

Abstract
The development of voltage-sensitive fluorescent probes suggests fluorescence lifetime as a promising readout for electrical activity in biological systems. Existing approaches fail to achieve the speed and sensitivity required for voltage imaging in neuroscience applications. Here we demonstrate that wide-field electro-optic fluorescence lifetime imaging (EO-FLIM) allows lifetime imaging at kHz frame acquisition rates, spatially resolving action potential propagation and sub-threshold neural activity in live adult Drosophila. Lifetime resolutions of < 5 ps at 1 kHz were achieved for single cell voltage recordings. Lifetime readout is limited by photon shot noise and the method provides strong rejection of motion artifacts and technical noise sources. Recordings revealed local transmembrane depolarizations, two types of spikes with distinct fluorescence lifetimes, and phase locking of spikes to an external mechanical stimulus.
Introduction
Recording the electrical activity of neurons at high spatial and temporal resolution is central to understanding brain function. Fluorescent probes of calcium activity enable optical studies of large neuron populations in vivo (1,2). However, the response time of calcium indicators is much slower than the underlying electrical signals. Fluorescent voltage sensors are a complementary approach, providing direct readout of neuron membrane potential with the capability to resolve action potentials. While voltage probes have rapidly developed with a variety of genetically-encoded (3–7) and chemical dyes (8) in use, there remain significant challenges to their application in vivo due to low sensitivity, rapid photo-bleaching, and motion artifacts. To achieve high speed recording and sufficient signal to noise, most voltage probes use fluorescence intensity to read out an underlying sensing mechanism based on absorption, Förster resonance energy transfer (FRET), or quenching.
We applied an alternative strategy for detecting fast probe dynamics based on lifetime imaging (9). Voltage sensors employing FRET and quenching mechanisms modulate the probe’s non-radiative decay rate, intrinsically connecting fluorescence intensity with nanosecond excited state lifetime. Lifetime is a promising readout for voltage imaging, especially due to its capability to provide an absolute indication of membrane potential (10). Recent results have validated fluorescence lifetime for static measurements of membrane potential in vitro (11). However, existing lifetime detectors, e.g. single photon counters or cameras with modulated pixels (12), fall short of the requirements for detecting fast dynamics and imaging neurons in vivo, either due to their limited photon throughput or prohibitively high noise.
EO-FLIM Technique
We demonstrate a new approach using EO-FLIM, an all-optical technique for lifetime imaging based on nanosecond gating of a wide-field image (13, 14). EO-FLIM allows efficient photon collection and is compatible with detection on high-speed, low-noise scientific cameras. With this method, we achieved lifetime imaging of action potentials in vivo.
EO-FLIM enables significant suppression of intensity artifacts, allowing robust imaging in the presence of tissue motion and fluctuations in illumination intensity. Such artifacts are ubiquitous in recordings of neural activity from awake, behaving animals (15, 16). This has two consequences: 1) it enables faithful recording of sub-threshold voltage waveforms and 2) it improves the signal-to-noise ratio by suppressing high-frequency intensity noise. These follow from the fact that in EO-FLIM, lifetime is estimated from the ratio of a pair of simultaneously recorded intensity channels deriving from a common optical source. In conventional approaches, corrections for intensity noise involve ratios of measurements which are non-simultaneous and easily corrupted by high-frequency noise or slower motion artifacts. Typically optical sensors of neuron activity are reported by ΔF/F, referencing fast intensity changes (ΔF) to a non-simultaneous, average fluorescence baseline (F). Fluorescence lifetime is capable of reading out an intensity-optimized sensor with improved temporal noise performance and long term stability without sacrificing acquisition speed.
We implemented our approach through incorporation of a Pockels cell into the fluorescence detection path of a standard epifluorescence microscope (Figs. 1(A), S1, and Methods). The Pockels cell design was optimized for resonant drive and wide-field imaging, incorporating thermal control and transverse crystal geometry to cancel off-axis birefringence. A high voltage modulation was applied to the Pockels cell using a resonant transformer (Figs. S2 and S3), resulting in a fast polarization rotation which was synchronous with the excitation pulses from a 100 picosecond laser source. Fluorescence from the sample was first polarized, then polarization modulated by the Pockels cell, and finally split by a polarizing beamsplitter into two wide-field images on a scientific CMOS camera corresponding to gated (G) and ungated (U) intensity. These two images encoded nanosecond time information in their intensity ratio. Since gating was performed with a beamsplitter, it was possible to capture the entire fluorescence decay with photon efficiency limited by optical coatings. Here, we modulated one input polarization and discarded half of the available signal on a first polarizer. This can be avoided in the future through addition of a second beam path (13). The fluorescence decay at each pixel was convolved with the temporal gating function of the Pockels cell and then sampled at a single modulation phase relative to the excitation laser [Fig. 1(B)]. Each image thus provided a lifetime estimate for every pixel in parallel at the framerate of the scientific camera. When imaging genetically targeted neurons in vivo, this allowed for 1 kHz frame rate recordings with a lifetime sensitivity of 2.53 ± 0.48 ps with 0.7 – 1.4 × 107 detected photons per frame [Fig. 1(C)], representing a significant improvement in throughput over previous FLIM recordings of cellular dynamics. EO-FLIM approaches fundamental sensitivity limits for estimating lifetimes between 1 and 4 nanoseconds (Fig. S2).
