

Abstract
A 25‐year‐old man was diagnosed with diabetic ketoacidosis (DKA) at the onset of fulminant type 1 diabetes. After acute‐phase DKA treatment including placement of a central venous catheter, a massive deep vein thrombosis (DVT) and pulmonary embolism (PE) were detected on hospital day 15. His protein C (PC) activity and antigen levels were low even 33 days after completing the DKA treatment, indicating partial type I PC deficiency. Severe PC dysfunction, due to overlapping of partial PC deficiency and hyperglycemia‐induced PC suppression, concomitant with dehydration and catheter treatment, may have induced the massive DVT with PE. This case suggests that anti‐coagulation therapy should be combined with acute‐phase DKA treatment in patients with PC deficiency, even those who have been asymptomatic. As patients with partial PC deficiency should perhaps be included among those with severe DVT complications of DKA, venous thrombosis should always be considered as a potential complication of DKA.
Keywords: Deep vein thrombosis, Diabetic ketoacidosis, Protein C
We report a case of diabetic ketoacidosis (DKA) at the onset of fulminant type 1 diabetes, who developed massive deep vein thrombosis with pulmonary embolism after the acute‐phase DKA treatment, it being subsequently recognized that he had type I protein C partial deficiency. Our case highlights that venous thrombosis should always be considered as a potential complication of DKA.
INTRODUCTION
Fulminant type 1 diabetes (FT1D) is characterized by an extremely rapid and almost complete destruction of β‐cells, and its diagnostic criteria include diabetic ketoacidosis (DKA) 1 . Deep vein thrombosis (DVT), especially the resultant pulmonary embolism (PE), is rarely described, but can be a life‐threatening complication of DKA 2 , 3 . DKA leads not only to dehydration and hyperglycemia, but also to coagulation abnormalities. Among them, the protein and the activity levels of protein C (PC), also known as factor XIV, reportedly decrease under hyperglycemic conditions 4 , 5 . PC is a vitamin K‐dependent anticoagulant enzyme, the activated form of which inactivates factors V and VIII.
PC deficiency is a heritable or acquired risk factor for thrombophilia, with presentations varying from asymptomatic to venous thromboembolism, and even neonatal purpura fulminans. This disease is classified into two types: type I, a quantitative abnormality in which PC activity and antigen levels are decreased, and type II, a qualitative abnormality in which PC activity is decreased but antigen levels are maintained 6 . We report a case with DKA at the onset of FT1D, who developed massive DVT with PE after the acute‐phase DKA treatment. He was subsequently diagnosed as having type I PC deficiency.
CASE REPORT
A 25‐year‐old man presented to a hospital with a 2 day history of epigastric pain, nausea, and headache. His serum amylase was elevated (369 U/L), and computed tomography (CT) showed diffuse parenchymal enlargement and an indistinct pancreatic margin. He was diagnosed as having acute pancreatitis and hospitalized. He received intravenous therapy containing gabexate mesylate. On admission, the serum glucose was normal (116 mg/dL), but 3 days later, had risen to a high level (over 600 mg/dL). He was suspected to have DKA and was transferred to our hospital.
Arterial blood gas analysis showed pH 6.985 and 7.4 mEq/L. His serum glucose was 1,004 mg/dL with relatively low HbA1c (6.1%). He had an extremely low serum C‐peptide level (0.09 ng/mL) and 3+ urinary ketones, indicating DKA. A hemodialysis catheter was placed in his right femoral vein for 2 days to provide renal replacement therapy, because of a high potassium level (7.6 mmol/L) and dehydration. An anti‐coagulant, nafamostat mesylate, was intravenously administered during renal replacement therapy. His urinary C‐peptide excretion was 1.4 μg/day, fasting and 6‐min post‐glucagon‐stimulated serum C‐peptide levels were 0.08 ng/mL and 0.11 ng/mL, respectively (Table 1). These data met the diagnostic criteria for FT1D. After continuous and intravenous insulin infusion, the therapy was switched to multiple daily insulin injections (MDII), ultimately achieving good control of blood glucose levels.
Table 1.
