

Abstract
MRPL39 encodes one of 52 proteins comprising the large subunit of the mitochondrial ribosome (mitoribosome). In conjunction with 30 proteins in the small subunit, the mitoribosome synthesizes the 13 subunits of the mitochondrial oxidative phosphorylation (OXPHOS) system encoded by mitochondrial Deoxyribonucleic acid (DNA). We used multi-omics and gene matching to identify three unrelated individuals with biallelic variants in MRPL39 presenting with multisystem diseases with severity ranging from lethal, infantile-onset (Leigh syndrome spectrum) to milder with survival into adulthood. Clinical exome sequencing of known disease genes failed to diagnose these patients; however quantitative proteomics identified a specific decrease in the abundance of large but not small mitoribosomal subunits in fibroblasts from the two patients with severe phenotype. Re-analysis of exome sequencing led to the identification of candidate single heterozygous variants in mitoribosomal genes MRPL39 (both patients) and MRPL15. Genome sequencing identified a shared deep intronic MRPL39 variant predicted to generate a cryptic exon, with transcriptomics and targeted studies providing further functional evidence for causation. The patient with the milder disease was homozygous for a missense variant identified through trio exome sequencing. Our study highlights the utility of quantitative proteomics in detecting protein signatures and in characterizing gene-disease associations in exome-unsolved patients. We describe Relative Complex Abundance analysis of proteomics data, a sensitive method that can identify defects in OXPHOS disorders to a similar or greater sensitivity to the traditional enzymology. Relative Complex Abundance has potential utility for functional validation or prioritization in many hundreds of inherited rare diseases where protein complex assembly is disrupted.
Introduction
Oxidative phosphorylation (OXPHOS) is the main metabolic pathway through which the majority of Adenosine triphosphate (ATP) is generated in eukaryotic cells (1). The OXPHOS system comprises five large protein complexes (I–V) embedded in the inner mitochondrial membrane (IMM). Of the 91 proteins that form the OXPHOS complexes, 13 are encoded by the mitochondrial DNA (mtDNA), which also encodes 22 tRNAs and 2 rRNAs required for translation by mitochondrial ribosomes (mitoribosomes) (2). Mitoribosomes are 55S nucleoprotein complexes that associate with the IMM for co-translational membrane insertion of the 13 mtDNA encoded proteins (3). They contain 82 mitoribosomal proteins (MRPs) which form two protein subunits: a 39S large subunit (mtLSU) composed of 52 MRPs (MRPLs) and a 28S small subunit (mtSSU) composed of 30 MRPs (MRPSs), as well as 16S and 12S mitochondrial rRNAs, and mitochondrial tRNAVal (4–7). All MRPs are encoded by the nuclear genome and imported into mitochondria for mitoribosome biogenesis (6). The mtLSU catalyzes peptide bond formation and attaches the ribosome to the IMM, whereas the mtSSU decodes the mRNA and recruits the correct aminoacyl tRNA to the translating ribosome (7). Mitoribosomes show substantial differences from their bacterial and cytoplasmic counterparts to accommodate the specialized requirements of protein synthesis within mitochondria including a higher protein: Ribonucleic acid (RNA) ratio and 36 mitochondria-specific proteins. The increased protein mass (~1 MDa) creates an extensive protein–protein interaction network, stabilizes mitoribosomal structures, protects the rRNA molecule from oxidative stress, attaches the mitoribosome to the IMM, and provides a hydrophobic passage for nascent peptides to emerge from the polypeptide tunnel exit into the IMM, among numerous other functions (6).
To date, 14 MRP genes including 4 MRPL genes have been implicated in mitochondrial disease [MRPS2 (8), MRPS7 (9), MRPS14 (10), MRPS16 (11), MRPS22 (12), MRPS23 (13), MRPS25 (14), MRPS28 (15), MRPS34 (16), PTCD3 (17), MRPL3 (18), MRPL12 (19), MRPL24 (20) and MRPL44 (21)]. Pathogenic variants in these genes (where tested) typically cause mitoribosomal subunit instability, as shown by either quantitative proteomics (16,17), sucrose gradient sub-fractionation (10,14–16,19–21), complexome profiling (8), quantitative PCR (qPCR) experiments showing increase or decrease in 12S:16S rRNA ratio and/or immunoblotting of multiple proteins from each subunit (8–10,14–16,19–21). Mitoribosomal instability causes dysfunctional mitochondrial translation, defective OXPHOS biogenesis and a resultant combined OXPHOS deficiency (22). Like other mitochondrial disorders, defects in these genes have been associated with a broad spectrum of nonspecific clinical features (23), ranging from severe fatal infantile multisystemic diseases [such as Leigh syndrome (16) or fatal neonatal lactic acidosis (11)] to cardiomyopathy (21,24) or primary ovarian insufficiency (25).
This report describes three unrelated individuals with biallelic pathogenic variants in the mitochondrial large ribosomal protein subunit gene, MRPL39 (MIM: 611 845), which has not previously been associated with human disease. Two individuals had a severe, lethal, infantile-onset form of the disease within the Leigh syndrome spectrum (MIM: 256 000) (26), whereas the third had a milder presentation of hypertrophic cardiomyopathy, lactic acidosis, attention-deficit/hyperactivity disorder (ADHD) and survival into adulthood. MRPL39 was identified in these individuals through the application of multi-omics technologies, including quantitative proteomics, exome and genome sequencing and RNA sequencing (RNA-seq). In two of the affected individuals, one of the causative variants was deep-intronic and thus not detected in first-round exome sequencing, substantiating the value of utilizing quantitative proteomics in gene prioritization and validation in exome-negative patients.
Results
Individual 1 (P1) was part of a previous cohort of patients with Leigh syndrome spectrum (individual LS19) (27). She remained undiagnosed after mtDNA sequencing and singleton research exome sequencing (ES) with analysis of genes associated with mitochondrial diseases, Leigh syndrome, or encoding known mitochondrial proteins (Fig. 1) (16,28). No rare (<0.005 minor allele frequency), protein-altering, biallelic, or X-linked variants were identified in these genes. Individual 2 (P2) underwent clinical trio ES in combination with mtDNA-massively parallel sequencing (mtDNA-seq) (29,30) as part of the Australian Genomics—Acute Care Genomics Flagship (31), which was uninformative (Fig. 1). P1 and P2 were both born to non-consanguineous parents of European background, presenting early in life with congestive cardiac failure, increased lactates, seizures, apnea, poor feeding and global developmental delay, leading to early death (< 1 year of age), refer to Supplementary Material, Case Reports for greater detailed clinical descriptions. They both showed normal OXPHOS enzyme activities in skeletal muscle, whereas P1 also had normal liver OXPHOS enzyme activities but showed decreased levels of complexes I and IV in fibroblasts (Table 1).
Figure 1.
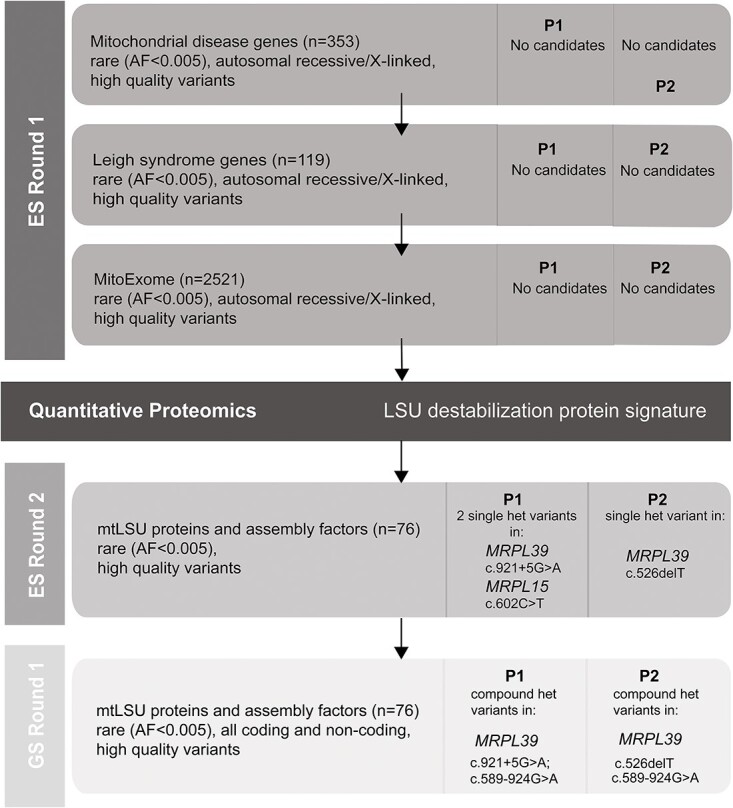
Flowchart illustrating the filtering strategy and chronological steps used to analyze exome and genome sequencing data for P1 and P2. Each horizontal bar represents a step in the analysis process based on the gene list used and contains information on the filter parameters used and the details of any candidate genes (gene, inheritance, variant) identified for P1 and P2. The vertical columns indicate whether exome (ES) or genome (GS) sequence was utilized. The black horizontal bar represents the stage at which quantitative proteomics was used to identify candidate disease genes. MitoExome refers to genes encoding all known and candidate proteins targeted to the mitochondria primarily based on the MitoCarta3.0 inventory (72). AF: allele frequency.
Table 1.
