

ABSTRACT
Cancer chemotherapy relies on a high ratio of toxicity toward cancer cells vs. nonmalignant cells, making it desirable to protect normal cells. Among the nonmalignant cells, epithelia of the gut belong to the most vulnerable ones toward chemotherapeutics. Here, we use a murine intestinal organoid model to assess a strategy for protecting such epithelia against chemotherapy. Cell cycle progression was first stalled by palbociclib, a clinically established cyclin-dependent kinase 4/6 (CDK4/6) inhibitor. Washout of the drug allowed subsequent outgrowth of gut organoids. This transient cell cycle arrest conferred near-complete protection of the cells toward the nucleoside analogue gemcitabine. Moreover, pre-treatment with palbociclib protected the organoids against SN-38, the topoisomerase I-inhibiting metabolite of irinotecan, which is otherwise known for its severe gastrointestinal toxicities. In contrast, RB1-mutated cancer cells were not protected against gemcitabine or SN-38 when pre-treated with palbociclib. Taken together, these results outline a strategy for protecting nonmalignant cells against the toxicities of chemotherapeutics commonly used to treat advanced colorectal and pancreatic cancer. We propose that this strategy is particularly promising to protect the gut when treating RB1-deficient tumors that fail to arrest the cell cycle in response to CDK4/6 inhibitors.
KEYWORDS: intestinal organoids, CDK4/6, irinotecan, gemcitabine, palbociclib, cyclotherapy
Introduction
Cancer chemotherapy relies on a suitable balance of therapeutic efficacy against tumor cells vs. toxicity toward healthy tissue, often referred to as the therapeutic index. Thus, strategies for improvement not only include enhanced aggressiveness against the tumor but also better protection of normal cells.
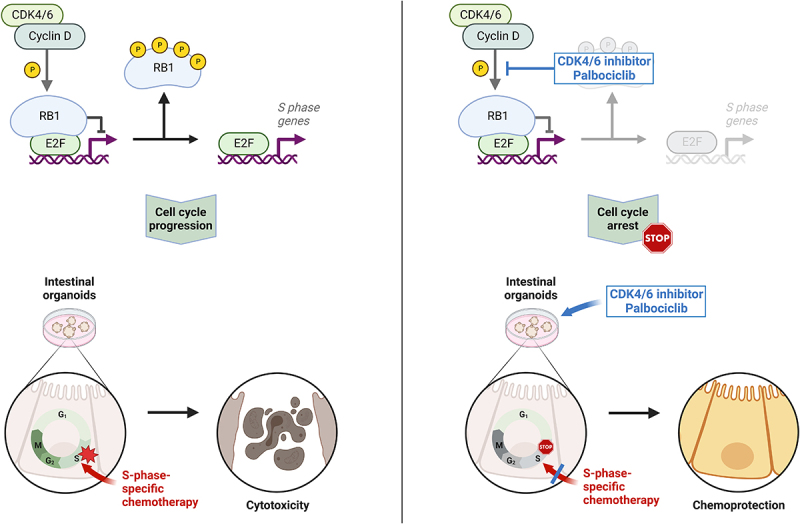
An important approach to achieve such protection takes advantage of the cell cycle specificity of many cancer drugs. Typically, cancer drugs preferentially eliminate cells that go through S or M phase of the cell cycle, i.e. DNA replication or mitosis. Thus, a way to protect normal tissue can consist in a transient arrest in the cell cycle. As long as cancer cells within the same patient continue cycling, this can improve the therapeutic index of chemotherapy. Such strategies designated “cyclotherapy” [1], rely on genetic differences between tumor cells and healthy tissue. For instance, many tumors lack functional p53. Thus, activating p53 by inhibition of its antagonist Mdm2 can arrest the cell cycle in most normal cells, whereas p53-mutant tumor cells keep cycling and remain sensitive toward drugs that act during S or M phase. In this way, p53-proficient, normal cells are protected against the nucleoside analogue gemcitabine but also against the mitotic spindle cell poison paclitaxel [2–5].
Inhibitors of the cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) represent clinically established oncotherapeutics, especially for treating breast cancer [6,7]. Palbociclib is the most widely used drug of this class [8,9]. CDK4/6 inhibition prevents the phosphorylation of the retinoblastoma protein (pRb). Unlike the phosphorylated protein, hypophosphorylated pRb is active in binding the transcription factors of the E2F family, thus repressing E2F-responsive genes [10–12]. Many such genes, e.g. PCNA or E2F1 itself, contribute to cell cycle progression. Therefore, CDK4/6 inhibition mainly arrests cells in G1 phase, reducing the proportion of cells that go through S and M phases. However, cells carrying deletions of RB1, the gene that encodes pRb, e.g. most small cell lung cancers (SCLCs), respond poorly to CDK4/6 inhibition and typically continue cycling in the presence of such drugs [13]. Also, cervical cancer cells that have incorporated the pRb antagonist E7, derived from human papillomaviruses (HPV), fail to arrest in response to CDK4/6 inhibition [14]. This provides a window of opportunity for protecting normal cells by CDK4/6 inhibitors, when using chemotherapeutics that act in S or M phase. Since cancer cells with RB1 mutations will continue cycling in this scenario, they are expected to remain susceptible toward chemotherapy. A number of studies have provided proof of this principle [15–18].
Perhaps, not surprisingly, normal tissues with a high proportion of proliferating cells are most vulnerable toward the toxicities of chemotherapeutics. These include the hematopoietic system, the epithelia of the gut and hair follicles. Among those, toxicities to hematopoiesis, i.e. myelosuppression, have gathered most attention in clinical and basic research [19]. However, the destruction of gut epithelia can give rise to dose-limiting and life-threatening side effects of chemotherapeutics [20–22]. It is therefore of high importance to evaluate strategies for avoiding intestinal toxicities during cancer therapy.
One of the chemotherapeutics with most severe intestinal toxicity is irinotecan, a drug currently used for treating colon cancer, but also studied to treat cancers of the lung, the cervix and the pancreas [22–28]. Irinotecan is first metabolized to become the active compound SN-38 [28]. SN-38 then inhibits topoisomerase I [29,30]. This enzyme removes torsional stress from the DNA during replication, by temporarily introducing a single-strand break. This allows the DNA to twist around the remaining strand. After re-ligation, the double stranded DNA is restored along with the relaxation of torsion. Irinotecan not only inhibits the re-ligation but also induces permanent association of topoisomerase I with the DNA, forming an obstacle to replication forks [31]. For these reasons, irinotecan is by far the most active in S phase, with multiple DNA replication forks acting in parallel. This raises the perspective of avoiding the intestinal toxicity of irinotecan by prior treatment with a cell cycle-arresting agent.
Like irinotecan, gemcitabine (2′-desoxy-2′,2′-difluorocytidine) is an anti-cancer drug that is active when cells are in S phase. It is a nucleoside analogue, acting on DNA synthesis, both by inhibition of ribonucleotide reductase and by false incorporation into nascent DNA [32]. We have shown previously that the activation of p53 by the Mdm2-inhibitor Nutlin-3a confers protection of cells against gemcitabine [2]. Gemcitabine is widely used to treat pancreatic cancer [33–35], but its potential use against colorectal cancer (CRC) is also under study [36–43]. This raises the question whether cell cycle arrest might represent a strategy to protect the intestine against the toxicities of gemcitabine.
One step toward studying the toxic impact of chemotherapy on gut epithelia consists in the establishment of suitable model systems. In particular, organoids of the intestine represent a convenient but authentic model system to evaluate drug toxicities toward the gut, especially when testing various drug concentrations and their combinations at multiple time points [44–46]. These organoids display a spherical shape, with the basolateral side of the epithelium oriented toward the culture medium and the apical portion toward the lumen. Toxic effects can then be studied by evaluating the shape and size of the organoids, by assessing cell viability and death, and by preparing tissue sections with subsequent staining and/or immunodetection [47].
Here, we show that the CDK4/6 inhibitor palbociclib only transiently reduces the outgrowth of intestinal organoids, by arresting the cell cycle progression within them. Palbociclib, thus, protects the cells of organoids against the toxic effects of gemcitabine as well as irinotecan. This raises the perspective of using CDK inhibitors to prevent the intestinal toxicities of chemotherapeutics that act in a cell cycle-specific manner.
Materials and methods
Preparation and cultivation of small intestinal organoids
We have previously described detailed methods to prepare intestinal organoids [47]. In brief, 8–20 weeks old male C57BL/6N mice were used to prepare organoids from healthy small intestinal epithelium. The intestinal tissue was thoroughly washed and incubated in EDTA/PBS before mechanical dissociation of the stem cell-containing crypts. Crypts were then plated in ice-cold Matrigel and cultured in small intestinal organoid culture medium (advanced DMEM F-12, supplemented with 20% Noggin conditioned medium, 10% Rspondin-1 conditioned medium, 50 ng/mL rmEGF, 80 µM N-Acetyl-L-Cysteine, N-2 and B-27, HEPES, Penicillin-Streptomycin and GlutaMAX). Organoids were cultivated at 37°C with 5% CO2. Medium changes were performed every 2–3 d, and organoids were passaged every 4–7 d.
Modified HEK293T cells expressing Noggin or Rspondin-1 were cultured to produce the required conditioned media for the small intestinal organoid culture medium [47].
Cancer cell cultivation
The human pancreatic cancer cell Panc02.03 and colonic cancer cells SW837, both with RB1 mutations [48–50], were cultured in RPMI1640 (ATCC-modified) and RPMI1640 (Invitrogen), respectively. The medium was supplemented with 10% (SW837) or 15% (Panc02.03) fetal calf serum and a penicillin-streptomycin antibiotic mix (all Invitrogen). SW837 additionally received glutamine (Invitrogen), and Panc02.03 additionally received insulin. The cells were cultivated at 37°C with 5% CO2.
Treatment of organoids and cancer cells
Cancer cells and organoids were treated with palbociclib (PD 0332991 isethionate, Sigma-Aldrich), gemcitabine (GEMCITABIN Actavis 40 mg/ml, PUREN Pharma), cisplatin (Cisplatin NeoCorp 1 mg/ml, HEXAL) or SN-38 (Selleck Chemicals). Palbociclib, gemcitabine and cisplatin were dissolved and diluted in aqueous solution, and SN-38 was dissolved in DMSO (AppliChem).
