

Abstract
The irreversible aggregation of proteins or peptides greatly limits their bioavailability; therefore, effective inhibition using small molecules or biocompatible materials is very difficult. Human calcitonin (hCT), a hormone polypeptide with 32 residues, is secreted by the C‐cells of the thyroid gland. The biological function of this hormone is to regulate calcium and phosphate concentrations in the blood via several different pathways. One of these is to inhibit the activity of osteoclasts; thus, calcitonin could be used to treat osteoporosis and Paget's disease of the bone. However, hCT is prone to aggregation in aqueous solution and forms amyloid fibrils. Salmon and eel calcitonin are currently used as clinical substitutes for hCT. In a previous study, we found that the replacement of two residues at positions 12 and 17 of hCT with amino acids that appear in the salmon sequence can greatly suppress peptide aggregation. The double mutations of hCT (DM hCT) also act as good inhibitors by disrupting wild‐type hCT fibrillization, although the inhibition mechanism is not clear. More importantly, we demonstrated that DM hCT is biologically active in interacting with the calcitonin receptor. To further understand the inhibitory effect of DM hCT on hCT fibrillization, we created four relevant peptide fragments based on the DM hCT sequence. Our examination revealed that the formation of a helix of DM hCT was possibly a key component contributing to its inhibitory effect. This finding could help in the development of peptide‐based inhibitors and in understanding the aggregation mechanism of hCT.
Keywords: amyloid formation, human calcitonin, mutations, osteoporosis, peptide‐based inhibitors
1. INTRODUCTION
Calcitonin is a thyroid hormone (Hirsch et al., 1963). Its sequence was first determined in 1968. Later, several other calcitonin variants derived from porcine, bovine, and salmon were also found and sequenced (Copp et al., 1962). Human calcitonin (hCT) and calcitonin from other species are generally 32 residues in length and contain a disulfide bridge from Cys‐1 to Cys‐7. Sequence alignment of the most known calcitonin suggests that residues 8 to 32 are very different among species, but the residues appearing in the N‐terminal loop are largely conserved (Brewer Jr & Ronan, 1969; Niall et al., 1969; Potts Jr et al., 1968). Most calcitonin variants have been considered to aggregate into amyloid fibrils, although their sequences are distinct (Arvinte et al., 1993; Gilchrist & Bradshaw, 1993; Siligardi et al., 1994). Amyloid is a typical supramolecular fibrillar structure in which protein or peptide monomers are stacked along the fibril axis. Each monomer adopts a β‐stand secondary structure (Chiti & Dobson, 2006). Such amyloid architecture not only appears in aggregated calcitonin peptides but has also been observed in numerous peptides such as amyloid β, α‐synuclein, and human islet amyloid polypeptide (Benson et al., 2018; Harrison et al., 2007). These peptides are commonly implicated in human diseases. The process of peptide aggregation has been shown to be toxic to peripheral cells, although the mechanism of toxicity is still not elucidated, and toxic species have not been fully identified (Kayed & Lasagna‐Reeves, 2013; Reiss et al., 2018). To date, aggregated calcitonin has not been shown to have similar toxicity‐causing behaviors as other amyloidogenic peptides. Localized deposits of calcitonin amyloids are commonly found in human medullary thyroid carcinoma, which may overexpress these hormone peptides (Khurana et al., 2004). The mechanism of hCT amyloid formation has not yet been fully elucidated. Based on solid‐state nuclear magnetic resonance spectroscopy, Naito's group revealed some structural information about the conformational change of hCT during aggregation (Kamihira et al., 2000). First, the hCT monomer was suggested to consist of an N‐terminal loop, central helix, and C‐terminal random coil. The central helix region starts from residues 10 to 18. During oligomerization, the central helix may convert into a β‐hairpin, while the N‐ and C‐terminal regions remain the same. This new β‐strand may incorporate more residues from the C‐terminal disordered region and gradually become fibrillar with rich β‐sheet structures. At pH 7.5, because of the two oppositely charged residues (Asp‐15 and Lys‐18), fibrils are considered to display antiparallel β‐stacking (Kamihira et al., 2003). Compared with hCT, salmon calcitonin (sCT) has a longer central helix with a C‐terminal region close to the central helix; the stable α‐helix region from residues 9–19 of sCT may play a crucial role in maintaining low aggregation propensity of sCT (Amodeo et al., 1999; Andreotti et al., 2006).
The physiological roles of calcitonin are mainly skeletal protection and hypercalcemia prevention (Austin & Heath, 1981; Talmage et al., 1980). The most prominent role of calcitonin is to inhibit osteoclasts by interacting with the calcitonin receptor widely expressed on the osteoclast membrane (Holtrop et al., 1974). Therefore, calcitonin can be used as a treatment for metabolic bone diseases such as osteoporosis and Paget's disease. However, hCT forms amyloid in aqueous solution, and the bioactivity of aggregated calcitonin is largely reduced (Arvinte et al., 1993; Cudd et al., 1995). Currently, sCT is the replacement of hCT, as a widely used active pharmaceutical ingredient. sCT has a low sequence identity with hCT, but its propensity to form amyloid fibrils is significantly less. The greatest concern of administrating sCT as medical treatment is its severe side effects, including anorexia, vomiting, and immune reactions (Lee et al., 2011). Therefore, the development of strategies to prevent hCT aggregation is important.
Previously, our group has shown that two residues of hCT, Tyr‐12, and Asn‐17, play crucial roles in inducing the fibrillization of hCT (Chen et al., 2019). When the residues at positions 12 and 17 in hCT were replaced with the amino acids present in sCT, these minor changes greatly reduced the amyloidogenicity of hCT but did not affect the physiological function. Moreover, Y12LN17H hCT (denoted as DM hCT) can serve as a peptide‐based inhibitor that suppresses amyloid formation induced by hCT. In this study, we aimed to understand how this variant obstructs hCT aggregation. This would improve our understanding of the development of peptide‐based inhibitors for hCT aggregation. Since the C‐terminal region of DM hCT remains the same as that of hCT and the role of the N‐terminal loop is not yet clear, we did not consider creating short peptides containing C‐terminal residues. Instead, we generated four N‐terminal DM hCT‐relevant peptides for this study. These four shorter peptides were DM 1–18, DM 1–22, DM 8–18, and DM 8–25 (Figure 1). By comparing the propensities of aggregation and helix formation among these short peptides, our findings suggest that helix–helix association would also be a key step for hCT fibrillization and segments 19–22 containing two aromatic side chains would facilitate helix–helix association. The model of helix–helix association has been proposed to be significant for other amyloidogenic proteins (Abedini & Raleigh, 2009a, 2009b; Ghosh et al., 2015; Liu et al., 2010), but it has not been discussed for hCT aggregation. However, the unstructured C‐terminal region from residues 23 to 31 was inferred to play a villous role obstructing hCT aggregation. Our data also revealed that the N‐terminal loop contributed to the stability of the central helix of hCT. Most importantly, we demonstrated that the helical structure of peptide inhibitors could help display an inhibitory effect on hCT aggregation. We believe that our observations combined with other structural information proposed for hCT aggregation will enhance our understanding of the details of hCT fibrillization.