Clinical, biochemical and genetic details of the individuals carrying deleterious variants in MRPL39
| Individual ID | P1 | P2 | P3 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Gender) | Female | Female | Male | |||||||||||||||
| Ethnicity | European (Australian) | European (Australian) | European (Danish) | |||||||||||||||
| Consanguinity | No | No | Yes | |||||||||||||||
| MRPL39 maternal allele | c.921+5G>A; p.(Ile257Hisfs*17) | c.589-924G>A; p.(Gln197Argfs*9) | c.896G>T; p.(Gly299Val) | |||||||||||||||
| MRPL39 paternal allele | c.589-924G>A; p.(Gln197Argfs*9) | c.526delT; p.(Ser176Leufs*8) | c.896G>T; p.(Gly299Val) | |||||||||||||||
| Inheritance pattern | Autosomal recessive | Autosomal recessive | Autosomal recessive | |||||||||||||||
| OXPHOS enzyme % activitiesa | CI | CII | CII + CIII | CIII | CIV | CI | CII | CII + CIII | CIII | CIV | CI | CII | CII + CIII | CIII | CIV | |||
| M | 107 | 92 | 68 | 60 | 94 | 104 | 67 | 66 | 52 | 89 | 204 | 247 | 157 | 50 | ||||
| L | 103 | 56 | 80 | 68 | ||||||||||||||
| F | 24 | 87 | 90 | 46 | 150 | 104 | ||||||||||||
| Age of onset | 6 weeks | 2 days | 15 months | |||||||||||||||
| Age last examined/age of death | Death at 7 months | Death at 11 months | Last seen at 19 years of age | |||||||||||||||
| Clinical summary | Leigh syndrome, hypotonia, failure to thrive, development delay, hepatomegaly, lactic acidosis, seizures, sepsis, vomiting, hypernatremia with depressed CNS function, bradycardia, recurrent apnea, congestive cardiac failure | Leigh syndrome spectrum, hypotonia, global developmental delay, failure to thrive, feeding difficulties, persistent irritability, bulbar palsy, colonic seizures, gastroesophageal reflux disease, elevated blood lactate, elevated blood alanine, elevated CSF alanine, mild lactate peak in MRI, apneic episodes, dysautonomia, left ventricular dysfunction, cardiac arrest | Hypertrophic cardiomyopathy, hypertonia, apathy, hypoglycemia with vomiting, migrainoid headaches, ADHD, learning disabilities and bilateral basal ganglia involvement, increased blood lactate and alanine | |||||||||||||||
M, skeletal muscle; L, liver; F, fibroblast.
aOXPHOS enzyme % activities for complexes I, II, II + III, III, IV (CI, CII,CII + III, CIII, CIV) relative to citrate synthase indicated as % of control mean activity for individuals P1 and P2 and as % of lowest control for individual P3. For P3, the activities in fibroblasts are expressed relative to complex II.
As these patients remained undiagnosed, quantitative proteomics was performed on whole-cell protein lysates from fibroblast cell lines obtained from P1, P2 and three normal controls. Decreased abundance of MRPLs, but not MRPSs, was detected for both P1 and P2 fibroblasts relative to controls (Fig. 2A and B; Supplementary Material, Table S1). Topographical heat mapping of the relative protein abundances onto the cryo-electron microscopy structure of the mitoribosome (6) shows a specific destabilization of the mtLSU (Fig. 2C and D). Relative complex abundance (RCA) analysis, which quantifies overall changes in OXPHOS complex abundance, shows significantly decreased amounts of complexes I, III and IV for both patients and complex V for P2 (Fig. 2E). These results prompted us to focus our exome reanalysis on genes associated with the assembly and structure of the large mitoribosomal subunit because specific destabilization of mitoribosomal subunits has previously been reported to be a consequence of defective MRPs, leading to defects in the translation of mtDNA encoded OXPHOS subunits found in complexes I, III, IV and V and subsequently OXPHOS complex assembly (8,9,12,16).
Figure 2.
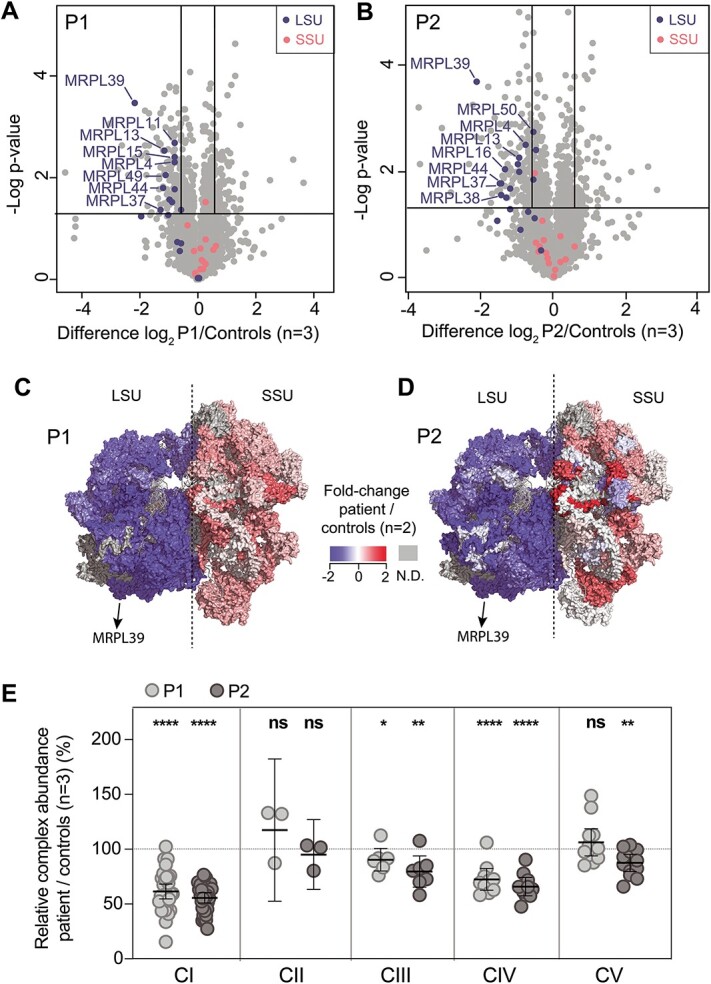
Quantitative proteomics identifies the decreased abundance of the large subunit of the mitoribosome and OXPHOS complexes in P1 and P2 fibroblasts. Volcano plot of label-free quantitative proteomics data from whole-cell fibroblasts from (A) P1, and (B) P2, depicting a specific decrease in the relative abundance of proteins of the large ribosomal subunit (LSU) in both cell lines compared to controls. mtLSU = large ribosomal subunit, mtSSU = small ribosomal subunit. The horizontal lines in A and B represent p = 0.05 and the vertical lines represent an equivalent fold-change of ±1.5. Topographical heat mapping of label-free quantitative proteomics data from isolated mitochondria in (C) P1, and (D) P2, relative to controls (n = 2) utilizing the cryo-electron microscopy structure of the mitoribosome (6) PDB: 3J9M), N.D. = not detected. (E) RCA analysis of OXPHOS complexes from whole-cell quantitative proteomics for P1 and P2 relative to controls (n = 3). The middle bar represents the mean value for the complex, whereas the upper and lower bars represent the 95% confidence interval of the mean value. Each dot represents a single protein. * = p < 0.05, ** = p < 0.01, **** = p < 0.0001 and ns = not significant represent P-value significances from paired t-test. CI = Complex I, CII = Complex II, CIII = Complex III, CIV = Complex IV, CV = Complex V.
The exome data for P1 and P2 were reanalyzed for all rare, protein-altering variants in mtLSU genes (without an inheritance filter). Two single heterozygous variants were identified in P1; one in MRPL15 (MIM: 611828) NM_014175.4:c.602C>T; p.(Pro201Leu) and the other in MRPL39 NM_017446.4:c.921+5G>A (Fig. 1). A single rare heterozygous paternally inherited indel in MRPL39 NM_017446.4:c.526delT; p.(Ser176Leufs*8) was identified in P2. The MRPL15:c.602C>T variant detected in P1 was absent from public databases [gnomAD v2 and v3 (32) and TopMed (33)] and predicted to be deleterious by in silico prediction tools [Combined Annotation Dependent Depletion (CADD) 25 (34), Eigen 9.7 (35), Revel 0.56 (36), PrimateAI 0.8 (37), SIFT_deleterious (38), MutationTaster_deleterious (39)]. polymerase chain reaction (PCR) amplification of full-length MRPL15 cDNA synthesized from RNA extracted from P1 fibroblasts grown with and without cycloheximide (CHX) to inhibit nonsense-mediated decay (NMD), did not indicate any alteration in MRPL15 splicing (Supplementary Material, Fig. S1A). Sanger sequencing confirmed that both alleles were expressed (Supplementary Material, Fig. S1B), making it unlikely that a pathogenic variant on the second allele was missed. Moreover, quantitative proteomics data showed MRPL15 protein to have a fold change reduction of ~1.7 in P1 relative to controls, similar to all other MRPLs in this individual except MRPL39, which has a fold change reduction of ~ 4.5 (Fig. 2A; Supplementary Material, Table S1). The MRPL39:c.921+5G>A variant in P1 is also predicted to be deleterious (CADD 16, Eigen 21, MutationTaster_deleterious) and result in aberrant splicing by abolishing the exon 8 donor site [SpliceAI 0.69 donor_loss (37), maximum entropy score or MES reduced 9.35 to 1.19 (40)]. It is listed in dbSNP (rs375392547) and is heterozygous in 4 individuals in gnomAD (2 of 240,488 alleles examined in v2.1.1 and 2 of 152 186 alleles examined in v3.1.2, no homozygotes observed). The MRPL39:c.526delT variant found in P2 (rs746607851) is heterozygous in 19 individuals in gnomAD v.3.1.2 (of 152 156 tested alleles) along with 30 heterozygous individuals in v2.1.1 (of 282 102 alleles), with no homozygotes identified. This variant is predicted to be deleterious (CADD score 23, MutationTaster_deleterious) and undergo NMD.
