

ABSTRACT
Here, we generated bispecific antibody (bsAb) derivatives that mimic the function of interleukin (IL)-18 based on single domain antibodies (sdAbs) specific to IL-18 Rα and IL-18 Rβ. For this, camelids were immunized, followed by yeast surface display (YSD)-enabled discovery of VHHs targeting the individual receptor subunits. Upon reformatting into a strictly monovalent (1 + 1) bispecific sdAb architecture, several bsAbs triggered dose-dependent IL-18 R downstream signaling on IL-18 reporter cells, as well as IFN-γ release by peripheral blood mononuclear cells in the presence of low-dose IL-12. However, compared with IL-18, potencies and efficacies were considerably attenuated. By engineering paratope valencies and the spatial orientation of individual paratopes within the overall design architecture, we were able to generate IL-18 mimetics displaying significantly augmented functionalities, resulting in bispecific cytokine mimetics that were more potent than IL-18 in triggering proinflammatory cytokine release. Furthermore, generated IL-18 mimetics were unaffected from inhibition by IL-18 binding protein decoy receptor. Essentially, we demonstrate that this strategy enables the generation of IL-18 mimetics with tailor-made cytokine functionalities.
KEYWORDS: Antibody engineering, bispecific antibody, cytokine mimetic, IL-18, IL-18 binding protein, IL-18BP, single domain antibody, surrogate agonist, valencies, VHH, yeast surface display
Introduction
Cytokines are potent immunomodulatory proteins with substantial therapeutic potential. Several such molecules, for instance, recombinant human (rh) IL-2 (aldesleukin) or (rh) TNF (tasonermin), have been granted marketing approval for the treatment of different diseases.1–3 However, the pleiotropic mechanism of action of many cytokines, their short half-life combined with often dose-limiting toxicities when administered systemically hamper their therapeutic applicability.2,4 To address these inherent limitations, ‘next-generation cytokines’ were generated.5 For instance, muteins of IL-2 that do not target the α-subunit of the IL-2 receptor (CD25) were engineered in order to eliminate the intrinsic bias of this cytokine for activating regulatory T cells (Tregs).6,7 To optimize the poor pharmacokinetics of cytokines, fusions with the (effector silenced or attenuated) Fc portion of IgGs8 and conjugates with polyethylene glycol were engineered.9 Moreover, bifunctional antibody cytokine fusion proteins were constructed aiming at accumulating the immunomodulatory function of cytokines at the site of disease.10,11 Another interesting approach regarding ‘next-generation cytokines’ relies on exploiting antibody-derivatives that mimic the function of cytokines, referred to as cytokine mimetics or surrogate cytokines.12 In this respect, bispecific antibody derivatives (diabodies) have been developed that dimerize the erythropoietin receptor (EpoR).13 Intriguingly, different diabodies induced differential EpoR phosphorylation ranging from weak and partial agonism to full agonism. In addition to this, single domain antibodies (sdAb)-based bispecifics were generated that activate signaling through the heterodimeric IL-2 receptor βγ (IL-2 Rβγ). These bsAbs mimic the function of IL-2, but without preferential activation of Tregs via IL-2 Rα binding.14 Garcia and coworkers recently described the engineering of VHH-derived surrogate agonists targeting IL-2 Rβγ, as well as type I IFN-mimicking bsAbs.15 Importantly, the group constructed surrogate agonists displaying functional diversification in terms of receptor downstream signaling compared to the natural cytokine. Moreover, also bsAbs were constructed triggering agonistic activity through binding to IL-2 Rβ and IL-10 Rβ, a receptor heterodimer that naturally does not exist. This clearly demonstrates that the modular assembly of bsAbs enables the generation of cytokine mimetics with tailor-made functionalities, which might be versatile building blocks for drug discovery.
Herein, we describe the generation of sdAb-derived bispecific surrogate agonists mimicking the functionality of IL-18 that are resistant to the decoy receptor IL-18BP. IL-18 is a proinflammatory cytokine belonging to the IL-1 family of cytokines that mediates signaling through heterodimerization of the receptor subunits IL-18 Rα and IL-18 Rβ.16,17 IL-18 stimulates IFN-γ production in innate lymphoid cells as well as antigen-experienced T cells in synergy with IL-12.18 Recombinant human (rh) IL-18 has been assessed in clinical trials and demonstrated a favorable toxicity profile, but limited efficacy as monotherapy.19–21 Physiologically, the activity of IL-18 is balanced via the high-affinity neutralizing and naturally occurring IL-18BP.17,18 After treatment with (rh) IL-18, substantially elevated levels of IL-18BP were found in the serum of patients.20,22 Ring and colleagues were able to show that IL18BP is expressed in the tumor microenvironment of several tumor types.23 Moreover, the authors found increased IL-18BP concentrations in the serum of patients with non-small cell lung cancer, which were further elevated post PD-1 or PD-L1 treatment. In the same study, the group engineered a decoy-resistant IL-18 mutein that still triggered IL-18 receptor activation. This next-generation IL-18 derivative showed superior antitumor efficacy in preclinical models as monotherapy and in combination with immune checkpoint inhibition.23
In this work, we present an alternative route to obtain IL-18 R agonists. This strategy relies on bispecific sdAb-based cytokine mimetics targeting IL-18 Rα and IL-18 Rβ. To this end, camelids were immunized with both receptor subunits and antigen-specific VHHs were identified by YSD.24 Several IL-18 R agonists with varying signaling capacities were obtained by combinatorial reformatting individual sdAbs into a strictly monovalent (1 + 1) bsAb architecture. By engineering of the overall design architecture with respect to valencies and to the spatial orientation of individual VHH-based paratopes within one molecule, IL-18 mimetics with tailor-made agonistic activities were retrieved. Essentially, generated bispecifics were tolerant to inhibition by IL-18BP receptor decoy (Figure 1A).
Figure 1.
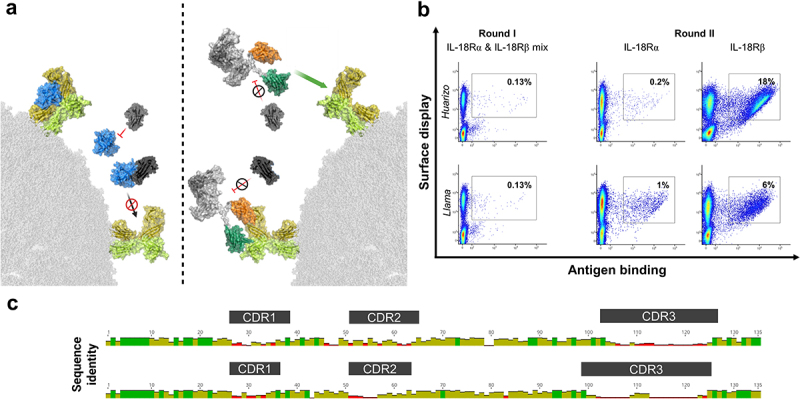
Overall strategy for the generation of tailor-made cytokine mimetics based on sdAb-derived bispecifics and YSD-enabled antibody discovery.
(A) Left: IL-18 (blue) triggers IL-18 R downstream signaling by consecutive binding to IL-18 Rα (yellow) followed by IL-18 Rβ (lime) recruitment. IL-18BP (dark gray) inhibits IL-18 by high-affinity binding and hence, by blocking the IL-18/IL-18 Rα interaction. Right: Camelid-derived sdAbs enable targeting IL18-Rα (orange) and IL-18 Rβ (green). Upon reformatting into an IgG-like bispecific, resulting cytokine mimetics cross-link the IL18R subunits and elicit down-stream signaling. Essentially, generated IL-18 mimetics are resistant to inhibition by IL-18BP. Structural visualization was generated with PyMOL software version 2.3.0, based on PDB entries 3WO4 and 7AL7, structural modeling as described in the methods section and modified using www.biorender.com. (B) Immunization of camelids followed by YSD facilitate the enrichment of sdAbs specific to (rh) IL-18 Rα and (rh) IL-18 Rβ. One sublibrary was generated for each specimen (huarizo and llama) and sorted separately. In the first round of selection, a mixture of both (rh) receptor subunits was exploited at a concentration of 250 nM. In the subsequent second sorting round, enriched libraries were sorted separately against each antigen at 250 nM. A two-dimensional sorting strategy was applied to select for full-length VHH display in addition to antigen binding. Percentage of cells in sorting gates are shown. Plots show 5 × 104 events. (C) Graphical alignments of 55 unique sdAb clones addressing IL-18 Rα (top) and 101 independent clones targeting IL-18 Rβ (bottom) retrieved from YSD library sorting. Complementarity-determining regions (CDRs) are highlighted. Red bars indicate high sequence diversity at the amino acid level and green bars represent high sequence conservation at a given position. Alignment conducted with MUSCLE alignment tool using Geneious Prime 2021.1.1.
