

Abstract
Spebrutinib is a new Bruton tyrosine kinase inhibitor developed by Avila Therapeutics and Celgene. Spebrutinib (SPB) is currently in phase Ib clinical trials for the treatment of lymphoma in the United States. Preliminary in-silico studies were first performed to predict susceptible sites of metabolism, reactivity pathways and structural alerts for toxicities by StarDrop WhichP450™ module, Xenosite web predictor tool and DEREK software; respectively. SPB metabolites and adducts were characterized in vitro from rat liver microsomes (RLM) using LC-MS/MS. Formation of reactive intermediates was investigated using potassium cyanide (KCN), glutathione (GSH) and methoxylamine as trapping nucleophiles for the unstable and reactive iminium, iminoquinone and aldehyde intermediates, respectively, with the aim to produce stable adducts that can be detected and characterized using mass spectrometry. Fourteen phase I metabolites, four cyanide adducts, six GSH adducts and three methoxylamine adducts of SPB were identified and characterized. The proposed metabolic pathways involved in generation of phase I metabolites of SPB are oxidation, hydroxylation, o-dealkylation, epoxidation, defluorination and reduction. Several in vitro reactive intermediates were identified and characterized, the formation of which can aid in explaining the adverse drug reactions of SPB. Several iminium, 2-iminopyrimidin-5(2H)-one and aldehyde intermediates of SPB were revealed. Acrylamide is identified as a structural alert for toxicity by DEREK report and was found to be involved in the formation of several glycidamide and aldehyde reactive intermediates.
Keywords: Spebrutinib, Bruton tyrosine kinase inhibitor, Metabolism, DEREK, Reactive intermediates, Trapping agents, StarDrop WhichP450™ module, LC-MS/MS
1. Introduction
Cancer is the second leading cause of death globally. In 2020, there were over 10 million deaths caused by cancer [1]. However, the treatment of cancer is associated with the adverse effects of anticancer agents. Despite the improved efficacy and increased patients’ survival rates of new therapeutic agents, adverse drug reactions are a major source of concern for both physicians and patients [2].
Bruton’s tyrosine kinase (BTK) is a cytoplasmic, non-receptor tyrosine kinase. BTK is an important target for the treatment of several types of cancer and autoimmune diseases since the discovery of its crucial role in B-cell receptor signaling pathways and the regulation of B-cell survival, activation, and proliferation. Many BTK inhibitors have been developed for the treatment of B-cell malignancies and autoimmune disorders. Based on their mode of binding to BTK, there are two types of BTK inhibitors: reversible and irreversible inhibitors. Irreversible inhibitors form a covalent bond with the amino acid residue Cys481 in the ATP-binding site of BTK. There are several BTK inhibitors currently in use or under development for the treatment of cancer and autoimmune diseases [3].
Spebrutinib (SPB) (N-[3-[[5-fluoro-2-[4-(2-methoxyethoxy) anilino] pyrimidin-4-yl] amino] phenyl] prop-2-enamide) is an orally bioavailable Bruton tyrosine kinase inhibitor developed by Avila Therapeutics and Celgene. Spebrutinib is currently in phase Ib clinical trial for the treatment of large B-cell lymphoma [4]. Upon administration, SPB targets and covalently binds to the amino acid residue Cys481 of BTK, thereby preventing its activity and leading to the inhibition of B cell receptor signaling and inhibition of cell proliferation of B-cell malignancies [3,5]. SPB binds covalently to Cys481 in the kinase domain of BTK using the electrophilic acrylamide moiety [6]. Chemical structure of SPB is shown in Fig. 1.
Fig. 1.
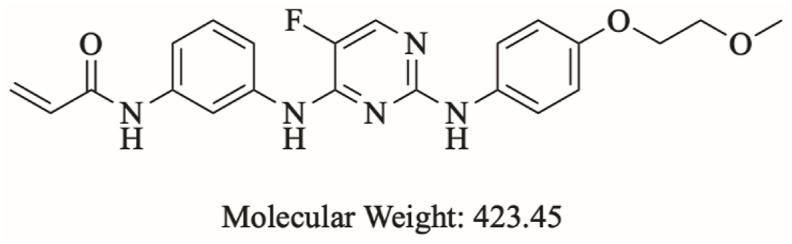
Chemical structure of spebrutinib.