The consequences of these MRPL39 variants were examined by PCR of cDNA synthesized from patients (P1 and P2) and control RNA extracted from fibroblasts grown with and without CHX. Gel electrophoresis of PCR products amplified with primers in exons 1 and 11 of the canonical MRPL39 transcript (NM_080794.4) showed an abundant band in P1 that was smaller than the control (Fig. 3A lanes 1 and 3). This band was stably expressed in the absence of CHX, whereas some additional less abundant larger-sized bands were stabilized with CHX. Sanger sequencing of the most abundant amplicon in P1 identified a transcript lacking exon 8, supporting in silico predictions that the c.921+5G>A variant abolishes the exon 8 donor site and creates a premature stop-codon, p.(Ile257Hisfs*17) (Fig. 3A and Supplementary Material, Fig. S2). As the predominant MRPL39 transcript expressed in fibroblasts (NM_017446.4) lacks exon 10 of the canonical transcript, this premature stop-codon introduced by the splicing defect lies less than 50 nucleotides upstream of the final exon-exon boundary, evading NMD (41). Amplification of P2’s cDNA identified an amplicon of comparable size to wild-type as well as a larger-sized band that was only stable in the presence of CHX (Fig. 3B lane 1). TOPO cloning and Sanger sequencing of the P2 CHX stabilized PCR products identified the presence of three transcripts: two prominent abnormal transcripts and a less abundant wild-type transcript. The larger abnormal transcript contained 94 bp of intron 5 sequence leading to a premature stop-codon p.(Gln197Argfs*9) (Fig. 3B and Supplementary Material, Fig. S5), whereas the other contained the c.526delT variant identified originally through ES (Fig. 3B). In the absence of CHX, a single band of the expected size was amplified (Fig. 3B lane 2). Sanger sequencing confirmed that this band only contained wild-type MRPL39 (Supplementary Material, Fig. S4) and that the two abnormal transcripts undergo NMD. Taken together, these results suggested that in both patients a second pathogenic MRPL39 allele was missed by ES. Genome sequencing (GS) revealed the presence of the same heterozygous deep intronic MRPL39 variant NM_017446.4:c.589-924G>A in both P1 and P2. This variant was heterozygous in 8 individuals in gnomAD v3.1.2 and 4 individuals in v2.1.1 (8 of 151 500 alleles and 4 of 31 106 tested, respectively), with no homozygotes observed and is listed in dbSNP (rs1209423257). The c.589-924G>A variant is predicted to alter splicing (SpliceAI: 0.340 donor-gain; NNSplice New donor score 0.8 (42); MES increased 3.09 to 7.88) and is located 3 bp downstream of the 94 bp intron 5 insertion identified in P2 by the cDNA studies (Fig. 3B). All three MRPL39 variants detected by ES and GS were validated by Sanger sequencing (Supplementary Material, Fig. S4), and segregation studies confirmed the variants to be biallelic, with the mutual c.589-924G>A being paternally inherited in P1 and maternally inherited in P2, whereas the c.921+5G>A variant was inherited from P1’s mother and the c.526delT variant identified in P2 was inherited paternally.
Figure 3.
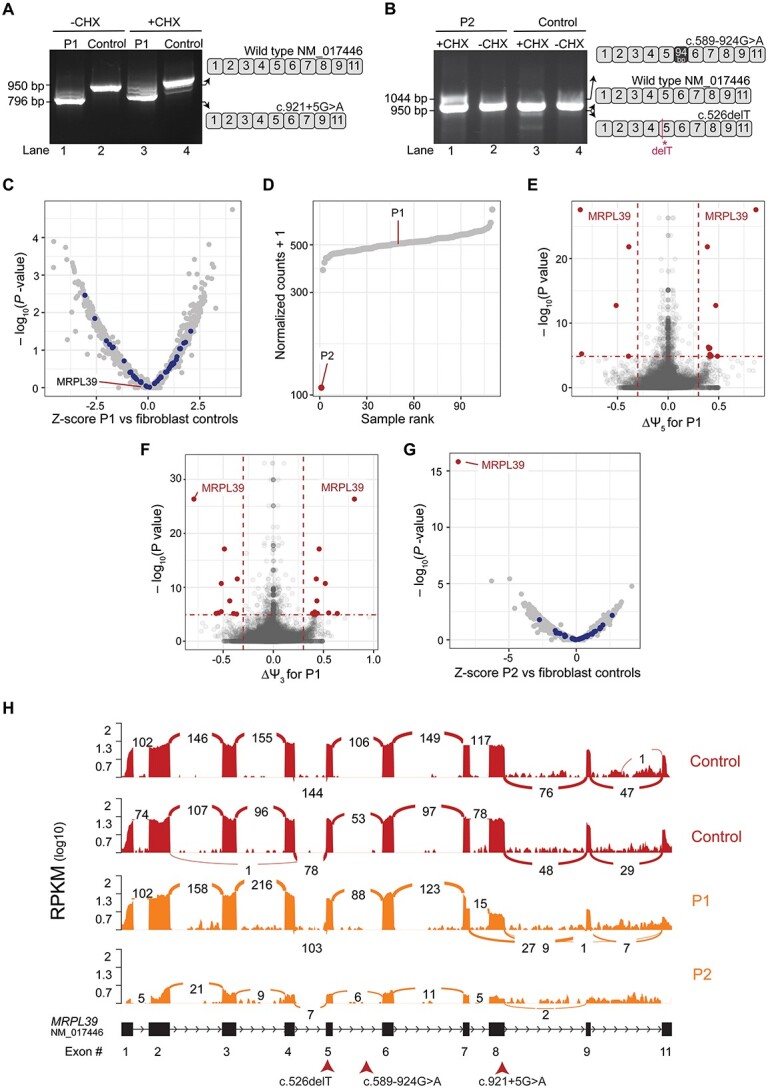
RNA-seq and cDNA studies show the deleterious effects of MRPL39 variants at a transcriptomic level. (A) cDNA analysis of P1 using RNA extracted from cycloheximide (CHX)-treated and -untreated fibroblasts. Gel electrophoresis of full-length PCR products amplified using cMRPL39_F (exon 1) and cMRPL39_R (exon 11) primers show aberrant and wildtype transcripts present. The wildtype and aberrantly spliced transcripts identified are depicted in the schematic diagram on the right. (B) cDNA analysis of P2 using RNA extracted from CHX-treated and -untreated fibroblasts as per A showing the aberrant and wildtype transcripts identified. The schematic diagram on the right depicts the aberrantly spliced transcripts identified in P2. (C) RNA expression volcano (significance -log10P value versus Z-score), a DROP analysis output, for P1 RNA-seq experiments performed on patient fibroblasts compared to GTEx fibroblast controls (n = 90) and patient fibroblast samples (n = 20). Each dot represents a transcript expressed in P1 compared to controls, showing normal expression for all the genes analyzed. MRPL gene transcripts are highlighted in blue, showing that they are expressed at normal levels. (D) RNA expression rank plot, generated from the DROP analysis pipeline, for P1 and P2 RNA-seq experiments performed on patient fibroblasts with normalized read counts for MRPL39 transcript plotted against all samples tested (90 GTEx controls + 21 patient fibroblast samples including both P1 and P2 samples) showing extremely low expression in P2 compared to all the other samples in the experiment and a normal expression level in P1. (E) Alternative accepter site usage (significance -log10P value versus effect ∆ψ5), a DROP analysis output, from P1 RNA-seq experiments revealed significant changes in acceptor site usage for MRPL39 depicted in red dots. Horizontal red dotted lines indicate the gene-level significance cutoff, and the vertical dotted lines represent the effect size cutoff. (F) Alternative donor site usage DROP analysis output (significance -log10P value versus effect ∆ψ3) for P1 showed a significant change in donor site usage for MRPL39 (red dots). (G) RNA expression volcano, significance -log10P value versus Z-score, for P2 with each dot representing a transcript expressed in P2 fibroblasts showing MRPL39 as a significant expression outlier compared to controls. All other MRPL genes, highlighted in blue, revealed normal expression levels. (H) MRPL39 sashimi plot of P1, P2 (orange) and two unaffected controls (red) illustrating the exon 8 skipping event in P1 and low expression in P2. RNA-seq read coverage is given as log10 RPKM (Reads Per Kilobase of transcript per Million mapped reads). Splicing is depicted by lines connecting exons with the number of split reads spanning a junction indicated on the line. A schematic of the MRPL39 transcript (NM_017446.4) expressed in fibroblasts along with the corresponding exon numbers are illustrated below the sashimi plot as well as the position of the patient variants indicated with arrows.
To further validate the effect of these variants at a transcriptomic level, RNA-seq was performed on RNA extracted from both P1 and P2 fibroblasts and analyzed using the DROP pipeline (43). For P1, no clear outliers in gene expression levels were detected across the subset of 6106 genes analyzed (Fig. 3C) compared with 109 fibroblast controls (19 additional patients and 90 GTEx controls) and MRPL39 expression levels were unremarkable (Fig. 3D). However, two significant aberrant splicing events related to the usage of the alternate donor (Exon 7 donor; ∆ψ5 = 0.87) and acceptor sites (Exon 9 accepter; ∆ψ3 = 0.8) were detected (Fig. 3E and F), in agreement with the exon 8 skipping event previously identified from cDNA studies. Most transcripts in P1 skipped exon 8, with only a small proportion of wild-type transcripts expressed (Fig. 3H). In P2, MRPL39 expression was markedly reduced and identified as a significant outlier compared to fibroblast controls (Fig. 3D and G). The RNA-seq data was also consistent with the presence of full-length transcripts at low abundance (Fig. 3H). In both P1 and P2, all other MRPL genes including MRPL15 were normally expressed at a transcript level, supportive of the destabilization of the mtLSU occurring at a protein level because of variants identified in MRPL39.