FIGURE 1.

Primary sequence of hCT, DM hCT, and DM hCT‐relevant peptide fragments. hCT, DM hCT, DM 1–18, and DM 1–22 contain a disulfide bridge between Cys 1 and Cys 7. DM hCT and its relevant peptide fragments have two mutation sites at residue 12 and 17 labeled in red. The C‐terminus of all peptides used in this study is amidated.
2. RESULTS AND DISCUSSION
2.1. Design and synthesis of four DM hCT‐relevant fragments
We designed four DM hCT‐relevant fragments and synthesized individual peptides, including wild‐type hCT and DM hCT. First, DM 1–18 and DM 1–22 were chosen to investigate the role of the C‐terminus, which was initially considered as an unstructured region. Later, DM 8–18 and DM 8–25 were also created to explore the role of disulfide bond bridges at the N‐terminus (Figure 1). In a previous report, it was found that sCT adopts a stable helical structure and its helical region is slightly longer than hCT (Andreotti et al., 2006). It was speculated that stable helices of sCT make it difficult to transform the structure into β‐sheets and exhibit low aggregation propensity. Our previous study also revealed that approximately 48% of sCT was estimated to exhibit a helical structure in the presence of 20% helix‐inducing agent, 2,2,2‐trifluoroethanol (TFE) (Chen et al., 2019). In the same buffer condition, DM hCT was approximately 31% helical, but hCT was still mostly random coil, suggesting that DM hCT was similar to sCT in preferring a helical structure. The conformational changes in DM hCT in a solution containing 20% TFE after 30 h of incubation were very small and did not affect amyloid formation. These observations illustrate that DM hCT may form a helical structure that is more stable than hCT. Therefore, although we do not have precise structural information for DM hCT, we assume that all these four DM hCT‐relevant fragments contain a central helical region 8–18 like hCT. Additionally, aromatic π‐π stacking interaction was also thought to be the major determinant between hCT and sCT (Itoh‐Watanabe, Kamihira‐Ishijima, Javkhlantugs, et al., 2013a; Itoh‐Watanabe, Kamihira‐Ishijima, Kawamura, et al., 2013b). hCT has four aromatic residues, Tyr‐12, Phe‐16, Phe‐19, and Phe‐22, as observed in the proposed β‐sheet region of the fibrillar state using NMR study (Itoh‐Watanabe, Kamihira‐Ishijima, Javkhlantugs, et al., 2013a; Itoh‐Watanabe, Kamihira‐Ishijima, Kawamura, et al., 2013b; Naito et al., 2004); therefore, we also prepared longer sequence peptides, DM 1–22 and DM 8–25, besides DM 1–18 and DM 8–22. These four peptides were synthesized via solid‐phase peptide synthesis, purified using high‐performance liquid chromatography (HPLC), and confirmed using mass spectrometry (Figure S1).
2.2. Thioflavin‐T assay to evaluate the aggregation property of individual peptides
We and other research labs commonly utilize a fluorescent probe, thioflavin‐T (ThT), to probe amyloid formation (Naiki et al., 1989). Unlike general protein aggregation, amyloid formation consists of rich β‐sheets (Sunde et al., 1997). When ThT binds to β‐sheet‐rich structures, it displays enhanced fluorescence and a characteristic red shift of its emission spectrum. Because ThT is not perfectly specific for amyloid, it may provide some biased results especially in the presence of other synthetic compounds and materials (Hudson et al., 2009). Data interpretation for ThT assays should be done carefully. Because we only used ThT co‐incubated with peptides whose absorbance range falls within the ultraviolet region, ThT is still the best probe to monitor amyloid structure in our examination. However, previously it was observed that, ThT does not bind to the fibrillar structure formed by certain peptide variants and fluoresce (Wong et al., 2016), thus the use of a transmission electron microscope (TEM) became a complementary tool for ThT assays. TEM was used to validate the status of the proteins. Here, we first tested the aggregation properties of four DM hCT‐relevant peptide fragments and compared them with hCT and DM hCT. As shown in Figure 2, we conducted a ThT assay using two peptide concentrations: 20 and 30 μM. Basically, the kinetics of protein aggregation measured under these two concentrations were quite similar suggesting we have conducted the experiments above critical aggregation concentration. As expected, compared with hCT, DM hCT showed a much longer lag time (40 h), indicating its low aggregation propensity. Among the four DM hCT relevant peptides, DM 1–22 was the only peptide that developed ThT fluorescence. The lag time of DM 1–22 aggregation was approximately 15–16 h and shorter than that of DM hCT suggesting that DM 1–22 is amyloidogenic with higher amyloidogenicity than that of DM hCT. To confirm this result, we monitored the conformational changes in DM 1–22, before and after incubation using circular dichroism (CD, Figure S2). When this peptide was freshly prepared, it was randomly coiled. After 42 h of incubation in microtubes, the CD spectra were significantly different from those recorded before incubation, and we did find amyloid fibrils formed by DM 1–22 in the TEM images (Figure S2F).
FIGURE 2.
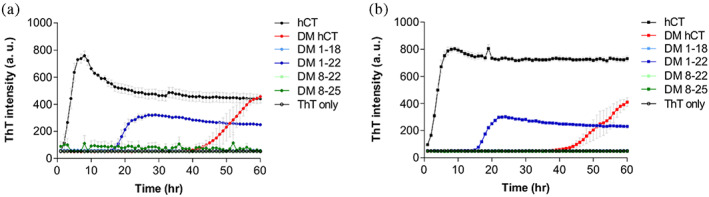
ThT monitored amyloid formation kinetics of hCT (black), DM hCT (red), DM 1–18 (light blue), DM 1–22 (blue), DM 8–22 (light green), and DM 8–25 (green) conducted at (a) 20 μM and (b) 30 μM, respectively, in 10 mM phosphate buffer at pH 7.4. Each condition was performed in triplicates.
