulated neuronal excitability to neuroanatomical defects, is not a prerogative of Na+ channels but, rather, a general feature of neuronal ion channels. Non-selective cation channels, such as the N-methyl-D-aspartate receptor, a ligand-gated channel broadly expressed in the brain, provide compelling examples of how channels can cause both electrical and anatomical disturbances (Endele et al., 2010; Hamdan et al., 2011; Carvill et al., 2013; Lesca et al., 2013; Lemke et al., 2014, 2013; Platzer et al., 2017; Fry et al., 2018). Mature N-methyl-D-aspartate receptors are heteromers composed of two GluN1 subunits and GluN2 and/or GluN3 subunits that, upon binding of glutamate and glycine, conduct Ca2+, Na+, and K+ ions that increase cellular excitability (Salussolia et al., 2011). Several studies have identified pathogenic variants in both GluN1 and GluN2 genes that alter the biophysical and pharmacological properties of the receptors (for example, they alter the ligand sensitivity) and simultaneously produce severe MCDs, primarily polymicrogyria (overfolded cerebral cortex) along with SDDs, ID, autism, and movement disorders. In a study examining the functional properties of pro-epileptic alpha1 subunit of γ-aminobutyric acid type-A receptor subunits present in a cohort of French Canadian families, Lachance-Touchette et al. (2014) found that those mutations altered the morphology of GABAergic boutons and spine formation. The authors concluded that a simple LOF model could not explain the pathogenic effects of different γ-aminobutyric acid type-A receptor mutations, which turned out to be specific to the mutation. Meganathan et al. (2021) examined the effects of copy number variants of the alpha-7 nicotinic acetylcholine receptor subunit (CHRNA7 gene) using induced pluripotent stem cells obtained from an autistic child (AP), his asymptomatic mother and unrelated controls. Remarkably, while the copy number variants were associated with dysregulated excitability in both AP and asymptomatic mother neurons, the AP cells alone exhibited other defects not imputable to the conducting functions of the channel (non-ionic functions), including increased proliferation and impaired differentiation, maturation, and migration. This finding suggests that those non-ionic defects, rather than altered excitability, were the primary cause of the autistic traits in the AP patient. A multi-center study focused on calcium channel Cav2.2 LOF (CACNA1B gene) variants linked to DEEs reported that all affected individuals presented with epileptic encephalopathy, SDDs, and a hyperkinetic movement disorder, while some patients also had postnatal microcephaly and hypotonia (Gorman et al., 2019). Knocking out Cav1.2 significantly impaired neurite outgrowth in mice (Ehlinger and Commons, 2017; Kamijo et al., 2018). Moreover, expression of a GOF Cav1.2 mutant found in Timothy syndrome (an autosomal dominant disorder characterized by physical malformations, neurological and developmental defects, and autism spectrum disorders) led to impaired radial migration of Layer II/III excitatory neurons, along with neuroanatomical alterations and differences in serotonin metabolism.
Gain-of-Function and Loss-of-Function K+ Channel Variants Are Implicated in Developmental Encephalopathy
M current (so-called due to its sensitivity to muscarinic agonists) is a voltage-dependent non-inactivating K+ current, that activates at sub-threshold potentials (around –70 mV) (Brown and Adams, 1980). M current repolarizes the membrane during the firing of APs and, thus, acts as a brake of neuronal excitability. The molecular correlates of the M current are the voltage-gated K+ channels of the sub-family 7, KCNQ2, KCNQ3, and KCNQ5, which form heteromers to form M channels (Wang et al., 1998; Lerche et al., 2000; Schroeder et al., 2000). Mutations in KCNQ2, KCNQ3, and KCNQ5 genes have been found to cause benign familial neonatal seizures and epileptic encephalopathies (Nappi et al., 2020). Given the role of M current as a brake of neuronal excitability, one would expect KCNQx LOF mutations to be associated with severe epilepsies and GOF mutations with more benign clinical manifestations. However, this turns out not to be the case (Miceli et al., 2015; Lehman et al., 2017; Millichap et al., 2017; Mulkey et al., 2017; Rosti et al., 2019; Sands et al., 2019; Nappi et al., 2022, 2020). For example, an international team led by Sands et al. (2019) functionally characterized KCNQ3 mutations found in the genomes of 11 DEE patients presenting SDDs and autistic features, with or without seizures. All variants gave rise to GOF mutants. Similarly, de novo LOF and GOF missense mutations in KCNQ5 were identified in patients with ID and other neurological abnormalities, with or without seizures (Lehman et al., 2017). A systematic review of the literature of K+ channel variants implicated in ID, including KCNMA1, KCNN3, KCNT1, KCNT2, KCNJ10, KCNJ6, KCNJ11, KCNA2, KCNA4, KCND3, KCNH1, KCNQ2, KCNAB1, KCNQ3, KCNQ5, KCNC1, KCNB1, KCNC3, and KCTD3, showed that the majority gave rise to mutants with both LOF and GOF properties, and only a small fraction of mutants with LOF or GOF attributes alone (Kessi et al., 2020). A review of 37 KCNMA1 alleles--KCNMA1 encodes the large conductance calcium-activated potassium channel--from 69 patients affected by severe encephalopathies presenting movement disorders, seizures, SDDs, and ID, highlighted the lack of apparent differences in the seizure phenotype between individuals carrying LOF or GOF variants (Miller et al., 2021). Most importantly, K+ channel gene variants can give rise to MCDs. One of these genes is KCNB1, which encodes a delayed rectifier and voltage-gated K+ channel expressed in the brain, pancreas, and other organs (Sesti et al., 2014). In addition to conducting an outward K+ current that modulates the excitability of pyramidal neurons in the cortex and hippocampus, KCNB1 (alias Kv2.1) possesses multiple non-ionic functions (Forzisi and Sesti, 2022). For example, KCNB1 forms non-conducting clusters that participate in the organization of points of contact between the endoplasmic reticulum and the plasma membrane (Trimmer, 1991; Scannevin et al., 1996; Murakoshi and Trimmer, 1999; Kirmiz et al., 2018; Schulien et al., 2020). Further, KCNB1 forms macromolecular complexes with integrins that will be discussed in some detail in this review (Yu et al., 2017). Genetic studies show that the severity of the clinical manifestations of KCNB1 encephalopathies is graded: typically, all patients present with SDDs and ID, and the majority (~80%) have seizures. In addition, roughly 30% of patients also have MCDs, including periventricular white matter abnormalities, progressive brain atrophy, and mild cerebellar volume loss, just to mention some (de Kovel et al., 2017; Latypova et al., 2017; Marini et al., 2017; Bar et al., 2020; Uctepe et al., 2022). Púa-Torrejón et al. (2021) performed a retrospective study of four patients from three families with KCNB1 encephalopathy. All individuals had mild-to-moderate ID. One patient displayed progressive cortical atrophy. Three of them developed epilepsy. Generally, patients presenting with magnetic resonance imaging abnormalities have a severe DEE phenotype. However, clinical cases of LOF or GOF KCNB1 variants giving rise to mild phenotypes, MCDs (periventricular heterotopia, polymicrogyria, and abnormal corpus callosum), and no seizures are also well documented (Veale et al., 2022; Hiraide et al., 2023).
K+ Channels Play a Crucial Role in Prenatal Neurodevelopment
Many hypotheses have been formulated to explain the apparent paradox of K+ channel GOF mutants producing severe encephalopathies with MCDs (Niday and Tzingounis, 2018). Models based on ionic mechanisms alone fall short of accounting for the lack of seizures and/or the MCDs in several patients. K+ channels are robustly expressed during prenatal brain development, and deficits in neuronal migration are a primary cause of malformations of cortical development associated with seizures. This supports an argument that one possible mechanism by which pathogenic K+ channel mutants cause severe encephalopathies is by impairing neonatal neurodevelopment (Moffat et al., 2015). The first evidence corroborating this hypothesis came from work from Bando et al. (2014), who showed that knockdown of 2 pore K+ channels KCNK2, KCNK9, or KCNK10, impaired the migration of cortical excitatory neurons destined to the Layer II/III during the formation of the neocortex. KCNK9 is not the only K+ channel that can give rise to a migratory phenotype. KCNB1, a channel we have previously seen having strong epileptic potential, was found to impair the migration of glutamatergic neurons in the neocortex (Bortolami et al., 2022). It turns out that KCNB1 channels control migratory processes through their non-ionic functions.
K+ Channels Form Macromolecular Complexes with Integrins
Ion channels barely exist as isolated entities in the lipid membrane. Usually, functional channels are heteromeric complexes containing both conducting (alpha) and accessory (beta, gamma, etc.) subunits (Abbott, 2021). In addition, channels can form macromolecular complexes with signaling molecules and receptors of different classes (Forzisi and Sesti, 2022). One common type of those complexes is macromolecules resulting from the assembly of integrins with K+ channels, dubbed integrin-K+ channel complexes or IKCs (Forzisi and Sesti, 2022). Integrins are one of the main transmembrane cell adhesion molecules and are known for their role in binding the extracellular matrix, to regulate the cell cycle, the organization of the actin cytoskeleton, cell proliferation, differentiation, apoptosis, the transport of receptors, and of intermembrane proteins. Accordingly, integrins and their signaling machinery have an established importance in determining neurodevelopmental processes by regulating the activities of cytoplasmic kinases and growth factor receptors and by controlling the organization of the actin cytoskeleton. For example, the interactions of integrins with Reelin direct the physiological cortical placement of pyramidal neurons in the developing neocortex (Dulabon et al., 2000). Integrins are essential for neuron-glia recognition processes that occur during embryonic neuronal development and are fundamental for the maintenance of neuronal migration along glial fibers (Anton et al., 1999). By retroviral transfer of antisense integrin α6 and α8 sequences in the developing chicken central nervous system, Zhang and Galileo (1998) showed that integrin α8 plays a main role in cell survival, while α6 is involved in the migration of tectal cells into specific laminae. Moreover, Marchetti et al. (2010), using in-utero electroporation to introduce short hairpin RNAs in the developing brain, demonstrated the importance of α5β1 dimers to regulate the radial migration of cortical neurons. The cells that received electroporation exhibited abnormal morphology and accumulation in the pre-migratory region, underscoring the key role of integrins in the process of cortical layer fate determination.
