

Abstract
NLRP3 inflammasome activation is a highly regulated process for controlling secretion of the potent inflammatory cytokines IL-1β and IL-18 that are essential during bacterial infection, sterile inflammation, and disease including colitis, diabetes, Alzheimer’s disease, and atherosclerosis. Diverse stimuli activate the NLRP3 inflammasome and unifying upstream signals have been challenging to identify. Here, we report that a common upstream step in NLRP3 inflammasome activation is the dissociation of the glycolytic enzyme hexokinase 2 from the voltage-dependent anion channel (VDAC) in the outer membrane of mitochondria. Hexokinase 2 dissociation from VDAC triggers activation of inositol triphosphate receptors leading to release of calcium from the ER, which is taken up by mitochondria. This influx of calcium into mitochondria leads to oligomerization of VDAC, which is known to form a macromolecule size pore in the outer membrane of mitochondria that allows proteins and mitochondrial DNA (mtDNA), often associated with apoptosis and inflammation respectively, to exit the mitochondria. We observe that VDAC oligomers aggregate with NLRP3 during initial assembly of the multiprotein oligomeric NLRP3 inflammasome complex. We also find that mtDNA is necessary for NLRP3 association with VDAC oligomers. These data, together with other recent work, help to paint a more complete picture of the pathway leading to NLRP3 inflammasome activation.
One Sentence Summary:
Hexokinase release from mitochondria leads to formation of VDAC oligomers that recruit NLRP3 and initiates inflammasome assembly.
INTRODUCTION
Activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome in macrophages to stimulate processing and secretion of the potent inflammatory cytokine IL-1β is a key process in innate sensing of bacterial pathogens (1, 2) and is a significant process in diverse inflammatory diseases including diabetes (3), Alzheimer’s disease (4, 5), atherosclerosis (6), and colitis (7–10). We have previously observed that degradation of bacterial cell wall peptidoglycan inside phagosomes is an essential step in activating the NLRP3 inflammasome following phagocytosis of gram-positive bacteria (11). Further, we have observed that when bacterial cell wall peptidoglycan is degraded in phagolysosomes, large quantities of the sugar N-acetylglucosamine are liberated and inhibit hexokinase, causing dissociation of the enzyme from outer membranes of mitochondria. We also found that dissociation of hexokinase from mitochondria was sufficient to trigger NLRP3 inflammasome assembly and activation (12). However, how hexokinase release from mitochondria subsequently triggers NLRP3 activation is not known.
NLRP3 activation involves assembly of a large oligomeric protein complex including NLRP3, apoptosis-associated speck-like protein (ASC), NIMA-related kinase 7 (NEK7), and caspase-1 (1, 13). Caspase-1 is cleaved into its active form in this complex and subsequently processes proteins including IL-1β, IL-18, and gasdermin D into their active forms. Strong acute activation of this pathway by common experimental NLRP3 activators such as extracellular ATP, nigericin, or silica particles requires potassium efflux from the cytosol and results in a gasdermin-dependent inflammatory form of cell death called pyroptosis (1, 13). However, not all NLRP3 activators are associated with pyroptosis or potassium efflux, and we have observed that NLRP3 activation by gram-positive bacterial peptidoglycan does not require potassium efflux and is not associated with cell death (12). Similarly, Gross et al. observed that small molecules that inhibit mitochondrial oxidoreductases and activate the NLRP3 inflammasome do so in a manner that does not require potassium efflux (14), while Hornung and colleagues have noted that potassium efflux and cell death are not associated with LPS-induced inflammasome activation in human monocytes (15). Also, Pearlman and colleagues have noted that activation of NLRP3 in neutrophils leads to gasdermin D activation but is not necessarily associated with cell death (16). Calcium movement has also been implicated in NLRP3 activation, with influx of extracellular Ca2+ and release of ER-linked Ca2+ stores being implicated (17). Again, different studies have come to different conclusions as to whether the role of Ca2+ is universally required for NLRP3 activation (13). Despite the extensive work done on the mechanism of NLRP3 inflammasome activation, a unifying model of upstream features of NLRP3 activation has been elusive.
Compelling evidence implicates a role for mitochondria in regulation of the NLRP3 inflammasome. Mitochondrial dysfunction (18), reactive oxygen, release of mitochondrial DNA (mtDNA) (19–22), exposure of cardiolipin on mitochondrial outer membranes (23–25), and viral activation of NLRP3 via sensing by mitochondrial antiviral signaling protein (MAVS) (26, 27) have all been linked to NLRP3 inflammasome activation. Further, mitochondria have been proposed as a site of initial inflammasome nucleation (28). Activating signals have been reported to drive relocation of NLRP3, in the cytoplasm and associated with endoplasmic reticulum (ER) membranes, to both mitochondria and mitochondria-endoplasmic reticulum (ER) contact sites called MAMs (18, 29). However, the details of how this might operate are unclear.
In our previous study, we found a role for the enzyme hexokinase (HK) in NLRP3 activation in response to bacterial peptidoglycan (12). HK is the first enzyme of the glycolytic pathway, and it converts glucose to glucose 6-phosphate. On the mitochondrial outer membrane, HK binds to the voltage-dependent anion channel (VDAC), a channel that, together with the adenine nucleotide translocase (ANT) in the inner mitochondrial membrane, regulates flow of metabolites, ions, and nucleotides in and out of the organelle (30). HK binding to VDAC is thought to provide it with preferential access to adenosine triphosphates (ATPs) transported by VDAC and ANT out of the mitochondria. The interaction of HK with VDAC modulates cellular metabolism, with the bound form favoring oxidative phosphorylation and the released form favoring the funneling of glucose-6-phosphate more into glycolysis leading to lactate production and the pentose phosphate pathway (31–34). Previous studies have suggested VDAC can oligomerize in response to mitochondrial signals promoting apoptosis to allow for the formation of a multimeric structure that can allow proteins and macromolecules to escape from the mitochondria (35–39). In some cases, these oligomers interact with proteins including Bax and Bid to promote apoptosis (34, 35, 37, 40). Recently, VDAC-bound hexokinase has been noted to localize primarily to MAMs, at least in cancer cells, and hexokinase release from these sites has been linked to increased mitochondrial Ca2+ caused by Ca2+ release from ER via inositol triphosphate receptors (IP3Rs) and by Ca2+ entry through plasma membrane (41).
In the present study, we find that the dissociation of hexokinase from mitochondria previously shown to be induced by peptidoglycan is also triggered, albeit by a different mechanism, by other common NLRP3 inflammasome activators including ATP, nigericin, and silica. Release of hexokinase allows for Ca2+ influx into the mitochondria and frees up VDAC to oligomerize. We find that blocking VDAC oligomerization with the inhibitor VBIT-4 reduces peptidoglycan and hexokinase release-induced maturation and secretion of IL-1β. In agreement with recent work by Xian et al., VBIT-4 also reduces maturation and secretion of IL-1β in response to ATP, nigericin and silica (21), placing hexokinase release as a common upstream factor. Knocking down any one of the three VDACs expressed by macrophages was not sufficient to inhibit inflammasome activation, however, knocking down any two reduced IL-1β secretion, suggesting that the VDACs form hetero-oligomeric structures important for NLRP3 inflammasome activation. We also find that VDAC oligomerization is followed by recruitment of NLRP3 into close association with VDAC and is accompanied by activation of inflammasomes. The association between VDAC and NLRP3 was impaired upon inhibition of VDAC oligomerization, as is IL-1β secretion. Finally, we find that mtDNA, which has previously been implicated in NLRP3 inflammasome activation, is necessary for NLRP3 association with VDAC oligomers, indicating that VDAC oligomers, together with mtDNA, collaborate to nucleate NLRP3 inflammasome assembly. Our data suggest that HK dissociation from VDAC is a proximal step in the NLRP3 inflammasome cascade allowing for VDAC oligomerization, NLRP3 oligomerization and inflammasome activation.
RESULTS
HK release from mitochondria is a common feature of NLRP3-inflammasome activation
As we have observed previously (12), when we treated mouse bone marrow-derived macrophages (BMDM) with particulate peptidoglycan (PGN), we observed an increase in the amount of hexokinase 2 (HK2) in the cytosol of the cells (Fig. 1A), consistent with its release from the mitochondrial outer membrane. We had previously observed that peptidoglycan-derived N-acetylglucosamine directly inhibits hexokinase enzymatic activity resulting in HK2 dissociation from the mitochondria and that the ability of peptidoglycan to activate the NLRP3 inflammasome was distinct from other activators in that it did not require influx of extracellular potassium. Further, we have observed that HK dissociation from mitochondria was sufficient to trigger NLRP3 inflammasome activation (12). We now report that canonical NLRP3 inflammasome activators that trigger potassium influx (42–44) including ATP, nigericin, and silica particles also drive dissociation of hexokinase from mitochondria into the cytosol (Fig. 1A). To directly visualize this release, we expressed GFP-tagged HK2 in BMDM and observed ATP-triggered release of the protein from mitochondria by live cell fluorescence imaging (Fig. 1B). Within 15 minutes of treatment with ATP there is a measurable increase in HK2 in the cytosol that is not associated with a breakdown of mitochondrial structure since there is not a concurrent release of cytochrome c into the cytosol (Fig. 1C). Levels of hexokinase 1 (HK1) in the cytosol are not affected (Fig. 1C), suggesting that this effect is specific to HK2. This may be related to the observation that HK1 has a higher affinity to VDAC1 than HK2 (32).
Fig. 1. Common NLRP3 inflammasome activators trigger HK release from mitochondria.
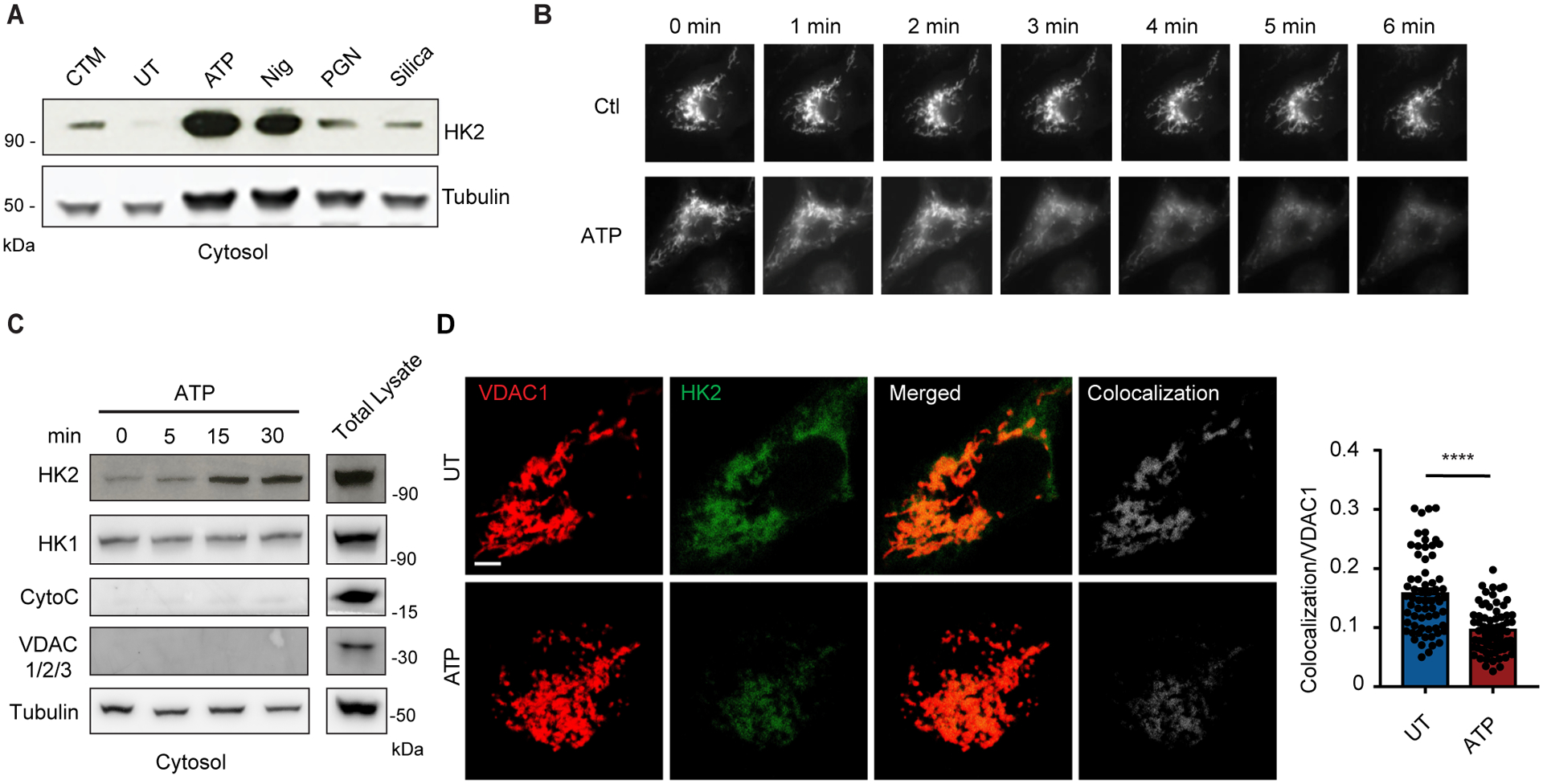
(A) Immunoblot of hexokinase 2 (HK2) and tubulin in cytosolic fractions from mouse BMDM primed with LPS for 3 hr and left untreated (UT) or activated with ATP (5 mM, 30 min), nigericin (Nig, 3 μM, 30 min), peptidoglycan (PGN, 40 μg/ml, 6 hr) or silica particles (20 μg/ml, 6 hr). Clotrimazole (CTM) treatment was used as a positive control for hexokinase release from mitochondria. (B) LPS-primed BMDM expressing HK2-GFP were imaged before and after treatment with 5 mM ATP as indicated. (C) Immunoblot time course of HK2 and hexokinase 1 (HK1) together with controls cytochrome c (CytoC), VDAC 1/2/3, and tubulin in cytosol fractions from BMDM stimulated with ATP (5 mM) for the indicated periods. (D) STED images of LPS-primed BMDM co-stained with antibodies to VDAC1 and HK2 and stimulated or not with ATP (5 mM, 1hr). Scale bar = 3 μm Colocalization was quantified in multiple cells (n = 65–59 cells across 5 independent experiments). Data are presented as means ± SEM. ****p < 0.0001 (unpaired t test).