For wash-out of drugs in organoid gel domes, treatment media were removed, pre-warmed PBS was added for 30 minutes (min) to infiltrate and wash the gel domes, including organoids. PBS was removed, and fresh small intestinal organoid culture medium was added. After 30 min incubation, the small intestinal organoid culture medium was renewed. Cancer cells were washed twice with pre-warmed PBS in-plate, and fresh culture medium was added.
Organoid growth quantification
Organoids were cultured and treated in black clear-bottom 96-well plates. Daily bright field images were taken using a semi-automated cell imager system (Celigo Image Cytometer, Nexcelom). For quantification, raw images were used and the identity of all images was blinded. Organoid growth was calculated as area covered by viable organoids. Each viable organoid was manually measured as an oval area using the software ImageJ, and the total area covered by viable organoids was determined for each well. The area of organoids on day 1 was set 100%, and organoid growth was normalized to day 1. Data were derived from three in-plate technical replicates.
Cancer cell growth assay
To evaluate the effect of combining gemcitabine or SN-38 with palbociclib, cancer cells were seeded in 96-well plates (Corning) and treated according to the experimental scheme. Cell confluency was measured using the Celigo Imaging Cytometer (Nexcelom) and calculated as percentage of covered vs. total area.
Cell viability assays
Cell viability measurements of organoids have been described in detail [47]. In brief, organoids were cultured in black clear-bottom 96-well plates. Upon drug treatments, the CellTiter-Glo 3D assay (Promega) was performed. Luminescence was measured using a DLReady Centro LB 960 luminometer (Berthold Technologies). ATP solutions with known concentrations were used as standards. Mean values from five in-plate replicates were taken from three independent biological replicates, which were then normalized to the untreated control condition.
Cancer cells were seeded and treated in white clear-bottom 96-well plates (Corning). Cell viability was assessed as above. Controls were set 100% and used as reference for each condition.
Propidium iodide/Hoechst 33,342 (PI/Hoechst) fluorescence staining
Propidium iodide (PI) and Hoechst 33,342 (Hoechst) staining for analysis and quantification of cell death in organoids was performed as previously described [47]. In brief, organoid culture was performed in black clear-bottom 96-well plates. After experimental treatments, 1 µg/mL PI and 10 µg/mL Hoechst, prediluted in PBS, were added to the medium. After 35 min incubation, fluorescence images were acquired using a Celigo Imaging Cytometer (Nexcelom). Images were blinded and analyzed using ImageJ. Organoids showing PI-negative, Hoechst-positive epithelial structures were considered viable, regardless of positive PI staining in other parts of the organoids. The mean percentages of viable organoid structures from five in-plate replicates were calculated.
Immunoblot analysis
Organoids were cultured and treated in 6-well plates. Gel domes were dissociated, collected and washed in ice-cold PBS. A final washing step was performed with Cell Recovery Solution (Corning). Organoids were lysed in standard RIPA buffer, composed of 1% sodium deoxycholate, 10 mM EDTA, 1% Triton X-100, 0.1% SDS, 150 mM sodium chloride, 20 mM Tris-HCl pH 7.5 with supplemented protease inhibitors (complete Mini EDTA-free Protease Inhibitor Cocktail, Roche) and phosphatase inhibitors (2 mM Imidazole, 1 mM sodium orthovanadate and 1 mM sodium fluoride). Protein concentrations were measured with a BCA protein assay (Pierce BCA Protein Assay Kit, Thermo Scientific). Equal protein amounts were loaded onto acrylamide gels for SDS gel electrophoresis with subsequent transfer onto nitrocellulose membranes. After blocking with 5% milk in TBS-T, membranes were incubated with the primary and secondary antibodies provided in Table 1. For chemiluminescent signal detection, Immobilon Western HRP Substrate Peroxide solution or SuperSignal West Femto Maximum Sensitivity Substrate were used.
Table 1.
Antibodies used in this study.
| Antibody | Manufacturer | Order No. | Applied dilution |
|---|---|---|---|
| Phospho-Rb (Ser807/811) (D20B12) ×P Rabbit mAb | Cell Signaling | 8516 | 1: 1000 for WB1: 500 for IHC |
| PCNA Polyclonal | Invitrogen | PA5–27214 | 1: 1000 for WB |
| Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb | Cell Signaling | 9664 | 1: 500 for WB |
| Anti-beta Actin | Abcam | 8227 | 1: 10000 for WB |
| Anti-KAP1 (phospho S824) | Abcam | 70369 | 1: 300 for WB |
| Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb | Cell Signaling | 9718 | 1: 1000 for WB1: 200 for IHC |
| Anti-GAPDH antibody [6C5] | Abcam | 8245 | 1: 15000 for WB |
| Anti-KAP1 antibody | Abcam | 10484 | 1: 500 for WB |
| Caspase-3 Antibody | Cell Signaling | 9662 | 1: 500 for WB |
| Rb (4H1) Mouse mAb | Cell Signaling | 9309 | 1: 500 for WB |
| RB1 Polyclonal antibody | Proteintech | 10048–2-Ig | 1: 500 for WB |
| Ki-67 rec Rabbit (SP6) | Invitrogen | MA5–14520 | 1: 500 for IHC |
| goat anti-rabbit IgG-HRP | Santa Cruz | 2004 | 1: 10000 for WB |
| goat anti-mouse IgG-HRP | Santa Cruz | 2005 | 1: 10000 for WB |
Note: WB, western blot, immunoblot; IHC, immunohistochemistry.
Quantitative real time PCR (Qrt-PCR)
For qRT-PCR analysis, organoids were cultured and treated in 12-well plates. mRNA was isolated using TRIzol. After mRNA quantification with a microvolume spectrophotometer (Nanodrop 1000 Spectrophotometer, Peqlab) and reverse transcription of 1 µg RNA (M-MuLV Reverse Transcriptase, NEB), the qRT-PCR analysis was performed using 75 mM Tris-HCl pH 8.8 (Roth), 20 mM (NH4)2SO4 (Roth), 0.01% Tween-20 (AppliChem), 3 mM MgCl2 (Sigma-Aldrich), 0.25% Triton X-100 (AppliChem), 0.2 mM dNTPs (Primetech), 20 U/ml Taq-polymerase (Primetech), 300 mM Trehalose (Roth) and SYBR Green (Invitrogen). Primers are listed in Table 2 and were used in a two-step protocol (2 min at 95°C pre-heating; 40 cycles at 95°C for 15 s followed by 60°C for 1 min). Each qRT-PCR was run as two in-plate replicates with additional no-template controls to exclude contaminations. Mean Ct-values of in-plate replicates were normalized to mean Ct-values of the housekeeping gene Rplp0 (36B4). Relative values are presented as ratio (2−ddCT) with reference to the untreated control.
Table 2.
Primers used in this study.
| Gene | Primer forward | Primer reverse |
|---|---|---|
| Rplp0 | 5‘−GCAGATCGGGTACCCAACTGTTG | 5‘−CAGCAGCCGCAAATGCAGATG |
| Ccnd1 | 5‘−GGAGCTGCTGCAAATGGAAC | 5‘−CAGTCCGGGTCACACTTGA |
| E2f1 | 5‘−AACTGGGCAGCTGAGGTGC | 5‘−CAAGCCGCTTACCAATCCC |
| Pcna | 5‘−AGTGGAGAGCTTGGCAATGG | 5‘−TCAGGTACCTCAGAGCAAACG |
Immunohistochemistry of organoids
For organoid collection, all used plasticware was coated with FBS to avoid organoid adhesion to the surfaces. Organoids were cultured and treated in 6-well plates. Gel domes were destroyed, and organoids were washed and collected in cold PBS. Cell Recovery Solution (Corning) was used as last washing step. Organoids were fixed in 4% paraformaldehyde in PBS overnight. For dehydration, organoids were washed in 70% ethanol, 10 µl of hematoxylin was added and organoids were resuspended in Histogel (Epredia Richard-Allan Scientific, Thermo Fisher Scientific). After paraffin embedding, immunohistochemical staining was performed as formalin-fixed paraffin-embedded (FFPE) samples. Antigen retrieval was done with Target Retrieval Solution High pH (Dako). Primary antibodies were diluted in Antibody Diluent (Zytomed, ZUC025–100) for 30 min at room temperature. Antibodies and dilutions are indicated in Table 1. The Dako EnVision+ System with HRP-Labelled Polymer was used for detection of primary antibodies. BrightDAB (WellMed) served as substrate for the horseradish peroxidase (HRP).
After hematoxylin counterstaining, slides were dehydrated in ethanol and xylol and embedded in mounting medium (Vitro-Clud, Langenbrinck).
After imaging by the ZEN imaging program, the identity of the images was blinded, and 10 fields of view per treatment condition (x40) were analyzed from two independent experiments. Image quantification was done using the ImageJ software. The color deconvolution tool was applied to set a threshold to the DAB staining intensity. Nuclei >50% above the threshold in the organoid structures were classified as stained and counted. The ratio of stained nuclei to all nuclei was calculated for each field of view, and mean values are shown.
Blinding of experimental conditions
The identities of images were blinded by assigning random words as file names (generated using https://www.randomlists.com/random-words?dup=false&qty=100) and then analyzing the images in alphabetic order.
Statistical analysis and visualization
Statistics of each experiment, such as numbers and forms of biological and technical replicates, location and dispersion parameters and the statistical tests used for significance, are provided in the figure legends.
The graphical abstract was created with BioRender.com. Statistical analysis was performed using GraphPad Prism 8.
Biological replicates are independent experiments.
Where data was normalized for better visualization, the mean value of all controls (untreated control; for mRNA expression analysis, 24 hrs untreated control; for organoid and cell growth analysis, day 1 as baseline) was set to 100%, and all other data were calculated and presented relative to this mean value.
Statistical significance was defined as p < 0.05.
Where Student’s t-test was performed, it was calculated as unpaired and two-tailed.
Ordinary two-way ANOVA was calculated comparing palbociclib pre-treated conditions to their respective controls with Dunnett’s multiple comparison test.