IKCs Cause Brain Abnormalities through Non-Ionic Mechanisms
One K+ channel known to form IKCs is KCNB1, which assembles with α5β5 integrin dimers in the neocortex (Figure 1; Yu et al., 2017; Bortolami et al., 2022). The first evidence that KCNB1 channels are implicated in migratory processes through integrins came from a study by Wei et al. (2008), who showed that KCNB1 acts to enhance cell adhesion and migration by favoring the phosphorylation of focal adhesion kinase (FAK) – an enzyme specifically activated by integrins. Those initial findings were later confirmed by an in vitro study examining the impact on cell migration and neurite outgrowth of IKCs formed with WT, named IKCKCNB1, and several KCNB1 mutants, including five variants found in children with DEE (Yu et al., 2019). IKCKCNB1 are multipronged macromolecules that regulate both ionic fluxes across the plasma membrane and intracellular biochemical events through the integrin signaling machinery. Thus, IKCKCNB1 signal via FAK, which autophosphorylates Tyr397, creating a binding site for Src family tyrosine kinases (Figure 1A; Wei et al., 2008; Yu et al., 2017). In turn, FAK-Src complexes connect IKCKCNB1 to mitogen-activated protein kinase (Ras-MAPK) pathways, to Akt signaling, and to the actin cytoskeleton through the recruitment of actin-binding proteins such as Vinculin, Talin, and Paxillin (Forzisi et al., 2022). All five KCNB1 DEE-susceptibility mutants were LOF: four did not conduct current and a fifth, R312H, exhibited a marked rightward shift (~40 mV) in the half-maximal voltage for activation, producing macroscopic currents significantly smaller than WT currents (Figure 1B; Yu et al., 2019). In contrast, the signaling of IKCs formed with the DEE mutants varied to a considerable extent: the more IKC signaling was impaired, the more their enhancing effect on cell migration and neurite outgrowth was diminished. These results prompted the novel concept that IKCKCNB1 may control processes such as cell migration and neurite outgrowth through their non-conducting functions (i.e., integrin signaling) rather than through their current. This notion was later tested using a knock in mouse model harboring the Kcnb1R312H gene variant, which was found in two children with DEE (de Kovel et al., 2017; Bortolami et al., 2022). The Kcnb1R312H mice exhibited a DEE-like phenotype characterized by frequent and spontaneous seizures, anxiety, and compulsive behavior. In addition, the glutamatergic neurons of Kcnb1R312H homozygotes displayed marked migratory defects. Significant numbers of neuronal progenies remained in the deep layers instead of migrating to the upper layers, indicating that IKCKCNB1 is required for neuronal migration. Deficits in neuronal migration are a primary cause of MCDs and can be associated with seizures (Moffat et al., 2015). Accordingly, in adult neocortices of Kcnb1R312H animals, the number of intracortically projecting neurons destined to the upper layers was markedly decreased because they remained stuck in the deep layers (Figure 1C). At the level of the single neuron, Kcnb1R312H cortical pyramidal cells had disrupted columnar organization of apical dendrites, that were either disoriented or reversed and presented excessive arborizations around the soma (Figure 1D). There was increased number of immature dendritic spines, and synaptic functionality was impaired (Figure 1E). The LOF attributes of R312H appear to be responsible for the severe seizure phenotype of the Kcnb1R312H animals, which was comparable between the heterozygotes and the homozygotes. In contrast, migratory and morphological defects were much more marked in the homozygotes compared to the heterozygotes. In parallel, the cascades engaged by IKCKCNB1 were significantly down-activated in the brains of the homozygotes, whereas they were normally active in the brains of the heterozygotes, pointing out to IKC signaling as the likely culprit for the neurodevelopmental phenotype. In fact, angiotensin II, a FAK activator, suppressed the morphological and synaptic defects of primary Kcnb1R312H cortical neurons (Polte et al., 1994; Bortolami et al., 2022). Conversely, the inhibition of FAK activity in WT neurons induced morphological and synaptic defects equivalent to those of Kcnb1R312H neurons. While the role of the non-ionic functions of IKCKCNB1 in the context of neurodevelopment appears to be sufficiently elucidated, the function of current remains poorly understood. One problem is that the interplay between ionic and non-ionic functions is synergetic and intrinsically linked in IKCKCNB1. Conformational changes in the KCNB1 channel associated with its conducting functions, such as opening and closing, are likely to be translated by the integrins into biochemical signals. Evidence suggests that the IKCKCNB1 current can enhance the biochemical cascades engaged by the complex (Yu et al., 2019). Further, the emergence of functional connectivity in the neocortex depends on neuronal migration (Evsyukova et al., 2013). Thus, it is tempting to speculate that during the formation of the neocortex, IKCKCNB1 sense and translate the spontaneous electrical activity of emergent circuits into biochemical signals that help guide their formation.
Figure 1.
