

Abstract
Therapeutic antibody engineering seeks to identify antibody sequences with specific binding to a target and optimized drug-like properties. When guided by deep learning, antibody generation methods can draw on prior knowledge and experimental efforts to improve this process. By leveraging the increasing quantity and quality of predicted structures of antibodies and target antigens, powerful structure-based generative models are emerging. In this review, we tie the advancements in deep learning-based protein structure prediction and design to the study of antibody therapeutics.
Keywords: Deep learning, antibody design, generative models, protein structure, protein structure design, protein sequence design, antigen
2. Introduction
Since the first FDA-approved antibody therapeutic for cancer in 1990, scientists have developed over 100 antibody-based therapeutics for various diseases across major human body systems, including infections, hematology, neurology, ophthalmology, metabolic and musculoskeletal diseases, and transplantation [1]. Antibodies have emerged as the realization of Paul Elrich’s vision of finding a ‘magic bullet’ medicine [2].
We live in a hostile environment filled with invading pathogens and we remain dependent on protection from the innate and adaptive immune systems. Antibodies are precisely targeted immune system proteins that evolve within our body to help stave off disease. The predominant class of human antibodies found in the list of approved monoclonal antibody therapeutics follow an IgG format, consisting of four chains (two heavy and two light), with variable domains containing the binding surface, or “paratope”, which binds with specificity to the target antigen, whose site of binding is called the “epitope”. The paratope typically is a subset of six distinct complementarity determining region (CDR) loops - three on the light chain (L1, L2, L3) and three on the heavy chain (H1, H2, H3), where H3 is significantly more variable than the remaining five canonical loops.
Antibody development typically begins with a therapeutic hypothesis and further engineering to support potential mechanisms of action for feasible clinical application [1]. The modern toolbox for antibody discovery includes robust routes to engineer human antibodies from other species using immunized animals or in vitro display technologies. This toolbox has been recently expanded to include deep learning (DL), which is the application of neural networks to “learn” the most important features from large amounts of data through the process of gradient descent and backpropagation.
In this review, we describe the rapid, DL-driven progress in antibody structure prediction and design, compare the performance of general protein-trained models versus antibody-specific models in antibody engineering tasks, and address the remaining challenges for de novo antibody design with desirable therapeutic properties. With improvements in antibody structure prediction methods, we argue that the future of generative models will incorporate significantly more synthetic (predicted) structures for better learning the therapeutic antibody manifold. For brevity, we will omit discussion of epitope- and paratope-specific prediction models, which have been covered in previous reviews [3] [4].
3. Antibody structure prediction
3.1. General protein structure prediction methods
The development of structure-based protein design methods relies on realistic protein structures for training, which are traditionally determined through costly and elaborate experimental processes such as x-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryoEM. The paradigm shift from slow energy-based models to fast neural networks has resulted in algorithms that can predict many more protein structures than currently available in the form of crystals, with a median accuracy of 2.1 Å [5]. Modern protein structure prediction pipelines primarily consist of four components: (i) An input protein representation coupled with a multiple sequence alignment (MSA) of homologous proteins to map evolutionary relationships between corresponding residues of genetically-related sequences, (ii) an algorithm to implicitly detect sequence-structure patterns, (iii) a module to convert the derived patterns into explicit 3D structure, and (iv) a physics-based refinement module [5]. AlphaFold2 (AF2) [6] and RosettaFold [7] were the first two DL methods with high accuracy in protein structure prediction.
3.2. Circumventing MSAs in antibody structure prediction
Due to the independent evolution of each antibody in a single organism, the relevant evolutionary histories for CDR H3 loop sequences are lacking, so the MSAs on CDR regions may not always be available or reliable [8]. Additionally, MSA-dependent tools suffer from longer run times, which can be impractical in a drug design pipeline that requires examining many antibodies in parallel. DeepH3 circumvented the use of MSAs by predicting structural restraints using a deep residual neural network and then relied on Rosetta to produce full atom structures [8]. SimpleDH3 performs better or on par to DeepH3, by using ELMo embeddings and forward and backward LSTM passes that directly output coordinates of backbone atoms in the H3 loop [9]. AbLooper uses an ensemble of five E(n)-equivariant graph neural networks (E(n)-EGNNs) trained in parallel to predict the position of backbone atoms in all six CDRs, with a confidence metric calculated as the deviation between the predicted structures of each of the five networks [10]. DeepAb extends the architecture of DeepH3 by incorporating an interpretable attention mechanism for the entire antibody Fv region [11]. DeepSCAb added side chain prediction to DeepAb, although it did not improve backbone prediction accuracy [12].
An advance in protein structure prediction was the incorporation of pre-trained language models, such as ESMFold’s use of ESM-2 which provides a rich embedded representation of protein sequences and a worthy substitution for MSAs [13]. For antibodies, IgFold [14]** leverages AntiBERTy’s [15] sequence embeddings to predict the atomic coordinates of antibody structures using triangular edge updates and invariant point attention. IgFold achieves accuracy comparable to AlphaFold’s predictions but with significantly faster computational speed due to the absence of time-consuming MSAs. tFold-Ab [16]**, xTrimoABFold [17], and ABodyBuilder2 [18] are three other models that use AlphaFold-like architectures but without the MSA searching component. tFold-Ab employs a simplified Evoformer stack to consider side-chain conformations, xTrimoABFold uses embeddings from AntiBERTy and a cross-modal homologous structure search algorithm to predict similar structures for similar antibody sequences, and ABodyBuilder2 is an antibody-specific version of the AF-Multimer’s [19] structure module with modifications such as using independent weightings of the eight sequential update blocks. RaptorX-Single is a single-sequence protein structure prediction pipeline consisting of a sequence embedding module that generates sequence embeddings of an input and its pair representation, an Evoformer module that iteratively updates the embeddings, and a structure module of IPA layers that outputs predicted atom 3D coordinates [20].
3.3. The top performing model for antibody-antigen docking targets at CASP15 was a general protein structure prediction method
The biannual Critical Assessment of Protein Structure Prediction (CASP) provides an opportunity to benchmark the accuracy of current structure prediction methods against a set of proteins, including immunoglobulins, whose experimentally-determined structures are unknown at the start of the event, and revealed afterwards. At CASP15 in 2022, many groups incorporated AF2 in some capacity, and differences in performance arose from constructing superior MSAs through manual homology searches. It was previously reported that a AF2 composite score of pLDDT and pTMscore enhanced antibody-antigen structure prediction, so it was no surprise AF2 would be competitive for immunoglobulin targets at CASP [21]. Wallner_TS used AF2 in action at CASP with AFSample [22], which performed best on immunoglobulin targets (specifically targets H1129, H1140, H1141, H1144, T1173o, and T1187o, with the 3rd highest overall z-score) by using a modified AlphaFold-Multimer. AFSample demonstrated improved prediction by (i) increasing the number of times the prediction is recycled in the network, (ii) randomly perturbing the input MSA, and (iii) dropping random nodes from the network at inference [23]. Notably, although the participants SHT, ClusPro, and Kozakov/Vajda [24] used antibody-specific models like DeepAb and ClusPro on antibody mode [25], AFSample, a general protein structure prediction method, surpassed in performance.
4. Antibody sequence design
4.1. Learning the language of antibodies
The similarities between human language and protein sequences make natural language processing (NLP) models a valuable tool in protein design tasks. Protein sequences and human language are both organized hierarchically, with amino acids and letters composing the basic building blocks that assemble into more complex structures. Protein fragments (secondary structures) can combine to form tertiary structures, just as words can form complex sentences. Amino acids interact with their sequential surroundings and distant (yet spatially close) parts of their chains, in the same way that words relate and interact with each other in human language [26]. ImmunoLingo aims to develop a rigorous set of linguistic rules for antibody sequences to guide the tokenization process and facilitate the design of language models [27]. This approach promotes the explicit learning of the compositional and structural semantics of antibody motifs and their binding to antigens. In contrast, foundation language models like GPT-3 do not rely on word or part-of-speech boundaries. Instead, these models operate on frequently occurring partial word fragments extracted from a predetermined corpus of text. It remains to be seen whether antibody language modeling will benefit from more explicit discretization, possibly diverging from recent advances in general linguistic and language structure analysis.