Results
Camelid immunization combined with YSD enables the isolation of sdAbs targeting IL-18 Rα and IL-18 Rβ
For the isolation of VHHs targeting IL-18 Rα and IL-18 Rβ, one llama and one huarizo (llama x alpaca) were immunized with a cocktail of the extracellular domains (ECDs) of both receptor subunits. Afterwards, for each specimen a YSD library was generated as described previously24,25 and both libraries were sorted individually. We applied a two-dimensional sorting strategy to select for full-length VHH display via hemagglutinin (HA) tag staining simultaneous to the binding functionality by exploiting an antigen concentration of 250 nM for each respective receptor subunit. In the first round of selection, we used a mixture of both receptor subunits (Figure 1B). In the second selection round, the enriched libraries were selected against both antigens separately and for each library and each receptor subunit we were able to enrich for an antigen-binding population (Figure 1B, Fig. S1). From the target-enriched libraries, 96 clones were sent for sequencing for each antigen. This resulted in 55 unique clones in total enriched for binding to IL-18 Rα and 101 unique clones for IL-18 Rβ, respectively (Figure 1C). By applying a clonotyping strategy based on complementarity-determining region-3 (CDR3) diversity, we selected 11 VHHs targeting each receptor subunit, respectively, for expression. To this end, we expressed all 22 sdAbs as one-armed (1 + 0) antibodies by using the strand-exchange engineered domain (SEED) technology, which relies on beta-strand exchanges of IgG and IgA CH3 constant domains, resulting in preferential heavy chain heterodimerization (Fig. S2A).26 sdAbs enriched for IL-18 Rα binding were grafted onto the hinge region of the AG chain of the SEEDbody and produced with a paratope-less GA chain, while sdAbs targeting IL-18 Rβ were fused to the GA chain and combined with a non-targeted AG chain. An effector-silenced derivative of the SEEDbody Fc region was exploited in order to abolish the potential of antibody-dependent cell-mediated cytotoxicity (ADCC) in primary cell assays. Following expression in Expi293™ cells and protein A purification, we assessed binding to the (rh) ECDs of IL-18 Rα and IL-18 Rβ by biolayer interferometry (BLI) using an antigen concentration of 100 nM (Fig. S2B). As expected, (given the low sequence identity of approximately 21% between IL-18 Rα ECD and IL-18 Rβ ECD), monospecific molecules obtained from the IL-18 Rα sorts only showed binding to IL-18 Rα ECD (ms_IL18R_VHHα1–11), but not to IL-18 Rβ ECD which was vice versa for monospecific clones selected from the IL-18 Rβ enrichments (ms_IL18R_VHHβ12–22). In addition to this, ms_IL18R_VHHα7 only displayed negligible binding to IL-18 Rα ECD, whereas IL-18 Rβ-directed paratope ms_IL18R_VHHβ19 exhibited no binding at all. Consequently, both sdAbs were excluded from further consideration.
Combinatorial reformatting of sdAbs targeting individual IL-18 receptor subunits as (1 + 1) bsAbs facilitates the identification of IL-18 receptor agonists
For functional characterization, all 10 VHHs targeting IL-18 Rα (engrafted to the AG chain of the SEED architecture) as well as all 10 sdAbs specific for IL-18 Rβ (fused to the GA chain) were recombined and expressed as bispecific SEEDbodies, resulting in 100 bispecific sdAb-based antibody derivatives. As initial cutoff in terms of biophysical properties, we only considered bispecifics for further investigation that comprised more than 86% target peak in analytical size exclusion chromatography (SEC) post-protein A purification, resulting in 12 molecules that were excluded. Interestingly, this encompassed all bsAbs harboring IL18R_VHHα11, indicating liabilities of the respective sdAb paratope. Notwithstanding, 88 VHH-derived bsAbs were assessed regarding their agonistic potential exploiting IL-18 reporter cells, stably expressing IL-18 Rα and IL-18 Rβ (HEK-Blue™ IL-18 cells). In an initial experiment, bsAbs and all monospecific SEEDbodies (either series ms_IL18R_VHHα1–11 or ms_IL18R_VHHβ12–22) were used at a concentration of 50 nM. Recombinant human (rh) TNF was used as negative control and (rh) IL-18 as positive control, showing a dose-dependent activation of IL-18 reporter cells (Fig. S3A). As expected, none of the monospecific SEEDbodies were able to agonize IL-18 reporter cells, whereas 44 of the 88 sdAb-based bispecifics induced NFκB activation that was at least 1.5-fold higher than for TNF at this fixed concentration (data not shown).
These molecules were further characterized exploiting the reporter cell assay in a dose-dependent manner. To this end, we also included IL18R_VHHα1β16, which did not show agonism potential but specific binding to the respective receptor subunits as negative control (Figure 2A, Fig. S3). Intriguingly, all 44 different bispecifics were able to agonize the IL-18 reporter cells to a certain extent, whereas IL18R_VHHα1β16 did not induce activation, as exemplarily shown for surrogate agonists IL18R_VHHα2β15, IL18R_VHHα8β15, IL18R_VHHα2β17, and IL18R_VHHα8β17 in Figure 2A. However, compared with (rh) IL-18, potencies (EC50 of NFκB reporter activation) as well as the magnitude (maximal NFκB activation) were attenuated for all bispecifics tested (Fig. S3A, B). According to their SEC profile and agonism potential, all bispecifics were ranked into four groups: molecules discarded due to low purity (target species < 86%; gray), functionally inactive bispecifics (red), minimally active agonists eliciting less than 15% of NFκB reporter activation normalized to (rh) IL-18 at 1 nM or EC50 higher than 0.1 nM (yellow) and moderately active surrogate agonists triggering either a normalized NFκB reporter activation of more than 15% relative to (rh) IL18 or displaying potencies of less than 0.1 nM (green, Figure 2B).
Figure 2.
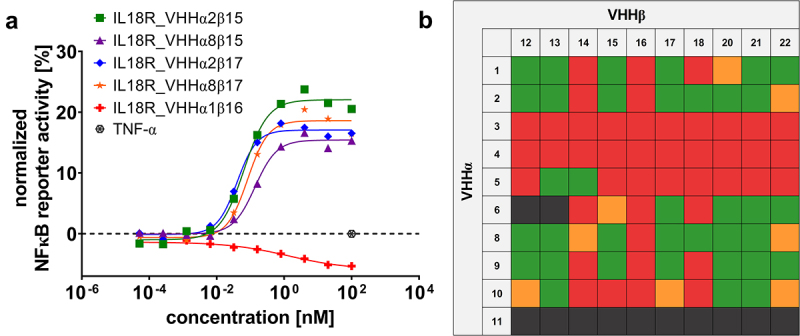
Combinatorial reformatting of monospecific (1 + 0) SEEDbodies into strictly monovalent (1 + 1) bsAbs enables the identification of IL-18 mimetics with attenuated capacities to trigger NFκB reporter activity on IL-18 reporter cells.
(A) HEK-Blue™ reporter cells were incubated with increasing concentrations of reformatted bsAbs, as exemplarily shown for IL18R_VHHα2β15, IL18R_VHHα8β15, IL18R_VHHα2β17, IL18R_VHHα8β17 and IL18R_VHHα1β16. Secreted embryonic alkaline phosphatase activity was monitored by determining the OD640. Reporter activity was normalized to maximal IL-18 read-out. As negative control, (rh) TNF was used. Graph shows one respective screening experiment. (B) Heatmap of NFκB reporter activitation elicited by combinatorial reformatted (1 + 1) bsAbs. Molecules failing initial quality control (target monomer peak in SEC < 86% post-protein A purification given in dark gray, functionally inactive bsAbs shown in red, minimally active surrogate agonists (NFκB reporter activitation < 15% compared to (rh) IL-18 at 1 nM or EC50 ≥0.1 nM) in yellow and moderately active IL-18 mimetics (NFκB reporter activitation ≥ 15% compared to (rh) IL-18 at 1 nM or EC50 <0.1 nM) depicted in green.
Bispecific sdAb-derived IL-18 mimetics elicit dose-dependent release of IFN-γ in peripheral blood mononuclear cell-based assays
To evaluate the functionality of the engineered bispecific cytokine mimetics in primary cell assays, we stimulated human peripheral blood mononuclear cells (PBMCs) either with (rh) IL-18 or with a selection of 16 different surrogate agonists that showed at least some level of agonistic activity in the IL-18 reporter cell assay (ranging from minimally active to moderately active agonists) in combination with low dose (rh) IL-12. Again, IL18R_VHHα1β16 was included as a negative control. To this end, we exploited a fixed concentration of 100 nM of each of bsAbs scrutinized, whereas a concentration of 1 nM was used for (rh) IL-18 due to an anticipated much stronger potency. As functional read out, we used PBMC-derived production of IFN-γ (Figure 3A). Stimulation of PBMCs with (rh) IL-18 triggered a robust release of IFN-γ (mean release at 1 nM of 1640.7 pg/mL), whereas IFN-γ production was negligible for the treatment with IL18R_VHHα1β16 (mean release at 100 nM of 39.5 pg/mL). For the 16 bispecific surrogate agonists, we observed a quite diverse release of IFN-γ at 100 nM. While for five bsAbs (IL18R_VHHα5β13, IL18R_VHHα2β22, IL18R_VHHα6β22, IL18R_VHHα8β22, IL18R_VHHα10β22), the magnitude of IFN-γ production was not appreciably different from the negative control (IL18R_VHHα1β16), there was a trend for higher proinflammatory cytokine release for the vast majority of bispecific surrogate agonists tested. Most importantly, sdAb-based IL-18 mimetics IL18R_VHHα2β15, IL18R_VHHα2β17, and IL18R_VHHα8β17 evoked a statistically significant higher IFN-γ production compared to IL18R_VHHα1β16, with IL18R_VHHα2β15 triggering the most prominent release (mean release 1390.2 pg/mL). In parallel, we also assessed IFN-γ production of all the molecules without low-dose (rh) IL-12 (Fig. S4). Neither (rh) IL-18, nor the generated bispecifics triggered IFN-γ production in the absence of (rh) IL-12, which is in accordance with the finding, that IL-18 induces IFN-γ either with IL-12 or IL-15.17
Figure 3.