Fig. 1. Lifetime imaging of action potentials.
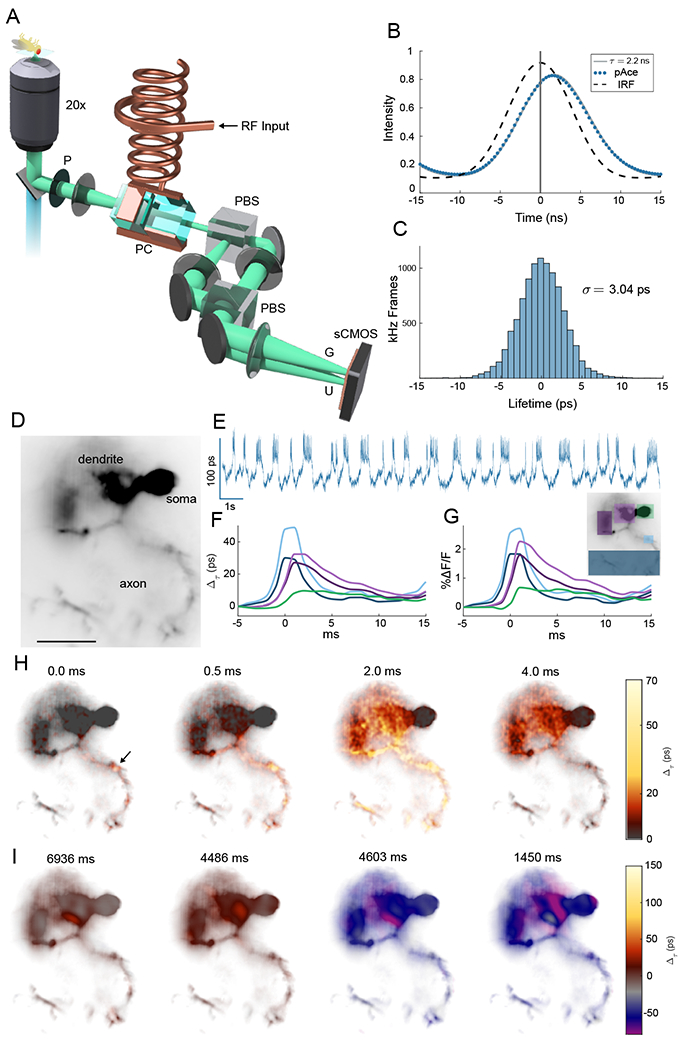
(A) Schematic of EO-FLIM microscope. Wide-field fluorescence images were modulated by a Pockels cell (PC) placed between crossed polarizers (P and PBS) and driven at 20 MHz by a high voltage resonant transformer. Two spatially offset output images were simultaneously captured after a second polarizing beamsplitter (PBS) on a sCMOS camera, corresponding to gated (G) and ungated (U) intensities. (B) Instrument response function (IRF) and fluorescence traces for the U channel were measured by varying the Pockels cell drive phase relative to the excitation laser. The pAce GEVI was fit to 2.2 ns lifetime. For kilohertz imaging, a single optimal phase point was captured (vertical line at 0 ns delay) and the G/U image intensity ratio was converted to a lifetime estimate (see also Fig. S1). (C) Histogram of measurements (highpass filtered) obtained at 1 kHz for a single neuron in vivo demonstrate a lifetime sensitivity of 3 ps (full trace in Fig. 2(A)) (D) Wide-field image of a neuron with structures indicated (scalebar 25 μm) (E) Whole cell lifetime trace resolves action potentials and sub-threshold transitions (F, G) Average spike shape is plotted in intensity and lifetime from color-coded regions (H) Frames from an interpolated lifetime movie demonstrate spike propagation, averaging the signal from ~300 individual spikes. The point of initiation is indicated by the arrow, and bidirectional propagation was observed both along the axon and backwards towards soma and dendrites (see Movies S1–S3). Spike propagation was also imaged directly without averaging in Movies S4 and S5. (I) Applying a 10 frame moving average allowed sub-threshold signals to be localized to neuron structures in Movies S6 and S7. Example frames demonstrate localization in the dendrite for both positive and negative sub-threshold signals.