This interesting phenomenon was unexpected. We attempted to infer the mechanisms of hCT aggregation using limited structural information by comparing the ThT results obtained in this study. First, the difference between DM 1–18 and DM 1–22 was the residue 19–22 (FHTF). This segment, containing two aromatic residues, may assist in the fibril formation of DM hCT 1–22. The importance of aromatic π‐π stacking interactions in hCT fibril formation has been demonstrated in previous studies (Itoh‐Watanabe, Kamihira‐Ishijima, Javkhlantugs, et al., 2013a; Itoh‐Watanabe, Kamihira‐Ishijima, Kawamura, et al., 2013b; Reches et al., 2002; Shtainfeld et al., 2010). Because all the central aromatics Tyr‐12, Phe‐16, and Phe‐19 in hCT covert to Leu in sCT, these residues initially attracted attention. The triple mutation of hCT to Leu residues causes fibril elongation at a significantly slower rate. Moreover, the hCT central pentapeptide fragment, DFNKF, was suggested to be essential for fibril formation. The iodinated analog DF(I)NKF(I) strongly promotes its ability to form amyloid fibrils (Bertolani et al., 2015). However, these investigations have not examined the role of Phe‐22. The comparison of ThT results of DM 1–18 and DM 1–22 is interesting. We now also recognize the importance of Phe‐22 in contributing to the amyloidogenicity of hCT. The existence of these four‐residue fragment, 19FHTF22 reverses the amyloidogenicity of short peptide fragments DM 1–18. Next, the comparison of the ThT results derived from DM hCT and DM 1–22 suggested that C‐terminal residues from Pro‐23 to Pro‐32, which were previously considered unstructured regions, obstructed fibril formation. DM hCT spent more time forming an active nucleus than DM 1–22. The lag time of DM hCT was almost double than measured for DM 1–22. In our previous study, we tested the amyloid‐forming ability of DM hCT and polar hCT (phCT) where we continued to develop peptide‐based inhibitors that have the highest sequence identity with wild‐type hCT (Chen et al., 2019). phCT was first designed by Motta's group according to the prediction results suggested by Waltz software, a computer algorithm trained from a large set of experimentally identified amyloid‐forming peptides (Andreotti et al., 2011). phCT has three additional mutational sites other than DM hCT at positions 26, 27, and 31 in the C‐terminus. However, we demonstrated that DM hCT was similar to phCT in many aspects, such as amyloidogenicity, extent of helical conformations induced by TFE, and the ability to inhibit wild‐type hCT amyloid formation. Thus, we conclude that the replacement of Tyr‐12 and Asn‐17 plays a key role in stabilizing hCT. It is only known that the remaining unchanged C‐terminus does not promote amyloid formation, and the role of the C‐terminus was not clear at that time. Here, a shorter hCT fragment with the FHTF sequence but without the C‐terminal region turns amyloidogenic, suggesting that the absence of an unstructured C‐terminal region makes peptides easy to self‐assemble.
However, DM 8–22 or DM 8–25 that lack an N‐terminal disulfide bond loop, were found to be unable to form amyloids during the monitored time course even though they all contain a central helical region and FHTF segment. So far, the importance of the N‐terminal loop of hCT has only been discussed and compared with the reduced form of hCT, but there is no solid conclusion regarding the same (Lantz et al., 2020; Renawala et al., 2021). Our experimental results clearly suggest that the N‐terminal loop assists fibril formation based on our observation of the difference between DM 1–22 and DM 8–22.
2.3. Amyloidogenic DM hCT 1–22 still significantly delays amyloid formation by hCT
The ultimate goal of this study was to understand how DM hCT affects hCT aggregation. As we understood the amyloidogenicity of individual peptide fragments, we next determined whether these DM hCT‐relevant peptide fragments inhibit hCT fibrillization. Here, we fixed the concentration of hCT at 20 μM in 50 mM phosphate buffer at pH 7.4, and then added two different doses (20 and 30 μM) of DM hCT or peptide fragments. ThT was still applied in this study to monitor the hCT aggregation (Figure 3). As expected, the condition containing hCT and DM‐hCT did not develop any ThT fluorescence within 50 h, suggesting that DM‐hCT suppressed hCT aggregation. Amyloid formation does occur when hCT is mixed with DM hCT after 50 h (Figure S3). However, we observed that the lag time for amyloid formation was significantly longer compared with hCT alone or DM hCT alone suggesting they mutually inhibit their own self‐assembly. In addition, preformed DM hCT fibrils cannot inhibit hCT amyloid formation instead that seeding effect was observed when hCT was mixed with small amount of DM hCT fibrils (Figure S3). With the addition of DM 1–18, DM 8–22, and DM 8–25, typical sigmoidal ThT curves with different lag times were obtained. However, the lag times measured here were all <8 h, indicating that these fragments delayed hCT aggregation but not in an efficient way. Conversely, DM 1–22 which was previously determined to form amyloid alone, significantly retarded the aggregation profile. The lag time of the 1:1 mixture of DM 1–22 and hCT was much longer than that of hCT alone. A higher doze of DM 1–22 showed a better inhibitory effect (Figure 3D). Previously, DM hCT and phCT were both found to be powerful fibril inhibitors (Chen et al., 2019). Initially, they were mutated and expected to convert into a low‐aggregation‐prone hCT analog. Because preformed hCT amyloid fibrils were able to induce DM hCT and phCT fibril formation but were unable to induce sCT, we reported that neither DM hCT nor phCT was non‐amyloidogenic. According to the test results shown in Figure 1, the aggregation properties of DM 1–18, DM 8–22, and DM 8–25 should be even lower than that of DM hCT. Here, we confirmed that amyloidogenic peptides DM hCT and DM 1–22, work as better inhibitors of hCT aggregation, suggesting that the first criterion of a good peptide‐based inhibitor of hCT aggregation is not its amyloidogenicity. In our previous report, non‐amyloidogenic sCT was also not able to block hCT fibril formation, although sequence diversity between sCT and hCT may rationalize this phenomenon (Chen et al., 2019). DM 1–18 and DM 1–22 are very similar in sequence but their inhibitory properties are quite different. The inhibitory effect of DM 1–22 on hCT aggregation was concentration‐dependent (Figure S4). Amyloidogenic DM 1–22 works as a good inhibitor.