Results
Intestinal organoids tolerate the treatment with the CDK4/6 inhibitor palbociclib
To assess the toxicity of cancer therapeutics on the gut mucosa, we prepared organoids of the murine small intestine. These organoids served as model systems for the response of gut epithelia toward drug treatment. Since we were planning to protect these cells against chemotherapeutics by CDK4/6 inhibition, we first needed to assess any potential toxicity of CDK4/6 inhibitors themselves. To do so, murine intestinal organoids were incubated with the CDK4/6 inhibitor palbociclib for 48 hrs, followed by further incubation with fresh and drug-free medium (Figure 1a). During and after treatment, we measured the size of the organoids, thus following their growth (Figures 1b and c). Within the first 3 d, palbociclib moderately slowed down the growth of the organoids (Figures 1b and c). Thereafter, however, the organoids resumed proliferation and displayed growth kinetics that hardly differed from untreated control organoids (Figure 1b). Furthermore, we did not observe any morphological signs of increased organoid damage (Figure 1c). We conclude that palbociclib merely induces a transient delay in growth but does not induce any prolonged toxicity on gut epithelia when applied for a 2 d interval.
Figure 1.
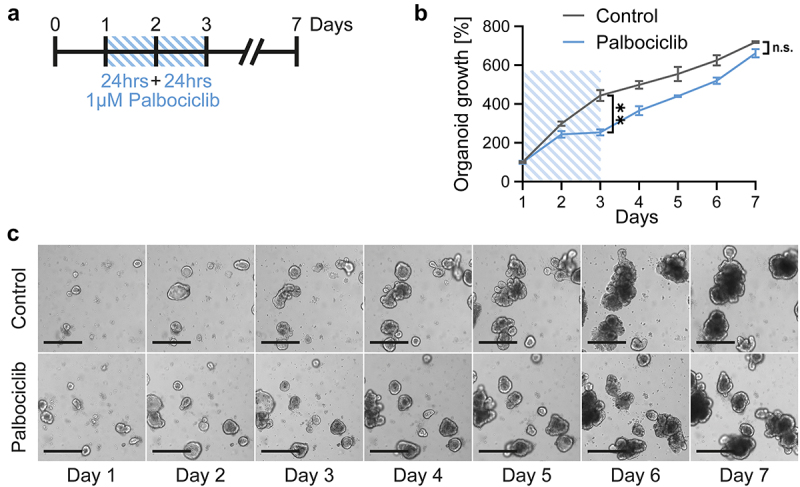
Intestinal organoids grow despite transient CDK4/6 inhibition.
Note: a. Treatment scheme. Organoids were seeded and cultured for 24 hrs, followed by treatment with 1 µM palbociclib for 48 hrs with drug renewal after 24 hrs (blue lines). On day 3, palbociclib was removed and organoids were further cultivated for 4 d.
b. Organoids were treated as described in a and images were taken daily. Organoid growth was quantified by taking bright field images and measuring of the area covered by organoids. Control, untreated samples. Mean of three in-plate replicates ± SEM. Organoid area on day 1 was set 100% and organoid growth was normalized to day 1. Student’s t-test. **p < 0.01; n.s. not significant.
c. Representative images from b were taken at the indicated time points. Accumulation of dead cells in the lumen of organoids (seen as a dark structures), but with an intact outer epithelial barrier (bright outer line), indicates normal organoid homeostasis. Scale bars, 300 µm.
Palbociclib reduces pRb phosphorylation, E2F target gene expression and cell proliferation in intestinal organoids
Next, we assessed the impact of CDK4/6 inhibition on the regulation of E2F-mediated transcription. The CDK4/6 target protein and E2F antagonist pRb were found phosphorylated to a lesser degree upon treatment of organoids with palbociclib, as revealed by immunoblot analysis of phosphorylated pRb (p-pRb) (Figures 2a and b). This led us to test if E2F target genes might be repressed by hypophosphorylated pRb, which generally displays higher activities as a repressor of transcription than its phosphorylated counterpart. In accordance, the levels of PCNA, an E2F target gene product, were found lower upon CDK4/6 inhibition, especially after 48 hrs of palbociclib treatment (Figure 2b). Moreover, the mRNA levels corresponding to E2f1 itself, a gene that can be induced by its own product [51], as well as Pcna, were diminished by CDK4/6 inhibition (Figure 2c). In contrast, the expression of Ccnd1 was upregulated by palbociclib, as previously described [52–55], reflecting cell cycle arrest in G1 (Figure 2c). The increased expression might be further explained by mTor- or MEK-dependent increased Ccnd1 expression upon treatment by palbociclib [56,57]. Finally, the appearance of p-pRb was also found diminished by palbociclib when assessed by immunohistochemistry analyses of the organoids (Figures 2d and e). Moreover, the levels of detectable Ki-67, a marker of cell proliferation, were strongly reduced (Figures 2f and g). This strongly suggests a lower percentage of proliferating cells. In conclusion, CDK4/6 inhibition by palbociclib reduces pRb phosphorylation and E2F-mediated transcription, as well as cell proliferation, in intestinal organoids.
Figure 2.
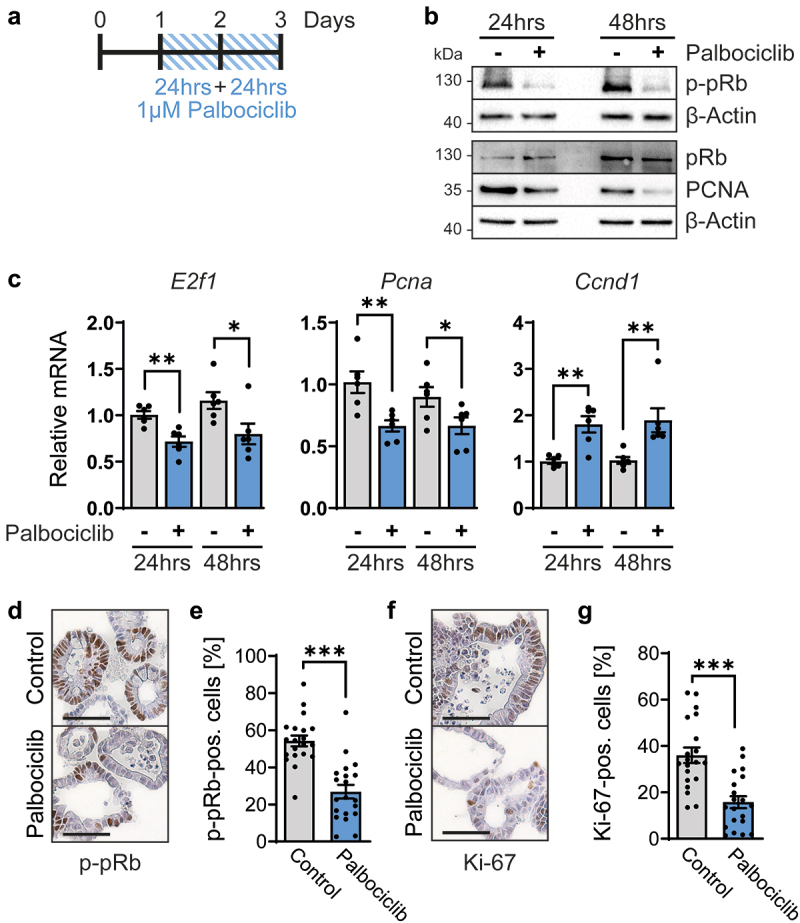
Palbociclib inhibits the cell cycle in intestinal organoids.
Note: a. Scheme of treatment. Organoids were cultured for 24 hrs, then treated with 1 µM palbociclib for 48 hrs, with a treatment renewal after 24 hrs (blue lines). After 24 hrs palbociclib (day 2) and 48 hrs palbociclib (day 3), organoids were analyzed.
b. Organoids were treated with 1 µM palbociclib (+) or left untreated (-) as described in a. Phosphorylated pRB (p-pRB), total pRb and PCNA were detected by immunoblot analyses. β-Actin was stained as a loading control. Representative blots are shown. For p-pRb and PCNA, n = 3, technical replicates from two independent experiments. For total pRb, n = 2, replicates from two independent experiments.
c. Organoids were treated as in a. mRNA levels of cell cycle-regulatory genes were determined by RT-PCR in untreated (-) and 1 µM palbociclib-treated (+) organoids. Expression levels of genes were normalized to Rplp0. Mean ± SEM (n = 6) of three independent experiments with two technical replicates each. Student’s t-test. *p < 0.05, **p < 0.01.
d, f. Representative immunohistochemistry to detect phosphorylated pRb (p-pRb) (d) and Ki-67 (f) in organoids treated as in a for 24 hrs. Control, untreated sample. Hematoxylin was used for counterstaining in blue. Scale bars, 50 µm.
e, g. Quantification of organoids from d and f. Organoids in 20 images (n = 20) from 2 independent experiments were counted for positive p-pRb staining. For each image, positive staining is shown as percentage of all cells. Mean ± SEM. Student’s t-test, ***p < 0.001.
Palbociclib protects intestinal organoids against the cytotoxicity of gemcitabine
Our earlier work revealed that the Mdm2 antagonist nutlin-3 confers protection of p53-proficient cells against gemcitabine [2], most conceivably by arresting the cell cycle and avoiding incorporation of gemcitabine into nascent DNA. Thus, we tested whether CDK4/6 inhibition might also ameliorate such toxicities of gemcitabine on gut epithelia. We treated intestinal organoids with palbociclib for 48 hrs (Figure 3a). After 24 hrs of palbociclib treatment, gemcitabine was added for 24 hrs in combination. Subsequently, all drugs were removed. Cell viability assays based on ATP content revealed a marked cytotoxic effect of gemcitabine alone, leaving less than 50% viability (Figure 3b). However, when the organoids had been pretreated with palbociclib, most cells were protected, with near-100% sustainable cell viability (Figure 3b). In contrast, when using cisplatin, which mediates DNA cross-links by cell cycle-independent mechanisms, the cells were not protected by palbociclib (Figure 3c). When evaluating organoids by cell death morphologies, marked protection against gemcitabine by palbociclib was observed again (Figures 3d and e). Upon treating organoids with these drug combinations (Figure 3a), we stained them with propidium iodide (red), a cell-impermeable dye visualizing compromised cell membrane integrity, and with Hoechst 33,342 (blue) to visualize the epithelial organoid lining [47] (Figure 3d). Propidium iodide entered cells with the concomitant loss of the outer epithelial layer upon treatment with gemcitabine alone, indicating cell death, whereas palbociclib pretreatment rescued organoids, as indicated by a blue Hoechst ring with little if any propidium iodide staining (Figure 3d). Accordingly, palbociclib-pretreated organoids re-grew upon gemcitabine treatment (Figures 3f and g) and they maintained their intact morphology (Figure 3g, higher magnifications). These findings strongly suggest that CDK4/6 inhibition protects normal gut epithelium against the toxicity of the S-phase specific drug gemcitabine, but not against cisplatin.