Laboratory results
| Unit | |||
|---|---|---|---|
| Blood chemistry | |||
| Glucose | 1,004 | mg/dL | |
| HbA1c | 6.1 | % | |
| Glycoalbumin | 23.5 | % | |
| Amylase | 136 | U/L | |
| Lipase | 190 | U/L | |
| T‐Bil | 0.5 | mg/dL | |
| γGTP | 21 | U/L | |
| AST | 17 | U/L | |
| ALT | 14 | U/L | |
| LDH | 175 | U/L | |
| BUN | 53 | mg/dL | |
| Cre | 2.04 | mg/dL | |
| UA | 12.6 | mg/dL | |
| TP | 7 | g/dL | |
| Alb | 4.5 | g/dL | |
| Na | 129 | mmol/L | |
| K | 7.6 | mmol/L | |
| Cl | 88 | mmol/L | |
| Ca | 8.9 | mg/dL | |
| CRP | 1.9 | mg/dL | |
| WBC | 17,100 | /μL | |
| Hb | 14.6 | g/dL | |
| Plt | 230 × 103 | /μL | |
| Urinalysis | |||
| Ketone body | 3+ | ||
| Protein | ± | ||
| Glucose | 4+ | ||
| Arterial blood gas analysis | |||
| Ph | 6.985 | ||
| PCO2 | 32.7 | mmHg | |
|
|
7.4 | mmol/L | |
| Base excess | −24.4 | mmol/L | |
| Insulin secretory ability | |||
| Serum CPR | 0.09 | ng/mL | |
| Urinary CPR (day 8) | 1.4 | μg/day | |
| Glucagon loading test (day 12) | |||
| CPR 0 min | 0.08 | ng/mL | |
| CPR 6 min | 0.11 | ng/mL | |
| Islet‐related autoantibody | |||
| Anti‐GAD antibody |
13.2 (day 1) 5.6 (day 29) |
U/mL | |
| Anti‐IA‐2 antibody | <0.4 | U/mL | |
| Anti‐Insulin antibody | <0.4 | % | |
| Anti‐ZnT8 antibody | <15.0 | U/mL | |
| HLA typing | |||
| HLA‐DRB1 | *15:02/*13:02 | ||
| HLA‐DQB1 | *06:01:01/*06:04:01 | ||
CPR, C‐peptide immunoreactivity; GAD, glutamic acid decarboxylase; HLA, human leukocyte antigen; IA‐2, insulinoma‐associated antigen‐2; ZnT8, zinc transporter 8.
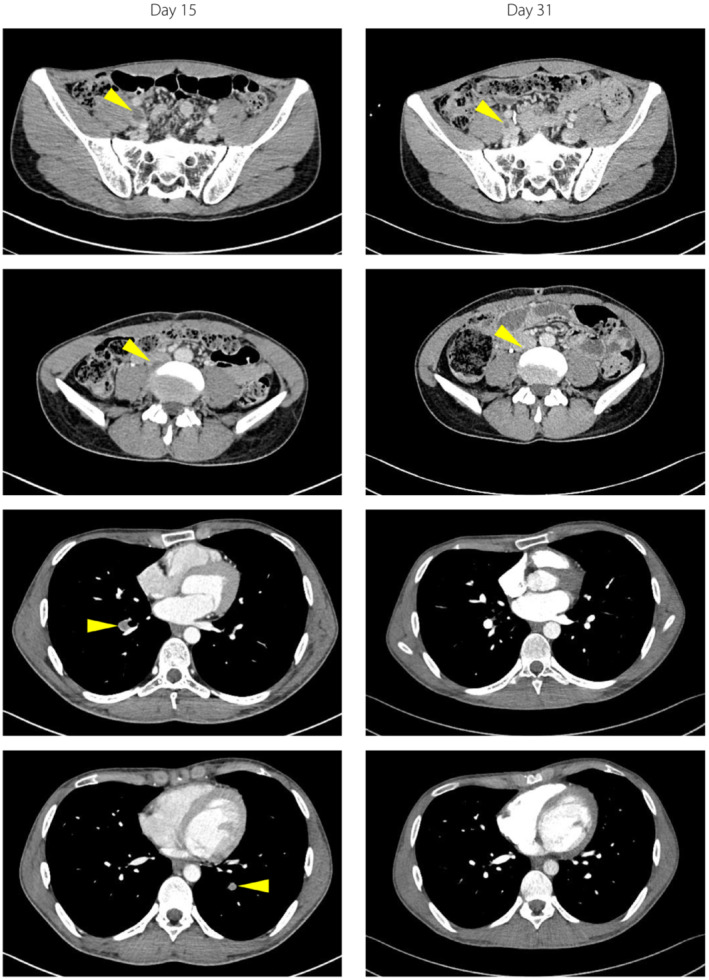
On hospital day 15, in the course of examinations to identify the cause of pancreatitis, a DVT and PE were detected coincidentally with no physical symptoms or vital sign abnormalities. The CT revealed a DVT extending from the right external iliac vein to the inferior vena cava (Figure 1). Blood tests showed elevated D‐dimer (2.5 μg/mL) and fibrin/fibrinogen degradation products (FDP; 5.9 μg/mL), and decreased PC activity (46%). Neither the patient nor any of his family members had had histories of thrombotic disorders, and the results of autoantibodies were negative (Table 2). We started anticoagulation therapy; heparin (5,000 units) was administered intravenously, followed by 30 mg of an oral direct factor Xa inhibitor, rivaroxaban. The CT images showed a tendency for amelioration of the DVT and PE. D‐dimer and FDP were also normalized. In contrast, PC activity (49%) and antigen (41%) levels were still low at day 33. These data allowed us to diagnose partial type I PC deficiency in this patient 6 .