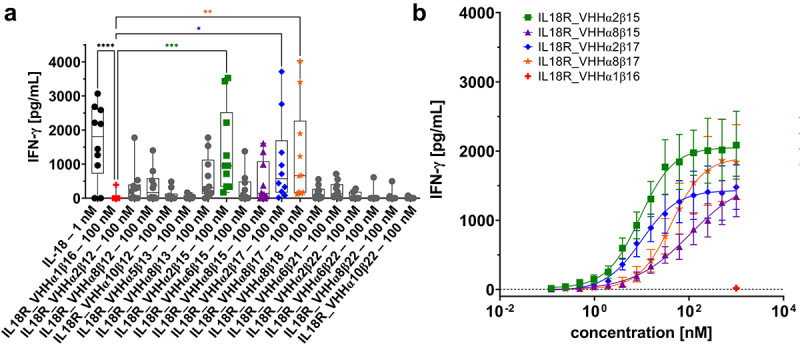
Bispecific (1 + 1) surrogate agonists trigger IFN-γ release on PBMCs isolated from healthy donors.
(A) IFN-γ production of PBMCs stimulated with bsAbs at a fixed concentration of 100 nM or with (rh) IL-18 at 1 nM. Experiments were performed in the presence of 10 ng/mL (rh) IL-12. Graph shows box and whisker plots as superimpositions with dot plots of IFN-γ release of 10 different donors. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.IL18R_VHHα1β16 was used as negative control (given in red). Four leading candidates used for further characterization shown in green, purple, blue, and orange. (B) TOP4 candidates evoke a dose-dependent IFN-γ read-out on PBMCs in the presence of low dose (rh) IL-12 (10 ng/mL). IL18R_VHHα1β16 as negative control shown in red was used at a fixed concentration of 1 µM. Mean values ± SEM of 10 independent experiments p[,//’/l;\]=\][l; are shown.
To further narrow down surrogate agonists for in-depth characterization, we only focused on the three molecules that triggered significant release of IFN-γ, as well as IL18R_VHHα8β15, which also elicited a moderate production of IFN-γ, albeit not being statistically significant. Remarkably, those four bispecifics were composed of different combinations of two sdAbs targeting IL-18 Rα (VHHα2 and VHHα8) as well as two VHHs specific for the IL-18 Rβ subunit (VHHβ15 and VHHβ17). For detailed characterization, we stimulated human PBMCs isolated from healthy donors with the four bispecific candidates using a range of different concentrations in combination with low dose (rh) IL-12 (Figure 3B). All four bispecific IL-18 mimetics triggered a dose-dependent release of IFN-γ with potencies (EC50 of IFN-γ production) of 9.1 nM to ~107 nM (Figure 3B, Table 1). Of note, IL18R_VHHα8β15 did not reach a maximum IFN-γ release at the highest concentration tested (1 µM). Hence, we have not been able to determine the EC50 of this bsAb accurately.
Table 1.
Binding kinetics and functional properties of the herein generated four leading bispecific IL-18 mimetics.
| Samples | KD IL-18 Rα [nM] | kon IL-18 Rα [1/Ms] | kdis IL-18 Rα [1/s] | KD IL-18 Rβ [nM] | kon IL-18 Rβ [1/Ms] | kdis IL-18 Rβ [1/s] | Competition vs. IL-18 on IL18Rα | EC50 IFN-γ Release [nM] |
|---|---|---|---|---|---|---|---|---|
| IL18R_VHHα2β15 | 5.2 | 1.36E + 05 | 7.00E–04 | 12.8 | 3.37E + 05 | 4.32E–03 | Yes | 9.1 |
| IL18R_VHHα8β15 | 27.7 | 3.31E + 05 | 9.17E–03 | 9.9 | 3.77E + 05 | 3.71E–03 | Yes | 106.9 |
| IL18R_VHHα2β17 | 4.1 | 1.48E + 05 | 6.09E–04 | 9.1 | 2.26E + 05 | 2.06E–03 | Yes | 9.9 |
| IL18R_VHHα8β17 | 35.6 | 3.11E + 05 | 1.11E–02 | 7.6 | 2.30E + 05 | 1.75E–03 | Yes | 42.7 |
Besides functional characterization, we also determined affinities of the four leading (TOP4) IL-18 mimetics against each receptor subunit (Table 1, Fig. S5). Affinities for binding to IL-18 Rα ranged from 5.2 nM for IL18R_VHHα2β15 to 35.6 nM for IL18R_VHHα8β17. Binding to the IL-18 Rβ subunit ranged from the single digit nanomolar range to binding in the lower double digit nanomolar range (for IL18R_VHHα2β15). Interestingly, there was a trend toward higher potencies and efficacies in terms of IFN-γ production for cytokine mimetics harboring a VHH with higher binding affinities to IL-18 Rα which were mainly driven by an improved off-rate (VHHα2 vs. VHHα8).
Antibody format engineering enables the generation of IL-18 cytokine mimetics with augmented and tailor-made functional properties
Next, we set out to further augment the functional properties of the herein generated bispecific surrogate agonists. To this end, we focused on IL18R_VHHα2β15, which triggered the most prominent production of IFN-γ on human PBMCs. In particular, we aimed at investigating the influence of paratope valencies, as well as the spatial orientation of individual paratopes within the overall antibody design architecture, on provoking a functional IFN-γ response. The different antibody designs are shown in Figure 4A. Besides the strictly monovalent (1 + 1) VHH SEED format exploited for initial IL-18 mimetic generation and characterization (IL18R_VHHα2β15), we used two additional designs. In 2020, our group described a VHH-based IgG-like bi- and multispecific antibody platform that relies on the replacement of the VH and the VL regions of a conventional antibody by two independently functioning VHH domains, resulting in a bispecific, tetravalent antibody derivative, herein referred to as single domain-based IgG (sdIgG).27 We engrafted the IL-18 Rα-specific VHHα2 onto the CH1 domain of the effector-silenced heavy chain (IgG1) and the IL-18 Rβ-targeting VHHβ15 onto the constant region of the lambda light chain (IL18R_sdIgGα2β15). Besides this IgG-like design, we also constructed tandem-VHH arrangements, grafted onto the hinge-region of an effector-silenced IgG1 Fc region (2 + 2). The two independent VHH domains were separated by a five amino acid Gly4Ser-linker. For this, we assessed both orientations, VHHα2 followed by VHHβ15 (from N-terminus to C-terminus, IL18R_tanVHHα2β15) and vice versa (IL18R_tanVHHβ15α2). In addition to these four molecules in total (including the initial 1 + 1 SEED design), all formats were also produced harboring the E340G mutation, that was first described by Parren and colleagues.28 This mutation enhances antibody hexamer formation on the target cell surface after antigen binding and was initially used to induce conditional complement-dependent cytotoxicity. In this regard, we speculated that on-target hexamerization of the different surrogate agonist formats would result in enhanced receptor clustering and consequently in improved IFN-γ response.
Figure 4.
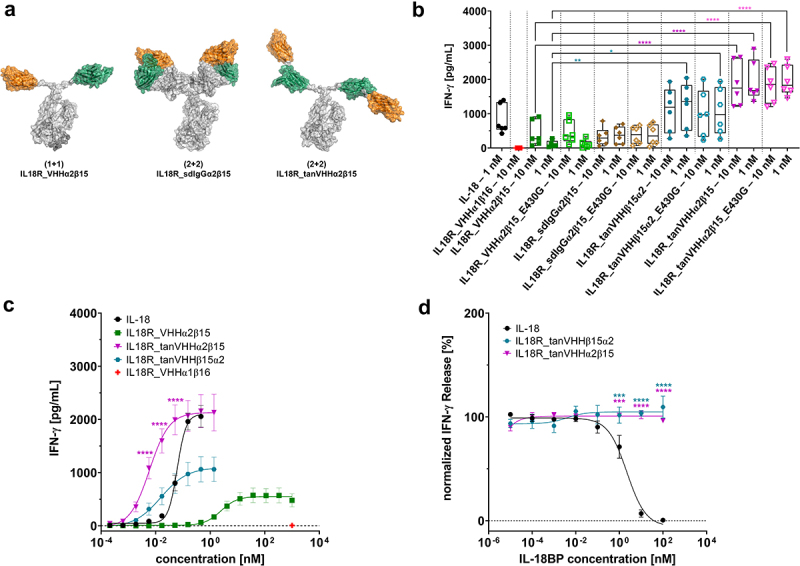
Antibody Engineering enables the generation of IL-18 mimetics with augmented agonism capacities.