Phase I clinical study of SPB was held between August 2011 and December 2013, this study enrolled and treated 113 patients with different types of B-cell malignancies at 13 centers in the US. For the overall patient population, grade 3-4 thrombocytopenia was observed in 8% of patients. Grade 3-4 neutropenia was observed in 16% of patients. The incidence of febrile neutropenia was 2%. The most common non-hematologic grade ≥3 TEAEs (Treatment Emergent Adverse Events) were pneumonia (4%) and hyperglycemia (3%). Common non-hematologic TEAEs of any grade were diarrhea (68%), fatigue (45%), nausea (35%), cough (27%), pyrexia (27%), and headache (25%). Only 5.3% of patients experienced ecchymosis and 2.7% had hematoma. Atrial fibrillation was observed in 4 of 113 patients, 3 of whom had a prior history. The proportion of patients who discontinued treatment due to Adverse effects was low (6.2%) [7,8].
Another phase I clinical trial is still held since 2014 for the assessment of SPB safety and efficacy in combination with investigational anticancer agents including rituximab, CC-122 and CC-223 for the treatment of diffuse Large B Cell Lymphoma (DLBCL) and Follicular Lymphoma. This clinical trial is sponsored by Celgene pharmaceuticals and includes 174 patients in multiple clinical centers. This study is estimated to be completed in April 2023 [9].
Due to the Adverse effects reported in the first clinical trials and the unexplainable length of the second clinical trials, an assessment of the toxicity and metabolism profile of this drug would be useful to understand the future of this drug and pinpoint any structural alerts that might be causing formation of harmful reactive intermediates.
2. Chemicals and methods
2.1. Chemicals and animals
All chemicals are analytical grade and solvents are HPLC grade. Sprague Dawley rats were used for rat liver microsomes (RLMs) preparation [10]. SPB was purchased from MedChemExpress company (Princeton, NJ, USA). Acetonitrile, formic acid, glutathione, methoxylamine and potassium cyanide were procured from Sigma-Aldrich company (St. Louis, MO, USA). Water (HPLC grade) was supplied by Milli-Q plus purification system (Billerica, MA, USA) that is available in-house. Sprague-Dawley rats were taken from experimental animal care center King Saud University (Riyadh, Saudi Arabia). Ethical approval for the Animal experiments were obtained from the Animal Ethics Committee at King Saud University (No. KSU-SE-22-83).
2.2. Chromatographic conditions
The separation of SPB and its related metabolites and adducts was done using an Agilent HPLC 1200 series (Agilent Technologies, Palo Alto, CA, USA). Mass Hunter software (Agilent Technologies, Palo Alto, CA, USA) was used to regulate the data acquisition and instruments. MS measurements were performed using a model 6320 Ion Trap (Agilent Technologies, USA) equipped with an electrospray ionization source (ESI). Electrospray ionization was performed at room temperature in positive ion mode. The nebulizer pressure was 60 psi, the dry gas was 10 L/min, the dry temperature was 350 °C, the trap drive level was 100% and the capillary temperature was 325 °C. The column used was an Eclipse plus C18 (4.6 × 150 mm, 3.5 μm) (Agilent Technologies, Palo Alto, CA, USA). LC separation was carried out using a mobile phase water with 1% formic acid (solvent A) and acetonitrile (v/v) (solvent B). Gradient chromatography (total run time of 55 min) was performed with water/acetonitrile mixture as the mobile phase at a flow rate of 0.5 mL/min. The program started with 95% mobile phase A, and then the amount of mobile phase B was increased from 5 to 75% within 25 min then kept at this percentage until minute 40 B% started to decrease to reach 5% at minute 55. Vials containing samples to be analyzed using HPLC were placed in the autosampler integrated in the HPLC machine, and 10 μl of a particular sample was injected into the HPLC system connected to the Agilent Ion Trap. The selected chromatographic conditions for the study of SPB metabolism and bioactivation are listed in Table 1.
Table 1.
Liquid chromatography and mass spectrometry selected parameters.
| Mobile phase | Binary system of 0.1% Formic acid in H2O (A) and ACN (B) | ESI source | Positive ESI | |
| High purity N2 gas Drying gas at 10 L/min with pressure (60 psi) | ||||