Figure 3.
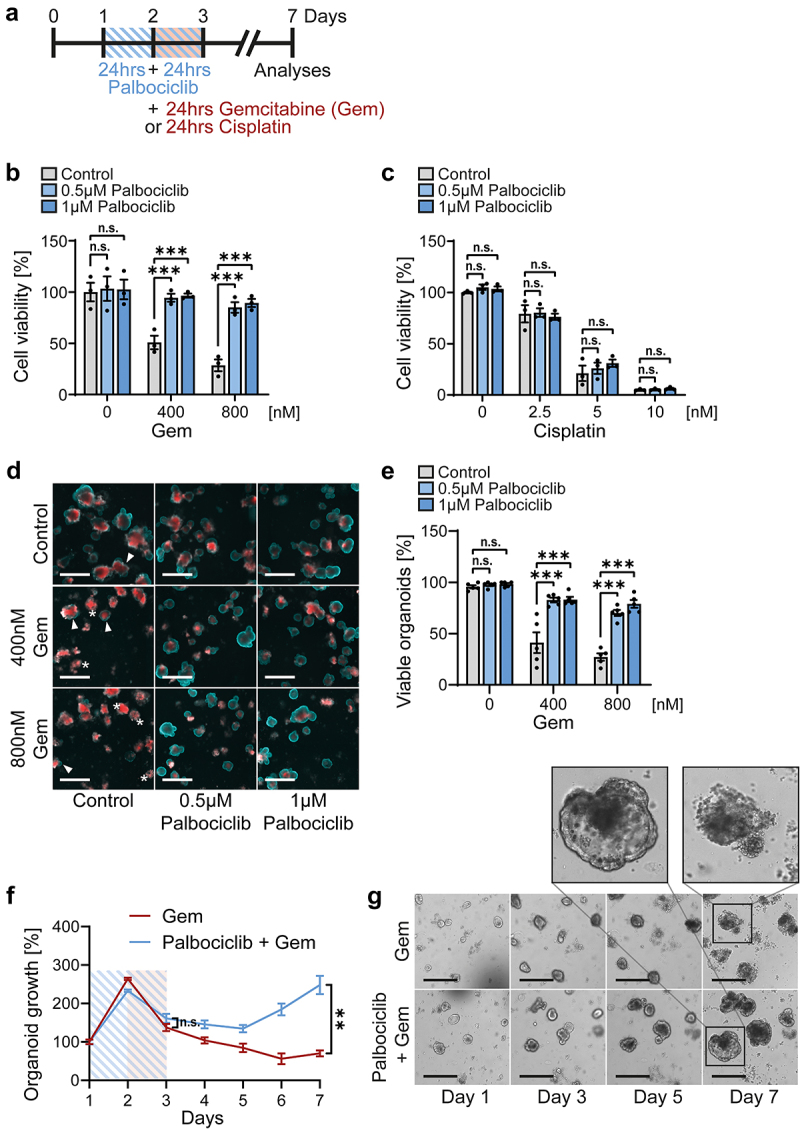
Intestinal organoids are protected against gemcitabine by CDK4/6 inhibition.
Note: a. Scheme of gemcitabine/palbociclib co-treatment of intestinal organoids. Twenty-four hours post-seeding, organoids were treated with palbociclib for 48 hrs (blue lines). For the final 24 hrs, gemcitabine (Gem) or cisplatin were added (red). On day 3, all treatments were removed, and organoids were further cultured for 4 d in their normal growth media. Analyses were performed on day 7.
b, c. Intestinal organoids were treated with gemcitabine (b) or cisplatin (c), with or without palbociclib pre-treatment, as described in a. Organoid viability was determined by CellTiter-Glo 3D assays on day 7. Mean ± SEM of three biological replicates. Untreated control (0 nM gemcitabine and 0 µM palbociclib) was set 100%. Two-way ANOVA with Dunnett’s multiple comparison test was performed to compare palbociclib pre-treated conditions to control, revealing that palbociclib provided protection against gemcitabine but not cisplatin. ***p < 0.001, n.s. not significant.
d. Representative propidium iodide (PI) and Hoechst staining to analyze organoid death. Organoids were treated as in a and, after a re-growth phase, were analyzed on day 7. Control, untreated samples. The membrane-impermeant dye propidium iodide (PI) only marks dead cells (red). The cell-permeable Hoechst dye served as counterstaining to visualize organoid structures (blue). Arrowheads indicate the accumulation of dead cells in the organoid lumen, seen as red staining in the middle of organoids, but with an intact outer epithelial barrier (blue line), showing normal organoid homeostasis representing viable organoids. Asterisks indicate dead organoids that lose the outer epithelial barrier (no blue ring staining), resulting in a diffuse PI staining. Scale bars, 500 µm. Representative images were shown to reveal the protective effect of palbociclib.
e. Quantification of d. Percentage of viable organoids shown as mean ± SEM of five in-plate replicates for each condition. Two-way ANOVA with Dunnett’s multiple comparison test, ***p < 0.001, n.s. not significant.
f. Organoids were treated with 800 nM gemcitabine (red line) or co-treated with 1 µM palbociclib and 800 nM gemcitabine (blue line) as described in a. Images were taken daily with bright field. Mean of three in-plate replicates ± SEM per condition. Organoid area on day 1 was set 100% and organoid growth was normalized to day 1, indicating more pronounced growth when organoids had been treated with palbociclib before exposure to gemcitabine. Student’s t-test. **p < 0.01, n.s. not significant.
g. Representative bright field images from f. Scale bars, 300 µm. The enlarged image sections show a destroyed organoid treated with gemcitabine alone, and an intact organoid that had been exposed to both palbociclib and gemcitabine.
Palbociclib fails to protect RB1-mutant cancer cells from gemcitabine
CDK4/6 inhibition induces pRb hypophosphorylation, and this mediates cell cycle arrest. In accordance, RB1-mutant pancreatic (Panc02.03) and colonic (SW837) cancer cells were not protected by palbociclib (Figure 4). Using the same treatment scheme as for organoids (Figure 4a), RB1-mutant cancer cells showed a sustained reduction in cell viability (Figure 4b) and cell growth (Figure 4c) upon different concentrations of gemcitabine, despite palbociclib pretreatment. Thus, palbociclib does not reduce the efficacy of gemcitabine against RB1-mutant cancer cells.
Figure 4.
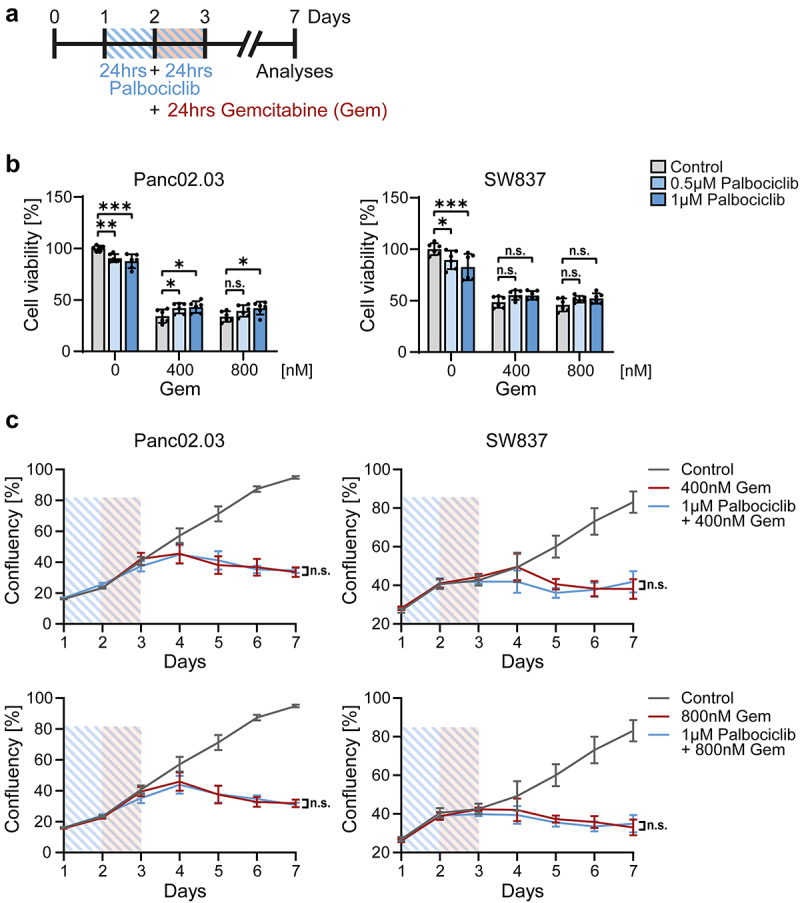
RB1-mutant cancer cells are not protected against gemcitabine by palbociclib.