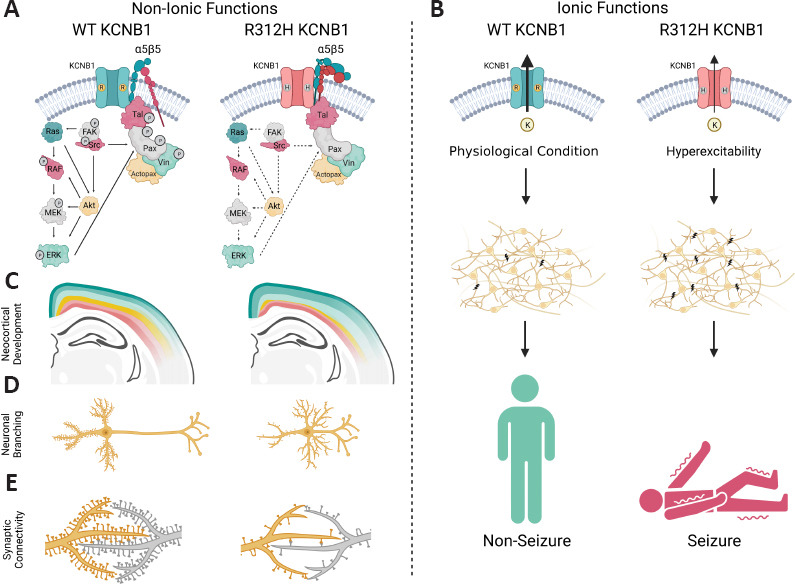
IKCs cause encephalopathy through ionic and non-ionic mechanisms.
(A) IKCs formed with integrin α5β5 dimers and KCNB1 WT (IKCKCNB1) channels recruit FAK via α5β5 integrins. FAK autophosphorylates Tyr397 (P) and then binds Src kinases. The newly formed FAK-Src dimers phosphorylate Paxillin that links the IKCKCNB1 to Ras-MAPK signaling pathways and to the actin cytoskeleton. Ras GTPases and FAK-Src dimers also cross-talk with PI3K/Akt signaling which phosphorylates several MAPK components. In turn, activated ERK phosphorylates Paxillin. In contrast, IKCs formed with R312H mutant channels (IKCR312H) fail to induce phosphorylation of the various signaling components including Vinculin, Talin-1, and Paxillin which however, still assemble with the complexes by binding to the cytoplasmic tail of integrin β5. (B) IKCs formed with either heteromeric WT/R312H or homomeric R312H channels conduct significantly smaller K+ currents compared to IKCs formed with WT channels alone. The LOF attributes of the mutant IKCs cause cortical hyperexcitability and seizures. (C) Along with impaired IKCR312H signaling, Kcnb1R312H mice, primarily the homozygotes, are affected by severe cortical neuroabnormalities, with many glutamatergic neurons destined to the Upper Layers misplaced in the Deep Layers (yellow). (D) The neurons of Kcnb1R312H mice develop shorter dendrites compared to WT neurons, and abnormal arborizations around the soma. (E) Synaptic functionality is significantly reduced in the brains of Kcnb1R312H homozygotes. This goes on par with the increased numbers of immature spines in the pyramidal neurons of those animals. α5β5: Integrin alpha5-beta5; Actopax: actopaxin; Akt: protein kinase B; ERK: extracellular signal-regulated kinase; FAK: focal adhesion kinase; KCNB1: voltage-gated potassium channel subfamily 2, member 1; MEK: mitogen-activated protein kinase kinase; P: phosphate; Pax: paxillin; RAF: rapidly accelerated fibrosarcoma kinase; Ras: rat sarcoma virus GTPase; Src: Src family kinase; Tal: talin-1; Vin: vinculin. Created with BioRender.com.
Conclusions
Epilepsy is one of the most common neurological disorders affecting tens of millions of individuals worldwide (GBD 2015 Disease and Injury Incidence and Prevalence Collaborators, 2016). Epilepsy reflects abnormal electrical activity and ion channels, given their role in modulating neuronal excitability, are one of the most common culprits--to the point that a specific term, channelepsy, has been coined (Pandolfo, 2011; D’Adamo et al., 2013). However, the fact that both LOF and GOF channel variants are associated with epilepsy syndromes and that pro-epileptic alleles of major ion channel families are often coupled to brain abnormalities, suggest that a picture in which dysfunctional ion channels contribute to epileptogenesis only by promoting hyperexcitability and seizures is too simplistic. Rather, it appears that ion channels can simultaneously affect the development and the excitability of the brain (Berg et al., 2010). This notion has received strong support from the recent finding that the K+ channels are key to landmark neurodevelopmental processes, including neuronal migration, neurite outgrowth, and synaptic formation, and when aberrant, can induce MCDs with strong ictogenic potential. Future studies will elucidate the molecular basis for these mechanisms in detail.
Acknowledgments:
We thank Diego Cotella and Elena Forzisi for the critical reading of the manuscript.
Footnotes
Funding: This work was supported by an NJ Governor’s Council for Medical Research and Treatment of Autism predoctoral fellowship (CAUT23AFP015) to AB, and a National Science Foundation grant (2030348) to FS.
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement: Not applicable.