Results
EO-FLIM was used to image a genetically encoded voltage indicator (GEVI) in Drosophila melanogaster. We expressed pAce positive polarity GEVI in a subtype of fly mushroom body output neuron (MBON), MBON-γ1pedc> αβ. pAce works via FRET and is a fusion of the bright fluorescent protein mNeonGreen with a voltage-sensitive opsin from Acetabularia (3). We surgically prepared Drosophila before imaging to provide optical access to the brain (17, 18). Action potentials and sub-threshold dynamics were readily resolved with action potentials corresponding to a 20-50 picosecond shift in the fluorescent lifetime [Fig. 1(D,E)]. Average spike readouts in lifetime and intensity for different neuron sub-regions were in strong agreement both in their relative timing and amplitudes [Fig. 1(F,G)].
The donor fluorescence lifetime of a FRET GEVI depends on radiative decay rate kf and the voltage sensitive non-radiative decay rate knr(V) associated with FRET as (10, 19). In pAce, the Ace opsin acts as acceptor and provides voltage sensitivity. The donor’s fluorescence intensity is directly quenched by FRET giving a signal where σ is the donor excitation cross section. For an ideal FRET process, one expects to find Δτ/τ = ΔF/F. pAce gave a linear but attenuated lifetime response of (0.70 ± 0.07)ΔF/F (Fig. S4). This may indicate components of the GEVI response in intensity, for example modulation of cross section σ, that did not affect lifetime readout.
Wide-field lifetime imaging correlates neuron activity with spatial structure. The point of action potential initiation in the axon is resolved along with bi-directional propagation along the axon and backwards towards the dendrite and soma (20). Action potentials were attenuated and broadened in the dendrites and soma [Figs. 1(F,G), S5, and S6]. Spike propagation is shown in Movies S1–S3 with still frames from Movie S1 displayed in Fig. 1(H), generated by spike triggered averaging over ~ 300 spikes and interpolating between frames. We also observed individual spikes and spike propagation in real time without temporal averaging (Movies S4 and S5). Comparison of recordings from multiple neuronal sub-regions revealed local depolarizations in the axons which fail to initiate action potentials across the entire cell. These were not resolved in intensity readout but are clearly visible in lifetime recordings (Figs. S5 and S6). Finally, by applying a 10 frame moving average, the spatial distribution of slower sub-threshold voltage signals could also be studied. Sub-threshold signals were often strongest and localized in the dendrites (Movies S6 and S7). Still frames from these movies are shown in Fig. 1(I) and Figs. S7 and S8.
By measuring a ratio of simultaneous intensities, EO-FLIM removes noise sources that are common mode to both the modulated channels. In Fig. 2 we show example recordings of MBON-γ1pedc> α/β neurons from six flies comparing intensity ΔF/F and lifetime readouts. In all recordings lifetime detection enhanced the signal-to-noise ratio (SNR) for action potential detection by ~ 2x. This SNR was quantified by comparing spike amplitude to the high frequency noise floor. We also analyzed traces using the spike detection fidelity d′, a discriminability index that quantifies spike detection by comparing the statistical distributions of spike amplitudes and background noise fluctuations (21). Lifetime detection improved d′ by 1.5 - 2.4 times in Fig. 2(A–E).
Fig. 2. Lifetime suppresses intensity noise and improves fidelity of sub-threshold recording.