HK2 associates with outer membranes of mitochondria through interaction of the final 15 amino acids of its N-terminal tail with voltage-dependent anion channel 1 (VDAC1) (34, 35, 45, 46). In resting cells, VDAC1 and HK2 colocalize by immunofluorescence microscopy (Fig. 1D), and treatment with ATP results in a marked decrease in that colocalization. Together, the data indicate that release of HK2 from the mitochondrial surface is a shared feature of common NLRP3 inflammasome activators, although the mechanisms driving that release might be different.
VDAC oligomerization is required for NLRP3 inflammasome activation
VDAC, an outer membrane channel, together with ANT in the inner mitochondrial membrane, regulates flow of metabolites, ions, and nucleotides in and out of the organelle. In some instances, particularly upon induction of apoptosis, VDAC1 has been observed to oligomerize and form a pore that can allow macromolecules through the outer mitochondrial membrane (47). VDAC is not absolutely necessary for formation of the permeability transition pore that participates in collapse of mitochondria during apoptosis but may play a role in the processes (35, 38, 39, 48, 49). We have observed that mitochondria do not collapse upon ATP-induced HK2 release from VDAC since cytochrome c remains associated with the organelle (Fig. 1C), but we nevertheless examined whether ATP could allow for VDAC oligomerization. Remarkably, we observed by crosslinking and immunoblotting that treatment of LPS-primed BMDM with ATP results in oligomerization of VDAC1 (Fig. 2, A and B).
Fig. 2. NLRP3 activators trigger VDAC oligomerization.
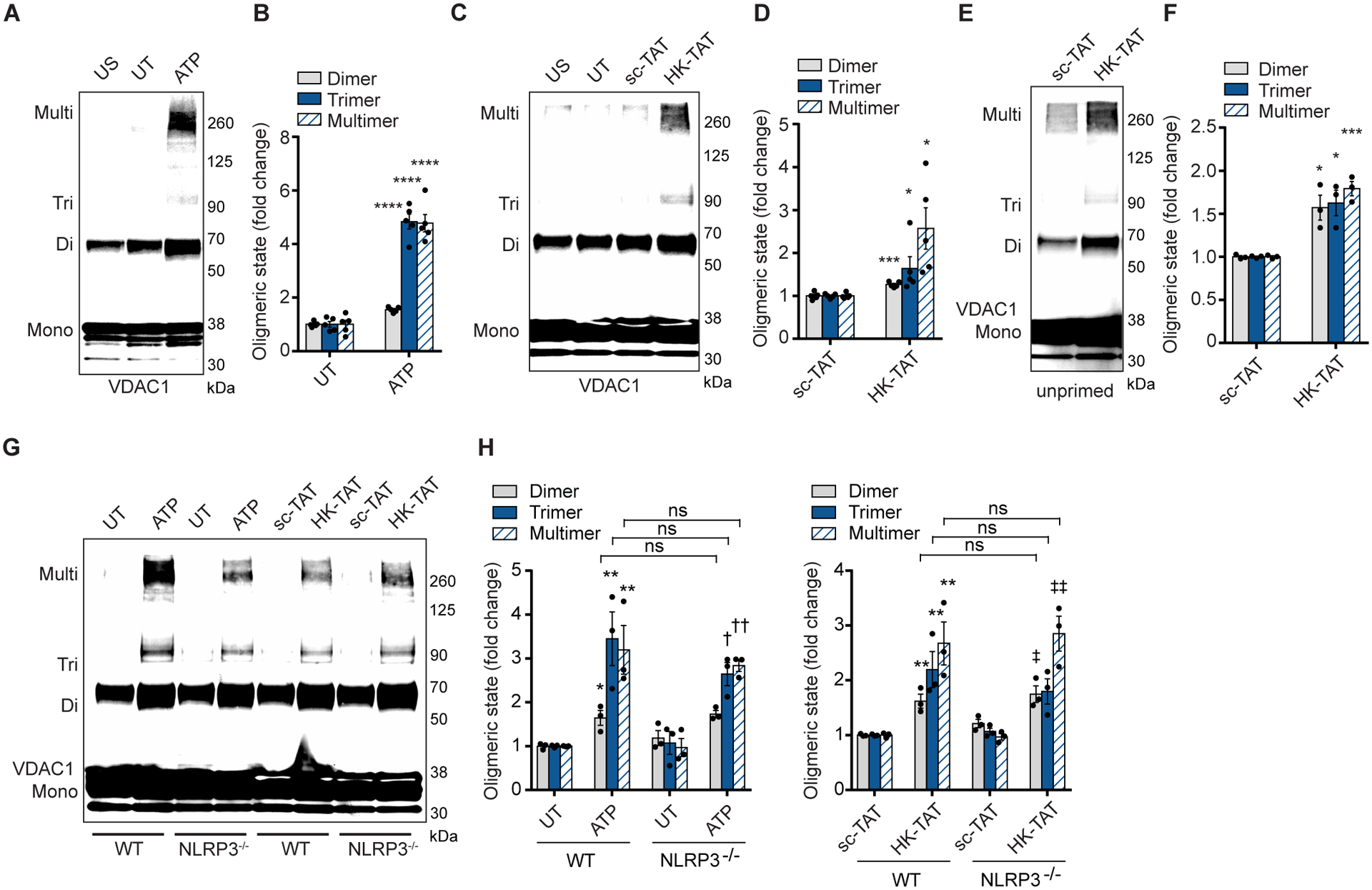
(A and B) Immunoblot of VDAC1 cross-linking in unprimed, unstimulated BMDM (US) or LPS-primed cells left untreated (UT) or stimulated with ATP (5 mM, 1hr) (A), and quantitative analysis of oligomers in multiple independent immunoblots (B) (n = 5). (C and D) Immunoblot of VDAC1 cross-linking assay as in (A) but stimulated with scramble peptide (sc-TAT) or with HK-TAT (20 μM, 2 hr) (C) and quantitative analysis of oligomers in multiple independent immunoblots (D) (n = 5). (E and F) Immunoblot of VDAC1 cross-linking assay as in (C), but in unprimed BMDM (E) and quantitative analysis of oligomers in multiple independent immunoblots (F) (n = 3). (G and H) Immunoblot of VDAC1 cross-linking assay in LPS-primed BMDM from wild type (WT) or NLRP3−/− mice stimulated as indicated (G) and quantitative analysis of oligomers in multiple independent immunoblots (H) (n = 3). Graphical data are presented as means ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001, †p < 0.05, ††p < 0.01 (compared to UT of NLRP3−/−), ‡p < 0.05, ‡‡p < 0.01 (compared to sc-TAT of NLRP3−/−). Comparisons were performed using unpaired t test (B, D, F) or one-way ANOVA with Bonferroni correction (H).
To clarify whether release of hexokinase from mitochondria is sufficient to promote VDAC oligomerization, we treated cells with HK-TAT, a membrane permeable form of a peptide mimicking the VDAC-binding domain of hexokinase that has been used experimentally by us and others to specifically drive release of hexokinase from VDAC into the cytosol and that we have found to be sufficient to trigger NLRP3 inflammasome activation in LPS-primed BMDM (12, 50, 51). HK-TAT-induced hexokinase dissociation from mitochondria proved to be sufficient to drive VDAC1 oligomerization (Fig. 2, C and D). A previous study has noted that VDAC oligomerization in the context of apoptotic stimuli can result from elevated expression of VDAC (52). We observed that treatment of cells with ATP or HK-TAT had no effect on the total cellular VDAC content (fig. S1). To better understand the placement of VDAC oligomerization in the sequence of events leading to NLRP3 inflammasome activation, we asked whether LPS priming of BMDM was necessary for VDAC oligomerization since priming is known to prepare BMDM by promoting expression of NLRP3 as well as its association with the mitochondrial outer membrane through association with cardiolipin (23–25). We observed that HK-TAT induced VDAC1 oligomerization in unprimed BMDM (Fig. 2, E and F), and that ATP and HK-TAT induced VDAC1 oligomerization normally in NLRP3−/− macrophages (Fig. 2, G and H) suggesting that VDAC1 oligomerization is upstream of NLRP3 inflammasome assembly.
To determine whether VDAC oligomerization is important for NLRP3 activation as a result of HK2 dissociation from mitochondria, we made use of VBIT-4, a cell-permeable inhibitor of VDAC oligomerization (53–55). VBIT-4 blocked oligomerization of VDAC1 in ATP-stimulated BMDM (Fig. 3, A and B). We further used nanoscale super-resolution imaging using τ-stimulated emission depletion (τ-STED) microscopy to directly observe clustering of VDAC1 on the surface of mitochondria (Fig. 3, C and D). ATP stimulated aggregation of VDAC1 into larger clusters, and this formation of larger clusters was blocked by VBIT-4, confirming that this measurement is a direct visualization of VDAC1 oligomerization. We observed that VBIT-4 blocks IL-1β production triggered by several common NLRP3 inflammasome activators in LPS-primed BMDM (Fig. 3, E and F), but not by the AIM2 inflammasome activator poly(dA:dT) (pdA:dT) (Fig. 3E), indicating that VDAC oligomerization is specifically required for NLRP3 inflammasome activation. To confirm that the blockade of IL-1β production by VBIT-4 is due to blocking NLRP3 inflammasome activation and not some other nonspecific effect on the cells, we observed that VBIT-4 blocked release of mature processed IL-1β into the culture supernatant (Fig. 3G) and activation of caspase-1 (Fig. 3, H to J), but did not impact TNF-α production (Fig. 3K) indicating a specific role in NLRP3 inflammasome activation. Finally, VDAC oligomerization has been extensively studied in response to several apoptosis inducing stimuli including staurosporine (STS) (35, 37, 56), and we hypothesized that these too might therefore activate the NLRP3 inflammasome. Indeed, we observed that STS triggers NLRP3-dependent IL-1β release from LPS-primed BMDM in a manner that is inhibited by VBIT-4 (Fig. 3, L and M). Together the data suggest that VDAC oligomerization is necessary for activation of the NLRP3 inflammasome.
Fig. 3. VDAC Oligomerization is required for NLRP3 inflammasome activation.