A third individual, P3, identified through GeneMatcher (44), was homozygous for a missense MRPL39 NM_017446.4:c.896G>T; p.(Gly299Val) variant identified by trio ES analysis. P3, now an adult, was born to first-cousin parents of European background who are both carriers of this MRPL39 variant. He presented at 15 months with repeated episodes of ketotic hypoglycemia with vomiting, apathy, hypotonia and migrainoid headache that required hospital treatment. Bilateral changes to the basal ganglia, lactic acidosis and hypertrophic cardiomyopathy were subsequently identified (Supplementary Material, Case Reports). Respiratory chain enzyme analysis of his skeletal muscle biopsy showed a combined OXPHOS complex I and IV enzyme deficiency, whereas his fibroblast cell line showed normal enzyme activities (Table 1). The MRPL39 c.896G>T variant is predicted to be pathogenic (SIFT_deleterious, MutationTaster_deleterious, Polyphen2.0_deleterious) and is not recorded in public databases (gnomAD or dbSNP). This variant was located 25 bp upstream from the donor site of exon 8; however, cDNA studies detected no alterations in splicing (Supplementary Material, Fig. S3). The p.Gly299 residue is highly conserved (Fig. 4A), with the change to a valine resulting in a Grantham score (45) of 109 and lies within a beta-sheet buried in the core of the protein as predicted by PSIPred and AlphaFold (46,47). According to these structures, the p.Gly299 residue interacts with a bulky phenylalanine residue at the 179th position in the adjacent beta sheet via hydrogen bonds. The introduction of a bulkier, more hydrophobic, and less flexible valine residue could disrupt these interactions and alter the backbone conformation of the peptide affecting the entire protein structure.
Figure 4.

The p.(Gly299Val) variant identified in individual P3 changes a highly conserved amino acid residue and results in selective destabilization of the mitochondrial large ribosomal subunit, in agreement with results seen in individuals P1 and P2. (A) Protein sequence alignment of MRPL39 with 11 of its homologs including Homo sapiens and 10 other vertebrate species. Asterisks (*) depict conserved amino acids and colons (:) depict semi-conserved residues. The p.Gly299 residue is changed to a valine in individual P3 and is indicated in bold and is 100% conserved in vertebrate species from humans to Danio rerio. (B) Representative immunoblots of large (MRPL) and small (MRPS) mitoribosomal proteins extracted from fibroblasts revealed a substantial decrease in MRPL levels in all three affected individuals including in MRPL39, whereas the MRPS levels remained comparable to controls. SDHA and VDAC1 represent loading controls for mitochondrial content. (C) Fibroblasts from a control individual and P1, P2 and P3 were transduced with wild-type MRPL39 cDNA. Representative SDS-PAGE immunoblot demonstrates an increase in protein levels of large mitoribosomal protein subunits, MRPL39 and MRPL28, in transduced patient fibroblasts relative to untransduced cells, whereas the small mitoribosomal protein subunit MRPS34 was unchanged. VDAC1 was used as a loading control. (D) Densitometry analysis revealed that the increase after transduction was significant in each patient. Results were normalized to VDAC1 and presented as the percent of average untransduced control cells. The data shown are the mean of three independent transfections ± SEM. **p < 0.002, ***p< 0.0002, ****p < 0.0001.
To verify the pathogenicity of the p.(Gly299Val) missense variant, immunoblotting was performed on protein lysates extracted from fibroblast cell lines from all three individuals and controls. Expression levels of all MRPL proteins analyzed (MRPL39, 9, 28, 44 and 45) were decreased in all three individuals while the MRPS proteins were expressed at levels either comparable to or elevated compared to that of control (Fig. 4B). These results were consistent with those obtained from quantitative proteomics for individuals P1 and P2 (Fig. 2), with both assays showing a preferential decrease in MRPLs. As P3 also has the same mtLSU protein signature on immunoblot as P1 and P2, which includes a decrease in MRPL39 levels, this provides support for the MRPL39 p.(Gly299Val) variant also being pathogenic through its effect on mtLSU stability or assembly.
As further support, we performed complementation studies to determine whether the expression of wild-type MRPL39 in patient cells could rescue the large mitoribosomal protein subunit defects. Fibroblasts from a control and all three patients were transduced with a lentiviral vector expressing wild-type MRPL39. The levels of mitoribosomal protein subunits MRPL39, MRPL28 and MRPS34 were examined by immunoblotting (Fig. 4C). Densitometry analysis confirmed a decrease in the large mitoribosomal protein subunit MRPL28 and MRPL39 levels in patient cells relative to control (Fig. 4D), which was able to be corrected using lentiviral-mediated expression of wild-type MRPL39 in all three patients. Thus, we conclude that the reduced abundance of these proteins in all three patients is because of the mutations in MRPL39. Lentiviral-mediated expression of MRPL39 in control cells caused a significant decrease in MRPL28 protein levels, suggesting a potential negative effect of overexpression in cells with a stable large mitoribosomal subunit, though not in contradiction with the rescue observed in cells from the patients.
Collectively, these data establish biallelic deleterious variants in MRPL39 as a cause of pediatric-onset mitochondrial disease.
Discussion
The specific role of MRPL39 in mitoribosomal function is uncertain, but it has homology to the C-terminal domain of threonyl-tRNA synthetase (48,49). MRPL39 has been proposed to be one of several MRPL rRNA binding proteins recruited to the mitoribosome during eukaryotic evolution and ultimately retained because of extensive protein–protein interactions having been established (50). Recent studies using pulse labeling coupled with quantitative proteomics identified MRPL39 as one of the early assembling proteins (51). Interestingly, most of the mitoribosomal proteins previously implicated in human disease also appear to be involved early in the assembly process of either the mtLSU or mtSSU (52); however, this may be because of ascertainment bias as they are all associated with severe clinical phenotypes. In mammals, MRPL39 along with MRPL44 and MRPL45 are located, in close proximity, at the polypeptide tunnel exit (50). In both human and yeast mitoribosomes, MRPL45 and Mba1 (the homolog to human MRPL45) facilitate ribosomal attachment to the inner mitochondrial membrane (3,53). Therefore, whereas its exact role is currently unclear, it could be hypothesized that human MRPL39 may facilitate early mtLSU assembly or stabilize the interaction between MRPL45 and the IMM.
At least one of the two splice-site variants identified in this study acts as a hypomorph, as wild-type transcripts were detected by RNA studies and immunoblotting showed residual protein of expected size, indicating that MRPL39 was not completely absent. In the previous reports of patients with pathogenic MRPL/S variants, it is interesting to note that no patients have biallelic null or loss-of-function (LoF) variants, with nonsense variants usually accompanied by a missense or a (potentially hypomorphic) splice-site variant, the one exception in the literature is a patient homozygous for a premature stop codon (c.331C>T; p.Arg111*) in MRPS16 (11). However, this mutation lies within the last exon resulting in a protein truncation of 27 amino acids rather than LoF, again supporting the likelihood that two null variants would be embryonic lethal.
We previously reported variants in the MRPS34 gene in patients within the Leigh syndrome spectrum causing translational defects and OXPHOS deficiencies (16). In that instance, quantitative proteomics on patient fibroblasts showed a specific loss of mtSSU proteins providing a mirror image to that observed here in MRPL39 patients (16). Additionally, several quantitative proteomics studies have recently been published that show characteristic protein signatures in patients with defects in one subunit or assembly factor that impact the stability of interacting proteins in multiprotein complexes. These include the OXPHOS complexes I (54–56) and IV (57), mtLSU (58) and this study, mtSSU (16,17) and the exocyst complex (59). Thus, quantitative proteomics has been shown to be a powerful tool in identifying characteristic ‘multiprotein complex destabilization’ signatures leading to genetic diagnoses. We developed RCA as a means to visualize these signatures (16). RCA quantification of the relative abundance of complexes based on quantitative proteomics data showed sensitivity in detecting combined OXPHOS deficiencies to a similar or greater extent than respiratory chain enzymology (RCE) assays, which is one of the current clinically accredited methods for assessing mitochondrial OXPHOS function. The two severely affected individuals (P1 and P2) presented in this report had no OXPHOS defect detected in patient muscle or liver biopsies using RCE. RCE in P1 fibroblasts did show a substantial but not definitive deficiency of CI and CIV. However, RCA plots from P1 and P2 fibroblasts were able to detect changes in the abundance of not only CI and CIV proteins but also CIII when compared to the RCE, suggesting greater sensitivity of RCA in detecting changes in protein abundance compared to enzyme deficiency from RCE assays. Furthermore, our clinically accredited RCE assay does not assess the activity CV, which was shown to be deficient in P2 fibroblasts using our proteomics-based RCA approach. Similar results were reported for an individual with a specific OXPHOS CI defect detected in fibroblasts by quantitative proteomics but found to be unremarkable using RCE (54). The statistical power of quantitative proteomics, allowing it to detect small changes in protein abundance with high confidence, stems from the detection of multiple peptides from thousands of proteins per cell. This power is even greater when analyzing protein complexes given that the stability of subunits within the 3000 to 4000 complexes found within human cells tends to be highly co-dependent (60,61) and approximately 45% of OMIM disease genes are part of a protein complex (62). A potential limitation of the technique, especially in detecting OXPHOS defects, is the possibility of reduced enzyme activity without a concurrent decrease in enzyme or complex abundance. As such, the technique should complement rather than replace existing approaches. Before it can be widely implemented as a clinical test, a systematic evaluation of proteomics' effectiveness in identifying OXPHOS disorders is necessary.
Transcriptomics is now accepted as a powerful and widely used adjunct in increasing the diagnostic yield of genomic sequencing, whereas quantitative proteomic approaches are not yet widely used in this context (63). Like RNA-seq, proteomics has potential utility in both the detection and interpretation of causal variants. We propose that proteomics and statistical approaches such as RCA analysis will provide a generic integrative approach to prioritize and validate candidate genes identified through genome sequencing and lead to rapid advancement for the diagnosis of hundreds of rare diseases.
This study underscores the importance of employing further investigations in cases where a single deleterious variant in a candidate recessive disease gene has been identified in ‘exome negative’ cases with high clinical suspicion. In particular, we demonstrate the power of harnessing several complementary omics technologies in parallel, such as GS, quantitative proteomics and RNA-seq, to move from variant prioritization and validation to disease gene discovery. Additionally, it highlights that normal muscle or liver OXPHOS RCE does not exclude the presence of a mitochondrial defect in individuals with predominantly neurological clinical presentations.