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Abbott GW. Control of biophysical and pharmacological properties of potassium channels by ancillary subunits. Handb Exp Pharmacol. 2021;267:445–480. doi: 10.1007/164_2021_512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anton ES, Kreidberg JA, Rakic P. Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- 3.Bando Y, Hirano T, Tagawa Y. Dysfunction of KCNK potassium channels impairs neuronal migration in the developing mouse cerebral cortex. Cereb Cortex. 2014;24:1017–1029. doi: 10.1093/cercor/bhs387. [DOI] [PubMed] [Google Scholar]
- 4.Bar C, Barcia G, Jennesson M, Le Guyader G, Schneider A, Mignot C, Lesca G, Breuillard D, Montomoli M, Keren B, Doummar D, Billette de Villemeur T, Afenjar A, Marey I, Gerard M, Isnard H, Poisson A, Dupont S, Berquin P, Meyer P, et al. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies:27 new patients and overview of the literature. Hum Mutat. 2020;41:69–80. doi: 10.1002/humu.23915. [DOI] [PubMed] [Google Scholar]
- 5.Barba C, Parrini E, Coras R, Galuppi A, Craiu D, Kluger G, Parmeggiani A, Pieper T, Schmitt-Mechelke T, Striano P, Giordano F, Blumcke I, Guerrini R. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014;55:1009–1019. doi: 10.1111/epi.12658. [DOI] [PubMed] [Google Scholar]
- 6.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, Moshé SL, Nordli D, Plouin P, Scheffer IE. Revised terminology and concepts for organization of seizures and epilepsies:report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 7.Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 8.Bortolami A, Yu W, Forzisi E, Ercan K, Kadakia R, Murugan M, Fedele D, Estevez I, Boison D, Rasin MR, Sesti F. Integrin-KCNB1 potassium channel complexes regulate neocortical neuronal development and are implicated in epilepsy. Cell Death Differ. 2022 doi: 10.1038/s41418-022-01072-2. doi:10.1038/s41418-022-01072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 10.Carvill GL, Regan BM, Yendle SC, O'Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, Sadleir LG, Turner SJ, Tsai MH, Webster R, Ouvrier R, Damiano JA, Berkovic SF, Shendure J, Hildebrand MS, Szepetowski P, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45:1073–1076. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 12.Corlew R, Bosma MM, Moody WJ. Spontaneous, synchronous electrical activity in neonatal mouse cortical neurones. J Physiol. 2004;560:377–390. doi: 10.1113/jphysiol.2004.071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crépel V, Aronov D, Jorquera I, Represa A, Ben-Ari Y, Cossart R. A parturition-associated nonsynaptic coherent activity pattern in the developing hippocampus. Neuron. 2007;54:105–120. doi: 10.1016/j.neuron.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 14.D'Adamo MC, Catacuzzeno L, Di Giovanni G, Franciolini F, Pessia M. K(+) channelepsy:progress in the neurobiology of potassium channels and epilepsy. Front Cell Neurosci. 2013;7:134. doi: 10.3389/fncel.2013.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Kovel CGF, Syrbe S, Brilstra EH, Verbeek N, Kerr B, Dubbs H, Bayat A, Desai S, Naidu S, Srivastava S, Cagaylan H, Yis U, Saunders C, Rook M, Plugge S, Muhle H, Afawi Z, Klein KM, Jayaraman V, Rajagopalan R, et al. Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. 2017;74:1228–1236. doi: 10.1001/jamaneurol.2017.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 17.Ehlinger DG, Commons KG. Altered Cav1.2 function in the Timothy syndrome mouse model produces ascending serotonergic abnormalities. Eur J Neurosci. 2017;46:2416–2425. doi: 10.1111/ejn.13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, Hellenbroich Y, Kalscheuer VM, Kohlhase J, Moog U, Rappold G, Rauch A, Ropers HH, von Spiczak S, Tönnies H, Villeneuve N, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 19.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 20.Evsyukova I, Plestant C, Anton ES. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol. 2013;29:299–353. doi: 10.1146/annurev-cellbio-101512-122400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forzisi E, Sesti F. Non-conducting functions of ion channels:the case of integrin-ion channel complexes. Channels. 2022;16:185–197. doi: 10.1080/19336950.2022.2108565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forzisi E, Yu W, Rajwade P, Sesti F. Antagonistic roles of Ras-MAPK and Akt signaling in integrin-K+channel complex-mediated cellular apoptosis. FASEB J. 2022;36:e22292. doi: 10.1096/fj.202200180R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fry AE, Fawcett KA, Zelnik N, Yuan H, Thompson BAN, Shemer-Meiri L, Cushion TD, Mugalaasi H, Sims D, Stoodley N, Chung SK, Rees MI, Patel CV, Brueton LA, Layet V, Giuliano F, Kerr MP, Banne E, Meiner V, Lerman-Sagie T, et al. De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain. 2018;141:698–712. doi: 10.1093/brain/awx358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorman KM, Meyer E, Grozeva D, Spinelli E, McTague A, Sanchis-Juan A, Carss KJ, Bryant E, Reich A, Schneider AL, Pressler RM, Simpson MA, Debelle GD, Wassmer E, Morton J, Sieciechowicz D, Jan-Kamsteeg E, Paciorkowski AR, King MD, Cross JH, et al. Bi-allelic loss-of-function CACNA1B mutations in progressive epilepsy-dyskinesia. Am J Hum Genet. 