Note: a. Schematic overview of drug treatment. After pre-culturing, pancreatic Panc02.03 and colonic SW837 cells (both carrying RB1-deletions) were treated with palbociclib for 48 hrs (blue lines), with co-treatment by gemcitabine (Gem) for the final 24 hrs (red rectangle). On day 3, all drugs were removed, and cells were cultured for 4 d in normal growth media.
b. Panc02.03 cells (left) and SW837 cells (right) were treated as described in a. Control, untreated cells. Cell viability was determined with CellTiter-Glo assays on day 7 to reveal only minor differences inferred by palbociclib pretreatment. Mean ± SEM from two biological replicates, each with three in-plate replicates (n = 6). Untreated control (0 nM gemcitabine and 0 µM palbociclib) was set 100%. Two-way ANOVA with Dunnett’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant.
c. Proliferation of Panc02.03 (left) and SW837 (right) cells. Cells were treated as in a. Cell confluency was measured daily via bright field imaging and calculated as percentage of covered area. Mean ± SEM from two independent experiments with three in-plate replicates each (n = 6). Control (gray line) was used for both gemcitabine concentrations. Blue lines, palbociclib for 48 hrs. Red rectangle, co-treatment with gemcitabine (Gem). Student’s t-test on day 7, n.s. not significant. Palbociclib did not significantly affect the sensitivity of the RB1-deficient cancer cells toward gemcitabine.
Palbociclib protects intestinal organoids against SN-38, the cytotoxic metabolite of irinotecan
Finally, we asked whether CDK4/6 inhibition might protect gut epithelia against the topoisomerase I inhibitor irinotecan. This was of interest for two reasons. Firstly, irinotecan is known for its strong side effects within the gastrointestinal tract, causing severe diarrhea and epithelial damage, which often leads to interruption or termination of treatment [26]. Secondly, irinotecan acts most strongly on cells in S phase, since it traps topoisomerase near DNA replication forks, resulting in DNA breaks and apoptosis [24,28]. Thus, we hypothesized that CDK4/6 inhibition might be suitable to diminish the toxic side effects of irinotecan. To test this, we pre-treated gut organoids with palbociclib, followed by parallel treatment with SN-38, the active metabolite of irinotecan (Figure 5a). Indeed, SN-38-induced apoptosis, revealed by cleaved/activated caspase-3, was reduced by a palbociclib pretreatment (Figure 5b). Next, we detected phosphorylated KAP1 and H2A×, two markers of the DNA damage response. These phosphorylations were markedly induced by SN-38, reflecting DNA damage (Figure 5b). Importantly, the DNA damage response was diminished by the parallel treatment with palbociclib (Figure 5b). Immunohistochemical staining for phosphorylated γ-H2A× confirmed the protective effect of palbociclib upon SN-38 (Figures 5c and d), strongly suggesting that CDK4/6 inhibition preserved DNA integrity in the presence of a topoisomerase inhibitor. Furthermore, staining by propidium iodide and Hoechst revealed severe toxicity of SN-38, which was substantially rescued by palbociclib again (Figures 5e and f). In parallel, SN-38 alone strongly reduced cell viability, as shown by ATP-based assays, whereas the pretreatment with palbociclib restored a substantial proportion of viability (Figure 5g). Finally, we assessed the long-term growth of treated organoids and found that palbociclib enabled them to resume growth 2 d after the end of SN-38 exposure; in contrast, SN-38 alone disabled their growth for at least 5 d (Figures 5h and i). Thus, palbociclib is an effective protector of intestinal organoids against SN-38.
Figure 5.
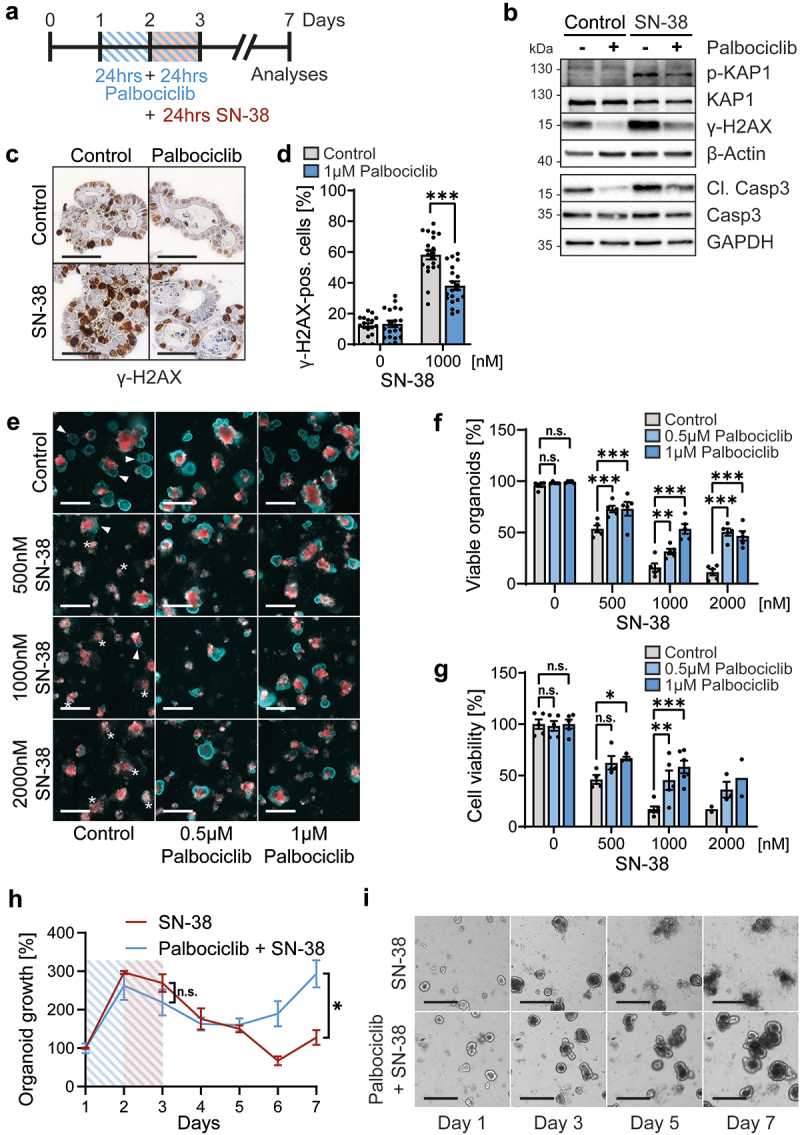
Palbociclib diminishes the cytotoxicity of SN-38, the active metabolite of irinotecan, against intestinal organoids.
Note: a. Treatment scheme. Organoids were pre-cultured for 24 hrs and then pre-incubated with palbociclib for 48 hrs. Palbociclib was renewed after 24 hrs (blue lines). For the final 24 hrs, SN-38 or DMSO was added (red rectangle). All treatments were removed on day 3, and organoids were further incubated for 4 d in their normal growth media. Analyses were performed on day 3 or day 7.
b. To reveal the DNA damage response, organoids were treated with drugs as described in a, and immunoblot analyses of the indicated proteins were performed on day 3. p-KAP1, γ-H2A× and cleaved Caspase-3 (Cl. Casp3) were all detected to reveal diminished SN-38-induced DNA damage by palbociclib. β-Actin or GAPDH, loading controls for the corresponding representative immunoblots. KAP1 and Caspase-3 (Casp3), as additional controls. n ≥ 2 from two biological replicates.
c. Representative immunohistochemical staining of γ-H2A× within intestinal organoids on day 3, again indicating protection by palbociclib. Organoids were handled as described in a. Hematoxylin served as counterstaining (blue). Scale bars, 50 µm.
d. Quantification of γ-H2A× staining from c. Nuclei staining positive for γ-H2A× staining is indicated as percentage of all nuclei. n ≥ 17 images were taken per condition from two independent experiments. Mean ± SEM. Student’s t-test. *** p < 0.001.
e. Intestinal organoids were treated as indicated in a. to reveal decreased damage to SN-38-treated organoids by pre-treatment with palbociclib. Control, DMSO treated samples. Visualization of organoid morphology using double staining with Propidium iodide (PI) (red) and Hoechst (blue). PI served as a marker for dead cells in unfixed organoid samples. Hoechst, counterstaining to visualize the organoid structure. Please note that accumulation of dead cells in the lumen of organoids (seen as red staining), but with an intact outer epithelial barrier (blue line), indicates normal organoid homeostasis. Thus, arrowheads represent viable organoids. A dead organoid loses the outer epithelial barrier, and PI staining also occurs there (asterisks). Scale bars, 500 µm.
f. Quantification of e. Percentage of viable organoids shown as mean ± SEM of n = 5 in-plate replicates. Two-way ANOVA with Dunnett’s multiple comparison test, **p < 0.01, ***p < 0.001, n.s. not significant.
g. Cell viability assays performed on organoids treated as in a on day 7, revealing protection of organoids by palbociclib against SN-38. n = 2–6 independent experiments for the indicated SN-38 concentrations, respectively. Normalization to control samples (DMSO treated). Data presented as mean ± SEM. When n ≥ 3, two-way ANOVA with Dunnett’s multiple comparison test was performed, *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant.
h. Organoids were treated as described in a with 1 µM palbociclib and 1 µM SN-38 (blue curve), or treated with 1 µM SN-38 alone (red curve). Bright field images for each condition were taken daily. Organoid growth was calculated as area covered by viable organoids. The area of organoids on day 1 was set 100% and organoid growth was normalized to day 1. Mean of three in-plate replicates ± SEM. Student’s t-tests for day 3 and day 7, *p < 0.05, n.s. not significant.
i. Representative bright field images from h. Scale bars, 300 µm.
Palbociclib does not reduce the toxicity of SN-38 against RB1-deficient cancer cells
RB1 mutated cancer cells were not protected by palbociclib upon SN-38 treatment (Figure 6). Pancreatic Panc02.03 cells and colonic SW837 cells showed a constant decrease in cell viability (Figure 6b) and cell growth (Figure 6c) despite palbociclib pretreatment compared to SN-38 alone, with three different concentrations of SN-38. Hence, CDK4/6 inhibition does not interfere with the therapeutic efficacy of irinotecan/SN-38 against RB1-deficient cancer cells.
Figure 6.