| 0.5 mL/min. | ||||
| Elution time: 55 min. | ||||
| Agilent Zorbax eclipse plus C18Column | Length | 150 mm | ESI temperature: 350 °C | |
| ID | 4.6 mm | Capillary voltage: 4000 V | ||
| Particle size | 3.5 μm | Modes | Mass scan and MS2 | |
| T | 22 ± 1 °C | Collision gas | High purity N2 | |
| Gradient elution system | Time in min. | % ACN | Analytes | SPB and its reactive metabolites |
| 0 | 5 | Mass parameters | Fragmentor voltage (FV): 145 V Amplitude: 1.25 V |
|
| 25 | 75 | |||
| 40 | 75 | |||
| 55 | 5 | |||
2.3. In silico prediction of SPB metabolites and structural alerts using WhichP450™ metabolism module and DEREK NEXUS module of StarDrop software
StarDrop software was used to predict the main sites of metabolism and to predict site lability specified by the composite site lability (CSL). This software contains the WhichP450 module which predicts regioselectivity of metabolism by different isoforms. This module concludes the findings by a pie chart of the most likely CYP450 isoform that has a major role in SPB metabolism. DEREK module was used to identify structural alerts in SPB that can cause certain toxicities.
2.4. In silico prediction of SPB reactive metabolites using XenoSite reactivity model
XenoSite web predictor has many different modules that helps in prediction of small molecule biochemistry; available free of charge at: http://swami.wustl.edu/xenosite. XenoSite reactivity module was used to predict possible sites of reactive intermediates. The software has the advantage of short run time. The chemical structure of SPB (SMILES format) was uploaded to the online website for the XenoSite reactivity module [11,12].
2.5. RLMs incubations
SPB was dissolved in dimethyl sulfoxide (DMSO). Protein concentration of the prepared RLMs was determined using Lowery method [13]. SPB (5 μM) was incubated with RLMs (1 mg/mL) in phosphate buffer (50 mM Na/K and 3.3 mM MgCl2) at pH 7.4. One mM NADPH was added to initiate the metabolic reaction. One mM of trapping agents KCN, GSH or methoxylamine was added in the experiments for the detection of iminium, iminoquinone and aldehyde reactive intermediates, respectively. The metabolic reactions were performed in a thermostatic shaking water bath (37 °C for 60 min). Two mL of ACN (ice cold) was added to stop the metabolic reaction by denaturation of enzymes protein. Centrifugation at 9000 g was done for 12 min at 4 °C to precipitate proteins. The clear supernatants were evaporated under a stream of nitrogen gas then reconstituted in mobile phase (50:50). The reconstituted samples were injected into the mass spectrometric system. Two controls were performed in the absence of RLMs or NADPH to verify that SPB phase I metabolites were metabolically generated [14].
2.6. Characterization of SPB reactive intermediates
Extracted ion chromatograms (EIC) and full mass range scan were utilized for characterization and localization of SPB metabolites in the extracts of various incubation mixtures, while generated fragment ions were utilized for reconstructing the chemical structure of SPB related metabolites.
3. Results and discussion
3.1. Results of in silico SPB metabolites prediction
StarDrop software predictions of SPB metabolism indicates the lability of each site with respect to metabolism by different isoforms of CYP450 enzymes. This indicates that C29 and C30 in the acrylamide group and C3 of the methoxy group at the other end of the molecule are predicted to be labile to metabolism by CYP450 enzymes especially CYP3A4 and CYP2D6. The composite site liability (CSL) is shown in the top-right of the metabolic landscape (Fig. 2A) and its value is 0.9951 which predicts that SPB is highly susceptible to metabolism by CYP450 isoforms. The results of the WhichP450™ module, concluded in the pie chart is predicting the most likely CYP450 isoforms that has a critical role in SPB metabolism (Fig. 2A). According to these results, CYP3A4 isoform has the major role in SPB metabolism. The software also predicted a list of phase I metabolites of SPB (Fig. 3).
Fig. 2.