(A) Schematic depiction of main different bispecific antibody architectures that were constructed within this work. Fusion of an anti-IL18Rα VHHα2 (orange) to the hinge region of the AG chain of the SEEDbody as well as engraftment an anti-IL18Rβ VHHβ15 (green) onto the GA chain results in the initially generated (1 + 1) format IL18R_VHHα2β15. Replacing the VH and VLλ of an effector silenced IgG by VHHα2 and VHHβ15, respectively, facilitates the generation of the IL18R_sdIgGα2β15 architecture (2 + 2). Within the IL18R_tanVHHα2β15 design (2 + 2), VHHα2 and VHHβ15 are arranged in tandem (from N-terminus to C-terminus) and separated by a five amino acid Gly4Ser linker. The tandem is fused to the hinge region of an effector silenced IgG1 Fc fragment. Of note, also the opposite orientation was constructed (VHHβ15 followed by VHHα2, IL18R_tanVHHβ15α2). In addition, all four molecules were also produced harboring the E430G mutation for on-target hexamerization. (B) Distinct surrogate agonist formats of the same paratopes (VHHα2 and VHHβ15) display differential properties in eliciting a functional IFN-γ response on human PBMCs isolated from healthy donors at fixed concentrations. Experiments were performed at two different concentrations (10 nM and 1 nM) in the presence of 10 ng/mL (rh) IL-12. Graph shows box and whisker plots as superimpositions with dot plots of IFN-γ release of six different donors. ****p < 0.0001, **p < 0.01, *p < 0.05. (C) Surrogate agonists arranged in tandem (IL18R_tanVHHα2β15 and IL18R_tanVHHβ15α2) elicit enhanced IFN-γ production in terms of potencies and magnitude on PBMCs isolated from healthy donors, resulting in a variant with increased potencies compared with (rh) IL-18. All experiments were performed in the presence of low dose (rh) IL-12 (10 ng/mL). IL18R_VHHα1β16 as negative control shown in red was used at a fixed concentration of 1 µM. Mean values ± SEM of 13 independent experiments are shown.****p < 0.0001(D) Potency augmented tandem IL-18 mimetics are resistant to inhibition by (rh) IL-18BP, whereas (rh) IL-18 is efficiently blocked from signaling. PBMCs of healthy human donors were stimulated either with (rh) IL-18 or IL18R_tanVHHα2β15 and IL18R_tanVHHβ15α2 at a fixed concentration of 0.5 nM in the presence of (rh) IL-12 (10 ng/mL) and different concentrations of (rh) IL-18BP. Five independent experiments were performed and mean values ± SEM are shown. ****p < 0.0001,***p < 0.001.
The different surrogate agonist formats were transiently produced in ExpiCHO™ cells and purified using protein A chromatography. Afterwards, aggregation propensities were determined by analytical SEC. Most of the different bispecific IL-18 mimetic designs showed quite favorable aggregation properties as indicated by SEC profiles above 90% target peak post-protein A purification. For two molecules with implemented E430G mutations (IL18R_VHHα2β15_E430G and IL18R_sdIgGα2β15_E430G), SEC purities were 79.3% and 82.6%, respectively. Hence, both molecules were further polished by preparative SEC, yielding final purities of 97.3% and 98.3% (Fig. S6). Final expression yields after protein A purification (and after preparative SEC) for the different designs ranged from 12 mg/L for IL18R_tanVHHβ15α2_E430G up to 375.2 mg/L for IL18R_tanVHHα2β15 (Table 2). Of note, IL18R_sdIgGα2β15_E430G precipitated significantly when stored at 4°C and needed to be purified via second step preparative SEC. Generally, we observed a trend toward lower expression for molecules harboring the E430G mutation. Interestingly, the different formats comprising the VHH tandem arrangements without the E430G amino acid exchanges displayed expression yields of more than 300 mg/L, indicating high production profiles for transient antibody expression. Additionally, the thermal stabilities of the engineered designs were monitored by measuring the Tonset representing the lowest temperature at which a protein starts to unfold. Generally, all molecules not harboring the E430G exchanges showed thermal stabilities (Tonset) above 50°C, indicating favorable biophysical properties, whereas all designs with incorporated E430G mutations showed diminished stabilities by 4°C − 8.6°C (Table 2, Fig. S7).
Table 2.
Biophysical, biochemical, and functional attributes of engineered cytokine mimetic formats.
| Samples | Final yield [mg/L] |
SEC purity after protein A [%] |
Final SEC purity after freeze thaw and polishing* [%] | Tonset [°C] | Mean IFN-γ Release [pg/mL] at 10 nM |
Mean IFN-γ Release [pg/mL] at 1 nM |
EC50 IFN-γ Release [nM] |
|---|---|---|---|---|---|---|---|
| IL18R_VHHα2β15 | 64.0 | 94.1 | 94.1 | 56.4 | 418.2 | 115.1 | 2.4 |
| IL18R_VHHα2β15_E430G | 91.6 | 79.3 | 97.3* | 49.7 | 466.4 | 114.9 | |
| IL18R_sdIgGα2β15 | 262.4 | 92.3 | 91.9 | 57.0 | 332.1 | 376.1 | |
| IL18R_sdIgGα2β15_E430G | 142.4 | 82.6 | 98.3* | 48.4 | 374.6 | 385.5 | |
| IL18R_tanVHHα2β15 | 375.2 | 95.5 | 95.2 | 54.3 | 1871.4 | 1946.0 | 0.006 |
| IL18R_tanVHHα2β15_E430G | 161.2 | 97.4 | 95.7 | 50.0 | 1845.9 | 1961.3 | |
| IL18R_tanVHHβ15α2 | 324.4 | 93.5 | 93.3 | 53.3 | 1116.5 | 1238.2 | 0.016 |
| IL18R_tanVHHβ15α2_E430G | 12.0 | 94.2 | 94.3 | 49.3 | 1017.5 | 1061.8 |
*Only for IL18R_VHHα2β15_E430G and IL18R_sdIgGα2β15_E430G preparative SEC was applied for polishing.
Subsequently, all different antibody designs based on VHHα2 and VHHβ15 were analyzed by means of triggering IFN-γ production on human PBMCs isolated from six distinct donors in total. The different formats were used at two different concentrations (10 nM and 1 nM). Intriguingly, profound differences were revealed for the tested design architectures (Figure 4B). The engineered sdIgG-based cytokine mimetic (IL18R_sdIgGα2β15) did not induce a substantially improved IFN-γ response compared to the initial (1 + 1) SEED design termed IL18R_VHHα2β15 (332.1 pg/mL and 376.1 pg/mL for IL18R_sdIgGα2β15 at 10 nM and 1 nM, respectively vs 418.2 pg/mL and 115.1 pg/mL). Hence, we de-prioritized this format for further analysis. In contrast to this, the stimulation of PBMCs with both tandem arrangements IL18R_tanVHHβ15α2 and IL18R_tanVHHα2β15 provoked a significantly augmented IFN-γ production, with IL18R_tanVHHα2β15 eliciting the strongest release (1871.4 pg/mL and 1946.0 pg/mL at 10 nM and 1 nM, respectively) within the set of engineered IL-18 mimetic architectures. In general, the implementation of the E430G amino acid exchanges did not seem to improve IFN-γ release. Consequently, we also discarded all formats harboring the E430G mutation for additional characterization.
Both formats comprising the VHH arranged in tandem (IL18R_tanVHHβ15α2 and IL18R_tanVHHα2β15), as well as the initial 1 + 1 SEED design (IL18R_VHHα2β15), were directly compared to (rh) IL-18 regarding their potential to elicit a functional IFN-γ response (Figure 4C, Table 2). In comparison with (rh) IL-18, the capacity of IL18R_VHHα2β15 to trigger IFN-γ release on PBMCs was clearly attenuated (EC50 of 2.4 nM for IL18R_VHHα2β15 vs 61 pM for (rh) IL-18). In contrast to this, the potential to evoke IFN-γ production of both mimetics harboring the tandem arrangements were substantially augmented (EC50 of 16 pM for IL18R_tanVHHβ15α2 and 6 pM for IL18R_tanVHHα2β15), resulting in molecules that were significantly more potent than (rh) IL-18. While potencies were fairly similar between both tandem formats, the magnitude IFN-γ release was quite different. In this regard, IL18R_tanVHHα2β15 caused a maximum release that was quite similar to (rh) IL-18 (2130 pg/mL vs 2097 pg/mL), whereas IL18R_tanVHHβ15α2 elicited a significantly reduced maximum IFN-γ production of 1095 pg/mL. Of note, we observed a strong hooking effect (bell-shaped curve) for (rh) IL-18, i.e., reduced IFN-γ release with high and increasing compound concentrations (Fig. S8). This effect was clearly not as pronounced for the different surrogate agonist formats tested.
To investigate whether biofunctional differences also translate into differential biochemical attributes, we performed BLI experiments aiming at evaluating target affinities and binding avidities of IL18R_VHHα2β15, IL18R_tanVHHα2β15 and IL18R_tanVHHβ15α2. For determining affinities, bispecific cytokine mimetics were captured on the sensor tips and the respective IL-18 receptor subunits were exploited as analytes (Supplementary Table S1). Compared with IL18R_VHHα2β15, binding affinities for the inner paratopes of tandem IL-18 mimetics were slightly reduced, whereas kinetics for the outer sdAb remained largely unaffected. To evaluate avidities, the respective receptor subunits were captured on the sensor tips and binding was assessed for the monovalent (for each target) bsAb IL18R_VHHα2β15 and for the bivalent (for binding to each receptor chain) surrogate agonists IL18R_tanVHHβ15α2 and IL18R_tanVHHα2β15 (Supplementary Table S1, Fig. S9). In this experimental setting, IL18R_VHHα2β15 displayed binding to both respective receptor chains in the lower double digit nanomolar range. In contrast to this, apparent binding affinities for both tandem IL-18 mimetics could not be determined, driven by the fact that no dissociation from the receptor subunits was observed. This gives strong evidence for high avidity binding of both tandem surrogate agonists.