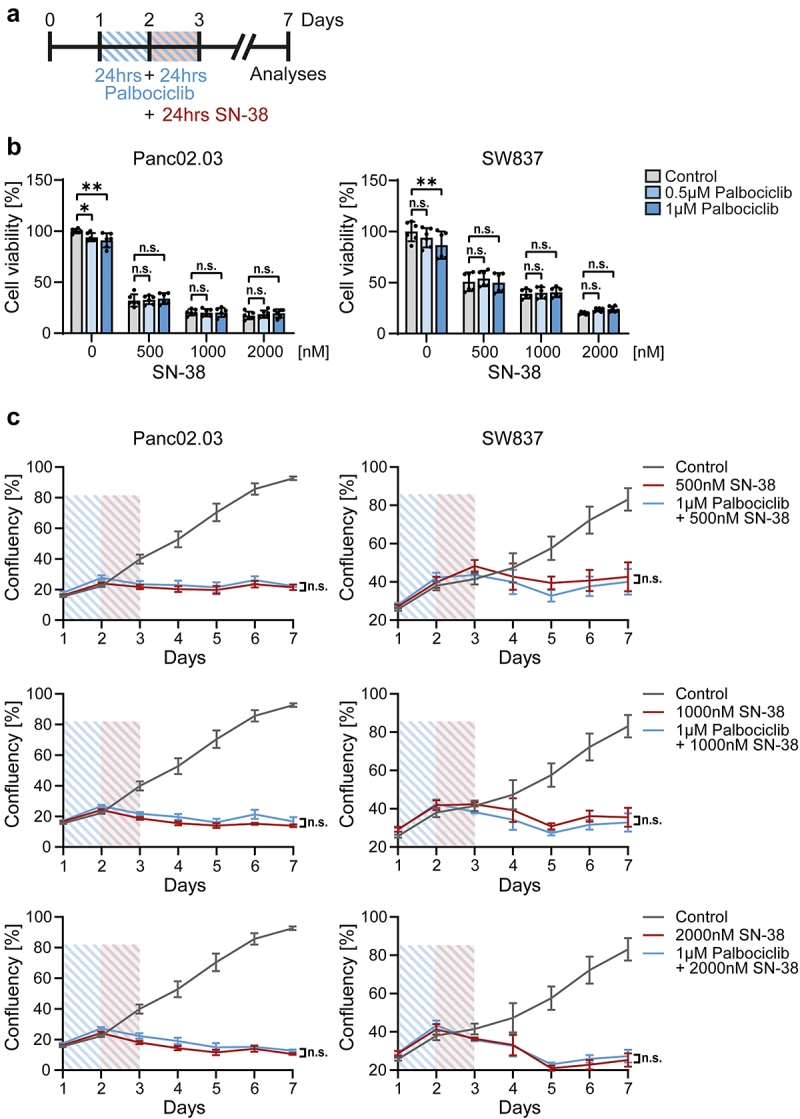
Palbociclib does not protect RB1-mutant cancer cells against SN-38.
Note: a. Scheme of co-treatment with SN-38 and palbociclib. RB1-mutant cancer cells were pretreated with palbociclib for 24 hrs with a renewal for 24 hrs (total 48 hrs, blue lines). For the final 24 hrs, SN-38 or DMSO was added (red box). On day 3, the drugs were removed and the cells were further cultured for 4 d, followed by analyses.
b. Panc02.03 (left) and SW837 (right) cells were handled as described in a to reveal that little or no protection by palbociclib was provided to them. On day 7, CellTiter-Glo assays were performed to assess cell viability. Mean ± SEM from two independent experiments, each with three in-plate replicates (n = 6). DMSO served as control treatment (0 nM SN-38 and 0 µM palbociclib) and was set 100%. Two-way ANOVA with Dunnett’s multiple comparison test, *p < 0.05, **p < 0.01, n.s. not significant.
c. Cancer cells were treated as in a. Daily measurements of cell growth of Panc02.03 (left) and SW837 (right) cells were performed via a bright field imaging system, indicating the lack of protection by palbociclib. The percentage of area covered by cells was calculated as cell confluency. Mean ± SEM from two biological replicates with ≥2 in-plate replicates (n ≥ 5). Control (DMSO treatment, gray line) was used for both SN-38 concentrations. Blue lines, palbociclib for 48 hrs. Red rectangle, co-treatment with SN-38. Student’s t-test on day 7, n.s. not significant.
Discussion
We found that, in an organoid model system of the intestine, short-term treatment with a CDK inhibitor is of no permanent harm to the epithelia, but it does cause a transient cell cycle arrest, protecting the cells against the toxicity of irinotecan or gemcitabine. This raises the perspective that CDK inhibitors might be usable to avoid intestinal toxicities of cell cycle-dependent chemotherapeutics.
The concept of cyclotherapy was proposed in 2002 [58] and refined ever since [1–5,16–18,59–69]. Some studies specifically addressed the bone marrow and the hematopoietic cells therein, representing a highly vulnerable fraction of cells during chemotherapy [16,64–69]. The options for protecting gut epithelia by inhibiting CDKs in the context of chemotherapy have not been evaluated in the past. Previous research has focused on radio- and chemoprotection of intestinal cells via induction of p53, while CDK inhibition to avoid intestinal toxicity has only been studied in the context of ionizing radiation [63,70–73]. However, at the same time, with our study, a protective effect of CDK4/6 inhibition against the toxicity of different chemotherapeutics was reported [74]. Both studies strongly underline that cell cycle inhibition is a promising route to protect normal intestinal epithelium.
An important caveat for the outlined strategy is that CDK inhibition should not interfere with the efficacy of chemotherapeutics on the tumor itself. This is most conceivable when treating tumors that lack the key target of CDKs, i.e. pRb. Besides retinoblastoma, a rare tumor species, bi-allelic losses of RB1 are most frequently found in SCLC, where they approach almost 100% [13]. However, more tumors show RB1 deletions or disabling point mutations at considerable frequencies. These include prostate cancer (5–25%), bladder cancer (15–25%), melanoma (4–15%) and advanced stages of colorectal cancer and pancreatic cancer (both approx. 5%) [48,49]. After assessing the RB1 status of such tumors, it might become possible to apply CDK inhibition for protecting normal tissues as part of a personalized cancer treatment regimen.
Besides RB1 deficiency, tumors that inactivate pRb by a viral antagonist are likely to be amenable to chemotherapy in combination with a cytoprotective CDK inhibitor. Cervical carcinoma represents the most prominent example for this situation, but the same is true for a fraction of head-and-neck cancers. The vast majority of these cancers display integrated portions of HPV genomes. This typically gives rise to the synthesis of HPV E7 proteins, which bind and inactivate pRb. Applying CDK inhibitors to such tumor cells does not lead to a cell cycle arrest, since even hypophosphorylated pRb no longer binds E2F proteins due to the competing interaction of E7 with pRb [75].
Other alterations associated with non-responsiveness of tumor cells to palbociclib include the lack of active CDK4, CDK6 alterations, strong expression of cyclin E1 or p16, or heterozygous RB1 defects. Such conditions can also lead to tumor cell proliferation despite the presence of pharmacological CDK4/6 inhibitors, leaving a window of opportunity for using the inhibitors for protecting non-cancerous tissues [52,76–80].
Recently, further chemoprotective concepts using CDK4/6 inhibitors even without stratifying for pRb inactivation have been proposed, such as the exploitation of their poor blood-brain barrier permeability [81].
Interestingly, CDK4/6 inhibitors can enhance the cytotoxicity of chemotherapeutics in some tumor species that preserve RB1 [82]. At first glance, this seems counterintuitive, but several explanations have been provided. Most importantly, CDK4/6 inhibition can compromise DNA repair machineries, in particular homologous recombination repair. This then leads to enhanced accumulation of DNA damage when CDK4/6 inhibitors are applied together with directly genotoxic agents [83–85]. Additional explanations for synergies between CDK inhibitors and chemotherapeutics include enhancement of the immune response against tumor cells [86], decreased glucose metabolism [87], inactivation of the tumor-promoting chaperone system [88] or impaired vascularization of the tumor) [89–91]. Of note, synergies between hypoxia and a CDK4/6 inhibitor were found in colon cancer cells [92], and this tumor species is most commonly treated with irinotecan. It is thus tempting to propose that CDK4/6 inhibitors might be cytotoxic toward the cells of colon cancers, in the context of hypoxia and irinotecan-treatment, while protecting normal gut epithelia from irinotecan-induced damage. The same might be true when treating colon cancer with other conventional chemotherapeutics, and along this line, an ongoing study uses a CDK4/6 inhibitor in parallel to the FOLFOXIRI scheme for treating metastatic colorectal cancer (ClinicalTrials.gov: NCT04607668).
Palbociclib is mostly used for reducing tumor growth [93]. However, for the purposes of myeloid cell protection, the CDK4/6 inhibitor trilaciclib has been evaluated within clinical studies [64–68]. Current studies combine conventional chemotherapeutics with trilaciclib for treating SCLC, triple-negative breast cancer, colorectal cancer and bladder cancer (ClinicalTrials.gov: NCT05071703, NCT04607668, NCT04799249, NCT05113966, NCT05112536, NCT04902885, NCT04887831). Our results suggest that CDK4/6 inhibition might not only serve to preserve hematopoiesis but also to avoid damage to the gastrointestinal epithelia in these patients.
Among established chemotherapeutics, irinotecan appears most prone to gastrointestinal toxicities. The clinical manifestation of this is diarrhea, with early and late diarrhea displaying different characteristics. Early diarrhea appears mostly due to increased cholinergic activity, whereas late and more severe diarrhea is caused by epithelial damage [24,27,94–98]. We thus expect that CDK inhibition is unlikely to avoid early diarrhea but might prevent intestinal epithelial damage. Furthermore, CDK inhibition should ameliorate bone marrow suppression and alopecia, other common side effects of irinotecan [99,100].
On top of irinotecan, other chemotherapeutics depend on the cell cycle and also display gastrointestinal toxicities [21]. Combination with CDK inhibitors might be suitable for avoiding damage to gastrointestinal epithelia in such settings as well. This includes etoposide, a topoisomerase II inhibitor used in the first-line therapy for SCLC. A frequent adverse effect of etoposide is diarrhea, which might be avoidable by CDK inhibition.