4.2. General sequence-based design methods
One of the first breakthrough protein language models was ESM-1b, a 650M parameter encoder transformer that has learned intrinsic biological properties from 250M protein sequences [28]. ESM-1b was later used for guided antibody affinity maturation, improving the binding affinities of four clinically relevant antibodies up to 7-fold and three unmatured antibodies up to 160-fold [29]. ProGen [30] is a 1.2B parameter model trained on 280M sequences, adapted from the CTRL model [31], and conditioned on taxonomic and keyword tags, providing the ability to generate sequences with controllable properties. ProtGPT2 is a transformer model with 738M parameters, based on the GPT-2 architecture, that can generate sequences in unexplored regions of protein space [32]. Along the theme of taxonomic tagging, The Manifold Sampler is a denoising autoencoder trained on 20M protein sequences with a function predictor trained on 0.5M labeled proteins [33].
4.3. Antibody-specific sequence-based design methods
The suite of ProGen2 [34] models have varying parameter sizes up to 6.4B, including ProGen2-OAS trained on 554M antibody sequences. The general-protein models outperformed the antibody-specific model, with the ProGen2-small model performing best in antibody binding fitness prediction, and ProGen2-xlarge performing best in antibody fitness prediction. These results suggest that functional antibody properties are informed by more than antibody sequences alone. The Manifold Sampler was also repurposed for antibody design by enabling multi-segment preservation [35] for the antibody framework region during CDR sequence sampling, and was capable of producing CDR3 designs within the observed length distribution of the training set [35].
Sapiens is a set of two BERT models trained separately (one on 20M heavy chain, the other on 19M light chain sequences) that produces sequences with improved humanness qualities [36]. IgLM [37]** is a left-to-right decoder-only transformer model trained on 558M antibody sequences for full-sequence antibody generation of various lengths or for targeted sequence infilling. IgLM can design CDR H3 loops with natural distributions of spatial aggregation propensity, solubility, and humanness. Both ESM-1b and IgLM demonstrate that larger models better capture data distributions: ESM-1b large (670M parameters) outperformed small (25M) and the 13M parameter version of IgLM outperformed IgLM-S (1.4M). Given the expansive sequence space of the CDR region, some groups have found it effective to use Bayesian optimization techniques, which balance exploration and exploitation by using a surrogate model that approximates the sequence design function. AntBO is a Bayesian optimization framework for in-silico design of CDR H3 capable of designing high-affinity sequences [38].
4.4. Antibody representation learning
Another general class of antibody language models attempt to implicitly learn metrics relevant in therapeutic development. AntiBERTy [15] is a BERT-based model trained on 558M antibody sequences, utilized for clustering antibodies into trajectories resembling affinity maturation and subsequently incorporated as a crucial component in IgFold [14]**. AntiBERTa [40] and AbLang [39] use the RoBERTa architecture, which trains on longer sequences of text than the original BERT model [43]. AntiBERTa is trained on 57M B-cell receptor (BCR) sequences (42M heavy, 15M light) that can trace the B cell origin of the antibody, quantify immunogenicity, and predict the antibody’s binding site [40]. AbLang is also a suite of two models for each chain and can restore the missing residues of antibody sequences more accurately and seven times faster than ESM-1b [39]. AbSci demonstrated antibody design with desirable therapeutic properties with a RoBERTa model trained on four datasets from OAS and fine-tuned with in-house affinity data [44]. They also introduced a “naturalness” metric that scores antibody variants for similarity to natural immunoglobulins based on pseudo-perplexity of CDRs in antibody heavy chains [45]. As an improvement to RoBERTa, DeBERTa incorporates disentangled attention, an enhanced mask decoder, and virtual adversarial training [46]. PARA is a DeBERTa model trained on only 18M human BCR sequences, yet outperforms AntiBERTy and AbLang on CDR H3 residue recovery [41]. Whether antibody representations are richer from antibody-specific or general encoder models is task-dependent: Previous work demonstrated that a BERT model trained on general sequences from Pfam [47] outperformed an antibody-specific BERT model in the antibody affinity binding prediction task [48], but similar work found that Ab-LMs were better in paratope prediction [49].
4.5. Fixed backbone sequence design
In drug discovery, pharmacologists collect multiple initial antibodies either from humanized mice or patients. Optimizing these antibodies for binding a particular antigen can be formulated as a fixed backbone design problem, where we constrain sampling to the antibody sequence given its backbone structure. The general architecture for fixed backbone design incorporates an encoder to represent geometric components of the protein as a rich set of features, which are then passed to a decoder for filling the backbone with suitable amino acids. Ingraham et al. [50] and proteinMPNN [51] use a graph-based, autoregressive model with message passing neural networks (MPNNs) [52] to capture higher-order dependencies between sequence and structure. Anand et al. use a 3D convolutional network that conditions on local backbone structure to learn residue-level patterns [53]. ESM-IF is a structure-to-sequence transformer tasked to recover the native sequence of the protein from the given backbone with invariant geometric input processing layers [54]. FvHallucinator is a sequence design model conditioned on structure that uses a hallucination framework for generating antibody Fv libraries [55]. The hallucination framework inverts DeepAb to find sequences matching a target structure. One application of fixed backbone design in biotechnology is to use a known antigen-bound mAb structure as a template to find diverse alternative binding sequences.
5. Antibody sequence and structure co-design
5.1. Iterative co-design methods alternate between designing sequence and structure
Unsupervised learning undoubtedly has provided major advances in antibody design and general protein design, given the lack of both crystal structures and sequences with reliable therapeutic fitness metrics. However, if the input is unannotated data of antibody sequences, the model must learn both structural syntactic rules and semantic mapping rules to perform antibody engineering predictions. Alternatively, if the input is already encoded for the structural interaction between CDRs, surrounding residues, and the antigen, the model only needs to learn the semantic mapping rules, which might be more interpretable for antibody design. Zaixiang et al. found that implanting a structural adapter into a protein LM endows it with improved sequence recovery [56]. Even for reinforcement learning algorithms like Q-learning which perform poorly in combinatorial optimization problems like antibody design, incorporating structural priors improves sequence sampling with higher binding energy for eight diverse target pathogens [57]. Antibody design models therefore should receive structural information as input, and recent design approaches usher the opportunity for co-designing antibody sequence and structure.
One of the first demonstrations of antibody CDR co-design is RefineGNN [58]**, a model that generates an antibody graph via an iterative refinement process while unraveling the sequence autoregressively. Although RefineGNN does not explicitly learn information about the antigen, the next iteration, AbDockGen [59], does. AbDockGen uses a hierarchical equivariant refinement network (HERN) for paratope docking and design by predicting the atomic forces and using them to refine an antibody-antigen complex in an iterative, equivariant fashion. Where both RefineGNN and AbDockGen suffer is the incurred computing cost and memory overhead due to unraveling the CDR sequence in an autoregressive fashion. To overcome this issue, Yang et al. developed a multi-channel equivariant attention network (MEAN) to co-design CDRs over three iterations, which is significantly less than RefineGNN which iterates over every residue in the CDR region [60]. Although requiring significantly less iterations than HERN, MEAN still operates autoregressively, which can be costly and propagate errors. Tie-Yan et al. offers a AbBERT model combined with GNN for one-shot, antigen-specific antibody co-design [61].