In conclusion, our study demonstrates that biallelic, deleterious variants in MRPL39 cause clinically heterogenous pediatric onset mitochondrial disease via destabilization of mtLSU proteins.
Materials and Methods
Ethics statement
Samples from probands and family members were obtained after receiving written, informed consent for diagnostic or research investigations from the respective responsible human ethics institutional review boards (P1 under HREC/RCH/34228 and HREC/16/RCHM/150, P2 HREC/16/MH251 and P3 under 3-3013-2462/1 and Rh-2018-20-6158) and research was conducted by the Declaration of Helsinki.
Mitochondrial respiratory chain enzyme assays
Spectrophotometric enzyme assays assessing mitochondrial respiratory chain and citrate synthase activities in cultured fibroblasts, skeletal muscle and liver biopsy were performed as described previously for individuals P1 and P2 (64). For individual P3, enzyme activities were performed in skeletal muscle and cultured skin fibroblasts as described (65).
Genomic sequencing and variant detection
For individual P1, DNA was extracted from skin fibroblasts and exome sequencing (ES) with parallel mtDNA sequencing was performed as published (as for subject 1 in reference) (16,28). FASTQ files were aligned to the UCSC GRCh38/hg38 human reference genome using BWA-MEM and then analyzed using GATK best practice guidelines, HaplotypeCaller, Variant Effect Predictor, and Seqr (66–71). A targeted analysis of 6106 HGNC genes was performed including known mitochondrial disease genes, candidate mitochondrial genes predicted or reported to be associated with mitochondrial function (72) and Mendeliome genes described to underlie disease per PanelApp Australia Mendeliome list v0.2814 (73). The possibility of incidental findings was minimized by excluding genes from the American College of Medical Genetics (ACMG) consensus statement (74).
Blood DNA was extracted, and trio ES testing in tandem with mtDNA-seq (29,30) was performed for individual P2 and her parents at a clinically accredited laboratory (Victorian Clinical Genetics Services, Melbourne, Australia), as previously described (29). Variant prioritization by a multidisciplinary team, including clinical geneticists and medical genomics scientists, was phenotype-driven as previously described (75,76). Research ES reanalysis was later performed as per individual P1 when she remained without an initial molecular diagnosis.
Trio ES was performed on DNA extracted from the blood of individual P3 and his parents at the molecular genetic laboratory at the Department of Genetics, Copenhagen University Hospital Rigshospitalet with variant prioritization restricted to known Mendeliome disease genes and the Mitocarta database of genes predicted to encode mitochondrial proteins (72,77).
PCR-Free whole genome sequencing (GS) was performed for individuals P1 and P2 fibroblast DNA using Illumina TruSeq DNA PCR-Free kit on an Illumina NovaSeq 6000 system with a targeted coverage of 30X (330 million reads). FASTQ files were analyzed using para bricks germline pipeline with reads aligned to the UCSC GRCh38/hg38 human reference genome following GATK4 best practice guidelines with data then analyzed using Seqr (67,71).
Molecular studies
All prioritized variants detected were independently validated by Sanger sequencing (Supplementary Material, Table S2 for details). For cDNA studies, cultured fibroblasts for all three individuals and control lines were grown in DMEM +10% FBS with and without CHX treatment, as described previously (78). Total RNA was extracted using the Illustra RNAspin Mini Isolation Kit (GE Healthcare) and synthesized into cDNA using the SuperScript III First-Strand Synthesis System (ThermoFisher Scientific) as per both manufacturer’s protocols and as described previously (78). To examine mRNA splicing in MRPL39 and MRPL15, PCR primers were designed to amplify the entire ORF from cDNA (Supplementary Material, Table S2 for details). PCR products amplified for P1 and P2 MRPL39 transcripts were cloned into the pCRTM2.1-TOPO® using the TOPO TA cloning kit (ThermoFisher Scientific). The vector was transformed into TOP10 competent cells (ThermoFisher Scientific), and individual colonies were examined and sequenced. All PCR products were analyzed on 1% agarose (Bioline) gels using DNA molecular weight marker VIII (Sigma-Aldrich) or 1 kb Plus DNA Ladder (ThermoFisher Scientific).
Quantitative proteomics
For diagnostic whole-cell proteomics, a total of 50 μg of protein from P1, P2 and three control fibroblasts was used; with P1 and P2 run as technical triplicates and the three independent controls runas singlicates. For proteomics on isolated fibroblast mitochondria used in the topographical heatmap, a total of 50 μg of P1 and P2 (in technical duplicates) and two independent controls (in singlicate) were used. Mitochondria were isolated as previously described (57,79). Protein amounts were estimated by Pierce BCA Assay Kit (ThermoFisher Scientific). Samples were solubilized in 1% (w/v) sodium deoxycholate (SDC; Sigma), 100 mM Tris pH 8.1, 40 mM chloroacetamide (Sigma) and 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP; BondBreaker, ThermoFisher Scientific) for 5 min at 99°C with 1500 rpm shaking followed by 15 min sonication in a water bath sonicator. Proteins were digested with trypsin (ThermoFisher Scientific) at a 1:50 trypsin:protein ratio at 37°C overnight. The supernatant was transferred to stage tips containing 3× 14G plugs of 3MTMEmporeTM SDB-RPS substrate (Sigma Aldrich) as described previously (80,81). Isopropanol 99% (v/v) and 1% (v/v) trifluoroacetic acid (TFA) were added to the tip before centrifugation at 3000 × g at room temperature. Stage tips were washed first with isopropanol (99%) plus TFA (1% v/v) solution and then subjected to an additional wash containing 0.2% (v/v) TFA. Peptides were eluted in 80% (v/v) acetonitrile (ACN) and 1% (w/v) ammonium hydroxide and then acidified to a final concentration of 1% TFA before drying in a CentriVap Benchtop Vacuum Concentrator (Labconco).
Peptides were reconstituted in 0.1% (v/v) TFA and 2% (v/v) ACN for LC–MS/MS analysis. Liquid chromatography (LC) coupled MS/MS was carried out on an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific) for diagnostic whole cell experiments and on an Orbitrap Eclipse for isolated mitochondria, both instruments equipped with a nanoESI interface in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC systems were equipped with an Acclaim Pepmap nano-trap column (Dionex-C18, 100 Å, 75 μm × 2 cm) and an Acclaim Pepmap RSLC analytical column (Dionex-C18, 100 Å, 75 μm × 50 cm). The tryptic peptides were injected into the trap column at an isocratic flow of 5 μL/min (Lumos) or 6 μL/min (Eclipse) of 2% (v/v) ACN containing 0.1% (v/v) formic acid for 5 min applied before the trap column was switched in-line with the analytical column. The eluents were 5% DMSO in 0.1% (v/v) formic acid (solvent A) and 5% DMSO in 100% (v/v) ACN and 0.1% (v/v) formic acid (solvent B). The flow gradient was (i) 0–6 min at 3% B, (ii) 6–95 min, 3–23% B, (iii) 95–105 min, 23–40% B, (iv) 105–110 min, 40–80% B, (v) 110–115 min, 80% B, (vi) 115–115 min ramp followed by, 80–3% and equilibrated at 3% B for 10 min before the next sample injection. The mass spectrometer was operated in positive-ionization mode with spray voltage set at 1.9 kV (Lumos) or 2.0 kV (Eclipse) and source temperature at 275°C. The mass spectrometers were operated in the data-dependent acquisition mode with MS spectra scanning from m/z 300–1600 (Lumos) or 375–1500 (Eclipse) at 120 000 resolution with an AGC target of 4e5. The ‘top speed’ acquisition method mode (3 s cycle time) on the most intense precursor was used whereby peptide ions with charge states ≥2–5 (Lumos) or ≥ 2–7 (Eclipse) were isolated with an isolation window of 1.6 m/z and fragmented with high energy collision (HCD) mode with a stepped collision energy of 35 ± 5% (Lumos) or 30 ± 5% (Eclipse). Fragment ion spectra were acquired in the Orbitraps at 15 000 resolution. Dynamic exclusion was activated for the 30 s.
Raw files were processed using the MaxQuant platform (v. 1.6.10.43) (82) and searched against UniProt human database (42 434 entries, June 2019) using default settings for a label-free quantification (LFQ) experiment with the following modifications: deamination (NQ) added as variable modification, ‘match between runs’ enabled with default settings, and ‘LFQ min. ratio count’ and ‘label min. ratio count’ set to 1. The proteinGroups.txt output from the search was processed in Perseus (v. 1.6.14.0) (83). Positive entries in ‘Only identified by site’, ‘Reverse’ and ‘Potential contaminant’ were removed before analysis. Log2-transformed LFQ intensities were grouped in control or patient groups and filtered to have at least 2 valid values in each group. Two-sample t-tests were performed between groups using P-value for truncation (threshold P-value = 0.05). Volcano plots were generated using a built-in scatter plot function with a P-value threshold set to 0.05 equivalent (−log10 = 1.301 and fold-change (difference) set to ±1.5 equivalent (log2 ± 0.585). For RCA, the difference between the patient and controls for each subunit was calculated with an R script. The mean and standard deviation were calculated for each protein within the complex, along with the 95% confidence interval based on the t-statistic for the complex. A paired t-test then calculated the significance (P-value) between the control and patient for each complex. For topographical mapping, we utilized data derived from isolated mitochondria but treated as above, with the differences in mean Log2 LFQ intensities between patient (P1 or P2) and controls (n = 2) plotted onto the mitoribosome structure (PDB id: 3J9M) (6) using a python script as previously described (80).