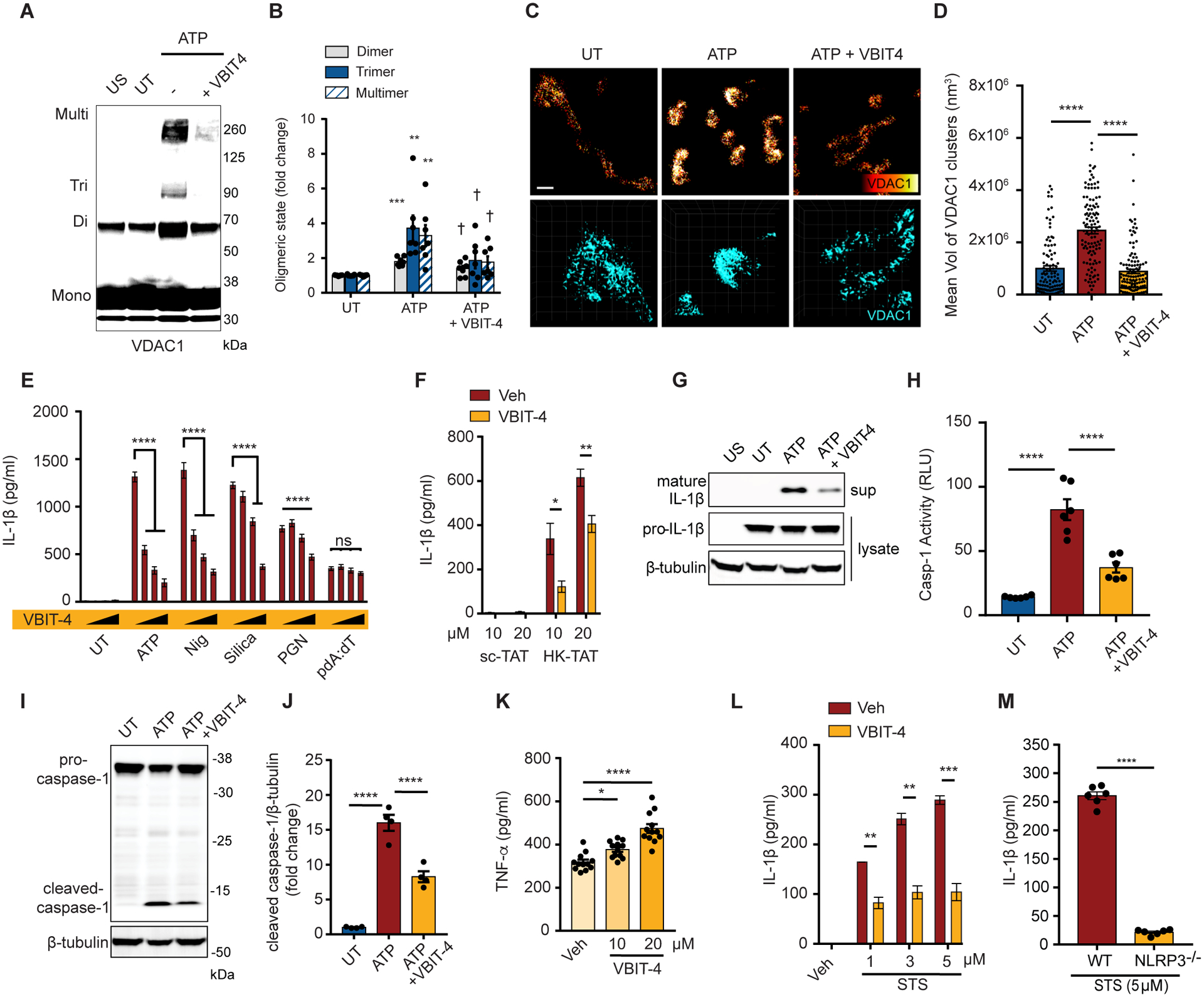
(A and B) Immunoblot of VDAC1 cross-linking in LPS-primed BMDM left untreated (UT) or stimulated with ATP (5 mM, 1 hr) with or without VBIT-4 (20 μM) (A), and quantitative analysis of oligomers in multiple independent immunoblots (B) (n = 7). (C) Highly magnified representative τ-STED images of VDAC1 on individual mitochondria in LPS-primed BMDM stimulated or not with ATP (30 min) with VBIT-4 as indicated. Upper panels scale bar 500 nm. Lower panels show 3D images used to calculate cluster volumes (grid unit = 200 nm). (D) Quantification of VDAC1 cluster size derived from images as in (C) (n = 109–121 mitochondria across 5 independent experiments) (E) IL-1β secretion measured by ELISA from LPS-primed BMDM stimulated as indicated (ATP (2 mM, 30 min), Nig (3 μM, 30 min), silica (20 μg/ml, 6 hr), PGN (40 μg/ml, 6 hr), or pdA:dT (1 μg/ml, 6 hr)) in the presence of increasing concentrations of VBIT-4 (5–20 μM) (n = 8 replicates across 3 experiments). (F) IL-1β secretion measured by ELISA from LPS-primed BMDM stimulated with sc-TAT or HK-TAT (2 hr) at the indicated concentrations in the presence of absence of VBIT-4 (n = 6). (G to J) LPS-primed BMDM were stimulated or not with ATP in the presence or absence of VBIT-4 as in (E). The presence of mature IL-1β in culture supernatants and pro-IL-1β in cell lysate was assessed by immunoblotting (G), activation of caspase-1 was assessed in cell pellets (n = 6) (H), cleaved caspase-1 in cell lysates was detected by immunoblotting (I) and quantified across multiple independent immunoblots (J) (n = 4). (K) BMDM were primed with LPS in the presence or absence of VBIT-4 and TNF-α production was assessed by ELISA (n = 12 across 3 experiments). (L) LPS-primed BMDM were treated with staurosporine (STS, 5 μM, 6 hr) in the presence or absence of VBIT-4, and IL-1β production was assessed by ELISA (n = 3). (M) IL-1β was measured in LPS-primed WT or NLRP3−/− BMDM treated with STS, and IL-1β production was assessed by ELISA (n = 6). Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (compared to UT if not otherwise indicated), †p < 0.05 (compared to ATP) Comparisons were performed by one-way ANOVA withTukey’s post hoc test (B, D, E, H, J, K) or unpaired t test (F, L, M).
Multiple VDAC proteins participate in NLRP3 activation
There are three isoforms of VDAC in mammalian cells; VDAC1, VDAC2, and VDAC3 (57). VDAC 1 is the most abundantly expressed in most cells types, and the different VDACs have both overlapping and nonredundant functions in cells (58). To investigate which of the VDACs are important for NLRP3 inflammasome activation, we performed knock-down of various combinations of VDACs using clustered regularly interspaced short palindromic repeats (CRISPR) gene editing in BMDM. We first knocked down VDAC1, VDAC3, or both in primary BMDM and confirmed by immunoblotting that protein expression was specifically decreased by their respective target guide-RNAs (Fig. 4A). Knockdown of these proteins did not adversely affect LPS priming of the cells since pro-IL-1β protein was induced as well or more (Fig. 4, A and B) and TNF-α protein was produced normally (Fig. 4C). Release of IL-1β into the culture supernatant after simulation with ATP or HK-TAT was unaffected by knockdown of VDAC1 or VDAC3 alone, however when VDAC1 and VDAC3 were suppressed together IL-1β release was significantly reduced, suggesting overlapping roles for VDAC 1 and VDAC3 in the process (Fig. 4D). Targeted reduction of VDAC1 and/or VDAC3 had no effect on IL-1β production in response to activators of other inflammasome including AIM2 (pdA:dT) or NLRC4 (Salmonella) (59) (Fig. 4D).
Fig. 4. Multiple VDACs participate in NLRP3 inflammasome activation.
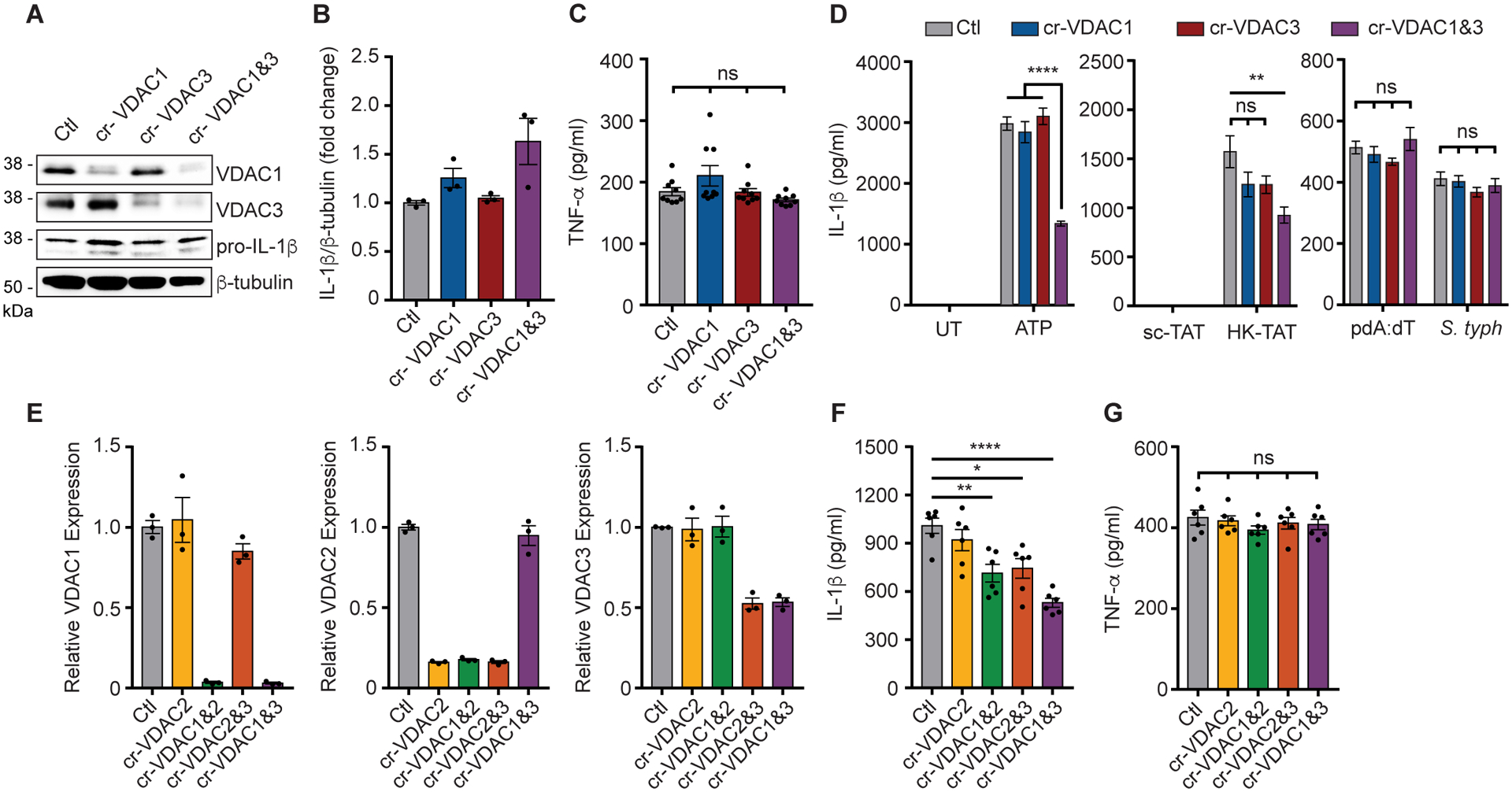
(A) Immunoblot of VDAC1, VDAC3, IL-1β, and β-tubulin from LPS-primed BMDM in which VDAC1 and/or VDAC3 are knock-downed using gRNAs (cr-VDAC1, cr-VDAC3, Ctl = control/no gRNA). (B) Combined quantitative analysis of pro-IL-1β protein expression across independent experiments (n = 3). (C) Secretion of TNF-α into the culture supernatant was measured by ELISA (n = 9 replicates across 3 experiments). (D) IL-1β secretion was measured by ELISA in culture supernatants of LPS-primed cells with ATP (n = 11 replicates across 3 experiments), HK-TAT (n = 6 replicates across 2 experiments), pdA:dT or infection with S. Typhimurium (n = 12 replicates across 3 experiments). (E) Relative mRNA expression of VDAC1, VDAC2, and VDAC3 was measured by RT-qPCR in BMDM in which VDACs were knock-downed as indicated (n = 3 biological replicates). (F) IL-1β secretion was measured by ELISA from primed BMDM with the indicated VDACs knocked down and induced with ATP (n = 6). (G) TNF-α secretion in primed BMDM in which the indicated VDACs are knocked down (n = 6). Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey’s post hoc test).
We were unable to find a good antibody to detect mouse VDAC2, so in an independent set of experiments, we knocked down VDAC2 alone or together with VDAC1 or VDAC3 and assessed mRNA knockdown by quantitative reverse transcription PCR (RT-qPCR) (Fig. 4E). Using LPS-primed BMDM with each of the VDACs knocked down, we again observed that single knockdown of VDAC2 had no effect on IL-1β production stimulated by ATP, but that IL-1β production was reduced when VDAC2 knockdown was combined with either VDAC1 or VDAC3 (Fig. 4F). Again, no combination of VDAC knockdown affected TNF-α production (Fig. 4G). Together, the data suggest that all 3 forms of VDAC can influence NLRP3 activation in primary macrophages.