5.2. Diffusion models can simultaneously design sequence and structure
Powerful new ways to design proteins have emerged by applying the same mechanistic tools behind text-to-image (DALL-E [62] and Stable Diffusion [63]), text-to-video, and video-to-video (Gen-1 and 2 [64]) to the design of proteins: Diffusion models. In image generation, diffusion models begin with grainy bits of static and gradually remove noise until a clear picture is formed. In the case of proteins, these diffusion models learn to generate new designs by denoising random conformations of proteins. Anand et al. [65] presented the first implementation of a diffusion model for protein structure, sequence, and rotamers, generating realistic proteins across a full range of domains in the PDB. Genie uses a generative model of protein structures that perform discrete-time diffusion using a cloud of oriented reference frames in 3D space [66]. Two prominent examples of general protein diffusion models are RFDiffusion [67]* and Chroma [68]*. RFDiffusion uses a fine-tuned version of RosettaFold to predict structure from sequence and for denoising a corrupted protein structure, coupled with proteinMPNN [51] for designing a sequence that best fits the predicted denoised structure. Chroma, on the other hand, uses a discrete component for denoising protein backbones while enforcing polymer physics and a separate component for designing a sequence for the denoised backbone. What makes Chroma and RFDiffusion particularly powerful is their programmability: Conditioning on a variety of features (including symmetry, shape, protein class, and natural language) enables them to produce high-quality, diverse, novel, and designable structures. ProteinGenerator is a sequence space diffusion model also based on the RosettaFold model, and capable of generating both sequence and structure [69]. DiffAb [70]** is the first antibody specific diffusion model that jointly models the sequences and structures of CDRs, by denoising residue identity, position, and orientation with equivariant neural networks while also conditioning on the 3D structure of the antigen. Recent work (FrameDiff) has put protein backbone diffusion models on a rigorous theoretical basis resulting in significantly smaller and faster models [71].
5.3. Jointly docking and designing antibody-antigen complexes
Although each of the aforementioned autoregressive and diffusion-based antibody co-design methods mentioned require a proposed docked position before designing the CDRs, dyMEAN proposes a complete end-to-end pipeline [72]. After inputting the antigen epitope structure and masked antibody sequence, dyMEAN iteratively updates via adaptive multi-channel message passing to predict a docked antibody-antigen complex and optimally designed CDR loops [72]. DockGPT is an encoder-decoder module that utilizes triangle multiplication, pair-based attention, and invariant point attention for docking and design [73]. DockGPT was fine-tuned to antibody-antigen complexes to enable de novo design of CDR loop regions, where the heavy and light chain coordinates are provided to the structure-decoder modules and the loops are missing to enable design [73]. Sculptor also approaches the epitope-specific design challenge with a generative model that jointly docks and designs a scaffold, while also incorporating loop conformational dynamics [74]*. Although Sculptor explores the conformational space of a single fold and uses a VAE as the generative model, the pipeline can be compatible with other generators (i.e. GANs and diffusion models), and trained on antibody chains [74]*.
6. Conclusion
Although monumental strides have been made in the therapeutic antibody design space since the application of deep learning-based computational approaches, there remain notable challenges to be overcome.
6.1. Expanding beyond the observed antigen landscape
A common challenge in deep generative learning is discovering modes of binding to a new antigen. RefineGNN demonstrated the capability of SARS-CoV-2 neutralization optimization, but this first required fine-tuning on the Coronavirus antibody database (CoVAbDab) [58]** [75]. One approach to reduce the data requirement may involve informing models with physical laws or energy functions, such as the Rosetta energy function. Wu et al. found that the training process of diffusion models for molecule generation could be steered with prior physics-based bridges improving generation quality and reducing sampling time, which may be a future direction for models like Chroma, RFDiffusion, FrameDiff, and DiffAb [76]. Alternatively if physics priors are less fruitful and access to the target distribution is not available (i.e. there is a lack of natural antibody sequences evolved for the target antigen), data augmentation must be considered.
The current antibody-specific co-design methods [58]** [59] [60] [61] [70]** [72] restrained their training data to the 3k crystal structures available in SAbDab [77]. Future antibody co-design methods may benefit significantly by incorporating data augmentation. Several prominent protein structure prediction methods have demonstrated their capabilities for large-scale prediction of protein structures for sequences that don’t have crystal structures, presenting an opportunity to develop an augmented synthetic antigen dataset. The AlphaFold Database [78] provides 200M predicted structures of protein sequences from UniProt and the ESM Metagonomic Atlas provides 617M metagenomic protein structures from the MGnify90 [79] database. Some existing models have taken advantage of AF structures for improving performance. IgFold used AF to predict the structure of 38,000 sequences from OAS [14]**, and ESM-IF augmented training data by nearly three orders of magnitude by predicting structures for 12M protein sequences from AF2 [54]. Antibody-specific synthetic databases exist as well. IgFold [14]** provides 104k non-redundant paired antibody sequences from OAS and a second set of 1.3M unique paired antibodies from human donors, collected by Jaffe et al. [80]. The software suite Absolut! [81] enables parameter-based unconstrained generation of synthetic lattice-based 3D antibody-antigen binding structures. Victor et al. demonstrated the power of Absolut! by generating a library of 1.1B antibody-antigen lattice-based structures with conformational paratope, conformational epitope, and affinity resolution [81].
6.2. Programmable therapeutic antibody engineering
The antibody design space is massive: When considering just the CDR region (~60 amino acids), there are ~2060 possible CDR loop combinations, which is more than the number of distinct proteins produced by extant organisms (~1012) [82]. Evidently, evolution has sparsely explored the full paratope domain, and powerful design tools are necessary to sample the subspace with optimized therapeutic properties - a subspace defined as the Pareto frontier [83]. Makowski et al. used linear discriminant analysis to predict continuous metrics strongly correlated with antibody specificity and affinity for variants of a clinical-stage antibody (Emibetuzumab) to a cancer stem cell marker (HGFR), and successfully identified variants with optimized on-target binding and minimized off-target binding. Bayesian optimization offers a sample-efficient framework for navigating the design space of biological sequences, and PropertyDAG builds upon this notion by identifying designs that are jointly positive of antibody properties [84]. Additionally, in antibody design some developability properties are orthogonal in nature: Wu et al. found that affinity maturation of an anti-respiratory syncytial virus antibody led to unwanted broad tissue binding and rapid clearance in cotton rats [85]. pcEBM addresses this by sampling new designs satisfying multiple properties of interest, even if they exhibit tradeoffs, by integrating multiple gradients within compositional energy based models [86]. The Smooth Discrete Sampler also takes advantage of the desirable properties of EBMs as well as improved sample quality of score-based models to propose antibody design, validating their method by expressing and purifying 270 of 277 single round proposed designs (97% success rate) with antibody-like properties [87]. Data augmentation of antibody biophysical properties using software tools will allow the exploration of generative models similar to Chroma that can create new antibodies based on functional programming instructions specific to therapeutic design. Instead of this bottom-up approach, engineering antibodies with desirable therapeutic properties may eventually become top-down: Hie et al. introduces a high-level programming language based on modular building blocks that demonstrated antibody functional site scaffolding for two antibody targets [88].
6.3. Closing remarks
Despite the considerable progress made in therapeutic antibody design, only a limited number of studies have presented experimental evidence. Among these studies, various techniques such as surface plasmon resonance [45], cryo-electron microscopy [51,67], size exclusion chromatography [67]*, and in vitro antibody expression [87] have been employed.
It is not yet clear when learning the entire protein landscape or specifically the antibody landscape is better for antibody engineering tasks, as in some cases general protein models perform better (e.g. ProGen-XL versus ProGen-OAS), and in others antibody-specific models supersede (e.g. AbLang versus ESM-1b). Rigorous design benchmark tests between design methods must be established to identify optimal approaches and drive innovation. DL approaches to antibody design will unveil an era of AI-driven therapeutic development - a day that may not lie far beyond our prediction horizon.
Fig. 1: Architecture of Antibody structure prediction models.