Figure 1.
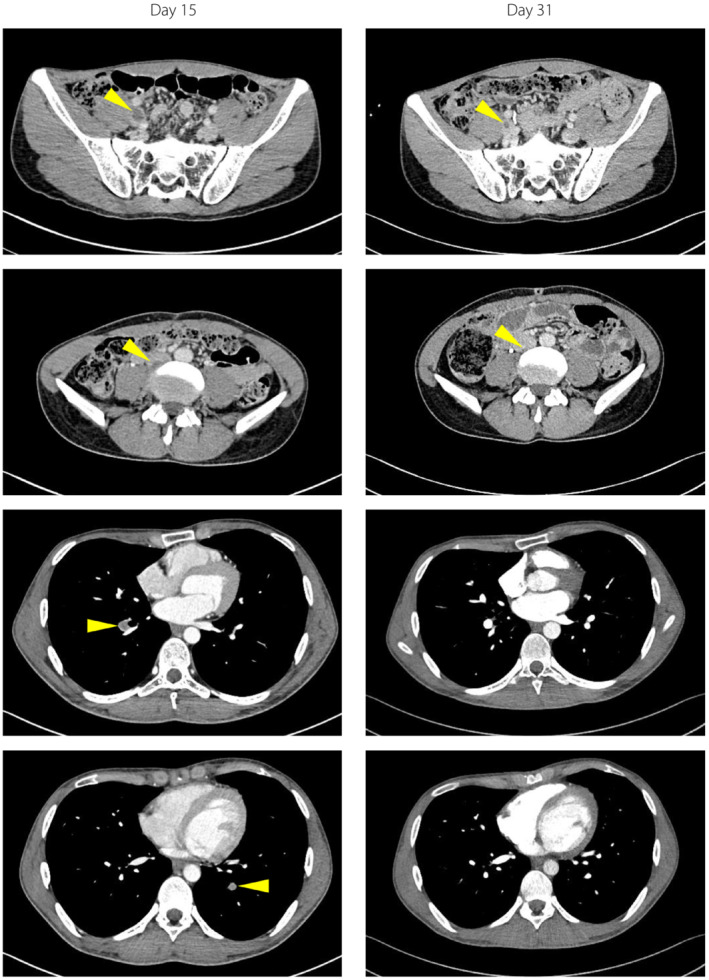
Contrast‐enhanced computed tomography images obtained on days 15 and 31. Yellow arrows indicate the thrombus.
Table 2.
Coagulation‐related laboratory results
| Unit | Normal range | ||
|---|---|---|---|
| D‐dimer |
0.5 (day 0) 2.5 (day 15) 0.6 (day 32) |
μg/mL | 0–0.90 |
| Fibrin/fibrinogen degradation products (FDP) |
≦2.5 (day 0) 5.9 (day 15) ≦2.5 (day 32) |
μg/mL | 0–4.90 |
| Protein C activity |
46 (day 16) 49 (day 33) |
% | 64–135 |
| Protein C antigen | 41 (day 33) | % | 70–150 |
| von Willebrand factor antigen |
≧201 (day 16) ≧201 (day 33) |
% | 50–150 |
| von Willebrand factor activity |
196 (day 16) 163 (day 33) |
% | 50–150 |
| Protein S antigen | 89 | % | 74–132 |
| PT | 83.3 | % | 70.10≦ |
| PT‐INR | 1.08 | INR | ≦1.15 |
| APTT | 34.4 | s | 29.6–40.8 |
| Thrombin‐antithrombin III complex | 3.9 | ng/mL | ≦3.0 |
| Prothrombin fragment F1 + 2 | 194 | pmol/L | 69–229 |
| Antithrombin III | 109 | % | 80–130 |
| Factor II activity | 107 | % | 74–146 |
| Factor X activity | 91 | % | 71–128 |
| Factor XIII antigen | 102 | % | 70–999 |
| Plasminogen | 88 | % | 80–130 |
| Antinuclear antibody | ≦1:40 | Titer | 0–1:79 |
| Anti‐cardiolipin antibody | 1 | U/mL | 0–9.9 |
| Anti‐cardiolipin β2‐glycoprotein I complex antibody | ≦1.3 | U/mL | 0–3.40 |
| Lupus anticoagulant | 1 | s | 0–1.30 |
DISCUSSION
This case exhibited DKA at the onset of FT1D preceded by acute pancreatitis. Although he had had neither past nor family histories of thrombotic disorders, DVT and PE developed after acute‐phase DKA treatment including central venous catheter placement for renal replacement therapy. His thrombosis extended from the right external iliac vein to the inferior vena cava, suggesting femoral vein catheterization to have triggered the DVT 2 .