FIGURE 3.

ThT monitored amyloid formation kinetics of hCT with DM hCT (red), with DM 1–18 (light blue), with DM 1–22 (blue), with DM 8–22 (light green), and with DM 8–25 (green). The concentration of hCT was fixed at 20 μM. DM hCT and its peptide fragments were added at (a) 20 μM and (b) 30 μM. (c, d) Analysis of the times needed to reach reaction plateau for each experimental condition. N/A represents no ThT intensity monitored within 50 h.
2.4. Helical conformation of peptide fragments induced by TFE and phospholipid
The formation and stability of the helical conformation are highly associated with the aggregation and bioactivity of calcitonin (Andreotti et al., 2006). The stable helix of sCT prevents the formation of amyloid and exhibits potent bioactivity. According to our previous report, the extent of helical conformations of DM hCT was estimated between hCT and sCT via CD measurement (Chen et al., 2019). DM hCT was considered bioactive through the measurement of stimulated cyclic adenosine monophosphate (cAMP) from peptide‐treated MCF‐7 cells (Martin et al., 1980). However, it is not known whether the inhibitory effect of DM hCT is also related to its helical conformation. Subsequently, we monitored the conformational changes of individual peptides at different percentages of TFE (Figure 4) and estimated the helix components using BeStSel (Micsonai et al., 2015, 2022; Figure 5). The most freshly prepared DM hCT and peptide fragments examined here are primary random coil structures with a low‐extent helical conformation. With the addition of TFE to the solution, the peptides were gradually induced to form helical structures (Figure S5). Among them, DM 8–22 and DM 8–25, without an N‐terminal loop, were the two peptide fragments that were most resistant to the formation of a helical conformation. Although they eventually adopted a helical conformation, a higher TFE percentage was required. We speculated that the N‐terminal loop might play a role in peptide folding or stabilization of the central helix. However, it was previously reported that the biological potency of calcitonin correlated well with its interaction with phospholipids, which assists the peptide in forming a helical structure (Epand et al., 1983). Therefore, we measured the CD spectra of these peptides again in the presence of anionic lipids and analyzed their helical components (peptide to lipid = 1:10, Figure 6). Interestingly, DM 1–22 was the one that had significant conformational change due to the presence of 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphorylglycerol (DMPG). For the other peptides, the helical percentage also increased, albeit to a minor extent (Figure S6).
FIGURE 4.

The CD spectra of (a) DM hCT, (b) DM 1–18, (c) DM 1–22, (d) DM 8–22, and (e) DM 8–25 prepared in 50 mM phosphate buffer at pH 7.4 with the presence of 0% to 40% TFE (form light color to dark color).
FIGURE 5.

Percent composition of helix estimated by CD fitting software, BeStStel for each condition shown in Figure 4.
FIGURE 6.

The CD spectra of (a) DM hCT, (b) DM 1–18, (c) DM 1–22, (d) DM 8–22, and (e) DM 8–25 prepared in 50 mM phosphate buffer at pH 7.4 without (black) and with 400 μM DMPG (red).
Based on these observations, we found that the inhibitory effect of peptide fragments tended to have a positive correlation with their propensity to form helical structures. DM 1–18 are worth further discussion. More DM 1–18 peptides would adapt helical structures with the addition of TFE, and the extent was similar to that measured for DM 1–22 under the same conditions, suggesting that both peptides are able to fold into a helical structure. However, only DM 1–22, not DM 1–18 was apparently helical‐dominant in the presence of DMPG. This may indicate that DM 1–18 does not strongly interact with DMPG or that the helical structure of DM 1–18 is less than DM 1–22. These two inferences are possible since DM 1–18 has less residues than DM 1–22. We speculated that DM 1–22 could disrupt hCT aggregation because its relatively stable helical structure would interact with the helical structure of hCT. We hypothesized that helix–helix association would be an important step in peptide‐inhibitor interactions. The same reasoning was used for the DM hCT. Since DM hCT also contains an unstructured C‐terminal region, which may make overall peptide assembly more difficult, the inhibitory effect of DM hCT may be better than DM 1–22. DM 1–18, DM 8–18, and DM 8–25, which are unable to adopt a stable helix and would not serve as good inhibitors of hCT aggregation.
2.5. Reexamination of the importance of helical conformation for the inhibitory effect
Amyloidogenic polypeptides were mostly disordered in an unaggregated state. In addition to calcitonin, important examples include islet amyloid polypeptide (IAPP) and amyloid beta (Aβ) peptide (Harrison et al., 2007; Murphy & Levine Iii, 2010; Westermark et al., 2011). These two peptides were probably the earliest evidence for the involvement of helical conformation during amyloid formation (Kirkitadze et al., 2001; Teplow et al., 2006; Williamson & Miranker, 2007; Yonemoto et al., 2008). Although the characterization of transient helical intermediates is not easy, an increase in helicity can be observed via CD before the appearance of β‐sheets and promoted by hexafluoroisopropanol (HFIP) and TFE (Buck, 1998; Kentsis & Sosnick, 1998). Thus, helix–helix association was considered an important step for oligomer formation and of course for amyloid formation (Abedini & Raleigh, 2009a, 2009b). Targeting helical intermediates is a good strategy to inhibit amyloid formation. For example, Hamilton et al. reported that oligopyridylamide‐based α‐helical mimetics were designed to prevent Aβ fibrillization. A tripyridylamide, namely ADH‐41, was shown to induce Aβ to form a strong α‐helical conformation, and such a stable helical structure prohibited amyloid formation (Kumar & Hamilton, 2017). On the other hand, the formation of amyloid fibrils by the mutant huntingtin protein (mHtt) was disrupted by incubating with 10‐residue polyproline PPII helices. The first 17 residues of the N‐terminal region of htt (Nt17) have been showed to adopt a helical conformation when interacting with a binding partner, including another Nt17 helix. The Nt17 helix–helix interaction is the driving force for peptide oligomerization (Arndt et al., 2020; Vieweg et al., 2021). The Nt17 − polyproline association replaced the original helix–helix interaction and led to the reduction of amyloid fibrils. Therefore, we aimed to examine whether this strategy is also applicable to perturb hCT fibrillation because we have similar inferences based on the results previously shown, and we believe that it would help us elucidate more details about the mechanism of hCT aggregation. Here, we used peptide fragments other than DM 1–22 in the following study because we noticed that the structures of DM 8–22 and DM 8–25 were also helix dominant when 30% TFE was present in solution. We monitored the kinetics of hCT aggregation with 30% TFE using the ThT assay and compared it with the hCT aggregation conditions with the addition of DM 8–22 or DM 8–25 (Figure 7). DM 8–22 and DM 8–25 had relatively low abilities to hinder hCT aggregation in phosphate buffer (Figure 3). Incubation of hCT with 30% TFE significantly prolonged the lag time. We speculated that the helical conformation of hCT was stabilized based on the observation of its CD spectra (Figure 4a) and made it more difficult for hCT to transform into β‐sheets. Interestingly, the inhibitory effect of DM 8–22 or DM 8–25 on hCT fibrillization under 30% TFE was also enhanced. We also incubated 20 μM hCT with 20 or 30 μM of peptide fragments. For DM 8–22, it was not possible to determine whether the inhibitory effect was concentration‐dependent, because DM 8–22 suppressed hCT aggregation very well within our monitored time window. No fibrils were observed under TEM (Figure 7b, c). For DM 8–25, the lag time of hCT aggregation was approximately doubled when 20 μM of the peptide fragment was applied. Amyloid fibrils were observed after 80 h of incubation (Figure 7e). When the concentration of DM 8–25 was increased to 30 μM, hCT aggregation was more effectively hindered. Overall, DM 8–22 performed its inhibition on hCT aggregation better than DM 8–25 which may also be related to their helical conformation. DM 8–22 had a higher helical component than DM 8–25 under conditions with 20%–40% TFE (Figure 5).