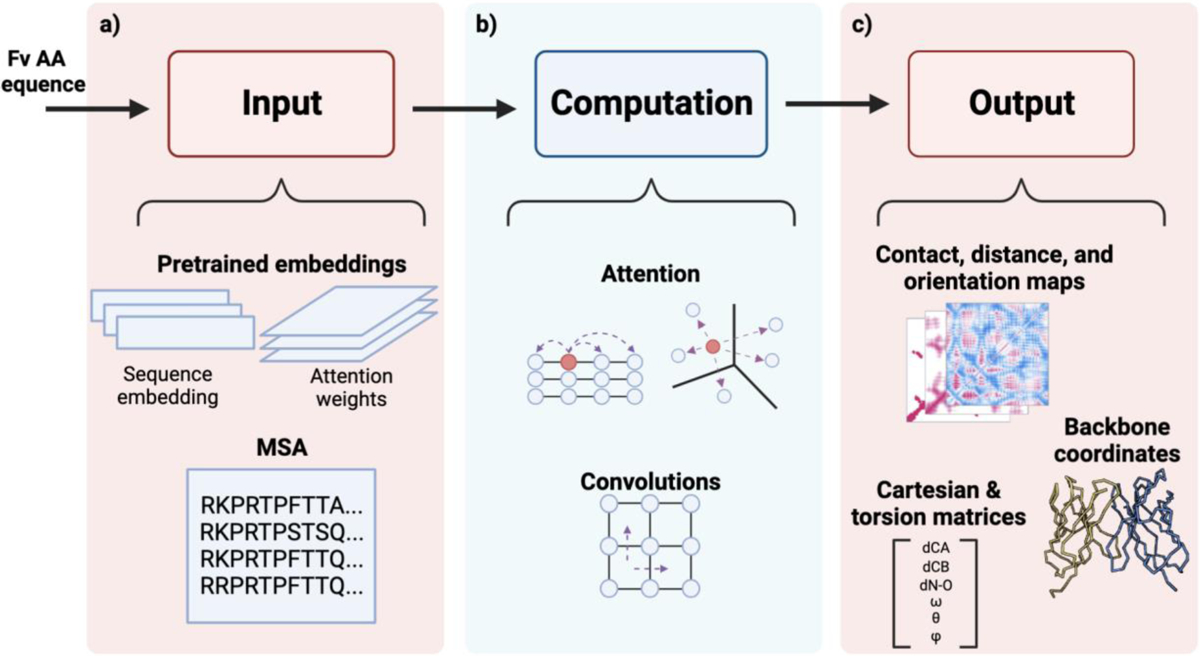
Antibody structure prediction pipelines are comprised of multiple modules, including inputs (embeddings from pretrained language models and (rarely) MSAs), computational algorithms (attention mechanisms and convolutions), and outputs (contact maps, distograms, orientograms, and backbone 3D structure).
Fig. 2: Schematic overview of sequence generation models in antibody design.
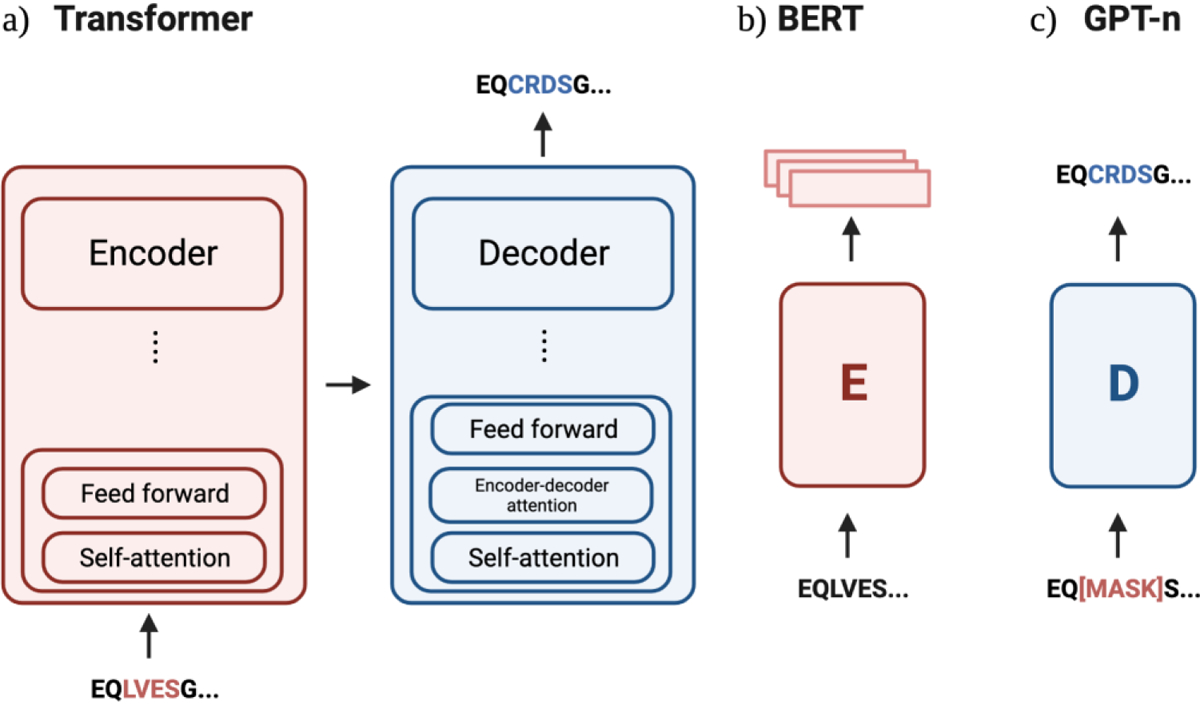
a) The original transformer architecture consisted of encoder and decoder models with stacks of six layers each. An example architecture that has both an encoder and decoder architecture is the Manifold Samplers [33,35]. b) BERT models are also known as autoencoders, and only use the encoder from the original Transformer. BERT-inspired protein models include AntiBERTy [15], AbLang [39], AntiBERTa [40], PARA [41], and the ESM suite [42] [13]. c) The GPT-n model contains only the decoder model, where the most prominent example in antibody design is IgLM [37]** and the ProGen suite [30,34].
Figure 3: Two distinct approaches to antibody sequence and structure co-design with conditioning on antigen structure.
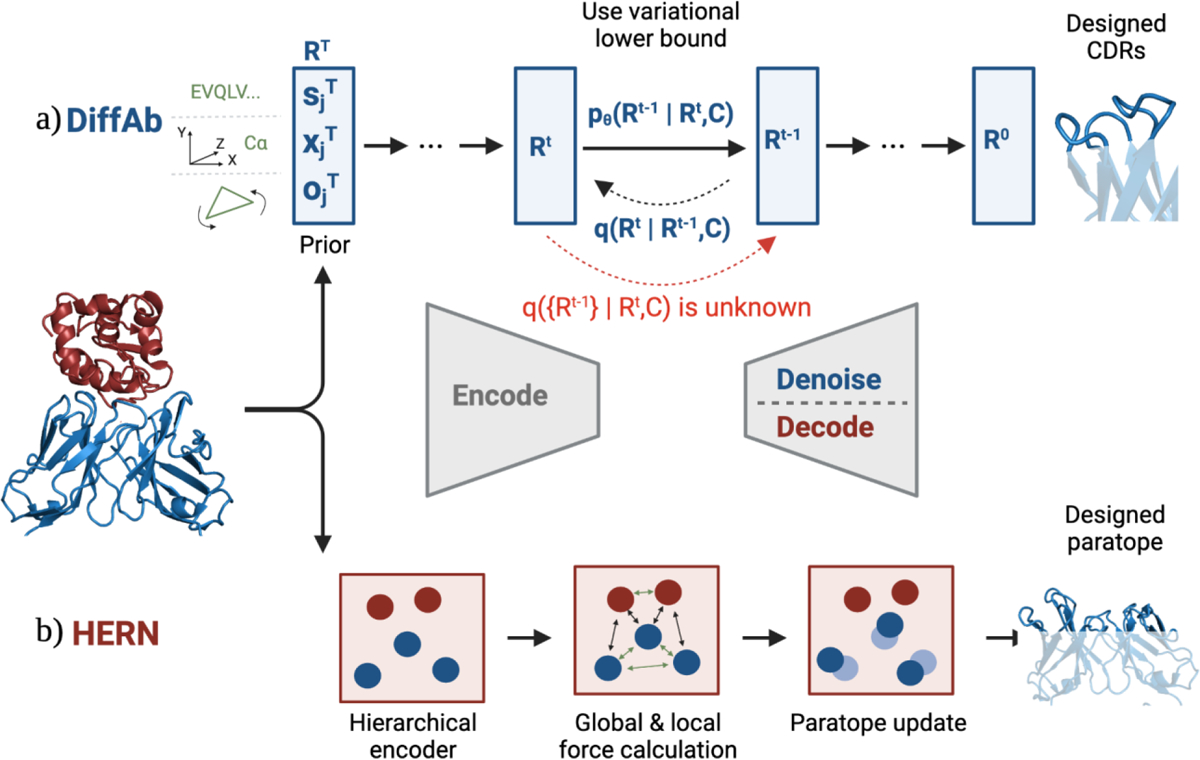
a) DiffAb parametrizes the distribution of the CDR’s sequences (s), positions (x), and orientations (o) for the next step of denoising. b) In each refinement step of HERN, the docking module encoders the residues and atoms into vector representations. It then computes the residue-level force between Cα atoms, and the local force between side chains. The paratope structure is then updated based on the predicted forces.