2019;104:948–956. doi: 10.1016/j.ajhg.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu X, Olson EC, Spitzer NC. Spontaneous neuronal calcium spikes and waves during early differentiation. J Neurosci. 1994;14:6325–6335. doi: 10.1523/JNEUROSCI.14-11-06325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerrini R, Conti V, Mantegazza M, Balestrini S, Galanopoulou AS, Benfenati F. Developmental and epileptic encephalopathies:from genetic heterogeneity to phenotypic continuum. Physiol Rev. 2023;103:433–513. doi: 10.1152/physrev.00063.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Park AR, Spiegelman D, Dobrzeniecka S, Piton A, Tomitori H, Daoud H, Massicotte C, Henrion E, Diallo O, S2D Group. Shekarabi M, Marineau C, Shevell M, Maranda B, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson MG, Milner LD, Landmesser LT. Spontaneous rhythmic activity in early chick spinal cord influences distinct motor axon pathfinding decisions. Brain Res Rev. 2008;57:77–85. doi: 10.1016/j.brainresrev.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hille B. Ionic channels of excitable membranes. 3rd ed. Sunderland, MA: Sinauer Associates; 2001. [Google Scholar]
- 31.Hiraide T, Akita T, Uematsu K, Miyamoto S, Nakashima M, Sasaki M, Fukuda A, Kato M, Saitsu H. A novel de novo KCNB1 variant altering channel characteristics in a patient with periventricular heterotopia, abnormal corpus callosum, and mild seizure outcome. J Hum Genet. 2023;68:25–31. doi: 10.1038/s10038-022-01090-5. [DOI] [PubMed] [Google Scholar]
- 32.Kamijo S, Ishii Y, Horigane SI, Suzuki K, Ohkura M, Nakai J, Fujii H, Takemoto-Kimura S, Bito H. A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J Neurosci. 2018;38:5551–5566. doi: 10.1523/JNEUROSCI.2357-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kandel ER, Schwartz JR, Thomas MJ. Principles of neuronal science. New York: McGraw-Hill; 2000. [Google Scholar]
- 34.Kessi M, Chen B, Peng J, Tang Y, Olatoutou E, He F, Yang L, Yin F. Intellectual disability and potassium channelopathies:a systematic review. Front Genet. 2020;11:614. doi: 10.3389/fgene.2020.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirkby LA, Sack GS, Firl A, Feller MB. A role for correlated spontaneous activity in the assembly of neural circuits. Neuron. 2013;80:1129–1144. doi: 10.1016/j.neuron.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirmiz M, Vierra NC, Palacio S, Trimmer JS. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum-plasma membrane junctions in mammalian brain neurons. J Neurosci. 2018;38:7562–7584. doi: 10.1523/JNEUROSCI.0893-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komuro H, Rakic P. Intracellular Ca2+fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 38.Lachance-Touchette P, Choudhury M, Stoica A, Di Cristo G, Cossette P. Single-cell genetic expression of mutant GABAA receptors causing human genetic epilepsy alters dendritic spine and GABAergic bouton formation in a mutation-specific manner. Front Cell Neurosci. 2014;8:317. doi: 10.3389/fncel.2014.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latypova X, Matsumoto N, Vinceslas-Muller C, Bezieau S, Isidor B, Miyake N. Novel KCNB1 mutation associated with non-syndromic intellectual disability. J Hum Genet. 2017;62:569–573. doi: 10.1038/jhg.2016.154. [DOI] [PubMed] [Google Scholar]
- 40.Lehman A, Thouta S, Mancini GMS, Naidu S, van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova R, CAUSES Study;EPGEN Study. Guella I, McKenzie MB, Datta A, Connolly MB, Kalkhoran SM, Poburko D, Friedman JM, Farrer MJ, Demos M, et al. Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am J Hum Genet. 2017;101:65–74. doi: 10.1016/j.ajhg.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, Geider K, Laube B, Schwake M, Finsterwalder K, Franke A, Schilhabel M, Jähn JA, Muhle H, Boor R, Van Paesschen W, Caraballo R, Fejerman N, Weckhuysen S, De Jonghe P, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. 2013;45:1067–1072. doi: 10.1038/ng.2728. [DOI] [PubMed] [Google Scholar]
- 42.Lemke JR, Hendrickx R, Geider K, Laube B, Schwake M, Harvey RJ, James VM, Pepler A, Steiner I, Hörtnagel K, Neidhardt J, Ruf S, Wolff M, Bartholdi D, Caraballo R, Platzer K, Suls A, De Jonghe P, Biskup S, Weckhuysen S. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann Neurol. 2014;75:147–154. doi: 10.1002/ana.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal m-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- 44.Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, Wright S, Tsintsadze V, Michel A, Doummar D, Lascelles K, Strug L, Waters P, de Bellescize J, Vrielynck P, de Saint Martin A, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;45:1061–1066. doi: 10.1038/ng.2726. [DOI] [PubMed] [Google Scholar]