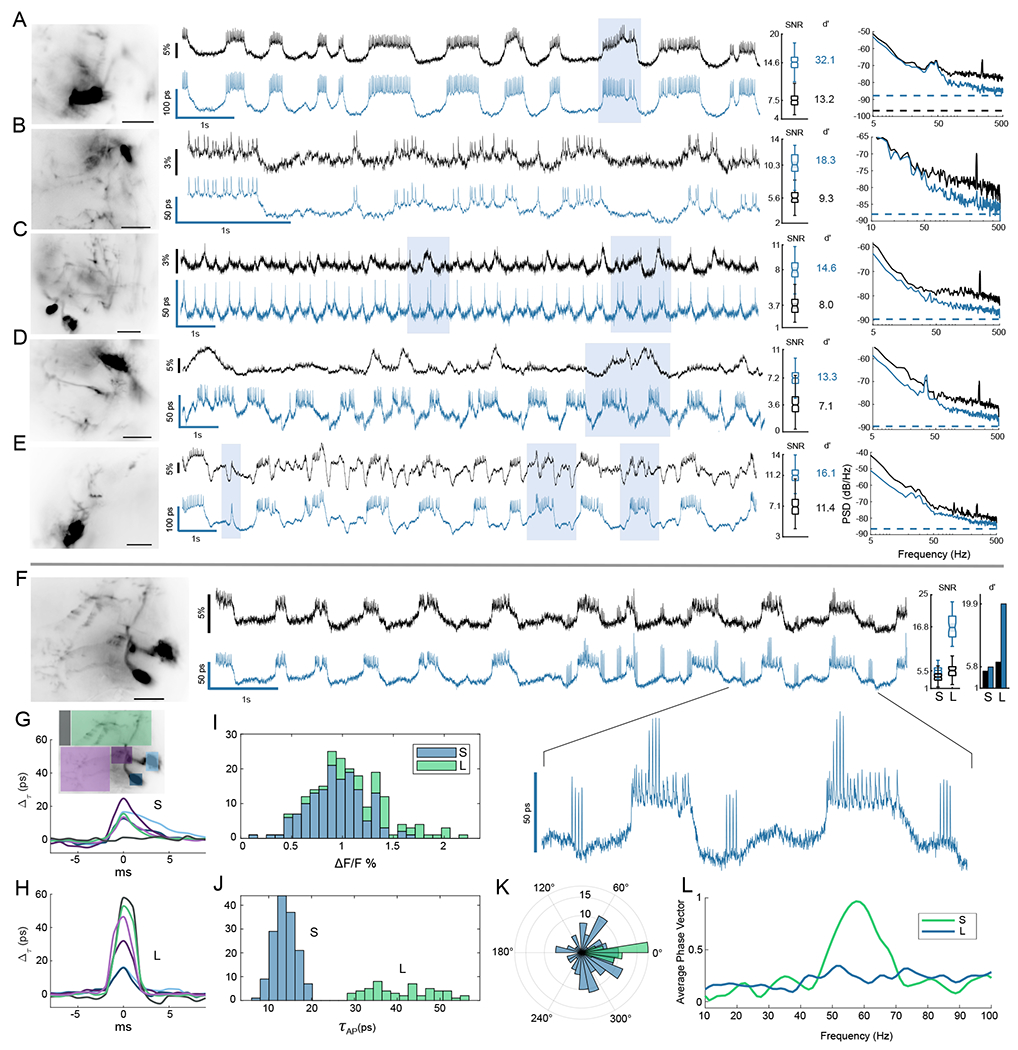
Six example MBON neurons are shown comparing lifetime (blue) to ΔF/F intensity recordings (black). Recordings were obtained by averaging over high-resolution images shown at left (scalebar 25 μm). Shaded boxes highlight some notable regions of the traces for improved lifetime readout. For each example, the distributions of spike SNR are compared for intensity and lifetime, with calculated spike detection fidelity d′ indicated. (A,B) Two examples of flies without motion demonstrate improvement of technical noise floor at high frequencies by up to 7 dB. The noise power spectra for the traces are compared at right, with dotted lines indicating the photon shot noise limits (C-E) Three examples of flies having low-frequency noise associated with motion artifacts. Lifetime improves noise power spectrum across temporal frequencies, rejecting intensity noise by up to 9 dB at low frequencies. See also further analysis in Fig. 3. (F-J) Lifetime provided an improved readout of two spike amplitudes in response to mechanical stimulus at 60 Hz. Large (L) spikes showed an enhanced lifetime responsivity and tripled detection SNR and d′ over the small (S) spikes. L spikes occured independent of sub-threshold waveform level but synchronized with spiking on plateaus in the inset. (G,H) Average spike waveforms for color-coded regions. The point of initiation for S spikes was a central region of the axon (consistent with Movies S1 and S2), while L spikes were diffuse and associated with background fluorescence. L spikes also correspond to local spikes in the dendrite and soma in (H). L spike background component possibly resulted from out of focus neurons (Movies S8 and S9). (I,J) Histograms of action potential amplitudes are compared. In intensity the L and S populations were not resolved and strongly overlap, but they were clearly separated in lifetime. Using lifetime to identify the spikes, the intensity histogram (I) is shaded with two colors to show overlapping populations. (K) A polar histogram demonstrates strong phase locking of the L spikes to mechanical stimulus using a bandpass filtered lifetime trace as phase reference. S spikes do not show phase locking. (L) The average phase vector length Σi cos(θi)/NAP is plotted vs. bandpass center frequency to show narrow-band locking response.