Table 1: Overview of deep learning models applied to antibody prediction and design tasks.
Structure prediction, fixed backbone design, sequence generation, and sequence and structure co-design models are included. For brevity, performance metrics are reported for primarily CDR3 and H3. It should be noted that performance metrics are taken directly from published results, and does not guarantee that methods within a particular subheader test against the same set of antibodies.
| Fv Structure prediction | |||
|---|---|---|---|
| Model | Architecture | Dataset | Performance (H3 RMSD) |
| AbLooper [10] | Five E(n)-EGNNs | 3k structures from SAbDab | 3.20 Å |
| DeepAb [11] | LSTM + residual NN | 118k sequences from OAS for LSTM, 1.7k structures from SAbDab | 3.28 Å |
| IgFold [14]** | AntiBERTy + Graph transformer + IPA | 4k crystals from SAbDab, 38k structures from AF | 2.99 Å |
| ABodyBuilder2 [18] | AlphaFold - Multimer | 4k crystals from SAbDab, 22k unpaired structures from AlphaFold | 2.81 Å |
| tFold-Ab [16]** | AlphaFold - Multimer | 7k paired crystals, 1k heavy only, 500 light only from SAbDab | 2.74 Å |
| xTrimoAbFold [17] | AlphaFold - Multimer | 18k BCR chains from PDB | 1.25 Å (for CDR3) |
| RaptorX-Single [20] | Sequence embedding module + Evoformer + IPA layers | 340k structures from PDB, fine-tuned on 5k heavy and light antibody chain structures from SAbDab | 2.65 Å |
| Fixed backbone sequence design | |||
| Model | Architecture | Dataset | Performance (H3 AAR) |
| Fv Hallucinator [55] | DeepAb | 11k immunoglobulin domains from antibody structure database AbDb/abYbank | 51% |
| Representation learning | |||
| Model | Architecture | Dataset | Performance (H3 AAR) |
| AntiBERTy [15] | BERT | 558M non-redundant sequences from OAS | 26.0% |
| AbLang [39] | RoBERTa | 14M heavy and 187k light sequences from OAS with 70% identity | 33.6% |
| AntiBERTa [40] | RoBERTa | 52.89M unpaired heavy and 19.09M unpaired light chains from OAS database | - |
| PARA | DeBERTa | 13.5M heavy and 4.5M light chains from OAS | 34.2% |
| Sequence generation | |||
| Model | Architecture | Dataset | Performance (H3 perplexity) |
| ProGen2-OAS [34] | Transformer decoder | 554M sequences from OAS (clustered at 85% sequence identity) | - |
| Manifold Sampler [33,35] | DAE | 20M from Pfam for DAE, 05M from Pfam for function predictor | - |
| IgLM [37]** | Transformer decoder | 558M sequences at 95% sequence identity | 4.653 |
| Sequence and structure co-design | |||
| Model | Architecture | Dataset | Performance |
| RefineGNN [58]** | GNN | 4994 antibody CDR H loops from SAbDab | H3 RMSD: 2.50 Å CDR AAR 35.37% PPL: H1: 6.09, H2: 6.58, H3: 8.38 |
| AbDockGen [59] | HERN | 3k Ab-Ag complexes from SAbDab after filtering structures without antigens and removing duplicates | AAR: 34.1% Contact AAR: 20.8% Designs with improved Edesign: 11.6% |
| MEAN [60] | E(3)-eGNNs | 3k complexes from SAbDab | H3 RMSD: 1.81 Å AAR: 36.77% ΔΔG: −5.33 |
| HMPN [61] | E(3)-eGNNs | 50M OAS Fv sequences for pretraining | H3 RMSD: 2.38 Å H3 AAR: 31.08% H3 PPL: 6.323 |
| DiffAb [70]** | DDPM | SAbDab structures higher resolution than 4A, H3 at 50% seq identity | H3 RMSD: 3.597 H3 AAR: 26.78% H3 ΔΔG: 23.63% |
| DockGPT [73] | Transformer encoder/decoder | 37k single chains from BC40 dataset, 33k general protein complexes from DIPS, 3k ab-ag complexes from SAbDab with < 40% sequence identity | H3 RMSD: 1.88 Å H3 PPL: 10.68 |
Acknowledgements
This work was supported by the Grant 5 R35 GM141881-03. We thank Jeff Ruffolo for helpful comments on the manuscript.
Michael Chungyoun reports financial support was provided by Johns Hopkins University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest, published within the period of review, have been highlighted as: * of special interest
** of outstanding interest
We strive to provide the reader with the most recent advancements in the field of antibody engineering. As such, a substantial amount of citations only accessible through bioRxiv and arXiv has been incorporated into this review. These papers have not completed formal peer-review and their findings should be considered preliminary.
Bibliography annotations
- [1].Carter PJ, Rajpal A, Designing antibodies as therapeutics, Cell. 185 (2022) 2789–2805. [DOI] [PubMed] [Google Scholar]
- [2].Strebhardt K, Ullrich A, Paul Ehrlich’s magic bullet concept: 100 years of progress, Nat. Rev. Cancer 8 (2008) 473–480. [DOI] [PubMed] [Google Scholar]
- [3].Akbar R, Bashour H, Rawat P, Robert PA, Smorodina E, Cotet T-S, Flem-Karlsen K, Frank R, Mehta BB, Vu MH, Zengin T, Gutierrez-Marcos J, Lund-Johansen F, Andersen JT, Greiff V, Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies, mAbs. 14 (2022). 10.1080/19420862.2021.2008790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hummer AM, Abanades B, Deane CM, Advances in computational structure-based antibody design, Curr. Opin. Struct. Biol 74 (2022) 102379. [DOI] [PubMed] [Google Scholar]
- [5].AlQuraishi M, Machine learning in protein structure prediction, Curr. Opin. Chem. Biol 65 (2021) 1–8. [DOI] [PubMed] [Google Scholar]
- [6].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature. 596 (2021) 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, Baker D, Accurate prediction of protein structures and interactions using a three-track neural network, Science. 373 (2021) 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ruffolo JA, Guerra C, Mahajan SP, Sulam J, Gray JJ, Geometric potentials from deep learning improve prediction of CDR H3 loop structures, Bioinformatics. 36 (2020) i268–i275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zenkova N, Sedykh E, Shugaeva T, Strashko V, Ermak T, Shpilman A, Simple End-to-end Deep Learning Model for CDR-H3 Loop Structure Prediction, arXiv [q-bio.BM]. (2021). http://arxiv.org/abs/2111.10656. [Google Scholar]
- [10].Abanades B, Georges G, Bujotzek A, Deane CM, ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation, Bioinformatics. 38 (2022) 1877–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ruffolo JA, Sulam J, Gray JJ, Antibody structure prediction using interpretable deep learning, Patterns (N Y). 3 (2022) 100406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Akpinaroglu D, Ruffolo JA, Mahajan SP, Gray JJ, Simultaneous prediction of antibody backbone and side-chain conformations with deep learning, PLoS One. 17 (2022) e0258173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, dos Santos Costa A, Fazel-Zarandi M, Sercu T, Candido S, Rives A, Language models of protein sequences at the scale of evolution enable accurate structure prediction, bioRxiv. (2022) 2022.07.20.500902. 10.1101/2022.07.20.500902. [DOI] [Google Scholar]
- [14]**.Ruffolo JA, Chu L-S, Mahajan SP, Gray JJ, Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies, bioRxiv. (2022) 2022.04.20.488972. 10.1101/2022.04.20.488972. [DOI] [PMC free article] [PubMed] [Google Scholar]; IgFold is a fast deep learning method for antibody structure prediction using a pre-trained language model (AntiBERTy) and graph networks to directly predict backbone atom coordinates end-to-end. IgFold predicts structures in less than one minute with similar or better quality than AlphaFold, and it has generated the largest database of over 1.3M synthetic antibody structures. https://github.com/Graylab/IgFold.