FIGURE 7.

(a) ThT monitored amyloid formation kinetics of hCT (black), hCT with 20 μM DM 8–22 (light green) and hCT with 30 μM DM 8–22 (green). (b, c) TEM images of hCT with 20 and 30 μM DM 8–22 at the end of kinetic experiments. Scare bar represents 200 nm. (d) ThT monitored amyloid formation kinetics of hCT (black), hCT with 20 μM DM 8–25 (light green) and hCT with 30 μM DM 8–25 (green). (e, f) TEM images of hCT with 20 and 30 μM DM 8–25 at the end of kinetic experiments. Scare bar represents 200 nm. hCT was prepared at 20 μM in each kinetic assay (10 mM phosphate buffer at pH 7.4 with 30% TFE).
3. CONCLUSION
The inhibition of hCT aggregation is an important goal for the development of hCT as a therapeutic drug. Inhibitors have traditionally been designed to delay the overall aggregation process; however, recent advances have suggested that strategies that would target mechanistic insights may be more effective. The microscopic steps underlying hCT aggregation have not yet been fully elucidated. Fortunately, however, some structural basis of hCT during peptide assembly was obtained based on solid‐state NMR spectroscopy (Kamgar‐Parsi et al., 2017; Kamihira et al., 2000). Thus, the hCT monomer that exhibits an N‐terminal loop, central α‐helix, and C‐terminal random coil is the most acceptable in this study field. Although the helix is considered to transform into a β‐hairpin during the early stages of oligomerization, important key clues to explain how primary nucleation occurs are still missing. If we could gain valuable information in this regard, the goal of hCT to serve as a pharmaceutical drug could possibly be realized. Previously, helix–helix association has been linked in many systems to provide stability through peptide–peptide or peptide‐membrane interactions. A number of amyloidogenic peptides have also been suggested to adopt a helical structure during aggregation (Abedini & Raleigh, 2009a, 2009b). Helix‐mediated association is considered to enhance the local concentration of an aggregation‐prone peptide and thus promote intermolecular β‐sheet formation. To identify the missing pieces in the mechanism of hCT aggregation, we attempted to resolve the inhibitory effect of a known peptide‐based inhibitor, DM hCT, from the point of view of the formation of helical structures.
Interesting findings were obtained from our tests of the four relevant DM hCT peptide fragments. DM 1–22 was the peptide that could form amyloid among the two C‐terminal truncated DM hCT peptide fragments synthesized in this study. Its speed in peptide assembly was much faster than that in DM hCT. According to the ThT and CD data derived from DM 1–22 and compared with those measured for hCT, DM hCT, and DM 1–18, we attempted to provide a model (Figure 8) to conjecture the process of hCT aggregation and how DM hCT retards hCT aggregation. First, CD was used to measure the formation of a helical structure of each peptide induced by TFE. DM 1–18, DM 1–22, and DM hCT showed more helical populations than hCT, suggesting that they are more likely to adopt a helical conformation (Figure S7). The stability of the helical conformation has been shown to be related to fibril‐forming ability based on the observation of sCT (Diociaiuti et al., 2011). We speculated that DM 1–22 would form amyloid due to three factors: the formation of a helical structure, the assistance of two aromatic residues from FHTH segments, and the elimination of disordered C‐terminus from residues to 23–31. Therefore, in the proposed model, we have a new sketch of hCT monomers, including an N‐terminal loop, a central helix, a segment with two aromatic side chains, and an unstructured C‐terminal. With the addition of DM hCT, a stable helical component may stabilize hCT via helix–helix association to retard its transformation into β‐sheets. We also examined hCT aggregation with DM 8–22 and DM 8–25 in the presence of 30% TFE, as it allowed us to examine the importance of the helical conformation of a good inhibitor. DM 8–22 and DM 8–25 both formed helical structures when the percentage of TFE was >30%. When we examined the hCT aggregation with 30% TFE, the lag time was significantly increased compared with that in pure buffer, indicating that hCT was stabilized, although its helical population only increased from 0.5% to 6%. We verified that the stability of the helical conformation of hCT affected its ability to form amyloids. Furthermore, DM 8–22 and DM 8–25 suppressed hCT aggregation under the conditions defined in this study. Inhibitors with helical conformations showed improved efficiency in inhibiting hCT aggregation. We concluded that helix–helix association may be a key step in hCT aggregation. Inhibition of hCT aggregation by DM hCT may also occur via this type of association. Furthermore, C‐terminal disordered domain of DM hCT also helps inhibit hCT fibrillization. Thus, DM hCT is so far the best peptide‐based inhibitor for hCT aggregation. The aggregation propensity of DM hCT was much lower than that of hCT, but higher than that of sCT. DM hCT can still form amyloids on hydrophobic surfaces. However, sCT did not affect hCT aggregation, suggesting that amyloidogenicity is not a determinant of a good inhibitor. The ability of DM hCT to form a helical structure and directly interact with hCT may work in concert to reduce hCT aggregation.