RNA sequencing (RNA-seq)
RNA was extracted from P1 and P2 fibroblasts using the miRNeasy Mini Kit (Qiagen), and RNA was treated with RNase-Free DNase (Qiagen). RNA quality and quantity were tested using TapeStation RNA ScreenTape analysis (Agilent) and Qubit RNA HS (ThermoFisher), respectively. P1 and P2 underwent RNAseq within a group of 21 undiagnosed patients with available fibroblast lines; included samples had an RNA integrity number of 9.3–10.0. The Kapa Biosystems mRNA Hyper Prep Kit (Roche), a strand-specific poly(A) mRNA capture protocol, was used to generate sequencing libraries from 500 ng RNA. Paired-end sequencing was performed at the Yale Center for Genome Analysis on an Illumina instrument with sequencing coverage of 50–100 million reads.
The Sequence Read Archive (SRA) toolkit was used to download GTEx data from the Database of Genotypes and Phenotypes. Given power analyses had shown that 50–60 samples were enough to enable the identification of rare outlier expression events (84), 100 fibroblast RNAseq samples, approximately 50:50 male and female were downloaded as SRA files. Fastq-dump was used to convert the SRA files to FASTQ format.
The analysis pipeline used for GTEx and TOPMed was implemented to process the patient and GTEx control RNAseq data (https://github.com/broadinstitute/gtex-pipeline). In brief, both patient and GTEx FASTQ files were aligned to hg38 human reference genome with GENCODE v26 annotations using STAR (v2.5.3a), with twopassMode = ‘Basic’ to enable detection of novel splice junctions. Picard (v2.9) was used to mark duplicate reads. Quality metrics were generated from STAR-aligned bam files by RNA-SeQC (v2.3.4), and PCA plots of gene expression were produced using gene coverage values calculated by RNA-SeQC.
PCA was used to identify outliers among GTEx control samples. These outliers were removed via the following filtering steps: (a) >45 million QC-passed reads, (b) > 18 000 genes detected and (c) manual identification of outliers on the PCA plot. This resulted in the removal of 10 GTEx samples, leaving 45 female and 45 male control RNA samples derived from fibroblasts.
Outlier expression, aberrant splicing, and mono-allelic expression were detected using the DROP (Detection of RNA Outliers Pipeline) pipeline v0.9.0 (https://github.com/gagneurlab/drop), with default settings for hg38. For the aberrant expression and splicing modules, the RNA STAR-aligned bam files from patient and GTEx control samples were provided as input. The mono-allelic expression module was performed using the RNA sequencing BAM files and corresponding exome VCF data for patients as input. For the mono-allelic expression analysis, a VCF file that only included autosomal variants not in linkage disequilibrium was also generated using SNPRelate and SeqArray R packages. The output data of all three modules was subsetted to the 6106 HGNC genes on the targeted gene list used in the previous ES analysis.
MRPL39 complementation
Lentiviral transduction of fibroblasts from all three patients as well as a control was performed using the MRPL39 open reading frame (ORF) (NM_017446.4) cloned into the lentiviral vector pF-5xUAS-MCS-SV40-puroGal4ERT2VP16 (GEV16)-W as previously described (78), following 21 days of selection, cells were harvested for SDS-PAGE Immunoblotting.
Immunoblotting
Whole-cell lysates were prepared from cultured fibroblasts grown in DMEM +10% FBS for P1, P2, P3 and controls. Cell pellets were resuspended in extraction buffer (1.5% n-Dodecyl-β-d-maltopyranoside, 25 mM HEPES and 100 mM NaCl), left on ice for 20 min and then centrifuged at 16000 rpm for 20 min at 4°C. Protein concentrations of supernatants were measured by BCA analysis (85), before further solubilized in sodium dodecyl sulfate (SDS)/glycerol solubilization buffer (125 mM Tris pH 8.8, 40% glycerol, 4% SDS, 100 mM DTT, 0.01% Bromophenol blue, protease inhibitor cocktail) before being analyzed by SDS-PAGE as described previously (78). SDS-PAGE gels were transferred onto polyvinylidene difluoride membrane and probed with primary antibodies raised against 5 mitoribosomal large subunit proteins, MRPL39 (1:10 000; Proteintech; 28 165-1-AP), MRPL44 (1:1000; Proteintech; 16 394-1-AP), MRPL9 (1:2400; Proteintech; 15 342-1-AP), MRPL45 (1:1000; Proteintech; 15 682-1-AP), MRPL28 (1:1000; Proteintech; 21 604-1-AP) plus 5 mitoribosomal small subunit proteins, MRPS34 (1:500; Sigma-Aldrich; HPA042112), MRPS17 (1:1000; Proteintech; 18 881-1-AP), MRPS7 (1:10 000; Proteintech; 26 828-1-AP), MRPS35 (1:500; Proteintech; 16 457-1-AP), MRPS16 (1:1000; Proteintech; 16 735-1-AP), as well as Total OXPHOS Human WB Antibody Cocktail (1:500; Abcam; ab110411). Primary antibodies raised against VDAC1 (1:10 000; MitoSciences; MSA05) and complex II subunit, SDHA (1:1000: Molecular Probes; A-11 142) were used as loading controls for mitochondrial protein content. Blots were incubated with anti-mouse or anti-rabbit IgG secondary antibodies (VWR International) and developed with Clarity Western ECL Substrate (Bio-Rad Laboratories) and visualized either using Hyperfilm ECL (GE Healthcare) or a ChemiDoc Imaging System (Bio-Rad Laboratories). Where quantification was required, relative band intensities were quantitated using Bio-Rad Image Lab software (version 6.1.0 build 7), followed by a two-way repeated-measures analysis of variance for comparisons of groups followed by post hoc analysis by the Bonferroni method to determine statistically significant differences.
Supplementary Material
Acknowledgements
This research was supported by grants and fellowships from the Australian National Health and Medical Research Council (1140851, 1164479, 1159456, 1155244, 2009732 plus 111353 to the Australian Genomics Health Alliance), plus grants from the US Department of Defense Congressionally Directed Medical Research Programs PR170396, the Australian Mito Foundation, the Vincent Chiodo Charitable Trust and the Victorian Government’s Operational Infrastructure Support Program. The Chair in Genomic Medicine awarded to JC is generously supported by The Royal Children’s Hospital Foundation. We acknowledge the Bio21 Mass Spectrometry and Proteomics Facility (MMSPF) for the provision of instrumentation, training, and technical support. The Yale Center for Mendelian Genomics (NIH M#UM1HG006504-05) is funded by the National Human Genome Research Institute and the National Heart, Lung, and Blood Institute. The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DHH is supported by a Melbourne International Research Scholarship and the Mito Foundation PhD Top-up Scholarship. VKM is an Investigator at the Howard Hughes Medical Institute.
Conflict of Interest statement. VKM is on the scientific advisory board of Janssen Pharmaceuticals and 5AM Ventures.
Contributor Information
Sumudu S C Amarasekera, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia.
Daniella H Hock, Department of Biochemistry and Pharmacology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, VIC 3010, Australia.
Nicole J Lake, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Genetics, Yale School of Medicine, New Haven, CT 06510 USA.
Sarah E Calvo, Broad Institute, Cambridge, MA 02142, USA; Howard Hughes Medical Institute and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Systems Biology, Harvard Medical School, Boston, MA 02446, USA.
Sabine W Grønborg, Department of Genetics, Copenhagen University Hospital Rigshospitalet, Copenhagen 2100, Denmark; Center for Inherited Metabolic Disease, Department of Pediatrics and Adolescent Medicine, Copenhagen University Hospital Rigshospitalet, Copenhagen 2100, Denmark.
Emma I Krzesinski, Monash Genetics, Monash Health, Melbourne, VIC 3168 Australia; Department of Paediatrics, Monash University, Melbourne, VIC 3168 Australia.
David J Amor, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia.
Michael C Fahey, Monash Genetics, Monash Health, Melbourne, VIC 3168 Australia; Department of Paediatrics, Monash University, Melbourne, VIC 3168 Australia.
Cas Simons, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia.
Flemming Wibrand, Department of Genetics, Copenhagen University Hospital Rigshospitalet, Copenhagen 2100, Denmark.
Vamsi K Mootha, Broad Institute, Cambridge, MA 02142, USA; Howard Hughes Medical Institute and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Systems Biology, Harvard Medical School, Boston, MA 02446, USA.
Monkol Lek, Department of Genetics, Yale School of Medicine, New Haven, CT 06510 USA.
Sebastian Lunke, Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia; Australian Genomics Health Alliance, Melbourne, VIC 3052, Australia; Department of Pathology, University of Melbourne, Melbourne, VIC 3010, Australia.
Zornitza Stark, Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia; Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia; Australian Genomics Health Alliance, Melbourne, VIC 3052, Australia.
Elsebet Østergaard, Department of Genetics, Copenhagen University Hospital Rigshospitalet, Copenhagen 2100, Denmark; Department of Clinical Medicine, University of Copenhagen, Copenhagen 2200, Denmark.
John Christodoulou, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia; Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia; Australian Genomics Health Alliance, Melbourne, VIC 3052, Australia; Discipline of Child & Adolescent Health, Sydney Medical School, University of Sydney, Sydney, NSW 2006, Australia.
David R Thorburn, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia; Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia; Australian Genomics Health Alliance, Melbourne, VIC 3052, Australia.
David A Stroud, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Biochemistry and Pharmacology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, VIC 3010, Australia; Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia.
Alison G Compton, Murdoch Children’s Research Institute, Royal Children's Hospital, Melbourne, VIC 3052, Australia; Department of Paediatrics, University of Melbourne, Melbourne, VIC 3010, Australia; Victorian Clinical Genetics Services, Murdoch Children’s Research Institute, Melbourne, VIC 3052, Australia.
Data and Code Availability
The ES, GS and full proteomics datasets have not been deposited in a public database because of privacy and ethical limitations but may be made available upon reasonable request.