A) Predicted metabolic sites for SPB by StarDrop WhichP450 module. B) Structural alerts of spebrutinib by DEREK module of StarDrop software. C) Predicted sensitive sites for bioactivation of spebrutinib by Xenosite web predictor. The red and green indicate high probability of bioactivation on these sites, whereas the faint blue color reveals the less probability to form reactive intermediates.
Fig. 3.

SPB phase I metabolites predicted by StarDrop software.
3.2. Results of in silico SPB toxicity and bioactivity prediction
DEREK software was used to assess the possible toxicities of SPB based on its chemical structure (Fig. 2B). SPB shows possibility of causing carcinogenicity (equivocal), chromosome damage (plausible) and skin sensitization (equivocal) due to the acrylamide moiety. It is also showing possibility of causing hepatotoxicity (plausible) and mitochondrial dysfunction (equivocal) due to the 4-oxy-aniline moiety and the adjacent atoms.
SPB reactivity was examined using reactivity module in Xenosite web page as shown in Fig. 2C. The predicted potential sites for bioactivation in SPB were the 2 double-bonded carbon atoms in the acrylamide group, the 4-oxy-aniline group, the α carbon atom adjacent to the nitrogen atom of pyrimidine ring and the carbon atom binding fluorine atom. The acrylamide group was the highest predicted site of SPB to undergo epoxidation (highlighted in green). It was also the highest predicted site of GSH binding (highlighted in red) (Fig. 2C).
3.3. Fragment ions study of SPB
SPB peak elutes at 20.1 min in product ion chromatogram (Fig. 4A). Dissociation of SPB ion at m/z 424 inside the collision cell produces five characteristic fragment ions at m/z 406, m/z 392, m/z 382, m/z 370 and m/z 366 (Fig. 4B). Interpretation of these fragments is shown in Fig. 4C.
Fig. 4.

A) Product ion chromatogram of SPB, B) Product ion mass spectrum of SPB, C) Proposed interpretation of SPB fragmentation.
3.4. Identification of SPB phase I metabolites
SPB incubation in RLM revealed the characterization of fourteen in vitro phase I metabolites (Fig. 5A). The proposed metabolic reactions included oxidation, hydroxylation, o-dealkylation, epoxidation, defluorination and reduction (Fig. 5B). Retention time and fragmentation of all metabolized are listed in table S1 in the supplementary data.
Fig. 5.

A) Proposed in vitro phase I metabolites of SPB. B) Proposed metabolic pathways of SPB.
3.4.1. Identification of M1
M1 (m/z 410) is proposed to be generated by o-demethylation of SPB. This metabolite’s peak elutes at 17.6 min in product ion chromatogram. Dissociation of M1 ion at inside the collision cell generates five fragment ions at m/z 392, m/z 366, m/z 356, m/z 348 and m/z338 (Fig. 6A). Interpretation of these fragments is shown in Fig. 6B. Predicted toxicities of M1 by DEREK Nexus module showed that demethylation created a new alert for carcinogenicity and skin sensitization (Fig. 6C).
Fig. 6.

A) Product ion mass spectrum of M1, B) Proposed interpretation of fragmentation of M1, C) Predicted new toxicities of M1 by DEREK Nexus module.
3.4.2. Identification of M2
M2 (m/z 426) is proposed to be generated by epoxidation of M1. This metabolite was predicted by StarDrop WhichP450 module. This metabolite’s peak elutes at 16.68 min in product ion chromatogram. Dissociation of M1 ion at inside the collision cell generates five fragment ions at m/z 408, m/z 390, m/z 384, m/z 372 and m/z 338 (Fig. 7A). Interpretation of these fragments is shown in Fig. 7B. Epoxidation caused several toxic alerts in DEREK Nexus module all plausible including Carcinogenicity, chromosome damage, mutagenicity, irritation of eye and skin, developmental toxicity, skin sensitization (Fig. 7C).
Fig. 7.

A) Product ion mass spectrum of M2, B) Proposed interpretation of fragmentation of M2, C) Precited epoxide toxicities by StarDrop DEREK Nexus module.
In summary, StarDrop software WhichP450 module and Xenosite have predicted the most vulnerable sites of metabolism in the structure of SPB. While the in-vitro results were in large agreement with the in-silico predictions, some results and sites of metabolism were not predicted by the used software at all. Five out of sixteen predicted by StarDrop software were confirmed in RLM incubations (Fig. 8).
Fig. 8.