Hexokinase dissociation from mitochondria activates release of ER calcium
It has previously been noted in HeLa cells that the HK2/VDAC complex is associated with inositol triphosphate receptors (IP3Rs) in endoplasmic reticulum (ER) membranes at sites where the ER and mitochondria are closely associated and that dissociation of HK2 from mitochondria activates IP3Rs (41). IP3Rs regulate release of calcium from ER stores, and we therefore wondered whether this calcium release could be responsible for VDAC oligomerization in response to NLRP3 activators. We observed that an inhibitor of IP3Rs, 2-aminoethoxydiphenyl borate (2-APB), blocks IL-1β secretion from LPS-primed BMDM activated with either ATP or HK-TAT (Fig. 5A) and that HK2 dissociation from mitochondria induced by HK-TAT is sufficient to trigger an elevation of mitochondrial calcium concentrations in LPS-primed BMDM that is inhibited by 2-APB (Fig. 5B). Calcium entry into mitochondria was not a consequence of NLRP3 activation since both ATP and HK-TAT induced mitochondrial calcium elevation in NLRP3-deficient macrophages (fig. S2, A and B). Calcium may enter mitochondria through the calcium uniporter on the inner mitochondrial membrane, which is inhibited by ruthenium red (RuR) (60, 61). RuR has also been implicated in direct binding to VDAC to regulate Ca2+ movement across the outer membrane (62). We observed that RuR inhibited ATP and HK-TAT stimulated IL-1β production from LPS-primed BMDM (Fig. 5C) as well as inhibited the elevation of mitochondrial calcium levels in response to these stimuli (Fig. 5, D and E). These data suggest that IP3R triggered calcium elevation may be required for VDAC oligomerization, and we found that indeed, VDAC oligomerization triggered by ATP or HK-TAT is blocked by 2-APB (Fig. 5, F and G). Finally, cyclophilin D has been identified as a key regulator of oligomerization of VDAC and ANT in formation of the permeability transition pore (PTP) in response to changes in the mitochondrial environment (63). Cyclophilin D is inhibited by cyclosporin A (CsA) and others have noted that CsA inhibits NLRP3 inflammasome activation by canonical NLRP3 activators, although the exact mechanism of this has not been clear (19, 21). We found that CsA also blocks inflammasome activation and IL-1β secretion in response to HK2 release from mitochondria induced by HK-TAT (fig. S2C).
Fig. 5. ER calcium stores drive VDAC oligomerization.
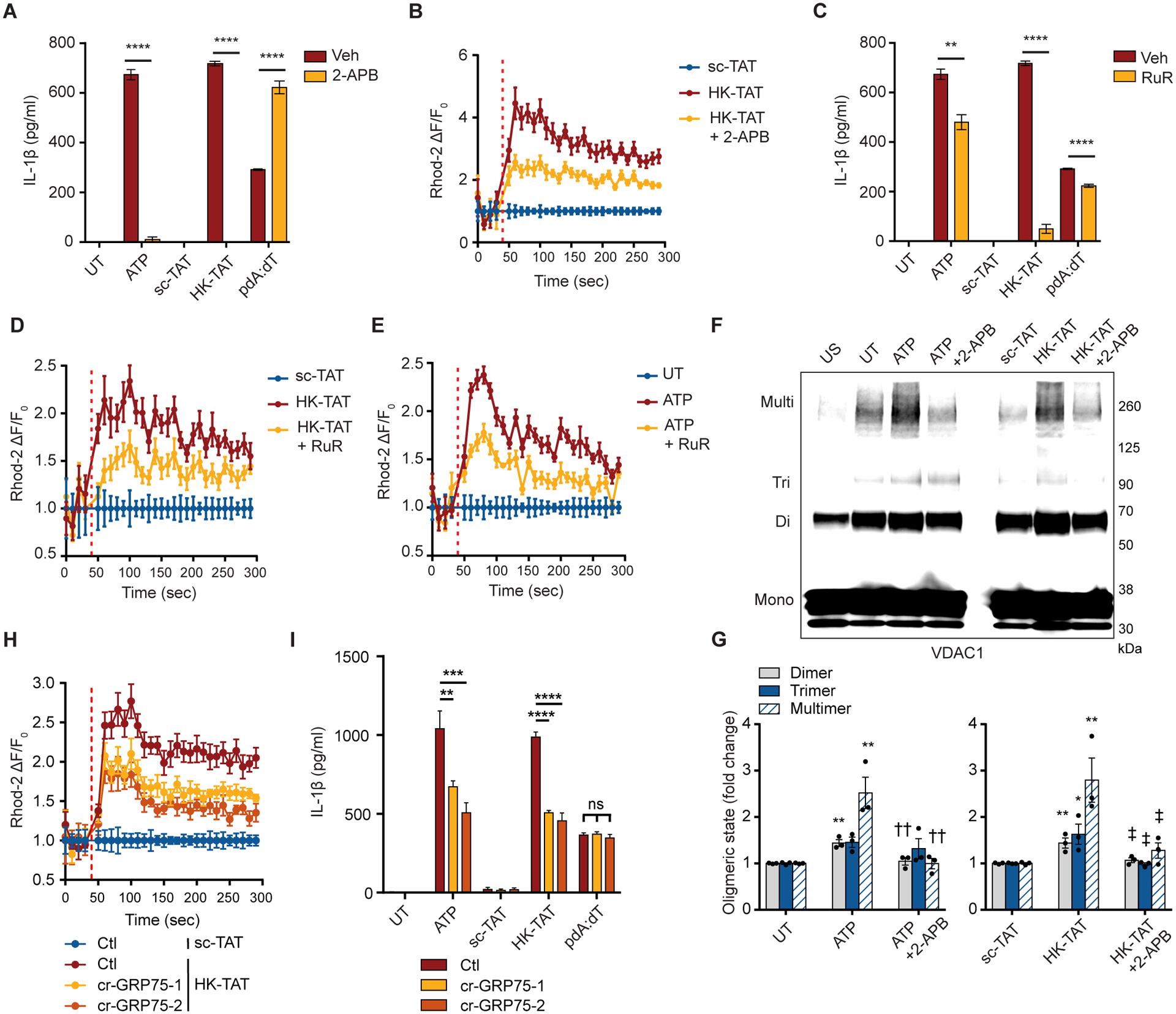
(A) LPS-primed BMDM were stimulated or not (Veh = vehicle control) in the presence or absence of the IP3R antagonist 2-APB (100 μM), and IL-1β production was assessed by ELISA (n = 3). (B) Primed cells were stimulated with HK-TAT in the presence of absence of 2-APB and mitochondrial Ca2+ levels were assessed by Rhod-2 fluorescence over time (n = 6). (C) IL-1β was measured as in (A) but in the presence or absence of ruthenium red (RuR, 30 μM) (n = 3). (D and E) Ca2+ was measured as in (B) but in cells stimulated with HK-TAT (D) or ATP (E) in the presence or absence of RuR (n = 6). (F and G) BMDM were left unprimed (US) or LPS-primed and treated (ATP, HK-TAT) or not (UT) in the presence or absence of 2-APB as indicated, and oligomerization of VDAC1 was assessed by immunoblot (F) and quantitative analysis of oligomers in multiple independent immunoblots (G) (n = 3). (H) Mitochondrial Ca2+ was measured as in (B) in HK-TAT stimulated cells in which GRP75 knocked down using two different gRNAs (cr-GRP75–1, cr-Grp75–2) (n = 6). (I) IL-1β was measured as in (A) in cells in which GRP75 was knocked down (n = 6). Data are presented as means ± SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001, †p < 0.05 (compared to ATP), ‡p < 0.05 (compared to HK-TAT) Comparisons were performed by unpaired t test (A, C) or one-way ANOVA with Bonferroni correction (G, I).
Calcium flow from the ER to mitochondria is thought to be facilitated by a protein complex containing VDAC, the IP3R, and glucose-regulated protein 75 (GRP75, product of Hspa9), and depletion of GRP75 was found to interfere with calcium transfer to mitochondria (64, 65). We therefore investigated whether calcium influx into mitochondria induced by hexokinase release from mitochondria was dependent on GRP75. CRISPR/Cas9 knockdown of GRP75 in BMDM (fig. S2D), suppressed calcium influx into mitochondria induced by HK-TAT (Fig. 5H) as well as stimulation of secretion of IL-1β (Fig. 5I). Together the data suggest that when NLRP3 activators trigger HK2 dissociation from mitochondria, this causes ER calcium stores to be released by IP3Rs, and the subsequent elevation of mitochondrial calcium levels triggers VDAC oligomerization.
To understand whether these observations linking calcium influx triggered oligomerization of VDAC are consistent for stimuli that trigger NLRP3 activation in distinct ways, we first investigated noncanonical activation via caspase-11 (15, 66, 67). We observed that IL-1β production in response to transfection with LPS was inhibited by VBIT-4 and 2-APB (fig. S3A), but there is only a partial impact on cytotoxicity induced by transfected LPS (fig. S3B). These data are consistent with the cytotoxicity being driven by initial caspase-11 gasdermin D cleavage that feeds into NLRP3/caspase-1 activation and IL-1β production (66, 67). We also examined the effects on NLRP3 activation downstream of imiquimod and CL097, which have been reported to activate NLRP3 independent of potassium efflux (68). We observed that IL-1β production in response to both stimuli was inhibited by VBIT-4 and 2-APB (fig. S3, C and D). We also investigated alternative NLRP3 activation upon treatment of human monocytes with LPS and observed that both VBIT-4 and 2-APB inhibited IL-1β produced by LPS-stimulated human monocytes (fig. S3E). These data imply that even when NLRP3 is activated by alternative or noncanonical stimuli there is still a role for VDAC oligomerization and mitochondrial Ca2+ uptake.
VDAC oligomers directly recruit NLRP3
We next wondered how VDAC oligomerization might be mechanistically linked to NLRP3-driven assembly of inflammasomes. Since inflammasome assembly is an oligomerization process itself, we hypothesized that VDAC oligomerization might be harnessed to promote assembly of the NLRP3 inflammasome, or that the oligomerized structure of VDAC might facilitate direct interaction with NLRP3. NLRP3 has been previously described to be associated with mitochondria (18, 23, 69, 70), and we therefore explored whether NLRP3 might directly interact with VDAC. Using a proximity ligation assay, we found that VDAC1 and VDAC3 are not in close proximity with NLRP3 in resting cells (Fig. 6, A and C), but the VDACs come into close proximity with NLRP3 upon stimulation of VDAC oligomerization with HK-TAT (Fig. 6, A to D). Association of NLRP3 with VDAC1 is also induced by ATP, and NLRP3 recruitment requires VDAC oligomerization since the association is inhibited by VBIT-4 (Fig. 6, E and F). The specificity of the VDAC1-NLRP3 proximity ligation signal was confirmed by the loss of ATP-induced proximity ligation signal in NLRP3-deficient cells (fig. S4A). We observed that VDAC oligomerization drives relatively large-scale recruitment of NLRP3 by simple co-localization using τ-STED-fluorescence lifetime imaging (FLIM), which has a spatial resolution of around 60 nm. While some colocalization of VDAC1 and NLRP3 is observed in resting cells, this colocalization is markedly elevated upon stimulation of oligomerization with ATP (Fig. 6, G and H). This colocalization is reduced to resting levels by blocking VDAC oligomerization with VBIT-4. ATP-induced colocalization was also lost in NLRP3-deficient cells, confirming the specificity of the assay (fig. S4, B and C). We further independently confirmed the close association of VDAC1 and NLRP3 using fluorescence acceptor photobleaching. In resting cells, there is a modest amount of fluorescence energy transfer (FRET) between antibody-labeled VDAC1 and NLRP3, but the efficiency of the FRET increases markedly upon stimulation of VDAC oligomerization with ATP (Fig. 6, I and J). The increased FRET is due to VDAC oligomerization since it is inhibited by VBIT-4. As an independent way to inhibit VDAC oligomerization, we treated cells with the IP3R inhibitor, 2-APB, that blocks VDAC oligomerization (Fig. 5F) and found that 2-APB also blocked VDAC association with NLRP3 as measured by proximity ligation (Fig. 6, K and L). Inhibition of NLRP3 association with VDAC by 2-APB was not due to a block in NLRP3 localization to the mitochondria since isolation of mitochondria revealed an equal amount of NLRP3 bound in HK-TAT-treated cells with or without 2-APB treatment (Fig. 6M). These results confirm previous observations that NLRP3 is recruited to mitochondria following LPS priming and before NLRP3 inflammasome activation (23, 70), and add that NLRP3 only associates with VDAC when VDAC oligomerizes.
Fig. 6. VDAC oligomers associate with NLRP3 during inflammasome activation.