- 45.Marchetti G, Escuin S, van der Flier A, De Arcangelis A, Hynes RO, Georges-Labouesse E. Integrin alpha5beta1 is necessary for regulation of radial migration of cortical neurons during mouse brain development. Eur J Neurosci. 2010;31:399–409. doi: 10.1111/j.1460-9568.2009.07072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marini C, Romoli M, Parrini E, Costa C, Mei D, Mari F, Parmeggiani L, Procopio E, Metitieri T, Cellini E, Virdò S, De Vita D, Gentile M, Prontera P, Calabresi P, Guerrini R. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol Genet. 2017;3:e206. doi: 10.1212/NXG.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meganathan K, Prakasam R, Baldridge D, Gontarz P, Zhang B, Urano F, Bonni A, Maloney SE, Turner TN, Huettner JE, Constantino JN, Kroll KL. Altered neuronal physiology, development, and function associated with a common chromosome 15 duplication involving CHRNA7. BMC Biol. 2021;19:147. doi: 10.1186/s12915-021-01080-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao P, Tang S, Ye J, Tang J, Wang J, Zheng C, Li Y, Feng J. Differential functional changes of Nav1.2 channel causing SCN2A-related epilepsy and status epilepticus during slow sleep. Front Neurol. 2021;12:653517. doi: 10.3389/fneur.2021.653517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci. 2015;35:3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller JP, Moldenhauer HJ, Keros S, Meredith AL. An emerging spectrum of variants and clinical features in KCNMA1-linked channelopathy. Channels (Austin) 2021;15:447–464. doi: 10.1080/19336950.2021.1938852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, Soldovieri MV, Ambrosino P, Shashi V, Mikati MA, Cooper EC, Taglialatela M. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia. 2017;58:e10–15. doi: 10.1111/epi.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moffat JJ, Ka M, Jung EM, Kim WY. Genes and brain malformations associated with abnormal neuron positioning. Mol Brain. 2015;8:72. doi: 10.1186/s13041-015-0164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grønborg S, Jiang YH, Joshi N, Kelly M, Koolen DA, Mikati MA, Park K, Pearl PL, Scheffer IE, Spillmann RC, Taglialatela M, Vieker S, Weckhuysen S, Cooper EC, Cilio MR. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia. 2017;58:436–445. doi: 10.1111/epi.13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+channel as a major component of the delayed rectifier K+current in rat hippocampal neurons. J Neurosci. 1999;19:1728–1735. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nappi M, Barrese V, Carotenuto L, Lesca G, Labalme A, Ville D, Smol T, Rama M, Dieux-Coeslier A, Rivier-Ringenbach C, Soldovieri MV, Ambrosino P, Mosca I, Pusch M, Miceli F, Taglialatela M. Gain of function due to increased opening probability by two KCNQ5 pore variants causing developmental and epileptic encephalopathy. Proc Natl Acad Sci U S A. 2022;119:e2116887119. doi: 10.1073/pnas.2116887119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nappi P, Miceli F, Soldovieri MV, Ambrosino P, Barrese V, Taglialatela M. Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflugers Arch - Eur J Physiol. 2020;472:881–898. doi: 10.1007/s00424-020-02404-2. [DOI] [PubMed] [Google Scholar]
- 57.Niday Z, Tzingounis AV. Potassium channel gain of function in epilepsy:an unresolved paradox. Neuroscientist. 2018;24:368–380. doi: 10.1177/1073858418763752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 59.Pandolfo M. Genetics of epilepsy. Semin Neurol. 2011;31:506–518. doi: 10.1055/s-0031-1299789. [DOI] [PubMed] [Google Scholar]
- 60.Platzer K, Yuan H, Schütz H, Winschel A, Chen W, Hu C, Kusumoto H, Heyne HO, Helbig KL, Tang S, Willing MC, Tinkle BT, Adams DJ, Depienne C, Keren B, Mignot C, Frengen E, Strømme P, Biskup S, Döcker D, et al. GRIN2B encephalopathy:novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J Med Genet. 2017;54:460–470. doi: 10.1136/jmedgenet-2016-104509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Polte TR, Hanks SK, Naftilan AJ. Focal adhesion kinase is abundant in developing blood vessels and elevation of its phosphotyrosine content in vascular smooth muscle cells is a rapid response to angiotensin II. J Cell Biochem. 1994;55:106–119. doi: 10.1002/jcb.240550113. [DOI] [PubMed] [Google Scholar]
- 62.Púa-Torrejón RC, González-Alguacil E, Soto-Insuga V, Moreno-Cantero T, Ortiz-Cabrera NV, Pérez-Poyato MS, Ruiz Falcó-Rojas ML, García-Peñas JJ. Variability of the clinical expression of KCNB1 encephalopathy. Rev Neurol. 2021;73:403–408. doi: 10.33588/rn.7312.2021267. [DOI] [PubMed] [Google Scholar]
- 63.Reynolds C, King MD, Gorman KM. The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol. 2020;24:117–122. doi: 10.1016/j.ejpn.2019.12.016. [DOI] [PubMed] [Google Scholar]
- 64.Richards K, Jancovski N, Hanssen E, Connelly A, Petrou S. Atypical myelinogenesis and reduced axon caliber in the Scn1a variant model of Dravet syndrome:An electron microscopy pilot study of the developing and mature mouse corpus callosum. Brain Res. 2021;1751:147157. doi: 10.1016/j.brainres.2020.147157. [DOI] [PubMed] [Google Scholar]