Noise power was also compared across temporal frequencies, demonstrating broad suppression of intensity noise in Fig. 2(A–E) and showing that EO-FLIM approaches the photon shot noise limit. To allow a direct comparison of noise power spectrum, the responsivity of spikes in intensity and lifetime channels were normalized. For flies not displaying much motion [Fig. 2(A,B)], lifetime primarily reduced technical noise at high frequencies that resulted from the excitation laser (4-7 dB). Even in these well-behaved examples lifetime readout resulted in improved SNR and detection fidelity d′.
Lifetime recordings also improved long-term stability in the voltage readout. Intensity-based voltage imaging often displays strong motion artifacts which degrade stability. In Drosophila, these artifacts result from movements such as extension of the proboscis. For flies displaying motion [Figs. 2(C–E) and S9], lifetime readout suppressed artifacts at low frequencies by up to 9 dB compared to intensity readout (here sub-threshold waveforms may only be resolved in lifetime). Fig. 3 quantifies trace stability by the spike amplitude distribution and the uniformity of threshold voltage level at action potential locations. Histograms of mean normalized spike heights and mean normalized sub-threshold level are plotted for each spike, showing that lifetime improves spike uniformity by up to 2.5 times and threshold uniformity by up to 5.8 times.
Fig. 3. Lifetime improves uniformity in action potential amplitude and threshold level.
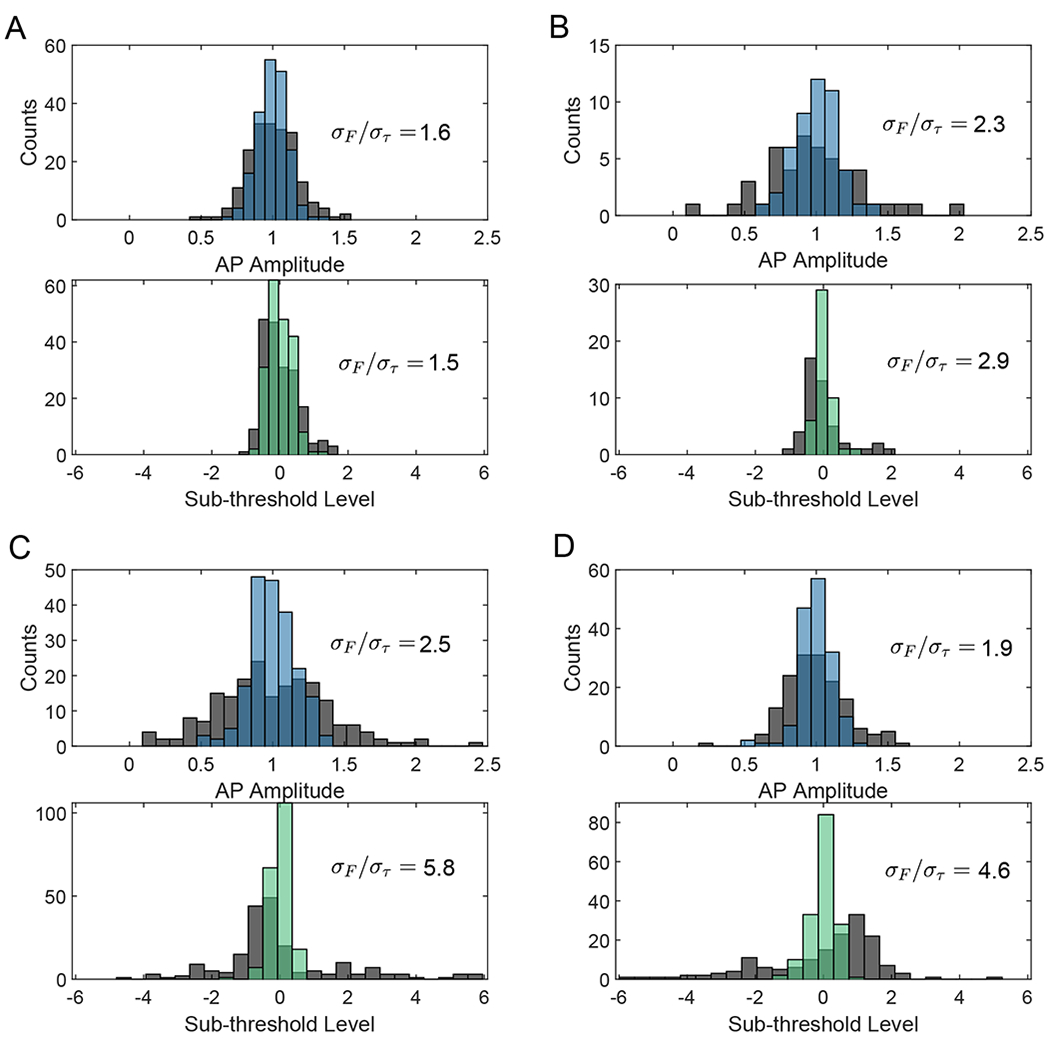
Histograms of action potential amplitudes (lifetime in blue) and action potential levels on the sub-threshold waveform (lifetime in green) are plotted for each activity trace, overlayed on the same histograms for intensity (grey). Action potential amplitudes are normalized to the mean. Sub-threshold level is also mean normalized as where L is the spike’s corresponding level on a low-pass filtered trace, and its distance is measured relative to the mean level of all other spikes . A perfectly uniform threshold would thus result in L = 0 for all spikes. In each histogram, the ratio of standard deviation between intensity and lifetime readouts σF/στ is given as a figure of merit for uniformity. Panes (A, B-D) correspond to panes (A, C-E) in Fig. 2 respectively.