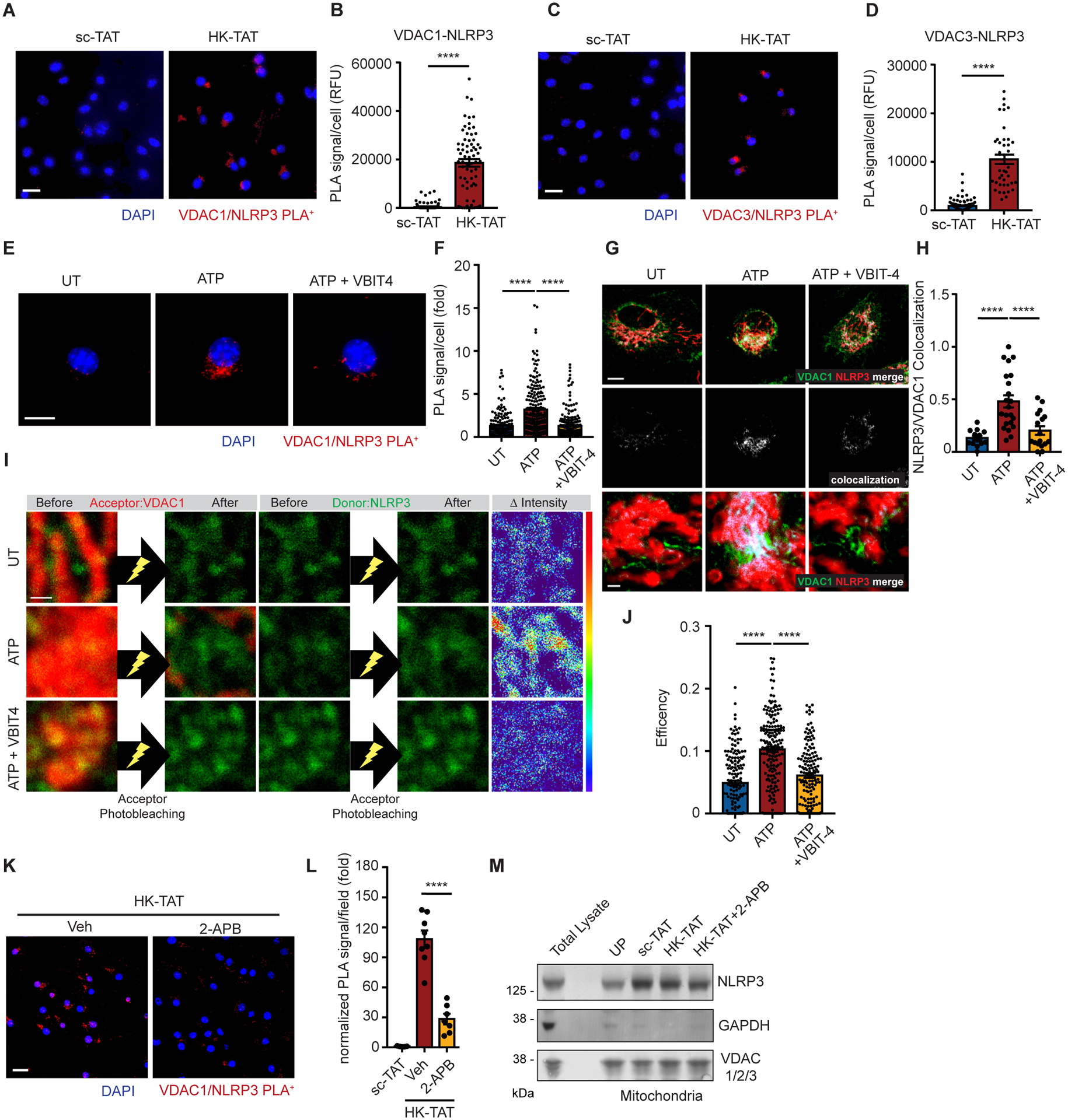
(A and B) LPS-primed BMDM were stimulated with sc-TAT or HK-TAT (20 μM, 2 hr), and close association between VDAC1 and NLRP3 was assessed by proximity ligation assay (A) (scale bar = 20 μm) and quantified (B) (n = 70–102 cells across 3 independent experiments). RFU = relative fluorescence units. (C and D) VDAC3-NLRP3 close association was assessed as in (A and B) (n = 42–109 cells across 3 independent experiments). (E and F) VDAC1-NLRP3 close association was assessed as in (A and B) (E) (scale bar = 10 μm) except cells were stimulated or not with ATP (2 mM, 30 min) (n = 162–236 cells across 5 independent experiments). (G and H) τ-STED-FLIM images of individual UT or ATP-treated BMDM (2 mM, 30 min) with or without VBIT-4 (20 μM, 1 hr) using antibodies against VDAC1 and NLRP3 (top row, scale bar = 5 μm), and colocalization between VDAC1 and NLRP3 is highlighted (middle row). More magnified regions (bottom row) highlight areas of colocalization (scale bar = 0.5 μm). Colocalization was quantified (H) (n = 13–22 cells across 3 independent experiments). (I and J) Acceptor photobleaching assay with UT or ATP-treated BMDM with or without VBIT-4 using antibodies against VDAC1 as the acceptor (red) and NLRP3 as a donor (green). Representative images (I) of VDAC1 and NLRP3 before and after acceptor photobleaching (left and middle, scale bar = 0.5 μm) and the amounts of changes of the intensity of donor shown by RGB spectrum (right). The degree of acceptor photobleaching was quantified (J) (n=150–181 photobleached areas from 16 cells across 3 experiments). (K and L) VDAC1-NLRP3 close association was assessed as in (A and B) in cells treated or not with the IP3R inhibitor 2-APB (n = 5–8 imaged areas normalized by DAPI count across 2 independent experiments) (scale bar = 20 μm). (M) Mitochondrial fractions from unprimed (UP) BMDM or from LPS-primed BMDM stimulated as indicated were assessed for the presence of NLRP3, VDAC1 and GAPDH by immunoblotting. A total lysate was included for antibody reference. Data are presented as means ± SEM. (F, J, L) ***p < 0.001, ****p < 0.0001 (one-way ANOVA, Tukey’s post hoc test). (B, D) ****p < 0.0001 (unpaired t test).
Mitochondrial DNA is necessary for NLRP3/VDAC aggregation
Finally, many investigators have noted that oxidized mitochondrial DNA (mtDNA) can leak out of mitochondria during inflammasome activation and that this mtDNA is important for activation of the NLRP3 inflammasome (19–21, 71, 72). We therefore wondered what role mtDNA might have when VDAC oligomerization is induced by hexokinase release. MtDNA can be depleted from cells by culturing in the presence of ethidium bromide (EtBr) (21, 72, 73). We therefore cultured cells with EtBr and confirmed depletion of mtDNA by qPCR (Fig. 7A). As expected, IL-1β production triggered by NLRP3 activators that drive hexokinase release from mitochondria and VDAC oligomerization including ATP, peptidoglycan, silica, and HK-TAT was reduced in cells depleted of mtDNA (Fig. 7B). Remarkably however, VDAC oligomerization occurred normally in cells depleted of mtDNA (Fig. 7, C and D), placing VDAC oligomerization upstream of a role for mtDNA in NLRP3 inflammasome activation. However, NLRP3 recruitment to VDAC oligomers, as measured by proximity ligation assay, was blocked in cells depleted of mtDNA by EtBr, but does not require ASC (Fig. 7, E and F). Association of NLRP3 with mitochondria increased with priming as seen previously (23), but in LPS-primed cells depleted of mtDNA there was no effect on NLRP3 recruitment to mitochondria (Fig. 7G).
Fig. 7. Mitochondrial DNA promotes NLRP3 association with VDAC oligomers.
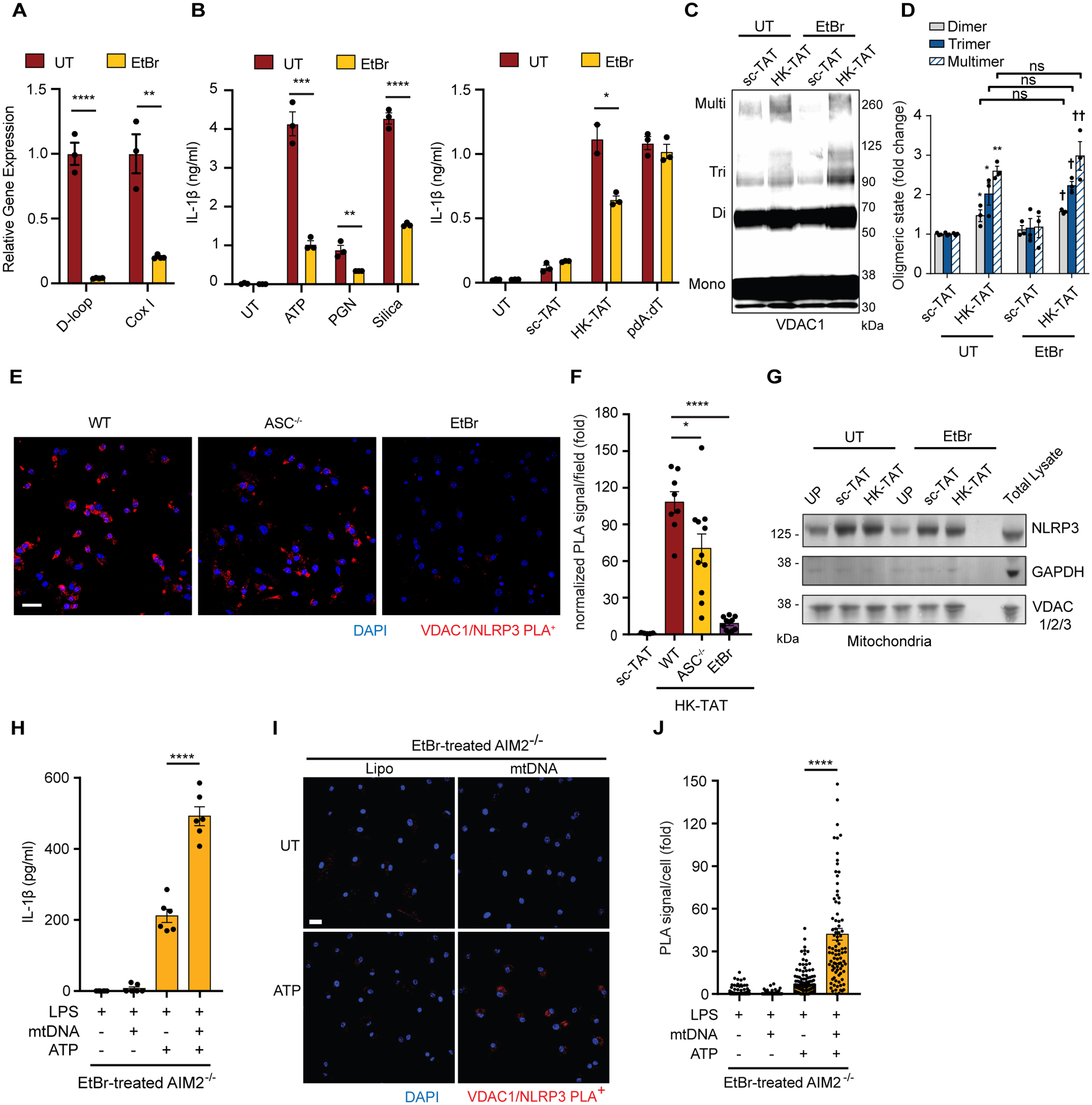
(A) BMDM were grown in the presence of ethidium bromide (EtBr) to deplete mitochondrial DNA (mtDNA), and depletion was confirmed by quantitative PCR of two regions of mtDNA (D-loop and CoxI) normalized to nuclear DNA (n = 3). (B) Ethidium bromide-treated BMDM (EtBr) were LPS-primed and stimulated with ATP (5 mM, 2 hr), PGN (40 μg/ml, 6 hr) or silica (20 μg/ml, 6 hr), or sc-TAT and HK-TAT (20 μM, 2hr), or pdA:dT (1 μg/ml, 6 hr), and IL-1β was measured by ELISA (n = 3). (C and D) BMDM treated or not with EtBr were LPS-primed and treated with sc-TAT or HK-TAT. Oligomerization of VDAC1 was assessed by immunoblot (C) and quantitative analysis of oligomers in multiple independent immunoblots (D) (n = 3). (E and F) LPS-primed WT, ASC−/− or EtBr-treated BMDM were stimulated with HK-TAT, and close association between VDAC1 and NLRP3 was assessed by proximity ligation assay (E) (scale bar 20 μm) and quantified (F) (n = 5–11 imaged areas normalized by DAPI count across 2 independent experiments). (G) NLRP3, GAPDH, and VDAC levels were assessed by immunoblotting of mitochondrial fractions from BMDM and EtBr-treated BMDM primed with LPS and stimulated as indicated or left unprimed (UP). (H) IL-1β was measured in EtBr-treated AIM2−/− BMDM primed with LPS and stimulated with or without mtDNA or ATP (n = 6). (I and J) LPS-primed EtBr-treated AIM2−/− BMDM were stimulated or not with ATP and close association between VDAC1 and NLRP3 was assessed by proximity ligation assay (I) (scale bar 20 μm) and quantified (J) (n = 90–176 cells across 3 independent experiments). Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, †p < 0.05 (compared to sc-TAT of EtBr-treated BMDM) Comparisons were made by unpaired t test (A, B, F) or one-way ANOVA with Bonferroni correction (D) or Tukey’s post hoc test (H, J).