FIGURE 8.

A schematic diagram of how DM hCT inhibits hCT aggregation. Formation of a stable helix for DM hCT would be an important step which allows it to interact with helical structure of hCT.
4. MATERIALS AND METHODS
4.1. Peptide synthesis and purification
Synthetic hCT, DM hCT, and its related variant fragments (DM 1–18, DM 1–22, DM 8–22, DM 8–25) were produced with the assistance of microwave peptide synthesizer. Fmoc‐Rink amide ProTide (0.16 mmol/g, CEM corporation) was chosen as a solid support and to provide amidated C‐terminus after peptide cleavage. A detailed description of peptide synthesis has been described elsewhere (Chen et al., 2019). In brief, the Fmoc protecting group was first removed from resin using 10% piperazine (w/v) solution prepared using ethanol and N‐methylpyrrolidone (10: 90) mixture. The carboxylic acid group of first amino acid after activation (0.25 M diisopropylcarbodiimide prepared in dimethylformamide) underwent a coupling reaction to form a new amide bond to attach on the reaction resin. Later, repeated actions (deprotection‐activation‐coupling) allowed the peptide to be synthesized from the C‐terminus to the N‐terminus. Finally, a cleavage reaction (cleavage cocktail including trifluoroacetic acid, water, triisopropylsilane, and 3,6‐dioxa‐1,8‐octanedithiol, 92.5: 2.5: 2.5: 2.5) was carried out to remove the peptide from the resin. Crude peptides were precipitated using cold ether after trifluoroacetic acid evaporated. hCT, DM hCT, DM 1–18, and DM 1–22 were further oxidized to form disulfide bonds between Cys1 and Cys7 using I2 dissolved in methanol. The peptides were purified by reverse‐phase HPLC using a Proto 300 C18 semi‐preparative column (4.6 mm × 250 mm, Higgins analytical). Two solutions were used: solution A consists of 100% H2O and 0.045% HCl (v/v) and solution B includes 80% acetonitrile, 20% H2O, and 0.045% HCl. Peak fractions were separately collected and lyophilized. The identity of the pure products was confirmed by checking the molecular weight using a Bruker matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry. The HPLC chromatograms and mass spectra of mentioned peptides were shown in Figure S1.
4.2. Sample preparation
About 0.1 mg peptide powder was first dissolved in 300 μl of 50% acetonitrile (ACN) solution and centrifuged at 15 000 rpm for 10 min. The supernatant was transferred into another microtube. 10 μl of each peptide ACN solution was utilized to determine protein concentration using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, USA). According to different experimental designs, peptide solution was dispended in an individual microtube or mixed with another peptide solution in a microtube. The solution was further freeze dried to remove organic solvent. The resulting peptide powder was treated with HFIP for 6 h and HFIP was removed again by lyophilizer. The treated peptide powder was dissolved in buffer to provide the desired concentration just prior to the experiment.
4.3. ThT kinetic assay
The amyloid formation by hCT alone, peptide fragments alone or peptide mixtures were monitored by using ThT assays. The peptide solution, in general, was prepared in 10 mM phosphate buffer with 10 μM ThT at pH 7.4. They were incubated in sealed 384‐well microplate (Greiner # 781076, 40 μl/per well) with agitation for 1 min every 1 h. Measurements were conducted using a multimode microplate reader (SpectraMax M2, Molecular Devices, USA) with excitation at 430 nm and emission at 485 nm. In order to ensure experimental consistency, each condition was tested in triplicates and at least using two batches of samples.
4.4. CD measurements
CD experiments were performed using a JASCO J‐715 CD spectrometer. The peptide solutions were prepared as previously described but with 50 mM phosphate buffer and different percentages of TFE. hCT was measured at 40 μM. Due to peptides that are different in length, they were prepared at different concentrations in order to keep the total number of amide bonds consistent and to ensure the collecting of good signals (DM hCT: 40 μM; DM 1–18: 69 μM; DM 1–22: 54 μM; DM 8–22: 74 μM; DM 8–25: 63 μM). Such preparation would be better for analyzing the composition of their secondary structures later by using BeStSel. In general, 1 mm path length quartz cell which carried 280 μl of peptide solutions was used for CD measurement. Spectra were recorded from 200 to 260 nm with 1 nm intervals at 25°C (scan speed 50 nm/min). The data were averaged from 10 scans and subtracted from the background spectrum. For samples containing DMPG, we need to first prepare the lipid vesicles beforehand. A detailed description of vesicle preparation has been described elsewhere (Hsieh et al., 2022).
4.5. TEM
TEM was performed in an instrumentation center at National Taiwan University using a Hitachi H‐7100 transmission electron microscope with an accelerating voltage of 120 kV. TEM samples were prepared from the same solutions used for ThT assays or CD measurements. Ten microliters of alcohol were first blotted on a carbon‐coated Formvar 300‐mesh copper grid and removed by filter paper. Later, 10 μl of solution transferred from other experiments were blotted on grid for 2 min, washed twice with H2O, and then negatively stained with saturated uranyl acetate for 1 min.
AUTHOR CONTRIBUTIONS
Ya‐Ping Chuang: Data curation (lead); investigation (lead); methodology (lead); formal analysis (lead); visualization (supporting). Yu‐Pei Chang: Data curation (supporting); visualization (supporting). Ling‐Hsien Tu: Conceptualization (lead); funding acquisition (lead); project administration (lead); visualization (lead); supervision (lead); writing–review and editing (lead).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interests.
Supporting information
DATA S1. Supporting Information
ACKNOWLEDGMENTS
This work was supported by the Ministry of Science and Technology, Taiwan (MOST 111‐2113‐M‐003‐009). The authors thank the Instrumentation Center at National Taiwan University for assistance with the TEM experiments and the Instrumentation Center at National Taiwan Normal University for assistance with the mass spectrometry.