With the improved readout stability afforded by lifetime recording, we observed two spike amplitudes in Fig. 2(F–L). The small amplitude spikes occured on top of sub-threshold voltage plateaus while large amplitude spikes were observed in bursts that were independent of sub-threshold voltage level. In this sample, the fly was mechanically stimulated near a resonance of the microscope stage with sound waves. Fig. 2(I,J) plot histograms of spike heights in both intensity and lifetime. The two spike populations were clearly distinguished using lifetime readout but were not separable using intensity. Here the large spikes showed enhanced lifetime responsivity (1.34 ΔF/F, compared to 0.68 ΔF/F for smaller spikes). The difference in responsivity may indicate kinetic differences in the GEVI response to different action potential waveforms (3). Spike triggered averages (Movies S8 and S9) showed that the L spikes originated diffusely across the image while the S spikes initiated in a local area of the axon as in Movies S1 and S2. The L spikes may be associated with out of focus neurons displaying off-target GEVI expression, and they were also synchronous with spiking of the targeted neuron as demonstrated in Fig. 2(F–H). The L spikes displayed strong phase locking in response to mechanical stimulus, while the S spikes did not. A histogram of phases is displayed in Fig. 2(K) where phase is determined by each spike’s location on a trace that has been filtered at the stimulus frequency. Narrow-band locking response is demonstrated by Fig. 2(L). Similar large amplitude spikes were also observed during the mechanical stimulus sweep shown in Fig. 4.
Fig. 4. Phase locking of spikes to direct mechanical stimulus.
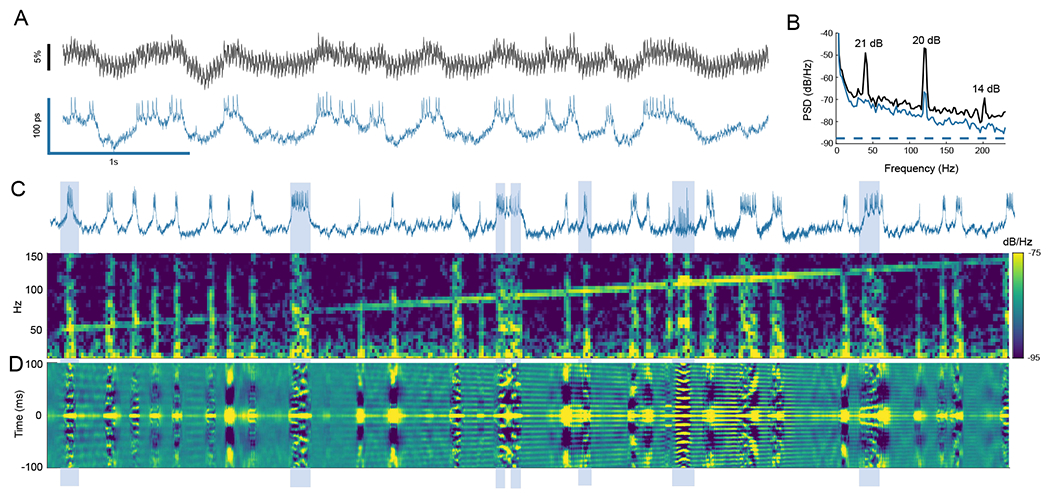
(A) Lifetime provided strong rejection of intensity noise associated with shaking the sample (40 Hz square wave, ~ 0.8 μm peak-to-peak). (B) Intensity noise at the stimulus frequency and second harmonic were attenuated by 21 and 20 dB. (C) A spectrogram of the lifetime trace is plotted as stimulus is swept from 50 to 150 Hz. Vertical lines of activity in the spectrogram correspond to spike bursts in the lifetime trace. Mechanical cross-talk is seen as the diagonal line sweep, and phase locking appears as increased frequency content at the stimulus frequency during spike bursts. (D) To show phase locking visually, a sliding window autocorrelation of the lifetime trace is plotted using a 150 ms window. Phase locking may be seen by observing alignment of autocorrelation peaks during activity bursts to the peaks resulting from mechanical cross-talk signal. Examples of bursts showing phase locking are highlighted.