To better understand the role of mtDNA in assembly of the NLRP3-VDAC complex, we tested whether delivery of mtDNA into the cytosol would be sufficient to drive the association. In wild-type cells transfection of mtDNA induced production of IL-1β in an AIM2-dependent manner (fig. S5) consistent with previous studies (74). Therefore, to further understand the role mtDNA in NLRP3 inflammasome activation we performed experiments in AIM2−/− BMDM. Transfection of mtDNA into LPS-primed AIM2−/− BMDM is not sufficient to drive IL-1β production (Fig. 7H and fig. S5) nor can mtDNA alone drive VDAC1-NLRP3 association (Fig. 7, I and J). However, in AIM2−/− BMDM that have been depleted of their endogenous mtDNA with EtBr, transfection of exogenous mtDNA enhanced IL-1β production (Fig. 7H) and restored VDAC1-NLRP3 association (Fig. 7, I and J) that was normally lost in mtDNA-depleted cells (Fig. 7, E and F). Together, the data suggest that NLRP3 is poised for inflammasome activation by associating with mitochondria, but that VDAC oligomerization drives assembly of a larger NLRP3 complex and leads to inflammasome activation. In addition, the NLRP3 recruitment to VDAC oligomers requires mtDNA.
DISCUSSION
The NLRP3 inflammasome is a signaling complex central to inflammation in defense against microbial infection as well as in sterile inflammation and multiple disease processes including atherosclerosis, myocardial infarction, steatohepatitis, and colitis among others (1, 13). Considerable progress has been made in mapping out the molecular signals and mechanisms that coordinate activation and assembly of this large multiprotein complex. However, given the diverse range of stimuli and signaling pathways that have been reported to trigger or influence NLRP3 inflammasome assembly, unifying factors controlling initial recruitment and assembly of the complex have been especially challenging to identify. In this study we find that a common step in NLRP3 inflammasome activation is the dissociation of hexokinase 2 from the outer mitochondrial membrane channel VDAC (fig. S6). Hexokinase dissociation from mitochondria activates IP3R signaling, initiating the release of calcium from the ER that flows into mitochondria. The data suggest that elevated matrix calcium drives oligomerization of VDAC on the outer membrane that, together with inner membrane partners, form pores, which is consistent with previous observations about VDAC oligomerization (47). This oligomerized VDAC, together with mtDNA fragments that have access to the outside of the mitochondria, perhaps through the open pore, recruits NLRP3 to a VDAC-NLRP3 complex on the mitochondrial surface that is important for assembly of the active NLRP3 inflammasome (fig. S6).
Mechanistically, the HK2/VDAC complex has been shown to localize to regions where mitochondria and ER membranes are tightly associated and interact directly with ER IP3Rs (41), and, consistent with our observations in macrophages, dissociation of hexokinase from this complex has been observed to be sufficient to activate IP3R-mediated calcium release and has been reported to contribute to altered VDAC conductance (45). In addition, GRP75 has been implicated in coupling ER IP3Rs to VDAC and facilitating calcium flow from ER stores into mitochondria (64, 65). Consistent with this, we find that GRP75 is required for elevation of mitochondrial calcium levels triggered by hexokinase release. Elevated matrix calcium has been well-characterized as a regulator of activation of the “mitochondria permeability transition pore” (PTP), an activity that is regulated by cyclophilin D, although the role of VDACs in this complex, once considered certain, is currently less clear (75). The mitochondrial permeability transition pore is a still incompletely characterized assembly of inner and outer membrane mitochondrial proteins that allows flow of low molecular weight solutes, including fragments of mtDNA, across the normally impermeable inner membrane. VDACs may function as a part of the pore, or as regulators and associated proteins. Nevertheless, we observe IP3R-regulated Ca2+ flux drives oligomerization of VDAC on the mitochondrial surface in response to hexokinase dissociation.
The Shoshan-Barmatz lab has done extensive work reporting that VDAC has the ability to oligomerize and generate a macromolecule-sized pore in the outer membrane of mitochondria that can pass mtDNA fragments (47). They observed in mouse embryo fibroblasts that short mtDNA fragments could cross mitochondrial membranes via oligomerized VDAC1 channels, were released into the cytosol where they triggered type I interferon signaling, and that VBIT-4 can inhibit this process (76). These data, together with the data presented here, are consistent with recent data from Xian et al. who found in macrophages that mtDNA release and subsequent type I interferon signaling in response to inflammasome activators is blocked by VBIT-4 (21). Also, consistent with our findings, Xian et al. noted that VBIT-4 inhibits NLRP3 inflammasome activation by canonical stimuli. Kim et al. observed that mtDNA binds directly to VDAC1 and promotes oligomerization of purified VDAC1, suggesting that production of mtDNA fragments itself might promote formation of VDAC1 oligomers (76). In contrast, both Xian et al. and our data suggest that depletion of mtDNA in macrophages does not reduce VDAC oligomerization in response to NLRP3 activators, pointing at other upstream activators of VDAC oligomerization. Our data suggest that release of hexokinase from VDAC in macrophages, together with the resulting IP3R-mediated Ca2+ elevation in mitochondria promotes the VDAC oligomerization (48).
Somewhat divergent from previous observations about the role of the three VDACs in NLRP3 inflammasome activation (18, 21), where individual knockdown of individual VDACs was sufficient to inhibit the NLRP3 inflammasome, we find that knockdown of one VDAC is not sufficient to block NLRP3 inflammasome activation. One possible explanation for the different data is likely a differential level of VDAC knockdown between experiments. We interpret the data to suggest that the overall amount of the three VDACs is what is important. However, previous studies did not perform combination knockdown of the VDACs, and our data clearly indicate that combined knockdown of any two VDAC impacted NLRP3 activation. Unlike previous studies, our knockdown data, as well as data showing NLRP3 and VDAC3 in close association, indicate that the different VDAC isoforms collaborate in influencing NLRP3 activation.
Shimada et al. initially observed that oxidized mtDNA was produced by cells stimulated with NLRP3 inflammasome activators and that oxidized mtDNA fragments directly interact with NLRP3 (19). Several studies have since reported that LPS priming leads to increased production of mtDNA fragments and that alterations in mitochondrial endonucleases and DNA repair machinery impact mtDNA production and NLRP3 inflammasome activation (19–21). Thus far the data support a model in which mtDNA needs to exit the mitochondria across both mitochondrial membranes to have an inflammatory impact. Experiments using the PTP inhibitor cyclosporin A implicate cyclophilin D and PTP in mtDNA crossing the inner mitochondrial membrane (19–21, 71, 72). Here we report together with recent work (21) that VDAC can oligomerize to form a macromolecule size pore allowing for mtDNA to transit the outer mitochondrial membrane. Since it has been reported that mtDNA can also bind to VDAC directly (76), together with our observation that oligomerized VDAC comes into close association with NLRP3, we examined whether mtDNA may play a role in NLRP3/VDAC aggregation. Depleting mtDNA did not block VDAC oligomerization but did block association of VDAC with NLRP3. Further, we found that transfecting mtDNA into the cytosol of cells depleted of mtDNA was not in itself sufficient to mobilized NLRP3 to bind VDAC or form an active NLRP3 inflammasome. However, while NLRP3-VDAC association was blocked in ATP-stimulated cells depleted of mtDNA, transfecting in mtDNA restored activation but only in the presence of ATP. This suggests a model in which VDAC plays two roles in NLRP3 inflammasome activation; first it oligomerizes to form a pore large enough for mtDNA fragments to exit the mitochondria, and second, it either binds to mtDNA allowing for a bridge that links VDAC oligomers to NLRP3 aggregation and initiation of inflammasome assembly or mtDNA binds to NLRP3 and stimulates its aggregation with VDAC and subsequent inflammasome activation.
Andreeva et al. have proposed in a cryo-EM structure analysis of recombinant proteins that NLRP3 exists in a “a 12- to 16-mer double-ring cage” that is membrane localized (they suggest the TGN) that is disrupted by NEK7 leading to active inflammasome assembly (77). Similarly, Hochheiser et al. suggested that the “inactive” form of NLRP3 is a decamer ring. In both cases, the “cage” formed is proposed to block pyrin domain interactions with downstream inflammasome components (78). We have not analyzed the multimeric structure of the NLRP3 associating with oligomerized VDAC. It is possible that NLRP3 monomer binding to oligomerized VDAC facilitates assembly of larger NLRP3 oligomers on their way to becoming active. It is also possible that binding of preformed “caged” NLRP3 oligomers to oligomerized VDAC facilitates a structural shift that exposes NLRP3 pyrin domains for binding to ASC and further inflammasome assembly/activation.
We note that many of our experimental interventions are incomplete in their ability to block NLRP3 activation. It is possible that the interventions (e.g., inhibitor treatments) are incomplete and thus leave some remaining activity, or that the interventions are completely effective but that another parallel pathway of NLRP3 activation exists that is unaffected. Our data are consistent with a large and growing body of work implicating mitochondria as a signaling nexus for NLRP3 inflammasome activation (13). However, additional studies have suggested that dissolution of the trans-Golgi network (TGN) and localization of NLRP3 to phosphatidylinositol 4-phosphate (PI4P) exposed on the dispersed vesicles facilitates NLRP3 inflammasome assembly (68, 79). Interestingly, the vesicles are also thought to interact with mitochondria via PI4P (80). Thus, it is possible that the TGN-associated NLRP3 activation pathway is parallel to, downstream of, or upstream of the VDAC oligomerization requirement explored in this study and in Xian et al. (21).
Hexokinase 2 has been widely studied as an anti-apoptotic and mitochondria-stabilizing protein, often overexpressed in tumor cells where it is thought to protect the cells from death (81). In this context, it is thought that interaction of HK with VDAC maintains VDAC in a “closed” state and prevents binding of pro-apoptotic Bcl family members Bax and Bid, which also interact with the same N-terminal tail of VDAC as HK2 (30, 34, 40, 56, 82–84). This relationship with regulation of cell death is intriguing given that inflammasome activation often, but not always, is associated with an inflammatory type of cell death called pyroptosis. Thus, hexokinase appears poised to influence the balance of multiple types of cell death and the inflammation that can result.
Hexokinase is fundamentally a metabolic enzyme, and yet we observe here that it is a nexus for inflammatory signaling in response to diverse stimuli and participates in setting up a cascade of events on the mitochondrial surface that promotes NLRP3 inflammasome activation. The events leading to initial aggregation of NLRP3 may be particularly attractive for designing strategies aimed at influencing the process, and the data here suggest novel approaches related to metabolic manipulations and targeting NLRP3 relocation across the mitochondrial surface. Healthy regulation of the NLRP3 inflammasome is important for diseases as wide ranging as diabetes, obesity, atherosclerosis, and inflammatory bowel disease.
MATERIALS AND METHODS
Study design
The aim of the study was to identify how hexokinase release from mitochondria leads to activation of the NLRP3 inflammasome. We used primary macrophages from wild type and genetically modified mice. We used biochemical, genetic, and microscopy approaches to test a model of inflammasome activation in which hexokinase release from the outer membrane led to changes in VDAC. All data were included, and no data or outliers were excluded. Sample size and experimental replicates are indicated in each figure caption. Unless otherwise stated all n= values represent technical replicates, and unless otherwise stated, all experiments were performed a minimum of 3 times with similar results. Investigators were not blinded to groups. The details of the reagents used are provided.
Animal experimental models
C57BL/6 mice and NLRP3−/− mice were purchased from Jackson Laboratory. ASC−/− and control mice kindly provided by Dr. Fayyaz Sutterwala and Dr. Suzanne Cassel. Aim2−/− mice were kindly provided by Dr. Christian Stehlik. Mice were housed under ad libitum access to food/water in specific pathogen-free conditions. Experimental cells were prepared from 6–12 week old female C57BL/6 mice. For knockout experiments, cells were prepared from 6–25 week old male and female mice with age and sex matched controls. All animal experimental procedures were conducted in accordance with Cedars-Sinai Medical Center Institutional Animal Care and Use Committee guidelines.