Chuang Y‐P, Chang Y‐P, Tu L‐H. Investigating the inhibitory property of DM hCT on hCT fibrillization via its relevant peptide fragments. Protein Science. 2023;32(8):e4711. 10.1002/pro.4711
Review Editor: Aitziber L. Cortajarena.
REFERENCES
- Abedini A, Raleigh DP. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng Des Select. 2009a;22(8):453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedini A, Raleigh DP. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys Biol. 2009b;6(1):15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodeo P, Motta A, Strazzullo G, Castiglione Morelli MA. Conformational flexibility in calcitonin: the dynamic properties of human and salmon calcitonin in solution. J Biomol NMR. 1999;13(2):161–74. [DOI] [PubMed] [Google Scholar]
- Andreotti G, Méndez BL, Amodeo P, Castiglione Morelli MA, Nakamuta H, Motta A. Structural determinants of salmon calcitonin bioactivity: the role of the leu‐based amphipathic α‐helix. J Biol Chem. 2006;281(34):24193–203. [DOI] [PubMed] [Google Scholar]
- Andreotti G, Vitale RM, Avidan‐Shpalter C, Amodeo P, Gazit E, Motta A. Converting the highly amyloidogenic human calcitonin into a powerful fibril inhibitor by three‐dimensional structure homology with a non‐amyloidogenic analogue. J Biol Chem. 2011;286(4):2707–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt JR, Chaibva M, Beasley M, Kiani Karanji A, Ghassabi Kondalaji S, Khakinejad M, et al. Nucleation inhibition of huntingtin protein (htt) by polyproline PPII helices: a potential interaction with the N‐terminal α‐helical region of Htt. Biochemistry. 2020;59(4):436–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvinte T, Cudd A, Drake AF. The structure and mechanism of formation of human calcitonin fibrils. J Biol Chem. 1993;268(9):6415–22. [PubMed] [Google Scholar]
- Austin LA, Heath H. Calcitonin: physiology and pathophysiology. New Engl J Med. 1981;304(5):269–78. [DOI] [PubMed] [Google Scholar]
- Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–9. [DOI] [PubMed] [Google Scholar]
- Bertolani A, Pirrie L, Stefan L, Houbenov N, Haataja JS, Catalano L, et al. Supramolecular amplification of amyloid self‐assembly by iodination. Nat Commun. 2015;6:7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer HB Jr, Ronan R. Amino acid sequence of bovine thyrocalcitonin. Proc Natl Acad Sci U S A. 1969;63(3):940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31(3):297–355. [DOI] [PubMed] [Google Scholar]
- Chen YT, Hu KW, Huang BJ, Lai CH, Tu LH. Inhibiting human calcitonin fibril formation with its most relevant aggregation‐resistant analog. J Phys Chem B. 2019;123(48):10171–80. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. [DOI] [PubMed] [Google Scholar]
- Copp DH, Cameron EC, Cheney BA, Davidson AGF, Henze KG. Evidence for calcitonin‐a new hormone from the parathyroid that lowers blood calcium. Endocrinology. 1962;70(5):638–49. [DOI] [PubMed] [Google Scholar]
- Cudd A, Arvinte T, Gaines Das RE, Chinni C, MacIntyre I. Enhanced potency of human calcitonin when fibrillation is avoided. J Pharm Sci. 1995;84(6):717–9. [DOI] [PubMed] [Google Scholar]
- Diociaiuti M, Gaudiano MC, Malchiodi‐Albedi F. The slowly aggregating salmon calcitonin: a useful tool for the study of the amyloid oligomers structure and activity. Int J Mol Sci. 2011;12:9277–95. 10.3390/ijms12129277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand RM, Epand RF, Orlowski RC, Schlueter RJ, Boni LT, Hui SW. Amphipathic helix and its relationship to the interaction of calcitonin with phospholipids. Biochemistry. 1983;22(22):5074–84. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, et al. Structure based aggregation studies reveal the presence of helix‐rich intermediate during α‐synuclein aggregation. Sci Rep. 2015;5:9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist PJ, Bradshaw JP. Amyloid formation by salmon calcitonin. Biochem Biophys Acta. 1993;1182(1):111–4. [DOI] [PubMed] [Google Scholar]
- Harrison RS, Sharpe PC, Singh Y, Fairlie DP. Amyloid peptides and proteins in review. Rev Physiol Biochem Pharmacol. 2007;159:1–77. [DOI] [PubMed] [Google Scholar]
- Hirsch PF, Gauthier GF, Munson PL. Thyroid hypocalcemic principle and recurrent laryngeal nerve injury as factors affecting the response to parathyroidectomy in rats. Endocrinology. 1963;73:244–52. [DOI] [PubMed] [Google Scholar]
- Holtrop ME, Raisz LG, Simmons HA. The effects of parathyroid hormone, colchicine, and calcitonin on the ultrastructure and the activity of osteoclasts in organ culture. J Cell Biol. 1974;60(2):346–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh IC, Chen T‐W, Chuang Y‐P, Lai Y‐J, Tu L‐H. Tyrosine 12 of human calcitonin modulates its amyloid formation, membrane binding, and bioactivity. Biochimie. 2022;197:121–9. [DOI] [PubMed] [Google Scholar]
- Hudson SA, Ecroyd H, Kee TW, Carver JA. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009;276(20):5960–72. [DOI] [PubMed] [Google Scholar]
- Itoh‐Watanabe H, Kamihira‐Ishijima M, Javkhlantugs N, Inoue R, Itoh Y, Endo H, et al. Role of aromatic residues in amyloid fibril formation of human calcitonin by solid‐state 13C NMR and molecular dynamics simulation. Phys Chem Chem Phys. 2013a;15(23):8890–901. [DOI] [PubMed] [Google Scholar]
- Itoh‐Watanabe H, Kamihira‐Ishijima M, Kawamura I, Kondoh M, Nakakoshi M, Sato M, et al. Characterization of the spherical intermediates and fibril formation of hCT in HEPES solution using solid‐state 13C‐NMR and transmission electron microscopy. Phys Chem Chem Phys. 2013b;15(39):16956–64. [DOI] [PubMed] [Google Scholar]
- Kamgar‐Parsi K, Tolchard J, Habenstein B, Loquet A, Naito A, Ramamoorthy A. Structural biology of calcitonin: from aqueous therapeutic properties to amyloid aggregation. Israel J Chem. 2017;57(7):634–50. [Google Scholar]