- [15].Ruffolo JA, Gray JJ, Sulam J, Deciphering antibody affinity maturation with language models and weakly supervised learning, arXiv [q-bio.BM]. (2021). http://arxiv.org/abs/2112.07782. [Google Scholar]
- [16]**.Wu J, Wu F, Jiang B, Liu W, Zhao P, tFold-Ab: Fast and Accurate Antibody Structure Prediction without Sequence Homologs. 10.1101/2022.11.10.515918. [DOI] [Google Scholar]; tFold-Ab is an end-to-end architecture that uses pre-trained language models for faster structure prediction and multi-level supervision for model training. It extracts sequence embeddings from the pre-trained ProtXLNet language model, which is then passed through an AF2 architecture with the standard Evoformer stack replaced and simplified to a single stack. tFold-Ab explicitly considers side-chain conformations, which can be critical for accurate CDR loop prediction.
- [17].Wang Y, Gong X, Li S, Yang B, Sun Y, Shi C, Wang Y, Yang C, Li H, Song L, xTrimoABFold: De novo Antibody Structure Prediction without MSA, arXiv [q-bio.QM]. (2022). http://arxiv.org/abs/2212.00735. [Google Scholar]
- [18].Abanades B, Wong WK, Boyles F, Georges G, Bujotzek A, Deane CM, ImmuneBuilder: Deep-Learning models for predicting the structures of immune proteins, bioRxiv. (2022) 2022.11.04.514231. 10.1101/2022.11.04.514231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D, Protein complex prediction with AlphaFold-Multimer, bioRxiv. (2022) 2021.10.04.463034. 10.1101/2021.10.04.463034. [DOI] [Google Scholar]
- [20].Jing X, Wu F, Xu J, RaptorX-Single: single-sequence protein structure prediction by integrating protein language models, bioRxiv. (2023) 2023.04.24.538081. 10.1101/2023.04.24.538081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gaudreault F, Corbeil CR, Sulea T, Enhanced antibody-antigen structure prediction from molecular docking using AlphaFold2, bioRxiv. (2022) 2022.12.26.521961. 10.1101/2022.12.26.521961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wallner B, AFsample: Improving Multimer Prediction with AlphaFold using Aggressive Sampling, bioRxiv. (2022) 2022.12.20.521205. 10.1101/2022.12.20.521205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M, ColabFold: making protein folding accessible to all, Nat. Methods 19 (2022) 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].ABSTRACT BOOK - Protein Structure Prediction CenterProceedings - Protein Structure Prediction Center, (n.d.). https://predictioncenter.org/casp15/doc/CASP15_Abstracts.pdf.
- [25].Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S, The ClusPro web server for protein-protein docking, Nat. Protoc 12 (2017) 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ferruz N, Höcker B, Controllable protein design with language models, Nature Machine Intelligence. 4 (2022) 521–532. [Google Scholar]
- [27].Vu MH, Robert PA, Akbar R, Swiatczak B, Sandve GK, Haug DTT, Greiff V, ImmunoLingo: Linguistics-based formalization of the antibody language, arXiv [q-bio.QM]. (2022). http://arxiv.org/abs/2209.12635. [DOI] [PubMed] [Google Scholar]
- [28].Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, Guo D, Ott M, Zitnick CL, Ma J, Fergus R, Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, Proc. Natl. Acad. Sci. U. S. A 118 (2021). 10.1073/pnas.2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hie BL, Xu D, Shanker VR, Bruun TUJ, Weidenbacher PA, Tang S, Kim PS, Efficient evolution of human antibodies from general protein language models and sequence information alone, bioRxiv. (2022) 2022.04.10.487811. 10.1101/2022.04.10.487811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Madani A, McCann B, Naik N, Keskar NS, Anand N, Eguchi RR, Huang P-S, Socher R, ProGen: Language Modeling for Protein Generation, arXiv [q-bio.BM]. (2020). http://arxiv.org/abs/2004.03497. [Google Scholar]
- [31].Keskar NS, McCann B, Varshney LR, Xiong C, Socher R, CTRL: A Conditional Transformer Language Model for Controllable Generation, arXiv [cs.CL]. (2019). http://arxiv.org/abs/1909.05858. [Google Scholar]
- [32].Ferruz N, Schmidt S, Höcker B, ProtGPT2 is a deep unsupervised language model for protein design, Nat. Commun 13 (2022) 4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gligorijević V, Berenberg D, Ra S, Watkins A, Kelow S, Cho K, Bonneau R, Function-guided protein design by deep manifold sampling, bioRxiv. (2021) 2021.12.22.473759. 10.1101/2021.12.22.473759. [DOI] [Google Scholar]
- [34].Nijkamp E, Ruffolo J, Weinstein EN, Naik N, Madani A, ProGen2: Exploring the Boundaries of Protein Language Models, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2206.13517. [DOI] [PubMed] [Google Scholar]
- [35].Berenberg D, Lee JH, Kelow S, Park JW, Watkins A, Gligorijević V, Bonneau R, Ra S, Cho K, Multi-segment preserving sampling for deep manifold sampler, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2205.04259. [Google Scholar]
- [36].Prihoda D, Maamary J, Waight A, Juan V, Fayadat-Dilman L, Svozil D, Bitton DA, BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning, MAbs. 14 (2022) 2020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shuai RW, Ruffolo JA, Gray JJ, Generative language modeling for antibody design. 10.1101/2021.12.13.472419. [DOI] [PMC free article] [PubMed] [Google Scholar]; The Immunoglobulin Language Model (IgLM) is a deep generative LM that generates synthetic libraries with generated variable-length spans (e.g. for CDR H3). Generated libraries have excellent developability metrics in-silico (low solubility, low thermal stability, high aggregation, and high immunogenicity can plague antibody libraries). https://github.com/Graylab/IgLM.