Summary of in-silico predicted metabolites of SPB and proposed metabolites found in-vitro; metabolites that were predicted in-silico using StarDrop software and were confirmed in RLM incubation are shown in the middle.
3.4.2.1. Defluorinated metabolites of SPB
Metabolites 10, 11 and 12 (Fig. 5) are proposed to be formed by defluorination of SPB. Fluorinated aromatic compounds are generally more resistant to defluorination by CYP450 enzymes than aliphatic compounds. However, metabolic defluorination by CYP450 enzymes does occur and is reported for some drugs [15]. Fluorine can be a cause of toxicity after it is liberated through oxidative dehalogenation. It is distributed in the hard and soft tissues in the body. A large fraction of fluorine ends up in bones and teeth. High levels of fluorine can lead to protein inhibition, free radical distribution, irregularities in metal homeostasis and tissue damage [16,17]. The formation of M10, M11 and M12 of SPB is a critical concern and could be the cause of toxicity of this drug.
3.5. Identification of iminium reactive intermediates using potassium cyanide as trapping agent
3.5.1. Identification of M15/KCN adduct
M15/KCN is proposed to form by the addition of cyanide group to SPB. M15/KCN (m/z 449) peak appeared at 19.09 min in product ion chromatogram. Dissociation of M15/KCN ion inside the collision cell produces five fragment ions at m/z 431 m/z 395, m/z 375 m/z 363 and m/z 257 (Fig. 9A). Interpretation of these fragments is shown in Fig. 9B.
Fig. 9.

A) Product ion mass spectrum of M15/KCN cyanide adduct, B) Proposed interpretation of fragmentation of M15/KCN.
3.5.2. Proposed bioactivation mechanism of SPB to iminium reactive intermediates
Fig. 10 shows the bioactivation pathway for SPB to iminium intermediates. The generation of M15/KCN, M16/KCN, M17/KCN and M18/KCN cyanide adducts (Fig. 11) revealed the formation of iminium unstable intermediates in the pyrimidine moiety of SPB. Metabolic hydroxylation at pyrimidine ring in SPB followed by the loss of one water molecule (dehydration) lead to the generation of iminium ion intermediates which are reactive and unstable that can be captured by cyanide as nucleophile forming stable adduct that can be characterized in mass spectrometry.
Fig. 10.
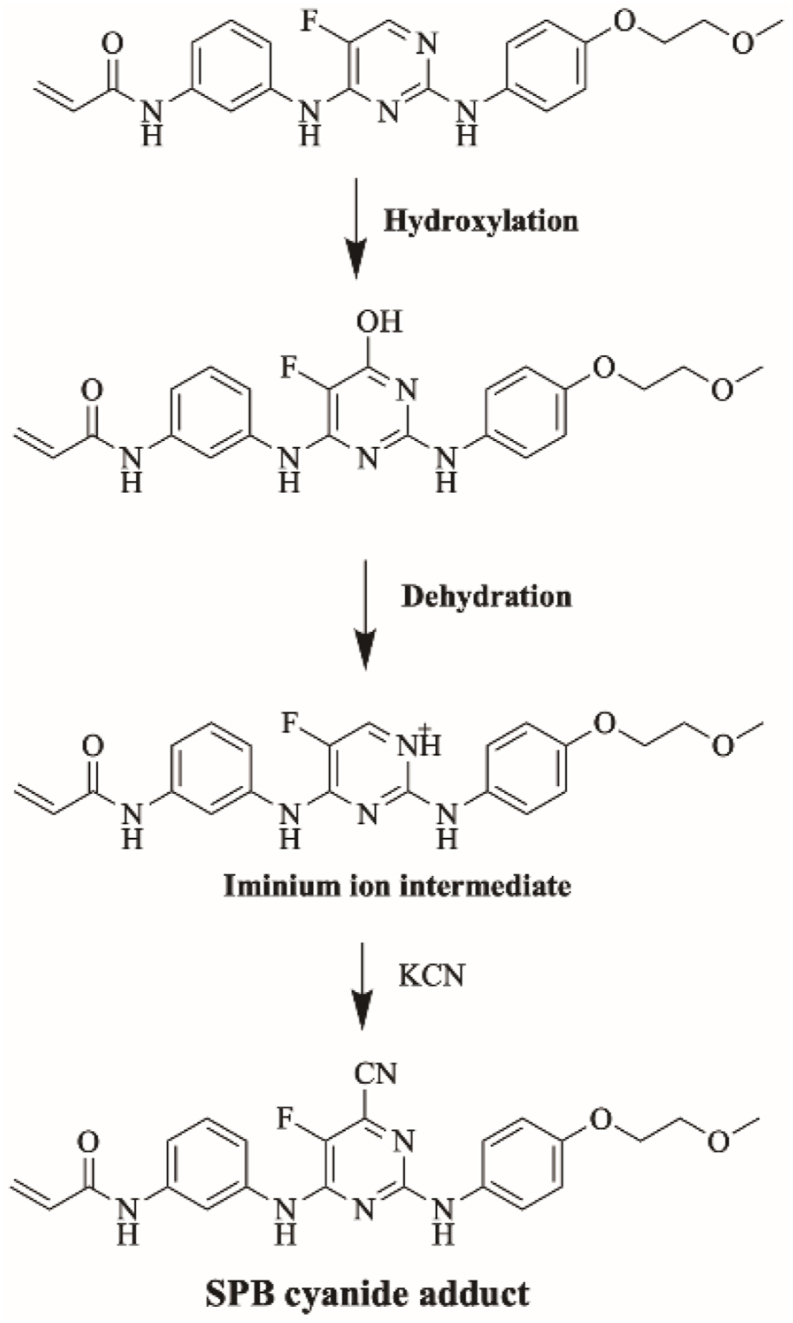
Proposed bioactivation pathways of SPB to iminium reactive intermediates trapped by potassium cyanide.
Fig. 11.