Web Resources
Seqr, https://seqr.broadinstitute.org/
UCSC Browser, https://genome.ucsc.edu/
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
GeneMatcher, https://www.genematcher.org
OMIM, https://www.omim.org/
Genomizer, https://genomizer.com
ClinVar Submission Number
Variants identified in this work are listed in ClinVar under accession numbers VCV001676671.1 (MRPL15 c.602C>T), VCV001676672.1 (MRPL39 c.921+5G>A), VCV001676673.2 (MRPL39 c.526delT) and VCV001676674.2 (MRPL39 c.589-924G>A) and VCV001525985.1 (MRPL39 c.896G>T).
References
- 1. Frazier, A.E., Thorburn, D.R. and Compton, A.G. (2019) Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J. Biol. Chem., 294, 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tang, J.X., Thompson, K., Taylor, R.W. and Oláhová, M. (2020) Mitochondrial OXPHOS biogenesis: co-regulation of protein synthesis, import, and assembly pathways. Int. J. Mol. Sci., 21, 3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itoh, Y., Andréll, J., Choi, A., Richter, U., Maiti, P., Best, R.B., Barrientos, A., Battersby, B.J. and Amunts, A. (2021) Mechanism of membrane-tethered mitochondrial protein synthesis. Science, 371, 846–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown, A., Amunts, A., Bai, X.-C., Sugimoto, Y., Edwards, P.C., Murshudov, G., Scheres, S.H.W. and Ramakrishnan, V. (2014) Structure of the large ribosomal subunit from human mitochondria. Science, 346, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greber, B.J., Boehringer, D., Leitner, A., Bieri, P., Voigts-Hoffmann, F., Erzberger, J.P., Leibundgut, M., Aebersold, R. and Ban, N. (2014) Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature, 505, 515–519. [DOI] [PubMed] [Google Scholar]
- 6. Amunts, A., Brown, A., Toots, J., Scheres, S.H.W. and Ramakrishnan, V. (2015) The structure of the human mitochondrial ribosome. Science, 348, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bieri, P., Greber, B.J. and Ban, N. (2018) High-resolution structures of mitochondrial ribosomes and their functional implications. Curr. Opin. Struct. Biol., 49, 44–53. [DOI] [PubMed] [Google Scholar]
- 8. Gardeitchik, T., Mohamed, M., Ruzzenente, B., Karall, D., Guerrero-Castillo, S., Dalloyaux, D., van den Brand, M., van Kraaij, S., van Asbeck, E., Assouline, Z. et al. (2018) Bi-allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am. J. Hum. Genet., 102, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Menezes, M.J., Guo, Y., Zhang, J., Riley, L.G., Cooper, S.T., Thorburn, D.R., Li, J., Dong, D., Li, Z., Glessner, J. et al. (2015) Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum. Mol. Genet., 24, 2297–2307. [DOI] [PubMed] [Google Scholar]
- 10. Jackson, C.B., Huemer, M., Bolognini, R., Martin, F., Szinnai, G., Donner, B.C., Richter, U., Battersby, B.J., Nuoffer, J.M., Suomalainen, A. and Schaller, A. (2019) A variant in MRPS14 (uS14m) causes perinatal hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features and neurological involvement. Hum. Mol. Genet., 28, 639–649. [DOI] [PubMed] [Google Scholar]
- 11. Miller, C., Saada, A., Shaul, N., Shabtai, N., Ben-Shalom, E., Shaag, A., Hershkovitz, E. and Elpeleg, O. (2004) Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol., 56, 734–738. [DOI] [PubMed] [Google Scholar]
- 12. Saada, A., Shaag, A., Arnon, S., Dolfin, T., Miller, C., Fuchs-Telem, D., Lombes, A. and Elpeleg, O. (2007) Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet., 44, 784–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kohda, M., Tokuzawa, Y., Kishita, Y., Nyuzuki, H., Moriyama, Y., Mizuno, Y., Hirata, T., Yatsuka, Y., Yamashita-Sugahara, Y., Nakachi, Y. et al. (2016) A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet., 12, e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bugiardini, E., Mitchell, A.L., Rosa, I.D., Horning-Do, H.T., Pitmann, A., Poole, O.V., Holton, J.L., Shah, S., Woodward, C., Hargreaves, I. et al. (2019) MRPS25 mutations impair mitochondrial translation and cause encephalomyopathy. Hum. Mol. Genet., 28, 2711–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pulman, J., Ruzzenente, B., Bianchi, L., Rio, M., Boddaert, N., Munnich, A., Rotig, A. and Metodiev, M.D. (2019) Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum. Mol. Genet., 28, 1445–1462. [DOI] [PubMed] [Google Scholar]
- 16. Lake, N.J., Webb, B.D., Stroud, D.A., Richman, T.R., Ruzzenente, B., Compton, A.G., Mountford, H.S., Pulman, J., Zangarelli, C., Rio, M. et al. (2017) Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and Leigh syndrome. Am. J. Hum. Genet., 101, 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borna, N.N., Kishita, Y., Kohda, M., Lim, S.C., Shimura, M., Wu, Y., Mogushi, K., Yatsuka, Y., Harashima, H., Hisatomi, Y. et al. (2019) Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics, 20, 9–25. [DOI] [PubMed] [Google Scholar]
- 18. Galmiche, L., Serre, V., Beinat, M., Assouline, Z., Lebre, A.S., Chretien, D., Nietschke, P., Benes, V., Boddaert, N., Sidi, D. et al. (2011) Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat., 32, 1225–1231. [DOI] [PubMed] [Google Scholar]
- 19. Serre, V., Rozanska, A., Beinat, M., Chretien, D., Boddaert, N., Munnich, A., Rotig, A. and Chrzanowska-Lightowlers, Z.M. (2013) Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim. Biophys. Acta, 1832, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nottia, M.D., Marchese, M., Verrigni, D., Mutti, C., Torraco, A., Oliva, R., Fernandez-Vizarra, E., Morani, F., Trani, G., Rizza, T. et al. (2020) A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol. Dis., 141, 104880. [DOI] [PubMed] [Google Scholar]
- 21. Carroll, C.J., Isohanni, P., Poyhonen, R., Euro, L., Richter, U., Brilhante, V., Gotz, A., Lahtinen, T., Paetau, A., Pihko, H. et al. (2013) Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet., 50, 151–159. [DOI] [PubMed] [Google Scholar]
- 22. Ferrari, A., Del'Olio, S. and Barrientos, A. (2021) The diseased mitoribosome. FEBS Lett., 595, 1025–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gorman, G.S., Chinnery, P.F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., Suomalainen, A., Thorburn, D.R., Zeviani, M. and Turnbull, D.M. (2016) Mitochondrial diseases. Nat. Rev. Dis. Primers., 2, 16080. [DOI] [PubMed] [Google Scholar]
- 24. Friederich, M.W., Geddes, G.C., Wortmann, S.B., Punnoose, A., Wartchow, E., Knight, K.M., Prokisch, H., Creadon-Swindell, G., Mayr, J.A. and Van Hove, J.L.K. (2021) Pathogenic variants in MRPL44 cause infantile cardiomyopathy due to a mitochondrial translation defect. Mol. Genet. Metab., 133, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen, A., Tiosano, D., Guran, T., Baris, H.N., Bayram, Y., Mory, A., Shapiro-Kulnane, L., Hodges, C.A., Akdemir, Z.C., Turan, S. et al. (2018) Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet., 27, 1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rahman, S., Thorburn, D. (2015) [Updated 2020 Jul 16] Nuclear Gene-Encoded LEIGH Syndrome Spectrum Overview. In Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M. and Amemiya, A. (Eds.), In GeneReviews(®)[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK320989/ [PubMed] [Google Scholar]
- 27. Rahman, S., Blok, R.B., Dahl, H.H., Danks, D.M., Kirby, D.M., Chow, C.W., Christodoulou, J. and Thorburn, D.R. (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol., 39, 343–351. [DOI] [PubMed] [Google Scholar]
- 28. Lake, N.J., Formosa, L.E., Stroud, D.A., Ryan, M.T., Calvo, S.E., Mootha, V.K., Morar, B., Procopis, P.G., Christodoulou, J., Compton, A.G. et al. (2019) A patient with homozygous nonsense variants in two Leigh syndrome disease genes: distinguishing a dual diagnosis from a hypomorphic protein-truncating variant. Hum. Mutat., 40, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akesson, L.S., Eggers, S., Love, C.J., Chong, B., Krzesinski, E.I., Brown, N.J., Tan, T.Y., Richmond, C.M., Thorburn, D.R., Christodoulou, J. et al. (2019) Early diagnosis of Pearson syndrome in neonatal intensive care following rapid mitochondrial genome sequencing in tandem with exome sequencing. Eur. J. Hum. Genet., 27, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rius, R., Compton, A.G., Baker, N.L., Welch, A.E., Coman, D., Kava, M.P., Minoche, A.E., Cowley, M.J., Thorburn, D.R. and Christodoulou, J. (2021) Application of genome sequencing from blood to diagnose mitochondrial diseases. Genes, 12, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lunke, S., Eggers, S., Wilson, M., Patel, C., Barnett, C.P., Pinner, J., Sandaradura, S.A., Buckley, M.F., Krzesinski, E.I., de Silva, M.G. et al. (2020) Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA, 323, 2503–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M. et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–d894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ionita-Laza, I., McCallum, K., Xu, B. and Buxbaum, J.D. (2016) A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet., 48, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ioannidis, N.M., Rothstein, J.H., Pejaver, V., Middha, S., McDonnell, S.K., Baheti, S., Musolf, A., Li, Q., Holzinger, E., Karyadi, D. et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B. et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 414–416. [DOI] [PubMed] [Google Scholar]
- 38. Ng, P.C. and Henikoff, S. (2001) Predicting deleterious amino acid substitutions. Genome Res., 11, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwarz, J.M., Cooper, D.N., Schuelke, M. and Seelow, D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 40. Yeo, G. and Burge, C.B. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol., 11, 377–394. [DOI] [PubMed] [Google Scholar]