The PC activity and its antigen are reportedly reduced in patients with insulin‐dependent diabetes compared with healthy controls 4 . In patients with type 2 diabetes as well, HbA1c levels are negatively associated with the plasma PC activity and antigen levels 7 . Notably, induction of hyperglycemia decreases both PC activity and antigen levels 4 . Accordingly, PC activity is reduced prior to and within 24 h in conditions of DKA, but soon normalizes after its treatment 5 . However, our case exhibited low PC activity and antigen levels even 33 days after receiving DKA treatment, when the blood glucose was well controlled with MDII therapy. At that time, he took an anticoagulant, rivaroxaban, which had no effect on his chromogenic PC assay 6 . Based on the diagnostic criteria, these laboratory data, including decreased PC activity/antigen during stable conditions, are sufficient to diagnose partial type I PC deficiency, even without genetic testing 6 . Furthermore, during acute‐phase DKA, the PC activity reportedly decreases by 20–35% 4 , 5 . Thus, severe PC dysfunction, due to overlapping of the partial PC deficiency and hyperglycemia‐induced PC suppression, may have induced the massive DVT with PE. Therefore, we emphasize that strict glycemic control should be the clinical aim in managing patients with both type 1 and type 2 diabetes presenting concomitantly with PC deficiency. Given that the patient had suffered FT1D and would thus be highly prone to hyperglycemia, anti‐coagulation therapy should be continued to prevent thrombotic events.
Our present case highlights that venous thrombosis should always be considered as a potential complication of DKA. The prevalence of homozygous PC deficiency is very low (1 in 500,000 to 1 in 750,000 births), while heterozygous PC deficiency is considerably more frequent (1 in 200 to 1 in 500 births). A possible reason for this very low frequency of homozygous PC deficiency is fetal demise and prenatal death 6 . Thus, further loss of PC activity due to hyperglycemia in patients with heterozygous PC deficiency is regarded as being very dangerous. Under DKA conditions, particularly, dehydration and catheterization may increase the risk of thrombosis. On the other hand, as illustrated by our patient, there are cases with partial PC deficiency in whom the condition would not initially be evident due to the absence of any history of thrombosis. Thus, this case suggests the necessity of anti‐coagulation therapy combined with acute‐phase DKA treatment in patients with PC deficiency, even in those who have been asymptomatic.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: N/A.
Informed consent: The patient provided written informed consent of this case report.
Registry and the registration no. of the study/trial: September 20, 2022, No. 27404.
Animal studies: N/A.
REFERENCES
- 1. Imagawa A, Hanafusa T, Awata T, et al. Report of the Committee of the Japan Diabetes Society on the research of fulminant and acute‐onset type 1 diabetes mellitus: new diagnostic criteria of fulminant type 1 diabetes mellitus (2012). J Diabetes Investig 2012; 3: 536–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gutierrez JA, Bagatell R, Samson MP, et al. Femoral central venous catheter‐associated deep venous thrombosis in children with diabetic ketoacidosis. Crit Care Med 2003; 31: 80–83. [DOI] [PubMed] [Google Scholar]
- 3. Scordi‐Bello I, Kirsch D, Hammers J. Fatal pulmonary thromboembolism in patients with diabetic ketoacidosis: a seven‐case series and review of the literature. Acad Forensic Pathol 2016; 6: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ceriello A, Quatraro A, Dello Russo P, et al. Protein C deficiency in insulin‐dependent diabetes: a hyperglycemia‐related phenomenon. Thromb Haemost 1990; 64: 104–107. [PubMed] [Google Scholar]
- 5. Carl GF, Hoffman WH, Passmore GG, et al. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res 2003; 29: 73–82. [DOI] [PubMed] [Google Scholar]
- 6. Dinarvand P, Moser KA. Protein C deficiency. Arch Pathol Lab Med 2019; 143: 1281–1285. [DOI] [PubMed] [Google Scholar]
- 7. Addai‐Mensah O, Annani‐Akollor ME, Nsafoah FO, et al. Effect of poor glycaemic control on plasma levels and activity of protein C, protein S, and antithrombin III in type 2 diabetes mellitus. PLoS One 2019; 14: e0223171. [DOI] [PMC free article] [PubMed] [Google Scholar]