Proposed SPB cyanide adducts.
3.6. Identification of glycidamide and 2-iminopyrimidin-5(2H)-one reactive intermediates using glutathione (GSH) as trapping agent
3.6.1. Identification of M19/GSH adduct
M19/GSH is proposed to form by the addition of GSH to the 2-iminopyrimidin-5(2H)-one reactive metabolite of SPB. M19/GSH (m/z 727) peak appeared at 21.2 min in fragment ion chromatogram. Dissociation of M19/GSH ion inside the collision cell produces three fragment ions at m/z 709 m/z 665, and m/z 598 (Fig. 12A). Interpretation of these fragments is shown in Fig. 12B.
Fig. 12.

A) Product ion mass spectrum of M19/GSH adduct, B) Proposed interpretation of fragmentation of M19/GSH.
3.6.2. Proposed bioactivation mechanism of SPB to glycidamide and 2-iminopyromidin-5(2H)-one reactive intermediates
Fig. 14 shows the proposed bioactivation pathways for SPB to glycidamide and 2-iminopyrimidin-5(2H)-one intermediates. The generation of different GSH adducts (Fig. 13) revealed the generation of glycidamide and 2-iminopyrimidin-5(2H)-one unstable intermediates at different sites of SPB. The de-fluorination followed by hydroxylation performed by CYP450 enzymes results in the formation of reactive 2-iminopyrimidin-5(2H)-one intermediates (M19, M20, M21 and M24).
Fig. 14.

Proposed bioactivation pathways of SPB to glycidamide and 2-iminopyrimidin-5(2H)-one reactive intermediates trapped by GSH.
Fig. 13.

Summary of proposed GSH adducts of SPB.
Two glycidamide reactive intermediates, M22 and M23, are formed at the acrylamide group of SPB. Hydroxylation of the acrylamide group by CYP450 enzymes results in the epoxide form of this moiety (glycidamide) which is very reactive and can binds to the proteins and DNA. These glycidamide reactive intermediates are further metabolized by epoxide hydrolase to glyceramide which can be trapped by GSH, specifically the sulfur atom in cysteine. Interestingly, acrylamide group is the site that binds covalently to Cys481 residue in the active site of BTK protein.
3.7. Identification of aldehyde reactive intermediates using methoxylamine as trapping agent
3.7.1. Identification of methoxylamine adduct M25/CH3ONH2
M25/CH3ONH2 is proposed to form by the addition of methoxylamine to the aldehyde reactive metabolite of SPB. M25/CH3ONH2 (m/z 427) peak appeared at 19.45 min in fragment ion chromatogram. Dissociation of M25/CH3ONH2 ion inside the collision cell produces six fragment ions at m/z 409, m/z 372, m/z 320, m/z 308, m/z 264 and m/z 247 (Fig. 15A). Interpretation of these fragments is shown in Fig. 15B.
Fig. 15.