- [38].Khan A, Cowen-Rivers AI, Grosnit A, Deik D-G-X, Robert PA, Greiff V, Smorodina E, Rawat P, Dreczkowski K, Akbar R, Tutunov R, Bou-Ammar D, Wang J, Storkey A, Bou-Ammar H, AntBO: Towards Real-World Automated Antibody Design with Combinatorial Bayesian Optimisation, arXiv [q-bio.BM]. (2022). http://arxiv.org/abs/2201.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Olsen TH, Moal IH, Deane CM, AbLang: an antibody language model for completing antibody sequences, Bioinform Adv. 2 (2022) vbac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Leem J, Mitchell LS, Farmery JHR, Barton J, Galson JD, Deciphering the language of antibodies using self-supervised learning, Patterns (N Y). 3 (2022) 100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao X, Cao C, Lai L, Pre-training with A rational approach for antibody, bioRxiv. (2023) 2023.01.19.524683. 10.1101/2023.01.19.524683. [DOI] [Google Scholar]
- [42].Wang W, Peng Z, Yang J, Single-sequence protein structure prediction using supervised transformer protein language models, bioRxiv. (2022) 2022.01.15.476476. 10.1101/2022.01.15.476476. [DOI] [PubMed] [Google Scholar]
- [43].Liu Y, Ott M, Goyal N, Du J, Joshi M, Chen D, Levy O, Lewis M, Zettlemoyer L, Stoyanov V, RoBERTa: A Robustly Optimized BERT Pretraining Approach, arXiv [cs.CL]. (2019). http://arxiv.org/abs/1907.11692. [Google Scholar]
- [44].Bachas S, Rakocevic G, Spencer D, Sastry AV, Haile R, Sutton JM, Kasun G, Stachyra A, Gutierrez JM, Yassine E, Medjo B, Blay V, Kohnert C, Stanton JT, Brown A, Tijanic N, McCloskey C, Viazzo R, Consbruck R, Carter H, Levine S, Abdulhaqq S, Shaul J, Ventura AB, Olson RS, Yapici E, Meier J, McClain S, Weinstock M, Hannum G, Schwartz A, Gander M, Spreafico R, Antibody optimization enabled by artificial intelligence predictions of binding affinity and naturalness, bioRxiv. (2022) 2022.08.16.504181. 10.1101/2022.08.16.504181. [DOI] [Google Scholar]
- [45].Shanehsazzadeh A, Bachas S, Kasun G, Sutton JM, Steiger AK, Shuai R, Kohnert C, Morehead A, Brown A, Chung C, Luton BK, Diaz N, McPartlon M, Knight B, Radach M, Bateman K, Spencer DA, Cejovic J, Kopec-Belliveau G, Haile R, Yassine E, McCloskey C, Natividad M, Chapman D, Stojanovic L, Rakocevic G, Yapici E, Moran K, Caguiat R, Abdulhaqq S, Guo Z, Klug LR, Gander M, Meier J, Unlocking de novo antibody design with generative artificial intelligence, bioRxiv. (2023) 2023.01.08.523187. 10.1101/2023.01.08.523187. [DOI] [Google Scholar]
- [46].He P, Liu X, Gao J, Chen W, DeBERTa: Decoding-enhanced BERT with Disentangled Attention, arXiv [cs.CL]. (2020). http://arxiv.org/abs/2006.03654. [Google Scholar]
- [47].Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA, Bileschi ML, Bork P, Bridge A, Colwell L, Gough J, Haft DH, Letunić I, MarchlerBauer A, Mi H, Natale DA, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A, InterPro in 2022, Nucleic Acids Res. 51 (2023) D418–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li L, Gupta E, Spaeth J, Shing L, Bepler T, Caceres RS, Antibody Representation Learning for Drug Discovery, arXiv [q-bio.QM]. (2022). http://arxiv.org/abs/2210.02881. [Google Scholar]
- [49].Wang D, Ye F, Hao Z, On Pre-trained Language Models for Antibody, bioRxiv. (2023) 2023.01.29.525793. 10.1101/2023.01.29.525793. [DOI] [Google Scholar]
- [50].Ingraham J, Garg V, Barzilay R, Jaakkola T, Generative models for graph-based protein design, Adv. Neural Inf. Process. Syst 32 (2019). https://proceedings.neurips.cc/paper/2019/hash/f3a4ff4839c56a5f460c88cce3666a2b-Abstract.html [Google Scholar]
- [51].Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, Wicky BIM, Courbet A, de Haas RJ, Bethel N, Leung PJY, Huddy TF, Pellock S, Tischer D, Chan F, Koepnick B, Nguyen H, Kang A, Sankaran B, Bera AK, King NP, Baker D, Robust deep learning based protein sequence design using ProteinMPNN. 10.1101/2022.06.03.494563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE, Neural Message Passing for Quantum Chemistry, in: Precup D, Teh YW (Eds.), Proceedings of the 34th International Conference on Machine Learning, PMLR, 06--11 Aug 2017: pp. 1263–1272. [Google Scholar]
- [53].Anand-Achim N, Eguchi RR, Mathews II, Perez CP, Derry A, Altman RB, Huang P-S, Protein Sequence Design with a Learned Potential. 10.1101/2020.01.06.895466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, Lerer A, Rives A, Learning inverse folding from millions of predicted structures, in: Chaudhuri K, Jegelka S, Song L, Szepesvari C, Niu G, Sabato S (Eds.), Proceedings of the 39th International Conference on Machine Learning, PMLR, 17--23 Jul 2022: pp. 8946–8970. [Google Scholar]
- [55].Mahajan SP, Ruffolo JA, Frick R, Gray JJ, Hallucinating structure-conditioned antibody libraries for target-specific binders, Front. Immunol 13 (2022) 999034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zheng Z, Deng Y, Xue D, Zhou Y, Ye F, Gu Q, Structure-informed Language Models Are Protein Designers, bioRxiv. (2023) 2023.02.03.526917. 10.1101/2023.02.03.526917. [DOI] [Google Scholar]
- [57].Cowen-Rivers AI, Gorinski PJ, Sootla A, Khan A, Furui L, Wang J, Peters J, Ammar HB, Structured Q-learning For Antibody Design, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2209.04698. [Google Scholar]
- [58].Jin W, Wohlwend J, Barzilay R, Jaakkola T, Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design, arXiv [q-bio.BM]. (2021) http://arxiv.org/abs/2110.04624. [Google Scholar]; RefineGNN’s novel iterative graph generation and accommodation method is capable in-silico of outperforming sequence-based (LSTM), autoregressive graph-based (AR-GNN), and physics-based (RosettaAntibodyDesign, RAbD) methods on three antibody generation tasks (structure prediction, antigen-binding antibody design, and SARS-CoV-2 neutralization optimization). https://github.com/wengong-jin/RefineGNN.
- [59].Jin W, Barzilay R, Jaakkola T, Antibody-Antigen Docking and Design via Hierarchical Equivariant Refinement, arXiv [q-bio.BM]. (2022). http://arxiv.org/abs/2207.06616. [Google Scholar]
- [60].Kong X, Huang W, Liu Y, Conditional Antibody Design as 3D Equivariant Graph Translation, arXiv [q-bio.BM]. (2022). http://arxiv.org/abs/2208.06073. [Google Scholar]
- [61].Gao K, Wu L, Zhu J, Peng T, Xia Y, He L, Xie S, Qin T, Liu H, He K, Liu T-Y, Incorporating Pre-training Paradigm for Antibody Sequence-Structure Co-design, (n.d.). 10.1101/2022.11.14.516404. [DOI] [Google Scholar]
- [62].Ramesh A, Dhariwal P, Nichol A, Chu C, Chen M, Hierarchical Text-Conditional Image Generation with CLIP Latents, arXiv [cs.CV]. (2022). http://arxiv.org/abs/2204.06125. [Google Scholar]
- [63].Rombach R, Blattmann A, Lorenz D, Esser P, Ommer B, High-Resolution Image Synthesis with Latent Diffusion Models, 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR). (2022). 10.1109/cvpr52688.2022.01042. [DOI] [Google Scholar]
- [64].Esser P, Chiu J, Atighehchian P, Granskog J, Germanidis A, Structure and Content-Guided Video Synthesis with Diffusion Models, arXiv [cs.CV]. (2023). http://arxiv.org/abs/2302.03011. [Google Scholar]
- [65].Anand N, Achim T, Protein Structure and Sequence Generation with Equivariant Denoising Diffusion Probabilistic Models, arXiv [q-bio.QM]. (2022). http://arxiv.org/abs/2205.15019. [Google Scholar]
- [66].Lin Y, AlQuraishi M, Generating Novel, Designable, and Diverse Protein Structures by Equivariantly Diffusing Oriented Residue Clouds, arXiv [q-bio.BM]. (2023). http://arxiv.org/abs/2301.12485. [Google Scholar]
- [67].Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, Ahern W, Borst AJ, Ragotte RJ, Milles LF, Wicky BIM, Hanikel N, Pellock SJ, Courbet A, Sheffler W, Wang J, Venkatesh P, Sappington I, Torres SV, Lauko A, De Bortoli V, Mathieu E, Barzilay R, Jaakkola TS, DiMaio F, Baek M, Baker D, Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models. 10.1101/2022.12.09.519842. [DOI] [Google Scholar]; RoseTTAFold Diffusion (RFdiffusion) is the first denoising diffusion model for protein design with experimental validation. By fine-tuning the RoseTTAFold structure prediction network on protein structure denoising tasks, RFdiffusion becomes a generative model of protein backbones. The authors experimentally characterized the structures and functions of hundreds of new designs, including protein binders, symmetric oligomers, enzyme active site scaffolding, and symmetric motif scaffolding for therapeutic and metal-binding proteins. https://github.com/RosettaCommons/RFdiffusion.