- 41. Lindeboom, R.G., Supek, F. and Lehner, B. (2016) The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet., 48, 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reese, M.G., Eeckman, F.H., Kulp, D. and Haussler, D. (1997) Improved splice site detection in Genie. J. Comput. Biol., 4, 311–323. [DOI] [PubMed] [Google Scholar]
- 43. Yépez, V.A., Mertes, C., Müller, M.F., Klaproth-Andrade, D., Wachutka, L., Frésard, L., Gusic, M., Scheller, I.F., Goldberg, P.F., Prokisch, H. et al. (2021) Detection of aberrant gene expression events in RNA sequencing data. Nat. Protoc., 16, 1276–1296. [DOI] [PubMed] [Google Scholar]
- 44. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grantham, R. (1974) Amino acid difference formula to help explain protein evolution. Science, 185, 862–864. [DOI] [PubMed] [Google Scholar]
- 46. Jones, D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol., 292, 195–202. [DOI] [PubMed] [Google Scholar]
- 47. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spirina, O., Bykhovskaya, Y., Kajava, A.V., O'Brien, T.W., Nierlich, D.P., Mougey, E.B., Sylvester, J.E., Graack, H.R., Wittmann-Liebold, B. and Fischel-Ghodsian, N. (2000) Heart-specific splice-variant of a human mitochondrial ribosomal protein (mRNA processing; tissue specific splicing). Gene, 261, 229–234. [DOI] [PubMed] [Google Scholar]
- 49. Greber, B.J. and Ban, N. (2016) Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem., 85, 103–132. [DOI] [PubMed] [Google Scholar]
- 50. Greber, B.J., Boehringer, D., Leibundgut, M., Bieri, P., Leitner, A., Schmitz, N., Aebersold, R. and Ban, N. (2014) The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature, 515, 283–286. [DOI] [PubMed] [Google Scholar]
- 51. Bogenhagen, D.F., Ostermeyer-Fay, A.G., Haley, J.D. and Garcia-Diaz, M. (2018) Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep., 22, 1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hock, D.H., Robinson, D.R.L. and Stroud, D.A. (2020) Blackout in the powerhouse: clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochemist, 477, 4085–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pfeffer, S., Woellhaf, M.W., Herrmann, J.M. and Förster, F. (2015) Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nat. Commun., 6, 6019. [DOI] [PubMed] [Google Scholar]
- 54. Helman, G., Compton, A.G., Hock, D.H., Walkiewicz, M., Brett, G.R., Pais, L., Tan, T.Y., De Paoli-Iseppi, R., Clark, M.B., Christodoulou, J. et al. (2021) Multiomic analysis elucidates complex I deficiency caused by a deep intronic variant in NDUFB10. Hum. Mutat., 42, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kremer, L.S., Bader, D.M., Mertes, C., Kopajtich, R., Pichler, G., Iuso, A., Haack, T.B., Graf, E., Schwarzmayr, T., Terrile, C. et al. (2017) Genetic diagnosis of mendelian disorders via RNA sequencing. Nat. Commun., 8, 15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumar, R., Corbett, M.A., Smith, N.J.C., Hock, D.H., Kikhtyak, Z., Semcesen, L.N., Morimoto, A., Lee, S., Stroud, D.A., Gleeson, J.G., Haan, E.A. and Gecz, J. (2022) Oligonucleotide correction of an intronic TIMMDC1 variant in cells of patients with severe neurodegenerative disorder. NPJ Genom. Med., 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hock, D.H., Reljic, B., Ang, C.S., Muellner-Wong, L., Mountford, H.S., Compton, A.G., Ryan, M.T., Thorburn, D.R. and Stroud, D.A. (2020) HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV. Mol. Cell. Proteomics, 19, 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Van Bergen, N.J., Hock, D.H., Spencer, L., Massey, S., Stait, T., Stark, Z., Lunke, S., Roesley, A., Peters, H., Lee, J.Y. et al. (2022) Biallelic variants in PYROXD2 cause a severe infantile metabolic disorder affecting mitochondrial function. Int. J. Mol. Sci., 23, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van Bergen, N.J., Ahmed, S.M., Collins, F., Cowley, M., Vetro, A., Dale, R.C., Hock, D.H., de Caestecker, C., Menezes, M., Massey, S. et al. (2020) Mutations in the exocyst component EXOC2 cause severe defects in human brain development. J. Exp. Med., 217, e20192040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Giurgiu, M., Reinhard, J., Brauner, B., Dunger-Kaltenbach, I., Fobo, G., Frishman, G., Montrone, C. and Ruepp, A. (2019) CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res., 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Drew, K., Lee, C., Huizar, R.L., Tu, F., Borgeson, B., McWhite, C.D., Ma, Y., Wallingford, J.B. and Marcotte, E.M. (2017) Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol., 13, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alston, C.L., Stenton, S.L., Hudson, G., Prokisch, H. and Taylor, R.W. (2021) The genetics of mitochondrial disease: dissecting mitochondrial pathology using multi-omic pipelines. J. Pathol., 254, 430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marwaha, S., Knowles, J.W. and Ashley, E.A. (2022) A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med., 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Frazier, A.E. and Thorburn, D.R. (2012) Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol. Biol., 837, 49–62. [DOI] [PubMed] [Google Scholar]
- 65. Wibrand, F., Jeppesen, T.D., Frederiksen, A.L., Olsen, D.B., Duno, M., Schwartz, M. and Vissing, J. (2010) Limited diagnostic value of enzyme analysis in patients with mitochondrial tRNA mutations. Muscle Nerve, 41, 607–613. [DOI] [PubMed] [Google Scholar]
- 66. Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Van der Auwera, G.A., Carneiro, M.O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., Jordan, T., Shakir, K., Roazen, D., Thibault, J. et al. (2013) From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M. et al. (2010) The genome analysis ToolKit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. DePristo, M.A., Banks, E., Poplin, R., Garimella, K.V., Maguire, J.R., Hartl, C., Philippakis, A.A., del Angel, G., Rivas, M.A., Hanna, M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McLaren, W., Gil, L., Hunt, S.E., Riat, H.S., Ritchie, G.R., Thormann, A., Flicek, P. and Cunningham, F. (2016) The Ensembl variant effect predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pais, L.S., Snow, H., Weisburd, B., Zhang, S., Baxter, S.M., DiTroia, S., O'Heir, E., England, E., Chao, K.R., Lemire, G. et al. (2022) seqr: a web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat., 43, 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rath, S., Sharma, R., Gupta, R., Ast, T., Chan, C., Durham, T.J., Goodman, R.P., Grabarek, Z., Haas, M.E., Hung, W.H.W. et al. (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res., 49, D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stark, Z., Foulger, R.E., Williams, E., Thompson, B.A., Patel, C., Lunke, S., Snow, C., Leong, I.U.S., Puzriakova, A., Daugherty, L.C. et al. (2021) Scaling national and international improvement in virtual gene panel curation via a collaborative approach to discordance resolution. Am. J. Hum. Genet., 108, 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stark, Z., Dashnow, H., Lunke, S., Tan, T.Y., Yeung, A., Sadedin, S., Thorne, N., Macciocca, I., Gaff, C., Melbourne Genomics Health, A et al. (2017) A clinically driven variant prioritization framework outperforms purely computational approaches for the diagnostic analysis of singleton WES data. Eur. J. Hum. Genet., 25, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stark, Z., Tan, T.Y., Chong, B., Brett, G.R., Yap, P., Walsh, M., Yeung, A., Peters, H., Mordaunt, D., Cowie, S. et al. (2016) A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med., 18, 1090–1096. [DOI] [PubMed] [Google Scholar]
- 77. Gronborg, S., Risom, L., Ek, J., Larsen, K.B., Scheie, D., Petkov, Y., Larsen, V.A., Duno, M., Joensen, F. and Ostergaard, E. (2018) A Faroese founder variant in TBCD causes early onset, progressive encephalopathy with a homogenous clinical course. Eur. J. Hum. Genet., 26, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Calvo, S.E., Compton, A.G., Hershman, S.G., Lim, S.C., Lieber, D.S., Tucker, E.J., Laskowski, A., Garone, C., Liu, S., Jaffe, D.B. et al. (2012) Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med., 4, 118ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Acin-Perez, R., Fernandez-Silva, P., Peleato, M.L., Perez-Martos, A. and Enriquez, J.A. (2008) Respiratory active mitochondrial supercomplexes. Mol. Cell, 32, 529–539. [DOI] [PubMed] [Google Scholar]
- 80. Stroud, D.A., Surgenor, E.E., Formosa, L.E., Reljic, B., Frazier, A.E., Dibley, M.G., Osellame, L.D., Stait, T., Beilharz, T.H., Thorburn, D.R. et al. (2016) Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature, 538, 123–126. [DOI] [PubMed] [Google Scholar]
- 81. Kulak, N.A., Pichler, G., Paron, I., Nagaraj, N. and Mann, M. (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods, 11, 319–324. [DOI] [PubMed] [Google Scholar]
- 82. Cox, J. and Mann, M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 83. Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M.Y., Geiger, T., Mann, M. and Cox, J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods, 13, 731–740. [DOI] [PubMed] [Google Scholar]
- 84. Brechtmann, F., Mertes, C., Matuseviciute, A., Yepez, V.A., Avsec, Z., Herzog, M., Bader, D.M., Prokisch, H. and Gagneur, J. (2018) OUTRIDER: a statistical method for detecting aberrantly expressed genes in RNA sequencing data. Am. J. Hum. Genet., 103, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Smith, P.K., Krohn, R.I., Hermanson, G.T., Mallia, A.K., Gartner, F.H., Provenzano, M.D., Fujimoto, E.K., Goeke, N.M., Olson, B.J. and Klenk, D.C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem., 150, 76–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ES, GS and full proteomics datasets have not been deposited in a public database because of privacy and ethical limitations but may be made available upon reasonable request.