Exploiting the mutation of cell cycle regulatory genes in cancer cells can serve to broaden the therapeutic window. Besides enhancing the cytotoxicity toward cancer cells, such strategies should include the protection of normal tissue, most conceivably by inducing a transient cell cycle arrest. Besides hematopoietic cells, the epithelia of the intestine may thereby become amenable to protection, thus increasing the options for cytotoxic tumor treatment, as well as improving the quality of patient life.
Acknowledgements
We thank Birgit Jünemann for her valuable technical support, John Wiedenhöft for statistical advice, and Katharina Ewers, Valentina Manzini, Tiago de Oliveira and Johannes Robert Fleischer for fruitful discussions and valuable advice.
Funding Statement
J. B. thanks the UMG Promotionskolleg, supported by the Else-Kröner-Fresenius-Foundation and the Jacob-Henle-Programm, and the Studienstiftung des Deutschen Volkes for their support .R. S.-H. is supported by the Deutsche Forschungsgemeinschaft (DFG) (SCHUH-3160/3-1). M. D. is supported by the Deutsche Forschungsgemeinschaft, the Deutsche Krebshilfe and the Wilhelm-Sanders-Stiftung.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Data are located in controlled access data storage at Gesellschaft für wissenschaftliche Datenverarbeitung mbH Göttingen (GWDG).
Ethics declaration
Mouse breeding, housing and handling were approved by state (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, LAVES, Germany) and institutional (Göttingen University Medical Center) committees, which ensured the conformity to the relevant regulatory standards. Mice were housed and handled under pathogen-free barrier conditions.
Author contributions
Conceptualization M. D.; Methodology J. B., L. C., T. I., A. D., L-C. C. and R. S-H.; Acquisition of data: all authors.; Analysis and interpretation of data: J. B.; L. C., T. I., A. D. and R. S-H.; Writing Original Draft J. B., R. S-H., M. D.; Writing Review & Editing: all authors; Final approval: all authors; Funding Acquisition R. S-H. and M. D.; Supervision L. C., R. S-H. and M. D.
References
- [1].van Leeuwen IMM. Cyclotherapy: opening a therapeutic window in cancer treatment. Oncotarget. 2012;3:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kranz D, Dobbelstein M. Nongenotoxic p53 activation protects cells against S-phase-specific chemotherapy. Cancer Res. 2006;66:10274–10280. [DOI] [PubMed] [Google Scholar]
- [3].van Leeuwen IMM, Rao B, Sachweh MCC, et al. An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells. Cell Cycle. 2012;11:1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Apontes P, Leontieva OV, Demidenko ZN, et al. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011;2:222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Carvajal D, Tovar C, Yang H, et al. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005;65:1918–1924. [DOI] [PubMed] [Google Scholar]
- [6].Eggersmann TK, Degenhardt T, Gluz O, et al. CDK4/6 inhibitors expand the therapeutic options in breast cancer: palbociclib, ribociclib and abemaciclib. BioDrugs Clin Immunother Biopharm Gene Ther. 2019;33:125–135. [DOI] [PubMed] [Google Scholar]
- [7].Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors: a 2022 update. Pharmacol Res. 2022;175:106037. [DOI] [PubMed] [Google Scholar]
- [8].Samuel Eziokwu A, Varella L, Lynn Kruse M, et al. Real-world outcomes of cyclin-dependent kinase inhibitors continued beyond first disease progression in hormone receptor-positive metastatic breast cancer. Clin Breast Cancer. 2021;21:205–209. [DOI] [PubMed] [Google Scholar]
- [9].Knudsen ES, Schultz E, Hamilton D, et al. Real-world experience with CDK4/6 inhibitors for metastatic HR+/HER2- breast cancer at a single cancer center. Oncology. 2022;27:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fischer M, Grossmann P, Padi M, et al. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fischer M, Schade AE, Branigan TB, et al. Coordinating gene expression during the cell cycle. Trends Biochem Sci. 2022 Dec;47(12):1009–1022. [DOI] [PubMed] [Google Scholar]
- [12].Uxa S, Bernhart SH, Mages CFS, et al. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019;47:9087–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu Y, Zhao R, Fang S, et al. Abemaciclib sensitizes HPV-negative cervical cancer to chemotherapy via specifically suppressing CDK4/6-Rb-E2F and mTOR pathways. Fundam Clin Pharmacol. 2021;35:156–164. [DOI] [PubMed] [Google Scholar]
- [15].Jin D, Tran N, Thomas N, et al. Combining CDK4/6 inhibitors ribociclib and palbociclib with cytotoxic agents does not enhance cytotoxicity. PLoS ONE. 2019;14:e0223555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].He S, Roberts PJ, Sorrentino JA, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med. 2017;9:eaal3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Purba TS, Ng’andu K, Brunken L, et al. CDK4/6 inhibition mitigates stem cell damage in a novel model for taxane-induced alopecia. EMBO Mol Med. 2019;11:e11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].DiRocco DP, Bisi J, Roberts P, et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol - Ren Physiol. 2014;306:F379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Balducci L, Falandry C, List A. New advances in supportive care: chemoprotective agents as novel opportunities in geriatric oncology. Curr Oncol Rep. 2022;24:1695–1703. [DOI] [PubMed] [Google Scholar]
- [20].Wardill HR, Bowen JM, Gibson RJ. Chemotherapy-induced gut toxicity: are alterations to intestinal tight junctions pivotal? Cancer Chemother Pharmacol. 2012;70:627–635. [DOI] [PubMed] [Google Scholar]
- [21].Boussios S, Pentheroudakis G, Katsanos K, et al. Systemic treatment-induced gastrointestinal toxicity: incidence, clinical presentation and management. Ann Gastroenterol. 2012;25:106–118. [PMC free article] [PubMed] [Google Scholar]
- [22].Parvez MM, Basit A, Jariwala PB, et al. Quantitative investigation of irinotecan metabolism, transport, and gut microbiome activation. Drug Metab Dispos Biol Fate Chem. 2021;49:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379:2395–2406. [DOI] [PubMed] [Google Scholar]
- [24].de Man FM, Goey AKL, van Schaik RHN, et al. Individualization of irinotecan treatment: a review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin Pharmacokinet. 2018;57:1229–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vogel A, Hofheinz RD, Kubicka S, et al. Treatment decisions in metastatic colorectal cancer - Beyond first and second line combination therapies. Cancer Treat Rev. 2017;59:54–60. [DOI] [PubMed] [Google Scholar]
- [26].Paulík A, Nekvindová J, Filip S. Irinotecan toxicity during treatment of metastatic colorectal cancer: focus on pharmacogenomics and personalized medicine. Tumori. 2020;106:87–94. [DOI] [PubMed] [Google Scholar]
- [27].Yue B, Gao R, Wang Z, et al. Microbiota-host-irinotecan axis: a new insight toward irinotecan chemotherapy. Front Cell Infect Microbiol. 2021;11:710945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kciuk M, Marciniak B, Kontek R. Irinotecan-still an important player in cancer chemotherapy: a comprehensive overview. Int J Mol Sci. 2020;21:4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Burris HA, Fields SM. Topoisomerase I inhibitors: an overview of the camptothecin analogs. Hematol Oncol Clin North Am. 1994;8:333–355. [PubMed] [Google Scholar]
- [30].Pommier Y, Pharm D, Fesen MR, et al. Cellular determinants of sensitivity and resistance to DNA topoisomerase inhibitors. Cancer Invest. 1994;12:530–542. [DOI] [PubMed] [Google Scholar]
- [31].Mei C, Lei L, Tan L-M, et al. The role of single strand break repair pathways in cellular responses to camptothecin induced DNA damage. Biomed Pharmacother Biomed Pharmacother. 2020;125:109875. [DOI] [PubMed] [Google Scholar]
- [32].Plunkett W, Huang P, Searcy CE, et al. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996;23:3–15. [PubMed] [Google Scholar]
- [33].Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet Lond Engl. 2017;389:1011–1024. [DOI] [PubMed] [Google Scholar]
- [34].Ouyang G, Liu Z, Huang S, et al. Gemcitabine plus cisplatin versus gemcitabine alone in the treatment of pancreatic cancer: a meta-analysis. World J Surg Oncol. 2016;14:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. [DOI] [PubMed] [Google Scholar]
- [36].Kim SH, Shin SJ, Kim SY, et al. Combining capecitabine, oxaliplatin, and gemcitabine (XELOXGEM) for colorectal carcinoma patients pretreated with irinotecan: a multicenter phase I/II trial. Cancer Chemother Pharmacol. 2012;69:91–97. [DOI] [PubMed] [Google Scholar]
- [37].Madajewicz S, Waterhouse DM, Ritch PS, et al. Multicenter, randomized phase II trial of bevacizumab plus folinic acid, fluorouracil, gemcitabine (FFG) versus bevacizumab plus folinic acid, fluorouracil, oxaliplatin (FOLFOX4) as first-line therapy for patients with advanced colorectal cancer. Invest New Drugs. 2012;30:772–778. [DOI] [PubMed] [Google Scholar]
- [38].Correale P, Botta C, Rotundo MS, et al. Gemcitabine, oxaliplatin, levofolinate, 5-fluorouracil, granulocyte-macrophage colony-stimulating factor, and interleukin-2 (GOLFIG) versus FOLFOX chemotherapy in metastatic colorectal cancer patients: the GOLFIG-2 multicentric open-label randomized phase III trial. J Immunother. 2014;37:26–35. [DOI] [PubMed] [Google Scholar]
- [39].Spindler KL, Pallisgaard N, Andersen RF, et al. Gemcitabine and capecitabine for heavily pre-treated metastatic colorectal cancer patients–a phase II and translational research study. Anticancer Res. 2014;34:845–850. [PubMed] [Google Scholar]
- [40].Lee K-W, Kim YJ, Lee K-H, et al. Phase II trial of gemcitabine plus UFT as salvage treatment in oxaliplatin, irinotecan and fluoropyrimidine-refractory metastatic colorectal cancer. Cancer Chemother Pharmacol. 2014;74:447–455. [DOI] [PubMed] [Google Scholar]
- [41].Passardi A, Fanini F, Turci L, et al. Prolonged pemetrexed infusion plus gemcitabine in refractory metastatic colorectal cancer: preclinical rationale and phase II study results. Oncology. 2017;22:886-e79. DOI: 10.1634/theoncologist.2017-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cahan B, Leong L, Wagman L, et al. Phase I/II trial of anticarcinoembryonic antigen radioimmunotherapy, gemcitabine, and hepatic arterial infusion of fluorodeoxyuridine postresection of liver metastasis for colorectal carcinoma. Cancer Biother Radiopharm. 2017;32:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baretti M, Karunasena E, Zahurak M, et al. A phase 2 trial of gemcitabine and docetaxel in patients with metastatic colorectal adenocarcinoma with methylated checkpoint with forkhead and ring finger domain promoter and/or microsatellite instability phenotype. Clin Transl Sci. 2021;14:954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- [45].Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. [DOI] [PubMed] [Google Scholar]