- [68].Ingraham J, Baranov M, Costello Z, Frappier V, Ismail A, Tie S, Wang W, Xue V, Obermeyer F, Beam A, Grigoryan G, Illuminating protein space with a programmable generative model. 10.1101/2022.12.01.518682. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chroma introduces a denoising diffusion process that respects the conformational statistics of polymer ensembles, an efficient neural architecture for molecular systems, and equivariant layers for efficiently synthesizing 3D structures of proteins. Chroma directly samples novel protein structures and sequences and can be conditioned to steer the generative process toward desired shapes or properties.
- [69].Lisanza SL, Gershon JM, Tipps S, Arnoldt L, Hendel S, Sims JN, Li X, Baker D, Joint Generation of Protein Sequence and Structure with RoseTTAFold Sequence Space Diffusion, bioRxiv. (2023) 2023.05.08.539766. 10.1101/2023.05.08.539766. [DOI] [Google Scholar]
- [70].Luo S, Su Y, Peng X, Wang S, Peng J, Ma J, Antigen-Specific Antibody Design and Optimization with Diffusion-Based Generative Models for Protein Structures. 10.1101/2022.07.10.499510. [DOI] [Google Scholar]; DiffAb is the first denoising diffusion model for antibodies, jointly modeling sequences and structures of complementarity-determining regions (CDRs) with equivariant networks. The model is capable of sequence-structure co-design, sequence design for given backbone structures, and antibody optimization. In-silico, DiffAb outperforms RosettaAntibodyDesign (RAbD) in binder design. https://github.com/luost26/diffab.
- [71].Yim J, Trippe BL, De Bortoli V, Mathieu E, Doucet A, Barzilay R, Jaakkola T, SE(3) diffusion model with application to protein backbone generation, arXiv [cs.LG]. (2023). http://arxiv.org/abs/2302.02277. [Google Scholar]
- [72].Kong X, Huang W, Liu Y, End-to-End Full-Atom Antibody Design, arXiv [q-bio.BM]. (2023). http://arxiv.org/abs/2302.00203. [Google Scholar]
- [73].Meenakshi DU, Nandakumar S, Francis AP, Sweety P, Fuloria S, Fuloria NK, Subramaniyan V, Khan SA, Deep learning and site-specific drug delivery, Deep Learning for Targeted Treatments. (2022) 1–38. 10.1002/9781119857983.ch1. [DOI] [Google Scholar]
- [74].Eguchi RR, Choe CA, Parekh U, Khalek IS, Ward MD, Vithani N, Bowman GR, Jardine JG, Huang P-S, Deep Generative Design of Epitope-Specific Binding Proteins by Latent Conformation Optimization, bioRxiv. (2022) 2022.12.22.521698. 10.1101/2022.12.22.521698. [DOI] [Google Scholar]; Sculptor is a deep generative design algorithm that creates epitope-specific protein binders by joint searching over positions, interactions, and generated conformations of a fold, using molecular dynamics to capture local conformational landscapes. The algorithm designed a multi-toxin binder against a conserved epitope on venom toxins implicated in neuromuscular paralysis, demonstrating epitope-targeted design.
- [75].Raybould MIJ, Kovaltsuk A, Marks C, Deane CM, CoV-AbDab: the coronavirus antibody database, Bioinformatics. 37 (2020) 734–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wu L, Gong C, Liu X, Ye M, Liu Q, Diffusion-based Molecule Generation with Informative Prior Bridges, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2209.00865. [Google Scholar]
- [77].Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, Shi J, Deane CM, SAbDab: the structural antibody database, Nucleic Acids Res. 42 (2014) D1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, Žídek A, Green T, Tunyasuvunakool K, Petersen S, Jumper J, Clancy E, Green R, Vora A, Lutfi M, Figurnov M, Cowie A, Hobbs N, Kohli P, Kleywegt G, Birney E, Hassabis D, Velankar S, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res. 50 (2021) D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mitchell AL, Almeida A, Beracochea M, Boland M, Burgin J, Cochrane G, Crusoe MR, Kale V, Potter SC, Richardson LJ, Sakharova E, Scheremetjew M, Korobeynikov A, Shlemov A, Kunyavskaya O, Lapidus A, Finn RD, MGnify: the microbiome analysis resource in 2020, Nucleic Acids Res. 48 (2020) D570–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jaffe DB, Shahi P, Adams BA, Chrisman AM, Finnegan PM, Raman N, Royall AE, Tsai F, Vollbrecht T, Reyes DS, Hepler NL, McDonnell WJ, Functional antibodies exhibit light chain coherence, Nature. 611 (2022) 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, Slabodkin A, Chernigovskaya M, Scheffer L, Smorodina E, Rawat P, Mehta BB, Vu MH, Mathisen IF, Prósz A, Abram K, Olar A, Miho E, Haug DTT, Lund-Johansen F, Hochreiter S, Haff IH, Klambauer G, Sandve GK, Greiff V, Unconstrained generation of synthetic antibody–antigen structures to guide machine learning methodology for antibody specificity prediction, Nature Computational Science. 2 (2022) 845–865. [DOI] [PubMed] [Google Scholar]
- [82].Huang P-S, Boyken SE, Baker D, The coming of age of de novo protein design, Nature. 537 (2016) 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- [83].Makowski EK, Kinnunen PC, Huang J, Wu L, Smith MD, Wang T, Desai AA, Streu CN, Zhang Y, Zupancic JM, Schardt JS, Linderman JJ, Tessier PM, Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space, Nature Communications. 13 (2022). 10.1038/s41467-022-31457-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Park JW, Stanton S, Saremi S, Watkins A, Dwyer H, Gligorijevic V, Bonneau R, Ra S, Cho K, PropertyDAG: Multi-objective Bayesian optimization of partially ordered, mixed-variable properties for biological sequence design, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2210.04096. [Google Scholar]
- [85].Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA, Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract, J. Mol. Biol 368 (2007) 652–665. [DOI] [PubMed] [Google Scholar]
- [86].Tagasovska N, Frey NC, Loukas A, Hötzel I, Lafrance-Vanasse J, Kelly RL, Wu Y, Rajpal A, Bonneau R, Cho K, Ra S, Gligorijević V, A Pareto-optimal compositional energy-based model for sampling and optimization of protein sequences, arXiv [cs.LG]. (2022). http://arxiv.org/abs/2210.10838. [Google Scholar]
- [87].Frey NC, Berenberg D, Kleinhenz J, Hotzel I, Lafrance-Vanasse J, Kelly RL, Wu Y, Rajpal A, Ra S, Bonneau R, Cho K, Loukas A, Gligorijevic V, Saremi S, Learning protein family manifolds with smoothed energy-based models, (2023). https://openreview.net/pdf?id=IilnB8jfoP9 (accessed May 22, 2023).
- [88].Hie B, Candido S, Lin Z, Kabeli O, Rao R, Smetanin N, Sercu T, Rives A, A high-level programming language for generative protein design, (n.d.). 10.1101/2022.12.21.521526. [DOI] [Google Scholar]