Bispecific IL-18 mimetics are resistant to inhibition by IL-18BP
Finally, we analyzed whether the herein generated IL-18 mimetics are affected and functionally inhibited by IL-18BP receptor decoy. To this end, we used the initially generated TOP4 cytokine mimetics, IL18R_VHHα2β15, IL18R_VHHα2β17, IL18R_VHHα8β17, and IL18R_VHHα8β15, as well as both tandem-engineered surrogate agonists IL18R_tanVHHβ15α2 and IL18R_tanVHHα2β15 (which are format engineered derivatives of IL18R_VHHα2β15). As determined by BLI, no binding interaction of (rh) IL-18BP was measurable to any of the IL-18 mimetics utilized (Fig. S10). Opposed to this, (rh) IL-18BP showed high-affinity binding to (rh) IL-18 (Fig. S10). Both IL-18BP and IL-18 Rα bind to IL-18 at an overlapping interface.23 Of note, all six surrogate agonists showed competitive binding with (rh) IL-18 for targeting IL-18 Rα, indicating a similar or at least a partially overlapping epitope on IL-18 Rα (Fig. S11 and Table 1). Ultimately, we evaluated the resistance of the engineered IL-18 mimetics to IL-18BP inhibition on PBMC stimulation. Therefore, we focused on both tandem IL-18 mimetics showing potencies at a similar level as (rh) IL-18 in provoking an IFN-γ response. Both cytokine mimetics and (rh) IL-18 were utilized at a fixed concentration of 0.5 nM combined with low dose (rh) IL-12 (10 ng/mL) and increasing concentrations of (rh) IL-18BP were titrated (Figure 4D). Contrary to (rh) IL-18, which was efficiently inhibited by (rh) IL-18BP (IC50 of 2.2 nM), both tandem IL-18 mimetics, IL18R_tanVHHα2β15, and IL18R_tanVHHβ15α2 triggered a robust IFN-γ release, regardless of the IL-18BP receptor decoy concentration tested.
Discussion
Cytokines are important signaling proteins that regulate immune function. Although many efforts have been made to harness cytokines for disease treatment, the therapeutic applicability has been limited,12 especially for indications in oncology. The pleiotropic mode of action of many cytokines and resulting ramifications thereof, for instance, dose-limiting toxicities or contrary functions on different immune cell subsets, are some of the main reasons for their restricted biomedical pertinence.29 To address some of the main limitations of conventional cytokine therapy, next-generation cytokine derivatives have been developed and currently many different molecules are being investigated in clinical trials.2,3
IL-18 is a proinflammatory cytokine promoting natural killer cell activation as well as effector T cell maturation and function.30 Consequently, IL-18 emerged as a promising potential therapeutic inducing antitumor immunity. IL-18 was assessed in several clinical trials either as single agent or as combination therapy.21,22,31–33 IL-18 therapy was well tolerated, but clinical efficacy has been limited. A possible explanation for the lack of therapeutic efficacy relies in the inhibition of IL-18 by the IL-18BP decoy receptor.17,34 IL-18BP binds to an overlapping site on IL-18 with IL-18 Rα, albeit with much higher affinities. Accordingly, it antagonizes signaling of IL-18 by blocking the IL-18 Rα interaction with the cytokine. Recently, Ring and colleagues described the generation of a decoy-resistant mutein of IL-18 showing great promise for anti-cancer therapy in preclinical models.23 This entity, ST-067, is currently being evaluated in a Phase 1/2 clinical study (NCT04787042) of patients with solid tumors.
Here, we present an alternative approach besides engineering IL-18 itself in order to tailor-make its biological functionality. To this end, we generated sdAb-based bispecifics mimicking IL-18 biology by cross-linking the IL-18 R subunits. For this, we immunized two camelids with a mixture of (rh) IL-18 Rα and (rh) IL-18 Rβ. Receptor subunit-specific VHHs were isolated using YSD as platform technology.35,36 We focused on sdAbs that, in addition to camelids, are also produced in sharks.37,38 Since the resulting paratopes are devoid of light chains, sdAbs afford the benefit of multiple reformatting options in a ‘plug-and-play’ manner involving beads-on-string assemblies or facile combinations with existing Fab-based paratopes for the generation of bispecifics.39,40 However, in contrast to the discovery of fully human antibodies, for instance, from transgenic rodents41 as well as from naïve or synthetic library approaches,42 camelid-derived VHHs typically must be humanized before being administered to patients. Several approaches have been described in this regard,43–45 and the fact that three VHH-based therapeutics have been granted marketing approval by different health authorities indicates that these entities are promising for biomedical applications.46–47,48
After YSD-enabled discovery of sdAbs targeting either (rh) IL-18 Rα or (rh) IL-18 Rβ, combinatorial bispecific reformatting of all paratopes in each possible combination enabled the identification of IL-18 R agonists. However, compared with (rh) IL-18, agonism potencies and efficacies were heavily attenuated. To augment capacities to trigger a functional IFN-γ release, we engineered paratope valencies and the spatial orientation of individual binding sites within a given molecule. We also applied Fc engineering by implementing the E430G mutation into each different antibody format.28 This mutation drives antibody hexamerization following target cell binding, and it was tempting to speculate that on-target multimerization of surrogate agonists evokes a more efficient receptor clustering. To our surprise, this amino acid exchange had no effect at all on any of the scrutinized mimetic designs. In addition to E430, additional mutations have been described that facilitate a stronger hexamerization of IgGs.49 It will be interesting to investigate how these mutations might affect cytokine-like functions of the herein generated IL-18 surrogate agonists.
Intriguingly, multivalent targeting of the receptor subunits by arranging individual VHHs in tandem substantially augmented potencies and the magnitude of IFN-γ release. Interestingly, the orientation of each sdAb within the tandem configuration significantly affected the potential to trigger IL-18 R agonism. When the IL-18 Rα-directed paratope was located at the outer N-terminal position within the tandem arrangement, significantly elevated levels of IFN-γ release were observed compared to the counterpart where the same paratope was located at the inner position. Interestingly, in an BLI experiment (Fig. S12), we determined a more pronounced interference pattern shift (indicating a higher association of the antigen) at a given antigen concentration, when the respective VHH was located at the outer position as compared with the inner position, indicating steric hindrance for the inner sdAb in the tandem arrangement. Apparently, steric hindrance for the IL-18 Rβ-specific VHH did not impede cytokine-like functions of the bsAb, while this was appreciable for the sdAb targeting IL-18 Rα. This gives some evidence that uncompromised binding to IL-18 Rα seems to be crucial for a more efficacious cytokine-like function. Essentially, we were able to generate cytokine mimetics with tailor-made capacities to mediate IFN-γ production, ultimately resulting in surrogate agonists with enhanced potencies compared to (rh) IL-18.
In addition, we profiled the generated TOP4 mimetics and engineered derivatives thereof in terms of IL-18BP inhibition. All generated bispecific surrogate agonists competed with (rh) IL-18 for binding to the (rh) IL-18 Rα subunit. However, as determined by BLI, we were not able to detect an interaction between any of the IL-18 mimetics with (rh) IL-18BP and even more importantly, engineered surrogate agonists were resistant to IL-18BP inhibition in their capacities to trigger a functional IFN-γ response in primary cell assays. In contrast to this, (rh) IL-18 was efficiently antagonized by IL-18BP. Essentially, the herein presented investigations support the notion that antibody engineering enables the generation of IL-18 mimetics with customized biological functionalities. It will be interesting to see whether these functionalities can be combined and synergize with other modes of action such as effector cell redirection50 as already demonstrated by Vivier and colleagues for a variant of IL-2.51
Materials and methods
Camelid immunization
For the immunization procedure, one llama (Lama glama) with an age of about 12.5 years and one male huarizo (Lama glama x Vicugna pacos) of about 11 years of age were immunized with a cocktail of recombinant human (rh) IL-18 Rα ECD as Fc-fusion (R&D Systems, catalog number: 816-LR) as well as (rh) his-tagged IL-18 Rβ ECD (Sino Biologics, catalog number: 10176-H08H). For administration, antigens were diluted to a stock concentration of 1 mg/mL in phosphate-buffered saline (PBS) and emulsified for initial immunization with Complete Freund’s Adjuvant or with Incomplete Freund’s Adjuvant for subsequent immunizations. Antigens were injected subcutaneously at three sites with 200 µg material in total (1:1 ratio of IL-18 Rα and IL-18 Rβ). This procedure was conducted four times over a period of 35 days, i.e., administration at d0, d14, d24 and d35. One week after final administration (d42), a volume of 150 mL blood was collected from each specimen for subsequent RNA extraction and cDNA synthesis. Of note, processes involving animals were performed at preclinics GmbH, Germany and in accordance with local regulations and animal welfare protection laws. Immunized animals remained alive after final blood collection.
Yeast strains and media
For antibody surface display, yeast Saccharomyces cerevisiae strain EBY100 (MATa URA3–52 trp1 leu2Δ1 his3Δ200 pep4:HIS3 prb1Δ1.6 R can1 GAL (pIU211:URA3)) (Thermo Fisher Scientific) was used. Cells were cultivated in yeast extract – peptone–dextrose (YPD) medium composed of 10 g/L yeast extract, 20 g/L peptone and 20 g/L dextrose, additionally supplemented with 10 mg/mL penicillin – streptomycin (Gibco). Cells harboring the library plasmids (pDisp) after gap repair cloning were cultivated in minimal synthetic defined (SD)-base (Takara Bio) medium supplemented with 5.4 g/L Na2HPO4 and 8.6 g/L NaH2PO4 H2O, also comprising the corresponding dropout mix (Takara Bio) composed of all essential amino acids except for tryptophan (−Trp) for selection. To induce antibody gene expression, dextrose was replaced by galactose as carbon source. Thus, cells were transferred into SG dropout medium (−Trp) consisting of SG-base medium (Takara Bio) and 10% (w/v) polyethylene glycol 8000 (PEG 8000).