- [46].Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–1597. [DOI] [PubMed] [Google Scholar]
- [47].Klemke L, Blume JP, Oliveira TD, et al. Preparation and cultivation of colonic and small intestinal murine organoids including analysis of gene expression and organoid viability. Bio-Protoc. 2022;12:e4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioportal. Sci Signal. 2013;6:l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994;8:1514–1525. [DOI] [PubMed] [Google Scholar]
- [52].Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Res. 2016;76:2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sathe A, Koshy N, Schmid SC, et al. CDK4/6 inhibition controls proliferation of bladder cancer and transcription of RB1. J Urol. 2016;195:771–779. [DOI] [PubMed] [Google Scholar]
- [54].Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5:6512–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Martin CA, Cullinane C, Kirby L, et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int J Cancer. 2018;142:2139–2152. [DOI] [PubMed] [Google Scholar]
- [56].Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle Georget Tex. 2013;12:3063–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell Cycle Georget Tex. 2002;1:375–382. [DOI] [PubMed] [Google Scholar]
- [59].Jackson RC, Veroli GYD, Koh S-B, et al. Modelling of the cancer cell cycle as a tool for rational drug development: a systems pharmacology approach to cyclotherapy. PLoS Comput Biol. 2017;13:e1005529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rao B, Lain S, Thompson AM. P53-Based cyclotherapy: exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. Br J Cancer. 2013;109:2954–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Leibowitz BJ, Yang L, Wei L, et al. Targeting p53-dependent stem cell loss for intestinal chemoprotection. Sci Transl Med. 2018;10:eaam7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Malhab LJB, Descamps S, Delaval B, et al. The use of the NEDD8 inhibitor MLN4924 (Pevonedistat) in a cyclotherapy approach to protect wild-type p53 cells from MLN4924 induced toxicity. Sci Rep. 2016;6:37775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang X, Zhu S, Qian L, et al. IL-1Ra selectively protects intestinal crypt epithelial cells, but not tumor cells, from chemotoxicity via p53-mediated upregulation of p21(WAF1) and p27(KIP1.). Pharmacol Res. 2014;82:21–33. [DOI] [PubMed] [Google Scholar]
- [64].Weiss JM, Csoszi T, Maglakelidze M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol. 2019;30:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tan AR, Wright GS, Thummala AR, et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2019;20:1587–1601. [DOI] [PubMed] [Google Scholar]
- [66].Daniel D, Kuchava V, Bondarenko I, et al. Trilaciclib prior to chemotherapy and atezolizumab in patients with newly diagnosed extensive‐stage small cell lung cancer: a multicentre, randomised, double‐blind, placebo‐controlled Phase II trial. Int J Cancer. 2021;148:2557–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hart LL, Ferrarotto R, Andric ZG, et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: results from a randomized, double-blind, placebo-controlled phase ii Study. Adv Ther. 2021;38:350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tan AR, Wright GS, Thummala AR, et al. Trilaciclib prior to chemotherapy in patients with metastatic triple-negative breast cancer: final efficacy and subgroup analysis from a randomized phase II study. Clin Cancer Res Off J Am Assoc Cancer Res. 2022;28:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bisi JE, Sorrentino JA, Roberts PJ, et al. Preclinical characterization of G1T28: a novel CDK4/6 inhibitor for reduction of chemotherapy-induced myelosuppression. Mol Cancer Ther. 2016;15:783–793. [DOI] [PubMed] [Google Scholar]
- [70].Pant V, Xiong S, Wasylishen AR, et al. Transient enhancement of p53 activity protects from radiation-induced gastrointestinal toxicity. Proc Natl Acad Sci. 2019;116:17429–17437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wei L, Leibowitz BJ, Wang X, et al. Inhibition of CDK4/6 protects against radiation-induced intestinal injury in mice. J Clin Invest. 2016;126:4076–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lee C-L, Oh P, Xu ES, et al. Blocking cyclin-dependent kinase 4/6 during single dose versus fractionated radiation therapy leads to opposite effects on acute gastrointestinal toxicity in mice. Int J Radiat Oncol Biol Phys. 2018;102:1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nag D, Bhanja P, Riha R, et al. Auranofin protects intestine against radiation injury by modulating p53/p21 pathway and radiosensitizes human colon tumor. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25:4791–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Xiang J, Wang H, Tao Q, et al. CDK4/6 inhibitor modulating active and quiescent intestinal stem cells for prevention of chemotherapy-induced diarrhea. J Pathol. 2023. DOI: 10.1002/path.6078 [DOI] [PubMed] [Google Scholar]
- [75].Gadsden NJ, Fulcher CD, Li D, et al. Palbociclib renders human papilloma virus-negative head and neck squamous cell carcinoma vulnerable to the senolytic agent navitoclax. Mol Cancer Res MCR. 2021;19:862–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Raspé E, Coulonval K, Pita JM, et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med. 2017;9:1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Paternot S, Raspé E, Meiller C, et al. Preclinical evaluation of CDK4 phosphorylation predicts high sensitivity of pleural mesotheliomas to CDK4/6 inhibition. Mol Oncol. 2022. DOI: 10.1002/1878-0261.13351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu X, Yang X, Xiong Y, et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat Cancer. 2021;2:429–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Palafox M, Monserrat L, Bellet M, et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER+ breast cancer. Nat Commun. 2022;13:5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Turner NC, Liu Y, Zhu Z, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor–positive metastatic breast cancer. J Clin Oncol. 2019;37:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Blagosklonny MV. Selective protection of normal cells from chemotherapy, while killing drug-resistant cancer cells. Oncotarget. 2023;14:193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Klein ME, Kovatcheva M, Davis LE, et al. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Pesch AM, Hirsh NH, Chandler BC, et al. Short term CDK4/6 inhibition radiosensitizes estrogen receptor positive breast cancers. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26:6568–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Salvador-Barbero B, Álvarez-Fernández M, Zapatero-Solana E, et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 2020;37:340–353.e6. [DOI] [PubMed] [Google Scholar]
- [85].Yi J, Liu C, Tao Z, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine. 2019;43:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ameratunga M, Kipps E, Okines AFC, et al. To cycle or fight—CDK4/6 inhibitors at the crossroads of anticancer immunity. Clin Cancer Res. 2019;25:21–28. [DOI] [PubMed] [Google Scholar]
- [87].Franco J, Balaji U, Freinkman E, et al. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016;14:979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Isermann T, Şener ÖÇ, Stender A, et al. Suppression of HSF1 activity by wildtype p53 creates a driving force for p53 loss-of-heterozygosity. Nat Commun. 2021;12:4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Cao J, Zhu Z, Wang H, et al. Combining CDK4/6 inhibition with taxanes enhances anti-tumor efficacy by sustained impairment of Prb-E2F pathways in squamous cell lung cancer. Oncogene. 2019;38:4125–4141. [DOI] [PubMed] [Google Scholar]
- [90].Azam SH, Porrello A, Harrison EB, et al. Quaking orchestrates a post-transcriptional regulatory network of endothelial cell cycle progression critical to angiogenesis and metastasis. Oncogene. 2019;38:5191–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Liu M, Xu S, Wang Y, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget. 2016;7:84951–84964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhang J, Zhou L, Zhao S, et al. The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell Cycle. 2017;16:1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Burne R, Balu S, Guérin A, et al. Comparison of healthcare resource utilization and costs of patients with HR+/HER2- advanced breast cancer treated with ribociclib versus other CDK4/6 inhibitors. J Med Econ. 2021;24:806–815. [DOI] [PubMed] [Google Scholar]
- [94].Saliba F, Hagipantelli R, Misset JL, et al. Pathophysiology and therapy of irinotecan-induced delayed-onset diarrhea in patients with advanced colorectal cancer: a prospective assessment. J Clin Oncol Off J Am Soc Clin Oncol. 1998;16:2745–2751. [DOI] [PubMed] [Google Scholar]
- [95].Xie R, Mathijssen RHJ, Sparreboom A, et al. Clinical pharmacokinetics of irinotecan and its metabolites in relation with diarrhea. Clin Pharmacol Ther. 2002;72:265–275. [DOI] [PubMed] [Google Scholar]
- [96].Ikuno N, Soda H, Watanabe M, et al. Irinotecan (CPT-11) and characteristic mucosal changes in the mouse ileum and cecum. J Natl Cancer Inst. 1995;87:1876–1883. [DOI] [PubMed] [Google Scholar]
- [97].Gandia D, Abigerges D, Armand JP, et al. CPT-11-induced cholinergic effects in cancer patients. J Clin Oncol Off J Am Soc Clin Oncol. 1993;11:196–197. [DOI] [PubMed] [Google Scholar]
- [98].Yumuk PF, Aydin SZ, Dane F, et al. The absence of early diarrhea with atropine premedication during irinotecan therapy in metastatic colorectal patients. Int J Colorectal Dis. 2004;19:609–610. [DOI] [PubMed] [Google Scholar]
- [99].Freites-Martinez A, Shapiro J, Goldfarb S, et al. CME Part 1: hair disorders in cancer patients. J Am Acad Dermatol. 2019;80:1179–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].van der Bol JM, Mathijssen RHJ, Loos WJ, et al. Cigarette smoking and irinotecan treatment: pharmacokinetic interaction and effects on neutropenia. J Clin Oncol. 2007;25:2719–2726. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Data are located in controlled access data storage at Gesellschaft für wissenschaftliche Datenverarbeitung mbH Göttingen (GWDG).