- 65.Rosti G, Tassano E, Bossi S, Divizia MT, Ronchetto P, Servetti M, Lerone M, Pisciotta L, Mancardi MM, Veneselli E, Puliti A. Intragenic duplication of KCNQ5 gene results in aberrant splicing leading to a premature termination codon in a patient with intellectual disability. Eur J Med Genet. 2019;62:103555. doi: 10.1016/j.ejmg.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 66.Salussolia CL, Prodromou ML, Borker P, Wollmuth LP. Arrangement of subunits in functional NMDA receptors. J Neurosci. 2011;31:11295–11304. doi: 10.1523/JNEUROSCI.5612-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sands TT, Miceli F, Lesca G, Beck AE, Sadleir LG, Arrington DK, Schönewolf-Greulich B, Moutton S, Lauritano A, Nappi P, Soldovieri MV, Scheffer IE, Mefford HC, Stong N, Heinzen EL, Goldstein DB, Perez AG, Kossoff EH, Stocco A, Sullivan JA, et al. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann Neurol. 2019;86:181–192. doi: 10.1002/ana.25522. [DOI] [PubMed] [Google Scholar]
- 68.Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS. Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+channel. J Cell Biol. 1996;135:1619–1632. doi: 10.1083/jcb.135.6.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies:Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–521. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- 71.Schulien AJ, Yeh CY, Orange BN, Pav OJ, Hopkins MP, Moutal A, Khanna R, Sun D, Justice JA, Aizenman E. Targeted disruption of Kv2.1-VAPA association provides neuroprotection against ischemic stroke in mice by declustering Kv2.1 channels. Sci Adv. 2020;6:eaaz8110. doi: 10.1126/sciadv.aaz8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schuster J, Laan L, Klar J, Jin Z, Huss M, Korol S, Noraddin FH, Sobol M, Birnir B, Dahl N. Transcriptomes of Dravet syndrome iPSC derived GABAergic cells reveal dysregulated pathways for chromatin remodeling and neurodevelopment. Neurobiol Dis. 2019;132:104583. doi: 10.1016/j.nbd.2019.104583. [DOI] [PubMed] [Google Scholar]
- 73.Sesti F, Wu X, Liu S. Oxidation of KCNB1 K(+) channels in central nervous system and beyond. World J Biol Chem. 2014;5:85–92. doi: 10.4331/wjbc.v5.i2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 75.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ, Jr, Clarkson RL, Sanders SJ, Bender KJ. The Autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron. 2019;103:673–685. doi: 10.1016/j.neuron.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spratt PWE, Alexander RPD, Ben-Shalom R, Sahagun A, Kyoung H, Keeshen CM, Sanders SJ, Bender KJ. Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 2021;36:109483. doi: 10.1016/j.celrep.2021.109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- 78.Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K. A missense mutation of the Na+channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Trimmer JS. Immunological identification and characterization of a delayed rectifier K+channel polypeptide in rat brain. Proc Natl Acad Sci U S A. 1991;88:10764–10768. doi: 10.1073/pnas.88.23.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uctepe E, Esen FN, Tümer S, Mancılar H, Yeşilyurt A. KCNB1 frameshift variant caused inherited intellectual disability, developmental delay, and seizure. Intractable Rare Dis Res. 2022;11:219–221. doi: 10.5582/irdr.2022.01096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Veale EL, Golluscio A, Grand K, Graham JM, Jr, Mathie A. A KCNB1 gain of function variant causes developmental delay and speech apraxia but not seizures. Front Pharmacol. 2022;13:1093313. doi: 10.3389/fphar.2022.1093313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 83.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits:molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 84.Wei JF, Wei L, Zhou X, Lu ZY, Francis K, Hu XY, Liu Y, Xiong WC, Zhang X, Banik NL, Zheng SS, Yu SP. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J Cell Physiol. 2008;217:544–557. doi: 10.1002/jcp.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu W, Gowda M, Sharad Y, Singh SA, Sesti F. Oxidation of KCNB1 potassium channels triggers apoptotic integrin signaling in the brain. Cell Death Dis. 2017;8:e2737. doi: 10.1038/cddis.2017.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu W, Shin MR, Sesti F. Complexes formed with integrin-alpha5 and KCNB1 potassium channel wild type or epilepsy-susceptibility variants modulate cellular plasticity via Ras and Akt signaling. FASEB J. 2019;33:14680–14689. doi: 10.1096/fj.201901792R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, Jansen AE, Verstraete L, Jacquinet A, Parrini E, Guerrini R, Fujiwara Y, Miyatake S, Ben-Zeev B, Bassan H, Reish O, Marom D, Hauser N, Vu TA, Ackermann S, et al. SCN3A-related neurodevelopmental disorder:a spectrum of epilepsy and brain malformation. Ann Neurol. 2020;88:348–362. doi: 10.1002/ana.25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J, Chen X, Eaton M, Wu J, Ma Z, Lai S, Park A, Ahmad TS, Que Z, Lee JH, Xiao T, Li Y, Wang Y, Olivero-Acosta MI, Schaber JA, Jayant K, Yuan C, Huang Z, Lanman NA, Skarnes WC, et al. Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep. 2021;36:109495. doi: 10.1016/j.celrep.2021.109495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Z, Galileo DS. Retroviral transfer of antisense integrin alpha6 or alpha8 sequences results in laminar redistribution or clonal cell death in developing brain. J Neurosci. 1998;18:6928–6938. doi: 10.1523/JNEUROSCI.18-17-06928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]