Plasmids for yeast surface display and library generation
Homologous recombination-based cloning, referred to as gap repair cloning, was utilized for the generation of the VHH libraries in yeast. For this, our group already described the specific PCR amplification of VHH fragments as well as library construction detailed elsewhere.24,52 In short, digestion of a stuffer sequence in the pDisp with BsaI allowed for subsequent genetic fusion of VHH library candidates in frame to Aga2p by gap repair cloning, enabling sdAb presentation on the surface of the yeast cell. Additionally, monitoring of proper full-length VHH presentation on yeast surface was realized by a HA epitope C-terminally linked to Aga2p on the pDisp backbone.
Library sorting
EBY100 cells were grown overnight in SD medium with dropout mix lacking tryptophan (−Trp) at 30°C and 120 rpm. Next day, cells were transferred into SG medium with dropout mix (−Trp) at 107 cells/mL to induce VHH surface expression and incubated for another 48 h at 20°C and 120 rpm. Fluorescence-activated cell sorting (FACS) was conducted on a BD FACSAria™ Fusion cell sorter (BD Biosciences) device. For library sorting purposes, full-length VHH surface expression was monitored by application of an Alexa Fluor 488 labeled mouse monoclonal anti-HA antibody (R&D Systems, catalog number: IC6875G, diluted 1:20). A two-dimensional sorting strategy was applied by simultaneous antigen-binding detection using indirect immunofluorescence staining with 250 nM (rh) his-tagged IL-18 Rα (Sino Biological, catalog number: 11102-H08H) and 250 nM (rh) his-tagged IL-18 Rβ ECD (Sino Biological, catalog number: 10176-H08H), respectively, for the first sorting round or separated (rh) IL-18 R subunits (250 nM) for the second round of sorting in combination with murine anti-his detection antibody (Penta His Alexa Fluor 647 Conjugate, Qiagen, catalog number: 35370, diluted 1:20, for sorting round I or Allophycocyanin anti-His Tag Antibody, BioLegend, catalog number: J095G46, diluted 1:20, for sorting round II), respectively (Figure 1B). Control samples of cells incubated with secondary labeling reagents and an unrelated antigen or cells incubated with secondary labeling only and untreated cells were used in every FACS analysis for adequate gate adjustment. After library sorting and sequencing, a clonotyping strategy was applied in which clones with less than three amino acid changes within CDR3 were considered to belong to the same sequence cluster.
Protein expression, purification, and analytics
After clone selection based on sequencing results obtained from FACS-enriched populations, VHH variants directed against IL18Rα ECD were N-terminally fused to the hinge region of Fc immune effector-silenced (eff-) SEED AG chains, while the VHHs targeting IL18Rβ ECD were fused accordingly to eff- SEED GA chains prior to cloning into pTT5 mammalian expression vector,53 enabling the production of eff-monospecific (IL18R_msVHH) and bispecific SEEDbodies (1 + 1 IL18R_VHHαβ). Of note, for Fc silencing, amino acid exchanges L234A, L235A, P329G were introduced.54 For monovalent (and monospecific) SEED expression, the VHH harboring SEED chain was paired with a paratope-less counterpart chain. For small-scale production of the proteins (25 mL scale), Expi293™ cells were transiently transfected with the respective pTT5 vectors according to the manufacturer’s instructions (Thermo Fisher Scientific) using a 2:1 plasmid ration (AG:GA). Six days post transfection the protein containing supernatants were harvested by centrifugation prior purification via MabSelect antibody purification chromatography resin (GE Healthcare). A buffer-exchange step to PBS pH 6.8 overnight using Pur-A-ExpiCHO™ Maxi 3500 Dialysis Kit (Sigma Aldrich) was followed by sterile filtration with Lyzer™-CL GV 0.22 μm centrifugal devices (Merck Millipore) and measurement of resulting molecule concentrations using Nanodrop ND-1000 (Peqlab). Protein purities were afterward determined by analytical SEC on a TSKgel UP-SW3000 column (2 µm, 4.6 × 300 mm, Tosoh Bioscience) using an Agilent HPLC 1260 Infinity system. 7.5 µg protein per sample were injected and run at a flow rate of 0.35 mL/min using 50 mM sodium phosphate, 0.4 M NaClO4 pH 6.3 as mobile phase.
Different antibody-derived cytokine mimetic formats were transiently expressed from ExpiCHO™ cells (Thermo Fisher Scientific) following a 11-day protocol at 250 mL scale according to the manufacturer’s instructions (Max Titer Protocol). The cultivation protocol included a temperature shift from 36.5°C to 32.0°C after addition of ExpiFectamine CHO Enhancer and first ExpiCHO Feed at day 1 while incubating at 5% CO2 and 80 rpm. Plasmids for transfections (pTT5 backbone) were used at 0.8 mg/L and were mixed 1:1 HC:LC (IgGs) or 2:1 AG:GA (SEEDs). Antibody-containing supernatants were harvested by centrifugation at 4000 × g for 20 min at 4°C and afterward sterile filtered. Protein purification was done by affinity capture on Protein A resin (HiTrap MabSelect SuRe, 5 mL) and acidic elution with 50 mM acetic acid pH 3.0 at 5 mL/min using an GE Healthcare ÄKTAxpress system followed by a desalting step at 10 mL/min (HiPrep 26/10 columns) into PBS pH 6.8. Protein purities were afterward determined by analytical SEC as described above. 7.5 µg protein per sample were injected and run at a flow rate of 0.35 mL/min using 50 mM sodium phosphate, 0.4 M NaClO4 pH 6.3 as mobile phase. For proteins with purities below 90%, a polishing step by preparative SEC (HiLoad 26/600 or 16/600 Superdex 200 pg) was applied. Final samples were sterile filtered over 0.2 µm, flash frozen in liquid nitrogen and stored at −80°C until further use. To assure no quality loss after freeze/thaw, a final purity determination by analytical SEC was done. Furthermore, thermal unfolding of the antibodies was assessed by differential scanning fluorimetry (DSF) on a Prometheus NT.PLEX nanoDSF instrument (NanoTemper). Samples were measured in duplicates using nanoDSF Standard Capillary Chips. A temperature gradient from 20°C to 95°C at a slope of 1°C/min was used while recording fluorescence at 350 and 330 nm. Tonset values were determined from the first derivative of the fluorescence ratio 350 nm/330 nm.
Biolayer interferometry
For all BLI measurements, the Octet RED96 system (ForteBio, Pall Life Science) using 25°C and 1000 rpm agitation settings was used. The data were fitted and analyzed with ForteBio data analysis software 8.0 using a 1:1 binding model after Savitzky – Golay filtering if needed. In order to determine binding properties of monospecific molecules, human IL-18 Rα-His (Sino Biological) or human IL-18 Rβ-His (Sino Biological) were loaded on anti-Penta His (HIS1K) biosensors at 3 μg/mL in PBS for 180 s, respectively, followed by 60 s sensor rinsing in kinetics buffer (KB; PBS + 0.1% Tween-20 and 1% bovine serum albumin, BSA). Afterwards, binding to monospecific IL18R_VHH SEEDbodies at 100 nM in KB was measured for 300 s followed by dissociation for 100 s in KB. In each experiment, one negative control using an irrelevant antibody and a second reference by incubating the IL-18 R subunits in KB instead of the monospecific SEEDbodies was measured.
Kinetic constants (KD) of bispecific molecules to determine binding affinities for their cognate receptors were measured by loading bispecific molecules on anti-human Fc (AHC) biosensors for 180 s at 5 µg/mL in PBS, followed by sensor rinsing in KB for 45 s and binding interaction assessment. To this end, association of (rh) IL18-Rα or (rh) IL-18 Rβ with concentrations from 100 nM to 12.5 nM (in KB) was recorded for 180 s prior dissociation in KB for 300 s for the TOP4 molecules. Binding affinities for the format engineered molecules were determined accordingly, except using slightly different concentrations ranging between 100 nM and 11 nM. The same assay settings were applied to determine avidity effects for the multivalent constructs, but utilizing HIS1K biosensors to immobilize polyhistidine-tagged (rh) IL18Rα-His or (rh) IL18Rβ-His and subsequently measuring the association of engineered tandem constructs and the parental molecule.
To illustrate that IL-18 R cytokine mimicking molecules are not affected by (rh) IL-18BP, (rh) IL-18BP His-Tag (Acro Biosystems) and (rh) IL-18BPa Fc Chimera (R&D Systems) were biotinylated with a 20-fold molar excess of EZ-LINK SULFO-NHS-BIOTIN reagent (Thermo Fisher) according to the manufacturer’s protocol and loaded on Streptavidin (SA) biosensors at 5 µg/mL for 180 s in PBS. After sensor rinsing in KB for 45 s, an association step using the TOP4 molecules and engineered tandem constructs was executed at 1000 nM for 180 s followed by dissociation in KB for 180 s. Kinetic measurement for human IL-18 vs. human IL-18BP was performed accordingly using decreasing concentrations of human IL-18 from 50 nM to 1.56 nM for 180 s followed by dissociation for 300 s in KB. Furthermore, to analyze competitive binding of our TOP4 molecules and engineered tandem constructs vs. human IL-18 on human IL-18 Rα, the surrogate agonists were loaded at 5 µg/mL in PBS for 180 s on AHC biosensors, followed by 45 s sensor rinsing in KB. Association of (rh) IL-18 Rα was measured for 300 s in KB at 200 nM prior to an additional association step with human IL-18 at 100 nM for another 180 s in KB. In parallel, control values using an unrelated antibody and controls using KB buffer were included.