Bone marrow-derived macrophage culture and stimulations
BMDM were prepared as described previously with recombinant human macrophage colony-stimulating factor (M-CSF, 50 ng/ml, PeproTech, 300–25) (12). Cells were plated at 5.0–10.0 × 105/ml and primed with 100 ng/ml LPS (S. minnesota, InvivoGen, tlrl-smlps) for 3–4 h. For inflammasome activation and IL-1β measurements, primed BMDM were treated by 2–5 mM ATP (Millipore-Sigma, A2383) for 30–120 min, 3–10 μM nigericin (Millipore-Sigma, 481990) for 30–120 min, N-terminal hexokinase peptide or scramble peptide fused to transactivator of transcription peptide (HK-TAT and sc-TAT respectively) (HK- MIASHLLAYFFTELN(beta-Ala)GYGRKKRRQRRRG, sc-ATAFLMEYNSHLFIL(beta-Ala)GYGRKKRRQRRRG) (United BioSystems) 20 μM for 2 h , 20–100 μg/ml silica (MIN-U-SIL 15, US Silica) for 6 h, 40 μg/ml S. aureus peptidoglycan (PGN) (Millipore-Sigma, 77140) for 6 h, 45 μg/ml imiquimod (InvivoGen, tlrl-imq) for 90 min, or 45 μg/ml CL097 (InvivoGen, tlrl-c97–5) for 90 min. pdA:dT (InvivoGen, tlrl-patn) 1 μg/ml in Opti-MEM (Gibco, 31985070) was mixed with Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) for 30 min at room temperature and added at final concentration of 66 ng/ml to cells for 6 h. Primed BMDM were infected by Salmonella typhimurium (S. typh, 200 multiplicity of infection) for 30 min and then the media were replaced with gentamicin-included media followed by 2 h of incubation. For noncanonical NLRP3 inflammasome activation, BMDM were primed with 1 μg/ml Pam3CSK4 (InvivoGen, tlrl-pms) for 4 h. Thirty min before stimulation, 10–40 μg/ml LPS in Opti-MEM was prepared by mixed with FuGENE transfection reagent (Promega, E5911) and added at final concentration of 0.667–2.667 μg/ml to cells for 16 h. For the inhibitor experiments, LPS-primed BMDM were then pretreated for 1h with 20 μM VBIT-4 (Selleckchem, S3544), 100 μM 2-aminoethoxydiphenyl borate (2-APB, Tocris Bioscience, 1224), 30 μM ruthenium red (RuR, Millipore-Sigma, 557450), 20 μM cyclosporin A (CsA, Millipore-Sigma, C3662) followed by addition of inflammasome activators as above.
CRISPR gRNA-Cas9 Neon transfection knockdown experiments
crRNAs (VDAC1: CATCAACCTCGGCTGTGACG, VDAC2: GTGGAACACCGATAACACTC, VDAC3: ACACCAACTTATTGCGACCT, GRP75–1 (Hspa9-1): TTTGTTGCGGCACATTGTCA, GRP75–2 (Hspa9-2): AAGACGCTTAGTAGCATAGA) were purchased from Integrated DNA Technologies. Duplex gRNAs were prepared by mixing equal amounts of 100 μM crRNAs and 100 μM tracrRNA (IDT, 1072533), and heating at 95° for 5 min. Transfection was performed with the Neon Transfection System (Thermo Fisher, MPK10096). The gRNAs were added into 100 μM Cas9 Electroporation Enhancer (IDT, 1075916) at 3:1 molar ratio and mixed with 62 μM Alt-R Cas9 enzyme (IDT, 1081059) to make RNP complex. 2.0 × 106 of BMDM resuspended in 100 μl of Neon buffer T were mixed with RNP complex and then electroporated at 1,900 V, 20 ms, and 1 pulse. Cells were stimulated as above after 48 hr.
ELISA
Cell supernatants were used for ELISA according to the manufacturer’s instructions (IL-1β: BioLegend, 432601 and R&D, DY401; IL-6 and TNF-α: Thermo Fisher, 88-7064-88 and 88-7324-88, respectively).
LDH assay
CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, G1780) was used for measuring lactate dehydrogenase (LDH) release following the manufacturer’s instructions.
Cell fractionation and HK dissociation assays
Mitochondria were isolated by resuspending cells in 1.5–2 ml of ice-cold cell disruption buffer (20 mM HEPES, 10 mM, 1.5 mM MgCl2, 1 mM EDTA, 250 mM sucrose, with fresh Sigma protease inhibitors). Cells were disrupted using nitrogen cavitation at 4°C for 5 min. Nuclei and cells were removed by centrifugation at 1000xg for 5 min at 4°C. Mitochondria were isolated by centrifugation at 12,000xg for 10 min at 4°C. Mitochondria were washed with cell disruption buffer and centrifuged 9,000xg for 10 min at 4°C. The mitochondria-enriched pellet was lysed in ice-cold RIPA buffer (50 mM Tris-HCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, protease and phosphatase inhibitor cocktails (Sigma), pH 7.4). Protein was quantitating using BCA Protein Assay (Pierce, 23227). In some cases, cytosol was isolated by adding 50 μg/ml digitonin to the cell disruption buffer and incubating with rocking for 10 min. Lysates were centrifuged at 10,000xg for 10 min, and supernatants were designated cytosol. If necessary, cytosol was concentrated using Amicon 0.5 ml 3kDa concentrators (Millipore). Protein was quantified using BCA Protein Assay or Coomassie Protein Assay (Thermo Fisher, 23236). Cytosol was mixed with Laemmli sample buffer with β-mercaptoethanol, boiled at 95° for 10 min and subject to immunoblotting.
Immunoblots and VDAC cross-linking assay
Lysates were prepared with NuPAGE LDS sample buffer (Thermo Fisher, NP0008) containing β-mercaptoethanol. For supernatants, cells were treated with serum-free Opti-MEM media. Supernatants for IL-1β were precipitated using StrataClean Resin (Agilent Technologies, 400714) and resuspended in Laemmli sample buffer (Bio-Rad, 1610747). Samples were boiled at 95° for 10 min. For cross-linking assay, 1 mg/ml cells in DPBS (pH 8.3) were incubated with 200 μM ethylene glycol bis(succinimidyl succinate) (EGS, Thermo Scientific, 21565) at 30° for 15 min. After adding LDS sample buffer, samples were boiled at 70° for 10 min and sonicated for 10 s twice. Samples were resolved on 4–12% polyacrylamide Bis-Tris gels (Thermo Fisher) and transferred to PVDF membrane (Merck Millipore) using a Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were incubated at 4° overnight with the following primary antibodies as indicated: VDAC1/Porin (1:1,000 unless otherwise stated, EPR10852(B), Abcam, ab154856), VDAC3 (1:1,000, Millipore-Sigma, SAB2108497), IL-1β (1:1,000, R&D systems, AF-401-NA), caspase-1 p10 (1:1,000, Santa Cruz, sc-514), β-tubulin (1:1,000, TUB 2.1, Millipore-Sigma, T-4026), hexokinase 2 (1:1,000, C64G5, Cell Signaling, 2867), hexokinase 1 (1:1,000, G-1, Santa Cruz, sc-46695), cytochrome c (1:500, A-8, Santa Cruz, sc-13156), GAPDH (1:1,000, 6C5, Santa Cruz, sc-32233), VDAC 1/2/3 (used for Fig. 1C, 6M and 7G only) (1:1,000, D73D12, Cell Signaling, 4661), ASC (1:1,000, Enzo, ADI-905-173-100), or NLRP3/NALP3 (1:1,000, Cryo-2, AdipoGen, 102514-284) and then at room temperature for 1 h with secondary antibodies coupled to either IR dye (1:15,000, Biotium or Licor) or HRP (1:5,000, Jackson Immunobiology). HRP secondaries were detected with ECL Pico Plus (Thermo Scientific). Immunoblots were imaged using Odyssey (Licor), Chemidoc (Bio-Rad), or film. Dimers, trimers, and multimers of VDAC1 were quantified using Image Lab software (Bio-Rad).
Cytochemistry and proximity ligation assay
Cells were plated on 12 mm round coverslips as described (12). Cells treated as indicated were rinsed with warmed DPBS and fixed with 4% paraformaldehyde for 15 min. 6 M guanidine hydrochloride or Tris buffer (pH 10.0, Abcam) were used to retrieve antigen for 10 min at room temperature or 95°, respectively. For permeabilizing and blocking, cells were treated with 0.1% Triton X-100 for 10 min followed by 1% BSA and 22.52 mg/ml of glycine in PBS with 0.1% Tween 20 for 40 min. Cells were incubated at 4° overnight with following primary antibodies as indicated: VDAC1/Porin (1:100, EPR10852(B), Abcam, ab154856), hexokinase 2 (1:100, OTI4C5, Bio-Rad, VMA00174), or NLRP3/NALP3 (1:100, Cryo-2, AdipoGen, 102514–284). Cells were incubated at room temperature for 1 h with Alexa 488- or 594-conjugated IgG (1:100, Thermo Fisher). ProLong Gold Antifade Mountant (Thermo Fisher) was used for mounting.
For proximity ligation (PLA) assay, fixed and permeabilized cells were blocked as described above and stained with VDAC1/Porin (1:100, EPR10852(B), Abcam, ab154856) and NLRP3/NALP3 (1:100, Cryo-2, AdipoGen, 102514–284). PLA assay was performed using Duolink In Situ PLA Kit (Millipore-Sigma, DUO92008, DUO92014) following the manufacturer’s instructions. PLA assay results were quantified by two approaches using ImageJ software. Integrated signal density was measured in individual cells and reported as “PLA signal/cell (RFU)” or as fold change compared to untreated control samples. Alternatively, PLA signal per field of view was measured normalized to DAPI signal per field and reported fold change compared to control samples and labeled “normalized PLA signal/field (fold)”.
τ-STED-FLIM imaging and acceptor photobleaching assay
A Stellaris 8 STED microscope (Leica) with a HC Plan-Apochromat CS2 100×/1.4 oil-immersion objective lens was used for τ-STED imaging and FLIM τ-STED imaging. The laser was aligned at 80% of power, and the pixel size was set to 18.94 nm. Z-stack images were acquired with a 0.3 μm interval. The volume of each VDAC1 cluster for a single mitochondrion was measured and then averaged. Each dot represents the average volume of each VDAC1 cluster in a single mitochondrion. VDAC1 cluster volume was measured using Imaris software (Bitplane). For acceptor photobleaching assays, regions of interest were randomly designated and acceptor-photobleaching was performed using Stellaris 8 STED microscope. The efficiency of donor fluorescence in photobleached areas were measured using Leica Application Suite X software (Leica). Images showing colocalization of VDAC1 and NLRP3 were extracted using ‘RG2B colocalization’ plugin of ImageJ. Integrated density of colocalized signals was normalized by integrated density of VDAC1 signals of each cell.
Caspase-1 Activity Assay
Caspase-Glo 1 Inflammasome Assay kit (Promega, G9951) was used for measuring the activity of caspase-1 following the manufacturer’s instructions.
GFP-HK2 Expression and Live Cell Imaging
All GFP-HK2 transduction of BMDM and live cell imaging were performed as described previously (12).
qPCR
RNA was extracted from cells using RNeasy kit (Qiagen) or DNA using QiaAMP kit (Qiagen) according to the manufacturer’s instructions. Gene expression was determined using SYBR Green (Bio-Rad) with gene specific primers (IDT). PrimeTime qPCR Primer Set Vdac1-Mm.PT.58.43905165, Vdac2- Mm.PT.58.30492179, Vdac3- Mm.PT.58.5914193, Hprt- (Fwd-AGGTTGCAAGCTTGCTGGT, Rvs-TGAAGTACTCATTATAGTCAAGG), D-loop- (Fwd-CCCAAGCATATAAGCTAGTAC, Rvs-ATATAAGTCATATTTTGGGGAACTAC), CoxI- (S598-CCAGTGCTAGCCGCAGGCAT, AS685- TTGGGTCCCCTCCTCCAGCGG), Gapdh-(Fwd-GGTCTACATGTTCCAGTATGACT, Rvs-GGGTCTCGCTCCTGGAAGAT). Data were collected on a qTOWER (Analytica Gena), analyzed by 2^-ΔCt, normalized to a housekeeping gene, and plotted as fold induction.
Mitochondrial Calcium Measurement
BMDM were primed with LPS for 3 h, washed with HBSS containing 25 mM glucose and 0.5% BSA once, and stained with a final concentration of 5 μM Rhod-2 AM (Thermo Fisher, R1245MP) in 0.04% F127 Pluronic (Thermo Fisher, P2443) in HBSS solution for 20 min at 37°. Cells were washed three times with phenol red free RPMI1640 culture media and stabilized for 30 min at 37°. Cells were treated as indicated and Rhod-2 AM fluorescence was read by CLARIOstar plate reader.