- Kamihira M, Naito A, Tuzi S, Nosaka AY, Saitô H. Conformational transitions and fibrillation mechanism of human calcitonin as studied by high‐resolution solid‐state 13C NMR. Protein Sci. 2000;9(5):867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamihira M, Oshiro Y, Tuzi S, Nosaka AY, Saitô H, Naito A. Effect of electrostatic interaction on fibril formation of human calcitonin as studied by high resolution solid state 13C NMR. J Biol Chem. 2003;278(5):2859–65. [DOI] [PubMed] [Google Scholar]
- Kayed R, Lasagna‐Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33:S67–78. [DOI] [PubMed] [Google Scholar]
- Kentsis A, Sosnick TR. Trifluoroethanol promotes helix formation by destabilizing backbone xxposure: Desolvation rather than native hydrogen bonding defines the kinetic pathway of dimeric coiled coil folding. Biochemistry. 1998;37(41):14613–22. [DOI] [PubMed] [Google Scholar]
- Khurana R, Agarwal A, Bajpai VK, Verma N, Sharma AK, Gupta RP, et al. Unraveling the amyloid associated with human medullary thyroid carcinoma. Endocrinology. 2004;145(12):5465–70. [DOI] [PubMed] [Google Scholar]
- Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β‐protein fibrillogenesis. J Mol Biol. 2001;312(5):1103–19. [DOI] [PubMed] [Google Scholar]
- Kumar S, Hamilton AD. α‐Helix mimetics as modulators of Aβ self‐assembly. J Am Chem Soc. 2017;139(16):5744–55. [DOI] [PubMed] [Google Scholar]
- Lantz R, Busbee B, Wojcikiewicz EP, Du D. Effects of disulfide bond and cholesterol derivatives on human calcitonin amyloid formation. Biopolymers. 2020;111(5):e23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SL, Yu LX, Cai B, Johnsons GR, Rosenberg AS, Cherney BW, et al. Scientific considerations for generic synthetic salmon calcitonin nasal spray products. AAPS J. 2011;13(1):14–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Prabhakar A, Aucoin D, Simon M, Sparks S, Robbins KJ, et al. Mechanistic studies of peptide self‐assembly: transient α‐helices to stable β‐sheets. J Am Chem Soc. 2010;132(51):18223–32. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Findlay DM, MacIntyre I, Eisman JA, Michelangeli VP, Moseley JM, et al. Calcitonin receptors in a cloned human breast cancer cell line (MCF 7). Biochem Biophys Res Commun. 1980;96(1):150–6. [DOI] [PubMed] [Google Scholar]
- Micsonai A, Moussong É, Wien F, Boros E, Vadászi H, Murvai N, et al. BeStSel: webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022;50(W1):W90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A, Wien F, Kernya L, Lee Y‐H, Goto Y, Réfrégiers M, et al. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci U S A. 2015;112(24):E3095–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Levine Iii H. Alzheimer's disease and the amyloid‐β peptide. J Alzheimers Dis. 2010;19(1):311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal Biochem. 1989;177(2):244–9. [DOI] [PubMed] [Google Scholar]
- Naito A, Kamihira M, Inoue R, Saitô H. Structural diversity of amyloid fibril formed in human calcitonin as revealed by site‐directed 13C solid‐state NMR spectroscopy. Magn Reson Chem. 2004;42:247–57. [DOI] [PubMed] [Google Scholar]
- Niall HD, Keutmann HT, Copp DH, Potts JT Jr. Amino acid sequence of salmon ultimobranchial calcitonin. Proc Natl Acad Sci U S A. 1969;64(2):771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts JT Jr, Niall HD, Keutmann HT, Brewer HB Jr, Deftos LJ. The amino acid sequence of porcine thyrocalcitonin. Proc Natl Acad Sci U S A. 1968;59(4):1321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reches M, Porat Y, Gazit E. Amyloid fibril formation by pentapeptide and tetrapeptide fragments of human calcitonin. J Biol Chem. 2002;277(38):35475–80. [DOI] [PubMed] [Google Scholar]
- Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer's disease. Rev Neurosci. 2018;29(6):613–27. [DOI] [PubMed] [Google Scholar]
- Renawala HK, Chandrababu KB, Topp EM. Fibrillation of human calcitonin and its analogs: effects of phosphorylation and disulfide reduction. Biophys J. 2021;120(1):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtainfeld A, Sheynis T, Jelinek R. Specific mutations alter fibrillation kinetics, fiber morphologies, and membrane interactions of pentapeptides derived from human calcitonin. Biochemistry. 2010;49(25):5299–307. [DOI] [PubMed] [Google Scholar]
- Siligardi G, Samori B, Melandri S, Visconti M, Drake AF. Correlations between biological activities and conformational properties for human, salmon, eel, porcine calcitonins and Elcatonin elucidated by CD spectroscopy. Eur J Biochem. 1994;221(3):1117–25. [DOI] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron X‐ray diffraction. J Mol Biol. 1997;273(3):729–39. [DOI] [PubMed] [Google Scholar]
- Talmage RV, Grubb SA, Norimatsu H, VanderWiel CJ. Evidence for an important physiological role for calcitonin. Proc Natl Acad Sci U S A. 1980;77(1):609–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, et al. Elucidating amyloid β‐protein folding and assembly: a multidisciplinary approach. Acc Chem Res. 2006;39(9):635–45. [DOI] [PubMed] [Google Scholar]
- Vieweg S, Mahul‐Mellier A‐L, Ruggeri FS, Riguet N, DeGuire SM, Chiki A, et al. The Nt17 domain and its helical conformation regulate the aggregation, cellular properties and neurotoxicity of nutant huntingtin exon 1. J Mol Biol. 2021;433(21):167222. [DOI] [PubMed] [Google Scholar]
- Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91(3):795–826. [DOI] [PubMed] [Google Scholar]
- Williamson JA, Miranker AD. Direct detection of transient α‐helical states in islet amyloid polypeptide. Protein Sci. 2007;16(1):110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AG, Wu C, Hannaberry E, Watson MD, Shea J‐E, Raleigh DP. Analysis of the amyloidogenic potential of pufferfish (Takifugu rubripes) islet amyloid polypeptide highlights the limitations of thioflavin‐T assays and the difficulties in defining amyloidogenicity. Biochemistry. 2016;55(3):510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemoto IT, Kroon GJA, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47(37):9900–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1. Supporting Information