Human IL-18 HEK reporter assay
To detect the activation of the NFκB – AP-1 pathway, the IL-18 HEK-Blue assay (InvivoGen, hkb-hmil18) was performed according to the manufacturer’s instructions. In brief, 5 × 104 cells were seeded into each well of a 96-well plate and stimulated with 50 nM of monospecific or bispecific IL18R_VHHαβ SEEDbodies for 24 h at 37°C and 5% CO2. Incubation of the HEK-Blue™ IL-18 cells with identical concentrations of TNF and IL-18 were used as negative and positive control. For EC50 determination, samples were titrated in a 1:5 serial dilution with concentrations ranging from 100 nM to 51.2 fM. Additionally, an IL-18 titration was performed in a 1:5 serial dilution with concentrations ranging from 100 nM to 0.02 fM. For background subtraction, cell culture medium only was measured as well. After 24 h 20 µl of cell culture supernatants were mixed with 180 μl QUANTI-Blue medium in a fresh 96-well plate and incubated for 3 h at 37°C and 5% CO2. Optical density was measured at 640 nm using a multi-mode microplate reader (Synergy HTX, BioTek).
IFN-γ release assay
To assess the induction of IFN-γ release, freshly isolated human PBMCs from healthy donors were used for all assays at a final concentration of 1 × 106 cells per well. Donors provided written informed consent. To isolate PBMCs, whole blood samples were processed according to StemCell Technologies’ SepMate PBMC Isolation protocol (2019) using SepMate-50 tubes (StemCell Technologies) and Lymphoprep medium (StemCell Technologies). Post isolation, PBMCs were cultivated in AIM V medium (Gibco) at 37°C and 5% CO2 in a humidity box for 24 h during functional assays using 96-well white-opaque cultivation plates (PerkinElmer). For initial cytokine release assays the PBMCs were stimulated with bispecific molecules at a fixed concentration of 100 nM or with 1 nM of (rh) IL-18 (R&D Systems). Stimulations were performed in the presence (10 ng/mL) or absence of (rh) IL-12 (R&D Systems), respectively. To determine EC50 values of the TOP4 molecules, PBMCs were stimulated with (rh) IL-12 (10 ng/mL) and the IL-18 mimicking molecules as 1:2 serial dilutions using concentrations ranging from 1 µM to 122 pM.
For direct comparison of the engineered constructs with (rh) IL-18, PBMCs were stimulated with two concentrations of 10 nM and 1 nM IL18R_VHH molecules or 1 nM for (rh) IL-18, all supplemented with 10 ng/mL (rh) IL-12. For subsequent determination of concentration-dependent IFN-γ induction of (rh) IL-18 and engineered constructs, PBMCs were stimulated with decreasing concentrations of engineered constructs (1 µM to 0.21 pM) and (rh) IL-18 (457 pM to 0.21 pM) as well as (rh) IL-12 (10 ng/mL). To analyze the effect of (rh) IL-18BP on the IL-18 R activation for generated constructs and for (rh) IL-18, PBMCs were stimulated with low dose (rh) IL-12 (10 ng/mL) and a fixed concentration of (rh) IL-18 (0.5 nM) or surrogate agonists (0.5 nM) in the presence of increasing concentration of (rh) IL-18BP ranging from 10 fM to 100 nM. A non-IL-18 R agonizing bispecific IL-18 mimetics serving as negative control and human PBMCs stimulated with (rh) IL-12 only were used as controls for every functional assay. Quantification of IFN-γ released by human PBMCs was performed using human cytokine HTRF kits (Cisbio) as previously described by our group.50 In brief, PBMCs were sedimented by centrifugation after 24 h incubation with the respective samples, and cytokine-containing supernatants were further processed according to the manufacturer’s instructions. Assay plates were measured in a PHERAstar FSX device (BMG Labtech). HTRF optical module that used an excitation at 337 nm and determined the emission at 620 nm and at 665 nm was used. Analyses and fitting of resulting data were facilitated by MARS software (v3.32, BMG) enabling a four-parameter logistic (4PL 1/y2) model fit of the standard curve following the kit manufacturer’s instructions allowing for subsequent IFN-γ quantification in the sample wells.
Molecular Modeling and structural visualization
Structural models of the VHH domains and constant regions of the different bispecific formats were generated using the antibody modeler tool in the molecular modeling software package MOE(Molecular Operating Environment 2020.09: Chemical Computing Group Inc.; 2020). VHHs domains were either directly fused to the constant regions or added via a Gly4Ser-linker using moe’s protein builder, followed by a conformational search of the linker and an energy minimization of the full constructs. Visualization of 3D structures and properties was done with PyMOL (The PyMOL Molecular Graphics System, Version 2.3.0 Schrödinger, LLC.).
Data processing and statistical analysis
Graphical and statistical analyses were conducted with GraphPad Prism 8 software. P-values were calculated utilizing appropriate ANOVA analyses and the Bonferroni test as recommended. p ≤ 0.05 was regarded as statistically significant.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- bsAb
bispecific antibody
- BLI
biolayer interferometry
- CDR
complementarity-determining region
- CH3
third constant Ig domain of the heavy chain
- ECD
extracellular domain
- EpoR
Erythropoietin receptor
- IFN
interferon
- IFN-γ
interferon-γ
- IL-2 Rα
interleukin-2 receptor α
- IL-2 Rβγ
IL-2 receptor βγ
- IL-10 Rβ
interleukin-10 receptor β
- IL-12
interleukin-12
- IL-15
interleukin-15
- IL-18
interleukin-18
- IL-18BP
interleukin-18 binding protein
- IL-18 Rα
interleukin-18 receptor α
- IL-18 Rβ
interleukin-18 receptor β
- NK cell
natural killer cell
- PBMC
peripheral blood mononuclear cell
- rh
recombinant human
- sdAb
single domain antibody
- SEC
size exclusion chromatography
- SEED
strand-exchange engineered domain
- TNF
tumor necrosis factor α
- Treg
regulatory T cell
- VH
variable domain of the heavy chain
- VHH
variable domain of the heavy chain of a heavy chain-only antibody
- VL
variable domain of the light chain
- YSD
yeast surface display
Supplementary Material
Acknowledgments
We thank Hannah-Melina Mayer, Kerstin Hallstein, Marion Wetter, Pia Stroh, Sigrid Auth, Gernot Musch, Markus Fleischer and Dirk Mueller-Pompalla for experimental support.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Disclosure statement
BL, PA, LU, LP and SZ filed a patent application based on this work. In addition, BL, PA, LU, LP, SK, SB, LT, VS, AE, and SZ are employees at Merck Healthcare KGaA. Besides, this work was conducted in the absence of any further commercial interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2236265
References
- 1.Propper DJ, Balkwill FR.. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237–13. doi: 10.1038/s41571-021-00588-9. [DOI] [PubMed] [Google Scholar]
- 2.Pires IS, Hammond PT, Irvine DJ. Engineering strategies for immunomodulatory cytokine therapies: challenges and clinical progress. Adv Ther. 2021;4(8):2100035. doi: 10.1002/adtp.202100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, Rodríguez-Ruiz ME, Ponz-Sarvise M, Castañón E, Melero I. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amin A, White RL. Interleukin-2 in renal cell carcinoma: a has-been or a still-viable option? J Ren Cancer VHL. 2014;1(7):74–83. doi: 10.15586/jkcvhl.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng X, Wu Y, Bi J, Huang Y, Cheng Y, Li Y, Wu Y, Cao G, Tian Z. The use of supercytokines, immunocytokines, engager cytokines, and other synthetic cytokines in immunotherapy. Cell Mol Immunol. 2022;19(2):192–209. doi: 10.1038/s41423-021-00786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. OncoImmunology. 2017;6(3):e1277306. doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolgin E. IL-2 upgrades show promise at ASCO. Nat Biotechnol. 2022;40(7):986–88. doi: 10.1038/s41587-022-01390-3. [DOI] [PubMed] [Google Scholar]
- 8.Jazayeri JA, Carroll GJ. Fc-based cytokines: prospects for engineering superior therapeutics. BioDrugs. 2008;22(1):11–26. doi: 10.2165/00063030-200822010-00002. [DOI] [PubMed] [Google Scholar]
- 9.Eliason JF. Pegylated cytokines: potential application in immunotherapy of cancer. BioDrugs. 2001;15(11):705–11. doi: 10.2165/00063030-200115110-00001. [DOI] [PubMed] [Google Scholar]
- 10.Murer P, Neri D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N Biotechnol. 2019;52:42–53. doi: 10.1016/j.nbt.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. doi: 10.1016/j.coi.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saxton RA, Glassman CR, Garcia KC. Emerging principles of cytokine pharmacology and therapeutics. Nat Rev Drug Discov. 2023;22(1):21–37. doi: 10.1038/s41573-022-00557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moraga I, Wernig G, Wilmes S, Gryshkova V, Richter CP, Hong W-J, Sinha R, Guo F, Fabionar H, Wehrman TS, et al. Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell. 2015;160(6):1196–208. doi: 10.1016/j.cell.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris KE, Lorentsen KJ, Malik-Chaudhry HK, Loughlin K, Basappa HM, Hartstein S, Ahmil G, Allen NS, Avanzino BC, Balasubramani A, et al. A bispecific antibody agonist of the IL-2 heterodimeric receptor preferentially promotes in vivo expansion of CD8 and NK cells. Sci Rep. 2021. [cited 2023 Feb 9];11(1):Available from https://www.nature.com/articles/s41598-021-90096-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yen M, Ren J, Liu Q, Glassman CR, Sheahan TP, Picton LK, Moreira FR, Rustagi A, Jude KM, Zhao X, et al. Facile discovery of surrogate cytokine agonists. Cell. 2022;185(8):1414–30.e19. doi: 10.1016/j.cell.2022.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in Health and Disease. Int J Mol Sci. 2019;20(3):649. doi: 10.3390/ijms20030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 Binding Protein. Front Immunol [Internet]. 2013;4. [cited 2023 Feb 9]. http://journal.frontiersin.org/article/10.3389/fimmu.2013.00289/abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura K, Bald T, Smyth MJ. Cancer‐killing, decoy‐resistant interleukin‐18. Immunol Cell Biol. 2020;98(6):434–36. doi: 10.1111/imcb.12359. [DOI] [PubMed] [Google Scholar]