To further demonstrate the noise rejection capabilities of lifetime detection, we placed the fly on a piezoelectric stage to provide mechanical shaking in the XY plane. This direct mechanical stimulus resulted in high levels of intensity noise that obscured neuron activity, but lifetime readout suppressed this noise by up to 21 dB in Fig. 4(A,B). Using this direct stimulus we could observe phase locked spiking behavior from 30 to >100 Hz in Figs. 4 and S10. In these figures, phase locking was observed through increased spectral power at the stimulus frequency in a spectral waterfall plot as the excitation frequency was swept. This phase locking may also be visualized using the autocorrelation of the lifetime trace [Fig. 4(C–D)]. These observations are consistent with previous studies on mechanical and auditory effects in Drosophila that identify a broad auditory response across the central brain (22) and responses to substrate vibrations (23, 24).
Discussion
EO-FLIM may be applied to both existing lifetime-sensitive probes and to donor readout of FRET-based biosensors. Standard FRET sensors are read out by the ratio of optical intensities in spectrally separated donor and acceptor channels (25), requiring an acceptor molecule with high quantum yield. Two-color readout frequently limits detection sensitivity due to spectral cross-talk and also prevents probe multiplexing (26). Lifetime measurement removes these limitations and allows quantitative FRET measurements using only the donor channel. In voltage imaging, lifetime will enable improved measurement of FRET-opsin (3, 7), hybrid FRET (6), and dye indicators (8). We also anticipate application to imaging calcium (27), neurotransmitters (28, 29), and cyclic AMP (30).
Use of GEVIs in vivo is often accompanied by a large fluorescence background that results from protein expression outside the cellular membrane (31) or leaky gene expression from non-targeted cells (32). This background signal had a different fluorescence lifetime and photobleaches at a different rate, resulting in slow drifts of the measured lifetime traces as shown in Fig. S11 (we expect in vitro studies will not be affected by such backgrounds). To measure absolute voltage, signal and background populations would need to be unmixed by discriminating between two closely spaced exponential decays. For this reason, we focus here on the improved stability and noise performance afforded by lifetime measurement rather than absolute quantification. In our current implementation, we measured the population weighted average lifetime by acquiring images at a single modulation phase. In the future, multiple modulation phases can be combined to unmix lifetime components and improve absolute measurement.
We have shown that EO-FLIM enables fluorescence lifetime imaging of neuron activity in vivo, overcoming the throughput and sensitivity limitations of existing FLIM techniques. We expect straightforward application to other systems, including mammalian brains which feature both larger neurons and action potentials compared to Drosophila (3, 7). Voltage imaging in neuroscience is one example application, but membrane potential is also broadly interesting throughout biology from bacteria (33, 34) and plants (35) to cardiac (36–38) and muscle tissue (39). The ability of EO-FLIM to strongly reject motion noise in vivo is relevant for brain and cardiac imaging applications where it is challenging to faithfully distinguish voltage dynamics from motion and hemodynamic artifacts (15, 16, 36, 37). It may become possible to perform voltage imaging during natural movements such as insect flight, or while tracking a freely moving organism (40). Further, recent advances in GEVI probes have enabled voltage imaging of populations of neurons (3, 5). Lifetime imaging will establish accurate long-term readout of sub-threshold activity across a neural circuit, allowing functional connectivity mapping where spike activity may be correlated with sub-threshold modulation of downstream neurons. Finally, using lifetime detection in combination with optogenetic tools (4, 41), it will be possible to improve techniques for targeted optical activation and control (42) of neuron membrane potential.