A) Product ion mass spectrum of M25/CH3ONH2 methoxylamine adduct, B) Proposed interpretation of fragmentation of M25/CH3ONH2.
3.7.2. Proposed bioactivation mechanism of SPB to aldehyde reactive intermediates
Fig. 16 shows the bioactivation pathway for SPB to aldehyde intermediates. The generation of SPB methoxylamine adducts (Fig. 17) revealed the generation of aldehyde unstable intermediates in the acrylamide site during in vitro metabolism of SPB. Metabolism at the acrylamide moiety leads to the generation of aldehyde reactive intermediate which are unstable that can be captured by methoxylamine forming stable adduct that can be characterized in mass spectrometry.
Fig. 16.
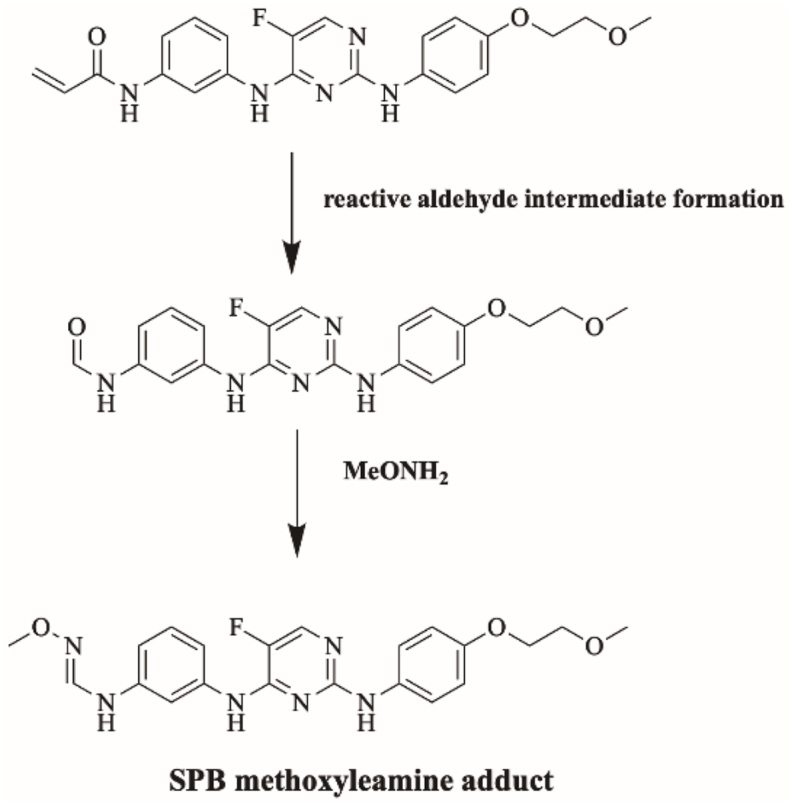
Proposed bioactivation pathways of SPB to aldehyde reactive intermediates trapped by methoxylamine.
Fig. 17.

Proposed SPB methoxylamine adducts.
Four iminium reactive intermediates, two glycidamide reactive intermediates, four 2-iminopyrimidin-5(2H)-one reactive intermediates and three aldehyde reactive intermediates of SPB were identified. The acrylamide moiety in SPB was proposed by DEREK Nexus toxicity module to cause carcinogenicity, chromosome damage and skin sensitization (Fig. 2B). This site binds covalently to Cys481 in the binding site of BTK and it is proposed that this moiety is bioactivated to glycidamide and formed two different reactive metabolites which were captured using GSH. Furthermore, the pyrimidine ring in the middle of SPB was also identified as a potential bioactivation site by Xenosite server (Fig. 2C). Four iminium and four 2-iminopyrimidin-5(2H)-one reactive intermediates were identified that were formed at this site. SPB and several phase I metabolites were bioactivated to eleven reactive intermediates which might be the cause of adverse effects (Fig. 18).
Fig. 18.