- 19.Atallah-Yunes SA, Robertson MJ. Cytokine Based Immunotherapy for Cancer and Lymphoma: Biology, Challenges and Future Perspectives. Front Immunol [Internet]. 2022;13. [cited 2023 Feb 9]. https://www.frontiersin.org/articles/10.3389/fimmu.2022.872010/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, Kathman S, Pandite LN, Oei C, Kirby LC, et al. Clinical and Biological Effects of Recombinant Human Interleukin-18 Administered by Intravenous Infusion to Patients with Advanced Cancer. Clin Cancer Res. 2006;12(14):4265–73. doi: 10.1158/1078-0432.CCR-06-0121. [DOI] [PubMed] [Google Scholar]
- 21.Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, Kathman SJ, Laubscher KH, Dar MM, Kirkwood JM. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer. 2009;115(4):859–68. doi: 10.1002/cncr.24100. [DOI] [PubMed] [Google Scholar]
- 22.Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, Bell WN, Thurmond LM, Weisenbach J, Dar MM. A Dose-Escalation Study of Recombinant Human Interleukin-18 Using Two Different Schedules of Administration in Patients with Cancer. Clin Cancer Res. 2008;14(11):3462–69. doi: 10.1158/1078-0432.CCR-07-4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou T, Damsky W, Weizman O-E, McGeary MK, Hartmann KP, Rosen CE, Fischer S, Jackson R, Flavell RA, Wang J, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583(7817):609–14. doi: 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth L, Krah S, Klemm J, Günther R, Toleikis L, Busch M, Becker S, Zielonka S. Isolation of Antigen-Specific VHH Single-Domain Antibodies by Combining Animal Immunization with Yeast Surface Display. Methods Mol Biol. 2020;2070:173–89. [DOI] [PubMed] [Google Scholar]
- 25.Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, Xiao Y, Krah S, Zaynagetdinov R, Lipinski B, et al. Multifunctional NK Cell–Engaging Antibodies Targeting EGFR and NKp30 Elicit Efficient Tumor Cell Killing and Proinflammatory Cytokine Release. J Immunol. 2022;209(9):1724–35. doi: 10.4049/jimmunol.2100970. [DOI] [PubMed] [Google Scholar]
- 26.Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo K-M, Huston JS. Seedbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies†. Protein Eng Des Sel. 2010;23(4):195–202. doi: 10.1093/protein/gzp094. [DOI] [PubMed] [Google Scholar]
- 27.Pekar L, Busch M, Valldorf B, Hinz SC, Toleikis L, Krah S, Zielonka S. Biophysical and biochemical characterization of a VHH-based IgG-like bi- and trispecific antibody platform. MAbs. 2020;12(1):1812210. doi: 10.1080/19420862.2020.1812210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, Horstman W, Engelberts PJ, Oostindie SC, Wang G, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol. 2016;14(1):e1002344. doi: 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue D, Hsu E, Fu Y-X, Peng H. Next-generation cytokines for cancer immunotherapy. Antib Ther. 2021;4(2):123–33. doi: 10.1093/abt/tbab014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holder PG, Lim SA, Huang CS, Sharma P, Dagdas YS, Bulutoglu B, Sockolosky JT. Engineering interferons and interleukins for cancer immunotherapy. Adv Drug Deliv Rev. 2022;182:114112. doi: 10.1016/j.addr.2022.114112. [DOI] [PubMed] [Google Scholar]
- 31.Robertson MJ, Kline J, Struemper H, Koch KM, Bauman JW, Gardner OS, Murray SC, Germaschewski F, Weisenbach J, Jonak Z, et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-hodgkin lymphoma. J Immunother. 2013;36(6):331–41. doi: 10.1097/CJI.0b013e31829d7e2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simpkins F, Flores A, Chu C, Berek JS, Lucci J, Murray S, Bauman J, Struemper H, Germaschewski F, Jonak Z, et al. Chemoimmunotherapy using pegylated liposomal doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase i dose-escalation study. Cancer Immunol Res. 2013;1(3):168–78. doi: 10.1158/2326-6066.CIR-13-0098. [DOI] [PubMed] [Google Scholar]
- 33.Robertson MJ, Stamatkin CW, Pelloso D, Weisenbach J, Prasad NK, Safa AR. A dose-escalation study of recombinant human interleukin-18 in combination with ofatumumab after autologous peripheral blood stem cell transplantation for lymphoma. J Immunother. 2018;41(3):151–57. doi: 10.1097/CJI.0000000000000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novick D, Kim S-H, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 Binding Protein. Immunity. 1999;10(1):127–36. doi: 10.1016/S1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 35.Doerner A, Rhiel L, Zielonka S, Kolmar H. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014;588(2):278–87. doi: 10.1016/j.febslet.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Valldorf B, Hinz SC, Russo G, Pekar L, Mohr L, Klemm J, Doerner A, Krah S, Hust M, Zielonka S. Antibody display technologies: selecting the cream of the crop. Biol Chem. 2022;403(5–6):455–77. doi: 10.1515/hsz-2020-0377. [DOI] [PubMed] [Google Scholar]
- 37.Zielonka S, Empting M, Grzeschik J, Könning D, Barelle CJ, Kolmar H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. MAbs. 2015;7(1):15–25. doi: 10.4161/19420862.2015.989032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Könning D, Zielonka S, Grzeschik J, Empting M, Valldorf B, Krah S, Schröter C, Sellmann C, Hock B, Kolmar H. Camelid and shark single domain antibodies: structural features and therapeutic potential. Curr Opin Struct Biol. 2017;45:10–16. doi: 10.1016/j.sbi.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 39.Lipinski B, Arras P, Pekar L, Klewinghaus D, Boje AS, Krah S, Zimmermann J, Klausz K, Peipp M, Siegmund V, et al. Nkp46 ‐specific single domain antibodies enable facile engineering of various potent NK cell engager formats. Protein Science [Internet]. 2023. [cited 2023 Feb 17]; Available from;32(3): https://onlinelibrary.wiley.com/doi/10.1002/pro.4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chanier T, Chames P. Nanobody engineering: toward next generation immunotherapies and immunoimaging of cancer. Antibodies. 2019;8(1):13. doi: 10.3390/antib8010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osbourn J, Groves M, Vaughan T. From rodent reagents to human therapeutics using antibody guided selection. Methods. 2005;36(1):61–68. doi: 10.1016/j.ymeth.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 42.Teixeira AAR, Erasmus MF, D’Angelo S, Naranjo L, Ferrara F, Leal-Lopes C, Durrant O, Galmiche C, Morelli A, Scott-Tucker A, et al. Drug-like antibodies with high affinity, diversity and developability directly from next-generation antibody libraries. MAbs [Internet]. 2021. [cited 2023 May 1];13(1):Available from https://www.tandfonline.com/doi/full/10.1080/19420862.2021.1980942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossotti MA, Bélanger K, Henry KA, Tanha J. Immunogenicity and humanization of single‐domain antibodies. FEBS J. 2022;289(14):4304–27. doi: 10.1111/febs.15809. [DOI] [PubMed] [Google Scholar]
- 44.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–84. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 45.Sulea T. Humanization of camelid single-domain antibodies. In: Hussack G, Henry K, editors. Single-Domain Antibodies. New York, NY: Springer US; 2022. pp. 299–312. [cited 2023 Mar 10]. InternetAvailable from: 10.1007/978-1-0716-2075-5_14. [DOI] [PubMed] [Google Scholar]
- 46.Duggan S. Caplacizumab: First Global Approval. Drugs. 2018;78:1639–42. doi: 10.1007/s40265-018-0989-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.SJ K. Ozoralizumab: First Approval. Drugs. 2023;83(1):87–92. doi: 10.1007/s40265-022-01821-0. [DOI] [PubMed] [Google Scholar]
- 48.Markham A. Envafolimab: First Approval. Drugs. 2022;82:235–40. doi: 10.1007/s40265-022-01671-w. [DOI] [PubMed] [Google Scholar]
- 49.van Kampen MD, Kuipers-De Wilt LHAM, van Egmond ML, Wilt LHAM K-D, van den Bremer ETJ, Wang G, Heck AJR, Parren P, Beurskens FJ, Schuurman J, et al. Biophysical characterization and stability of modified igg1 antibodies with different hexamerization propensities. J Pharmaceutical Sci. 2022;111(6):1587–98. doi: 10.1016/j.xphs.2022.02.016. [DOI] [PubMed] [Google Scholar]
- 50.Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, Oberg H-H, Krohn S, Boje AS, Gehlert CL, et al. Affinity Maturation of B7-H6 translates into enhanced NK cell–mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 Engagement. J Immunology. 2021;206(1):225–36. doi: 10.4049/jimmunol.2001004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demaria O, Gauthier L, Vetizou M, Blanchard Alvarez A, Vagne C, Habif G, Batista L, Baron W, Belaïd N, Girard-Madoux M, et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep Med. 2022;3(10):100783. doi: 10.1016/j.xcrm.2022.100783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benatuil L, Perez JM, Belk J, Hsieh C-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23(4):155–59. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 53.Durocher Y. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30(2):9e–9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457–66. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.