Mitochondrial DNA depletion, isolation, and transfer
For mitochondrial DNA depletion, BMDM were treated with culture media containing M-CSF and 900 ng/ml of ethidium bromide (EtBr) and it was replaced every 2 days for 2 weeks (21, 72, 73). mtDNA was quantified by SYBR® Green (Bio-Rad, 1725272) qPCR using D-loop (fwd: CCCAAGCATATAAGCAAGTAC and rev: GTAGTTCCCAAAATATGACTTATAT) and CoxI (fwd: TTCGGAGCCTGAGCGGGAAT and rev: ATGCCTGCGGCTAGCACTGG) on qTOWER qPCR machine (Analytik Jena) (12). Mitochondria from BMDM were isolated for immunoblotting and mtDNA isolation for transfection nitrogen cavitation to disrupt the plasma membrane and centrifugation to purify the mitochondria (85) followed by lysis in RIPA buffer or mtDNA isolation using QIAamp DNA Mini Kit (Qiagen, 51304). Isolated mitochondrial DNA was dissolved in water and stored at −20°. 3 μg/ml mitochondrial DNA in Opti-MEM was prepared by mixed with Lipofectamine 2000 for 30 min at room temperature and added to LPS-primed BMDM for 5 h followed by ATP treatment at final concentration of 2 mM for 1 h.
Human Blood-Derived Monocytes Isolation and Stimulation
Blood was collected from male and female healthy donors after informed consent by the Cedars-Sinai Medical Center MIRIAD Biobank in accordance with Cedars-Sinai Medical Center Institutional Review Board procedures. Peripheral blood mononuclear cells (PBMC) were separated using Ficoll-Paque (Cytiva). Monocytes were isolated using MojoSort Human CD14 Selection Kit (BioLegend, 480026) and an autoMACS Pro Separator (Miltenyi). Monocytes were stimulated with 2 μg/ml for 24h. IL-1β was measured by ELISA (BioLegend, 437004).
Statistical analysis
GraphPad Prism 8.0 software or Excel were used for data analysis. Data are presented as means ± SEM. Statistical significance was determined by t tests (two-tailed) or one-way or two-way analysis of variance (ANOVA) with multiple comparisons test for more groups. P < 0.05 was considered statistically significant where *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments:
Funding:
This study was supported by National Institutes of Health grants R01 AI148465 (AJW), R01 GM085796 (DMU), and R01 AI071116 (DMU).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data needed to support the conclusions of the paper are available in the main text or the supplementary materials. Materials are available upon request from the corresponding authors.
REFERENCES AND NOTES
- 1.Nozaki K, Li L, Miao EA, Innate Sensors Trigger Regulated Cell Death to Combat Intracellular Infection. Annu. Rev. Immunol 40, 469–498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tartey S, Kanneganti TD, Differential role of the NLRP3 inflammasome in infection and tumorigenesis. Immunology 156, 329–338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant R, Dixit V, Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol 4, 50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tzeng T-C, Golenbock D, NLRP3 inflammasome activation in Alzheimer’s disease (INC9P.446). J. Immunol 192, 188.185–188.185 (2014). [Google Scholar]
- 5.Zhang Y, Dong Z, Song W, NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target Ther 5, 37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heijden T. v. d., Kritikou E, Venema W, Duijn J. v., Santbrink P. J. v., Slütter B, Foks AC, Bot I, Kuiper J, NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E Deficient Mice. Arterioscler., Thromb., Vasc. Biol 37, 1457–1461 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP-Y, The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med 207, 1045–1056 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M, Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R, MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep 8, 8618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhen Y, Zhang H, NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol 10, 276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM, Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7, 38–49 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu NI, Coggeshall KM, Arditi M, Underhill DM, Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 166, 624–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanson KV, Deng M, Ting JP, The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S, Magnani G, Cikovic T, Hartjes L, Smollich J, Robertson AAB, Cooper MA, Schmidt-Supprian M, Schuster M, Schroder K, Broz P, Traidl-Hoffmann C, Beutler B, Kuster B, Ruland J, Schneider S, Perocchi F, Gross O, K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 45, 761–773 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V, Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 44, 833–846 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, Rathkey JK, Abbott DW, Wang K, Shao F, Catz SD, Dubyak GR, Pearlman E, N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun 11, 2212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T, Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A 109, 11282–11287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M, Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, Gottlieb RA, Chen S, Arditi M, Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res 119, e76–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS, Karin M, Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55, 1370–1385 e1378 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X.-j., Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M, New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, Gottlieb RA, Sutterwala FS, Cassel SL, Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol 200, 3047–3052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS, Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Wang T, He K, Xu M, Gong JP, Cardiolipin inhibitor ameliorates the non-alcoholic steatohepatitis through suppressing NLRP3 inflammasome activation. Eur. Rev. Med. Pharmacol. Sci 23, 8158–8167 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, Alnemri ES, The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol 191, 4358–4366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN, The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seoane PI, Lee B, Hoyle C, Yu S, Lopez-Castejon G, Lowe M, Brough D, The NLRP3–inflammasome as a sensor of organelle dysfunction. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alfonso-Loeches S, Urena-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C, Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell Neurosci 8, 216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klepinin A, Ounpuu L, Mado K, Truu L, Chekulayev V, Puurand M, Shevchuk I, Tepp K, Planken A, Kaambre T, The complexity of mitochondrial outer membrane permeability and VDAC regulation by associated proteins. J. Bioenerg. Biomembr 50, 339–354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts DJ, Miyamoto S, Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ 22, 248–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.John S, Weiss JN, Ribalet B, Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One 6, e17674 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Jesus A, Keyhani-Nejad F, Pusec CM, Goodman L, Geier JA, Stoolman JS, Stanczyk PJ, Nguyen T, Xu K, Suresh KV, Chen Y, Rodriguez AE, Shapiro JS, Chang HC, Chen C, Shah KP, Ben-Sahra I, Layden BT, Chandel NS, Weinberg SE, Ardehali H, Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell 82, 1261–1277 e1269 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM, Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol 8, 460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V, The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci 122, 1906–1916 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Betaneli V, Petrov EP, Schwille P, The role of lipids in VDAC oligomerization. Biophys. J 102, 523–531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keinan N, Tyomkin D, Shoshan-Barmatz V, Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol 30, 5698–5709 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varughese JT, Buchanan SK, Pitt AS, The Role of Voltage-Dependent Anion Channel in Mitochondrial Dysfunction and Human Disease. Cells 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zalk R, Israelson A, Garty ES, Azoulay-Zohar H, Shoshan-Barmatz V, Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J 386, 73–83 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsujimoto Y, Shimizu S, VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ 7, 1174–1181 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Ciscato F, Filadi R, Masgras I, Pizzi M, Marin O, Damiano N, Pizzo P, Gori A, Frezzato F, Chiara F, Trentin L, Bernardi P, Rasola A, Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca(2+) -dependent death of cancer cells. EMBO Rep 21, e49117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G, K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J, Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–1589 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Tapia-Abellán A, Angosto-Bazarra D, Alarcón-Vila C, Baños MC, Hafner-Bratkovič I, Oliva B, Pelegrín P, Sensing low intracellular potassium by NLRP3 results in a stable open structure that promotes inflammasome activation. Science Advances 7, eabf4468 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pastorino JG, Hoek JB, Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr 40, 171–182 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V, Voltage-dependent Anion Channel 1-based Peptides Interact with Hexokinase to Prevent Its Anti-apoptotic Activity*. J. Biol. Chem 284, 3946–3955 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Shoshan-Barmatz V, Shteinfer-Kuzmine A, Verma A, VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keinan N, Pahima H, Ben-Hail D, Shoshan-Barmatz V, The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 1833, 1745–1754 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Israelson A, Zaid H, Abu-Hamad S, Nahon E, Shoshan-Barmatz V, Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1. Cell Calcium 43, 196–204 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Tan VP, Smith JM, Tu M, Yu JD, Ding EY, Miyamoto S, Dissociation of mitochondrial HK-II elicits mitophagy and confers cardioprotection against ischemia. Cell Death Dis. 10, 730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A, Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One 3, e1852 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisthal S, Keinan N, Ben-Hail D, Arif T, Shoshan-Barmatz V, Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 1843, 2270–2281 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Ben-Hail D, Begas-Shvartz R, Shalev M, Shteinfer-Kuzmine A, Gruzman A, Reina S, De Pinto V, Shoshan-Barmatz V, Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem 291, 24986–25003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma A, Pittala S, Alhozeel B, Shteinfer-Kuzmine A, Ohana E, Gupta R, Chung JH, Shoshan-Barmatz V, The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: a potential therapeutic target. Mol. Ther 30, 726–744 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, Kang H, Brown AL, Park S-J, Xu X, Zandee van Rilland E, Kim MK, Cohen JI, Kaplan MJ, Shoshan-Barmatz V, Chung JH, VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q, Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 23, 1239–1247 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Messina A, Reina S, Guarino F, De Pinto V, VDAC isoforms in mammals. Biochim. Biophys. Acta 1818, 1466–1476 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Messina A, Reina S, Guarino F, De Pinto V, VDAC isoforms in mammals. Biochimica et Biophysica Acta (BBA) - Biomembranes 1818, 1466–1476 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Bryant C, Inflammasome activation by Salmonella. Curr. Opin. Microbiol 64, 27–32 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Bae JH, Park JW, Kwon TK, Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem. Biophys. Res. Commun 303, 1073–1079 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Woods JJ, Wilson JJ, Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol 55, 9–18 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Gincel D, Zaid H, Shoshan-Barmatz V, Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J 358, 147–155 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amanakis G, Murphy E, Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol 11, 595 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X, DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. U.S.A 116, 25322–25328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R, Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol 175, 901–911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA, Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM, Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Chen J, Chen ZJ, PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S, Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol 14, 454–460 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, Shin DM, Nguyen LT, Lee MS, Jin HS, Kim KK, Lee CH, Kim MH, Park SG, Kim JM, Choi HS, Jo EK, Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun 6, 6115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu Y, Huang Y, Chen M, Yang Y, Li X, Zhang W, Mitochondrial DNA in NLRP3 inflammasome activation. Int. Immunopharmacol 108, 108719 (2022). [DOI] [PubMed] [Google Scholar]
- 72.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M, New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warren EB, Aicher AE, Fessel JP, Konradi C, Mitochondrial DNA depletion by ethidium bromide decreases neuronal mitochondrial creatine kinase: Implications for striatal energy metabolism. PLoS One 12, e0190456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rathinam VAK, Zhao Y, Shao F, Innate immunity to intracellular LPS. Nat Immunol 20, 527–533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonora M, Giorgi C, Pinton P, Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol 23, 266–285 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, Kang H, Brown AL, Park SJ, Xu X, Zandee van Rilland E, Kim MK, Cohen JI, Kaplan MJ, Shoshan-Barmatz V, Chung JH, VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, Wu H, NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell 184, 6299–6312 e6222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, Latz E, Engel C, Geyer M, Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 604, 184–189 (2022). [DOI] [PubMed] [Google Scholar]
- 79.Schmacke NA, O’Duill F, Gaidt MM, Szymanska I, Kamper JM, Schmid-Burgk JL, Madler SC, Mackens-Kiani T, Kozaki T, Chauhan D, Nagl D, Stafford CA, Harz H, Frohlich AL, Pinci F, Ginhoux F, Beckmann R, Mann M, Leonhardt H, Hornung V, IKKbeta primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity 55, 2271–2284 e2277 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tabara LC, Morris JL, Prudent J, The Complex Dance of Organelles during Mitochondrial Division. Trends Cell Biol 31, 241–253 (2021). [DOI] [PubMed] [Google Scholar]
- 81.Ciscato F, Ferrone L, Masgras I, Laquatra C, Rasola A, Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci 22, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lauterwasser J, Todt F, Zerbes RM, Nguyen TN, Craigen W, Lazarou M, van der Laan M, Edlich F, The porin VDAC2 is the mitochondrial platform for Bax retrotranslocation. Sci. Rep 6, 32994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rostovtseva TK, Bezrukov SM, Hoogerheide DP, Regulation of Mitochondrial Respiration by VDAC Is Enhanced by Membrane-Bound Inhibitors with Disordered Polyanionic C-Terminal Domains. Int. J. Mol. Sci 22, 7358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V, Voltage-dependent Anion Channel 1-based Peptides Interact with Hexokinase to Prevent Its Anti-apoptotic Activity*. Journal of Biological Chemistry 284, 3946–3955 (2009). [DOI] [PubMed] [Google Scholar]
- 85.Gottlieb RA, Adachi S, Nitrogen cavitation for cell disruption to obtain mitochondria from cultured cells. Methods Enzymol 322, 213–221 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to support the conclusions of the paper are available in the main text or the supplementary materials. Materials are available upon request from the corresponding authors.