Supplementary Material
Funding:
We acknowledge funding from the Gordon and Betty Moore Foundation, the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0021976, NIH grant U01NS120822 (M.J.S. and Ganesh Vasan), and NSF NeuroNex grant DBI-1707261 (M.J.S. and K. Deisseroth). A.B. acknowledges support from the NSF Graduate Research Fellowship under grant 1656518 and the Stanford Graduate Fellowship.
Footnotes
Competing Interests: A.B. and M.K. are inventors on PCT/US2019/062640, US17/153438, and US17/898093.
Data Availability:
All data are available in the manuscript, the supplementary material, or deposited at Dryad (43). Code is available at Zenodo (44).
References and Notes
- 1.Kim TH, Schnitzer MJ, Cell 185, 9–41 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ, Nature Methods 10, 413–420 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Kannan M, et al. , Science 378, eabm8797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian H, et al. , 10.1101/2021.11.22.469481v1 (2021). [DOI] [Google Scholar]
- 5.Piatkevich KD, et al. , Nature 574, 413–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdelfattah AS, et al. , Science 365, 699–704 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Gong Y, et al. , Science 350, 1361–1366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu P, Miller EW, Accounts of Chemical Research 53, 11–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datta R, Heaster TM, Sharick JT, Gillette AA, Skala MC, Journal of Biomedical Optics 25, 071203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinks D, Klein AJ, Cohen AE, Biophysical journal 109, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazzari-Dean JR, Gest AM, Miller EW, eLife 8, e44522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raspe M, et al. , Nature Methods 13, 501–504 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Bowman AJ, Kasevich MA, ACS Nano 15, 16043–16054 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Bowman AJ, Klopfer BB, Juffmann T, Kasevich MA, Nature Communications 10, 4561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall JD, et al. , Cell 167, 1650–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creamer MS, Chen KS, Leifer AM, Pillow JW, PLOS Computational Biology 18, e1010421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinha S, et al. , Proceedings of the National Academy of Sciences of the United States of America 110, 18374–18379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C, et al. , Nature Communications 9, 872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clegg RM, Laboratory Techniques in Biochemistry and Molecular Biology 33, 1–57 (2009). [Google Scholar]
- 20.Huang C, et al. , https://assets.researchsquare.com/files/rs-1915648/v1_covered.pdf?c=1660253275 (2022). [Google Scholar]
- 21.Wilt BA, Fitzgerald JE, Schnitzer MJ, Biophysical Journal 104, 51–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pacheco DA, Thiberge SY, Pnevmatikakis E, Murthy M, Nature Neuroscience 24, 93–104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hehlert P, Zhang W, Göpfert MC, Trends in Neurosciences 44, 323–335 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Yan Z, Jan LY, Jan YN, Proceedings of the National Academy of Sciences of the United States of America 110, 13612–13617 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akemann W, et al. , Journal of Neurophysiology 108, 2323–2337 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Grant DM, et al. , Biophysical Journal 95, L69–L71 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Linden FH, et al. , Nature Communications 12, 7159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma P, et al. , 10.1101/2022.09.28.510014v1 (2022). [DOI] [Google Scholar]
- 29.Sistemich L, Galonska P, Stegemann J, Kruss S, Angewandte Chemie 62, e202300682 (2023). [DOI] [PubMed] [Google Scholar]
- 30.Klarenbeek J, Goedhart J, Van Batenburg A, Groenewald D, Jalink K, PLOS ONE 10, e0122513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Platisa J, Pieribone VA, Current Opinion in Neurobiology 50, 146–153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aso Y, et al. , eLife 3, e04577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prindle A, et al. , Nature 527, 59–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kralj JM, Hochbaum DR, Douglass AD, Cohen AE, Science 333, 345–348 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Serre NB, et al. , Nature Plants 7, 1229–1238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acker CD, Yan P, Loew LM, Progress in biophysics and molecular biology 154, 3–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee P, et al. , Cardiovascular Research 115, 1659–1671 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wikswo JP, Lin S, Abbas RA, Biophysical journal 69, 2195–2210 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hashemi NA, et al. , Proceedings of the National Academy of Sciences of the United States of America 116, 17051–17060 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen JP, et al. , Proceedings of the National Academy of Sciences of the United States of America 113, E1074–E1081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adam Y, et al. , Nature 569, 413–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergs AC, et al. , Nature Communications 14, 1939 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowman AJ, Huang C, Schnitzer MJ, Kasevich MA, 10.5061/dryad.nzs7h44wd (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman AJ, Huang C, Schnitzer MJ, Kasevich MA, 10.5281/zenodo.7706488 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript, the supplementary material, or deposited at Dryad (43). Code is available at Zenodo (44).