Bioactivation of SPB and some of its phase I metabolites to reactive intermediates. Reactive intermediates are shown in brackets, these are not isolated as they are but captured by relative trapping agents.
4. Conclusions
This study involved in silico and in vitro (RLMs) metabolite identification and characterization of SPB using LC-MS/MS. In silico results aimed at understanding the vulnerable sites of metabolism and bioactivation of SPB. Fourteen in vitro metabolites of SPB were identified (Fig. 5). Different pathways of phase I metabolism of SPB were proposed including oxidation, hydroxylation, o-dealkylation, epoxidation, defluorination and reduction. M10-12 involved defluorination which can be considered unusual pathway of phase I metabolism in aromatic compounds. These metabolites might be causing toxicities and need to be evaluated further. Formation of different reactive intermediates might be the cause of some of the adverse effects associated with SPB. In this study, in silico studies from different platforms identifying structural alerts and susceptible sites of metabolism were used as a guide for in vitro experiments. The use of such tools is very useful and saves scientist time and predict the possible structural causes of some of the drugs’ toxicities.
Ethics
Male Sprague-Dawley were maintained following the Animal Care Center instructions at King Saud University that were approved by Local Animal Care and Use Committee of KSU. The animal experimental procedure utilized in our current research was validated and approved by the King Saud University’s Ethics Review Committee (Number: KSU-SE-22-83).
Author contribution statement
Aishah Mohammed Alsibaee; Mohamed W. Attwa: Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Haya I Aljohar; Ali Saber Abdelhameed; Adnan A. Kadi - Conceived and designed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Data availability statement
Data included in article/supp. material/referenced in article.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to extend their appreciation to the Researcher Supporting Project Number (RSPD2023R750), King Saud University, Riyadh, Saudi Arabia for funding this research work.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.heliyon.2023.e17058.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Ferlay J., et al. Cancer statistics for the year 2020: an overview. Int. J. Cancer. 2021;149(4):778–789. doi: 10.1002/ijc.33588. [DOI] [PubMed] [Google Scholar]
- 2.Nurgali K., Jagoe R.T., Abalo R. Editorial: adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae? Front. Pharmacol. 2018;9:245. doi: 10.3389/fphar.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang C., et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur. J. Med. Chem. 2018;151:315–326. doi: 10.1016/j.ejmech.2018.03.062. [DOI] [PubMed] [Google Scholar]
- 4.Jerkeman M., et al. Targeting of B‐cell receptor signalling in B‐cell malignancies. J. Intern. Med. 2017;282(5):415–428. doi: 10.1111/joim.12600. [DOI] [PubMed] [Google Scholar]
- 5.Wishart D.S., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–d1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender A.T., et al. Ability of bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 2017;91(3):208–219. doi: 10.1124/mol.116.107037. [DOI] [PubMed] [Google Scholar]
- 7.Akinleye A., et al. Ibrutinib and novel BTK inhibitors in clinical development. J. Hematol. Oncol. 2013;6(1):59. doi: 10.1186/1756-8722-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown J.R., et al. Phase I study of single-agent CC-292, a highly selective Bruton's tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2016;101(7):e295–e298. doi: 10.3324/haematol.2015.140806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Profitós-Pelejà N., et al. Regulation of B-cell receptor signaling and its therapeutic relevance in aggressive B-cell lymphomas. Cancers. 2022;14(4) doi: 10.3390/cancers14040860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Dijk W., et al. A simple procedure for the isolation of lysosomes from normal rat liver. FEBS Lett. 1976;62(2):177–181. doi: 10.1016/0014-5793(76)80047-6. [DOI] [PubMed] [Google Scholar]
- 11.Hughes T.B., et al. Modeling reactivity to biological macromolecules with a deep multitask network. ACS Cent. Sci. 2016;2(8):529–537. doi: 10.1021/acscentsci.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes T.B., Miller G.P., Swamidass S.J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione. Chem. Res. Toxicol. 2015;28(4):797–809. doi: 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterborg J.H., Matthews H.R. Basic Protein and Peptide Protocols. 1994. The Lowry method for protein quantitation; pp. 1–4. [Google Scholar]
- 14.Kadi A.A., et al. Detection and characterization of ponatinib reactive metabolites by liquid chromatography tandem mass spectrometry and elucidation of bioactivation pathways. RSC Adv. 2016;6(76):72575–72585. [Google Scholar]
- 15.Park B.K., Kitteringham N.R., O'Neill P.M. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 16.Kyzer J.L., Martens M. Metabolism and toxicity of fluorine compounds. Chem. Res. Toxicol. 2021;34(3):678–680. doi: 10.1021/acs.chemrestox.0c00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston N.R., Strobel S.A. Principles of fluoride toxicity and the cellular response: a review. Arch. Toxicol. 2020;94(4):1051–1069. doi: 10.1007/s00204-020-02687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supp. material/referenced in article.