

Abstract
Microgels are the building blocks of microporous annealed particle (MAP) scaffolds which serve as a platform for both in vitro cell culture and in vivo tissue repair. In these granular scaffolds, the innate porosity generated by the void space between the interlinked microgels supports cell infiltration and migration. Controlling the void fraction and particle fraction is critical for MAP scaffold design, as porosity is a bioactive cue for cells. Spherical microgels can be generated on a microfluidic device for controlled size and shape then subsequently freeze-dried using methods that prevent fracturing of the polymer network. Upon rehydration, the lyophilized microgels lead to controlled particle fractions in MAP scaffolds. The implementation of these methods for microgel lyophilization has led to reproducible studies showing the effect of particle fraction on macromolecule diffusion and cell spreading. The following protocol will cover the microgel fabrication, lyophilization, and rehydration for controlling particle fraction in MAP scaffolds, as well as annealing the microgels through bio-orthogonal crosslinking for 3D cell culture in vitro.
SUMMARY:
Minimizing the variability in the particle fraction within granular scaffolds facilitates reproducible experimentation. This work describes methods for generating granular scaffolds with controlled particle fractions for in vitro tissue engineering applications.
INTRODUCTION:
Microporous annealed particle (MAP) gels are granular materials comprised of microgels (μgels) interlinked to form a bulk, porous scaffold. With the unique microarchitecture of these granular scaffolds, the innate porosity generated by the void space between interlinked spherical μgels supports accelerated cell infiltration and migration.1 The microgel building blocks of MAP scaffolds can be fabricated from both synthetic and natural polymers with chemical modifications.2 These methods specifically describe the use of μgels comprised of a hyaluronic acid (HA) backbone modified with functional norbornene (NB) handles. The NB functional handle on the HA polymer supports click chemistry reactions for i) forming μgels and ii) linking them together to generate MAP scaffolds.3, 4 Numerous schemes have been employed for linking the μgels together (i.e., annealing), such as enzymatic1, light-based5, 6, and additive-free click chemistry3, 7 reactions. The latter is described in this work, using the tetrazine-norbornene inverse electron demand Diels–Alder conjugation for interlinking the HA-NB μgels.
To fabricate MAP scaffolds, users first generate the μgel building blocks using reverse emulsions either in batch systems or within microfluidic devices, as well as via lithography, electrohydrodynamic spraying, and mechanical fragmentation.2 The production of spherical HA-NB μgels has been well described and previously reported using both batch emulsion2 and microfluidic droplet generation techniques8–11. In this work, spherical HA-NB μgels were generated on a flow-focusing microfluidic platform for controlled size and shape, as previously described.8–10 After purification, the μgels exist in an aqueous suspension and must be concentrated to induce a jammed state. When jammed, μgels exhibit shear-thinning properties which allow them to function as injectable, space-filling materials.1 One method of inducing a jammed state is to dry the μgels via lyophilization, or freeze-drying, then subsequently rehydrate the dried product in a controlled volume.12 Prior to microgel lyophilization, excess buffer was commonly removed from the microgel slurry via centrifugation over a strainer or with manual removal of the buffer from the microgel pellet either with aspiration or an absorbent material. Using centrifugation to dry the μgels generated a highly variable range of particle fraction and void fraction when making MAP scaffolds.12 Techniques for lyophilizing μgels have been described using 70% IPA for polyethylene glycol (PEG) μgels13, fluorinated oils for GelMa μgels14, and 70% ethanol for HA μgels12. This protocol highlights methods for freeze-drying spherical HA μgels using 70% ethanol, a standard laboratory reagent, to retain the original microgel properties during the drying process. The freeze-dried HA μgels can be weighed and rehydrated at user-defined weight percentages to control the final particle fractions in MAP scaffolds.12
The final step in MAP scaffold formation relies on annealing the μgels to create a bulk, porous scaffold.1 By utilizing native extracellular matrix components and employing bio-orthogonal annealing schemes, MAP scaffolds serve as a biocompatible platform for both in vitro cell culture and in vivo tissue repair.3 Through these approaches, MAP scaffolds can be fabricated from HA-NB building blocks with user-defined particle fractions for their employment in tissue engineering applications.12 The following methods will describe microfluidic production of HA-NB μgels followed by lyophilization and rehydration for controlling particle fraction in MAP scaffolds. Lastly, steps for annealing the μgels will be described using bio-orthogonal chemistry for 3D cell culture experiments in vitro.
PROTOCOL:
1. MICROFLUIDIC DEVICE FABRICATION
1.1. Soft lithography
NOTE: This protocol describes device fabrication of a flow-focusing microfluidic device design from de Wilson et al.9. However, this protocol can be used with any device design on an SU-8 wafer. The wafer can be taped to a Petri dish then needs to be silanized to prevent adherence of the PDMS to the wafer features.15
-
1.1.1.
Mix polydimethylsiloxane (PDMS) elastomer base to the curing agent at a 10:1 ratio. Pour the PDMS mixture onto the wafer and degas in a desiccator for approximately 30 minutes. Once all bubbles are gone, place in an oven at 60 °C for at least 2 hours to cure the PDMS.
-
1.1.2.
Use a knife to gently trace around the parameter of the device without cracking the wafer then carefully peel the PDMS off the wafer. Use a 1 mm biopsy punch to create the inlet and outlet channels.
NOTE: Be gentle when punching the microfluidic device. Tears or rips around the inlet or outlet channels can cause leaks during microgel production.
-
1.1.3.
Use tape to remove dust from the device on the feature side. Place the devices and clean glass slides on a hot plate at 135 °C for at least 15 minutes to remove moisture.
-
1.1.4.
In a fume hood, use a corona plasma gun on high on both the glass slides and devices (feature side exposed) for approximately 30 seconds, then quickly bond together. Gently apply pressure to ensure a good seal between the device and glass slide. Place the devices in a 60°C oven overnight to secure the bond.
2. MICROFLUIDIC PRODUCTION OF HYALURONIC ACID (HA) MICROGELS WITH NORBORNENE (NB) FUNCTIONAL HANDLES
2.1. HA-NB synthesis
NOTE: HA-norbornene (HA-NB) synthesis was adapted from Darling, et al.3 using 79 kDa sodium HA with molar equivalents of 1:1.5:2.5 of HA-repeat units to 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) to 5-norbornene-2-methylamine (NMA).
-
2.1.1.Weigh the reactants. Dissolve the HA at 20 mg/mL in 200 mM MES buffer (pH ~6) by stirring in a beaker or flask on a stir plate. Once dissolved, add the DMTMM to the HA solution and allow to react for approximately 20 min at room temperature.
-
2.1.2.
Add NMA dropwise to the HA/DMTMM solution. Add parafilm to the opening of the reaction vessel to minimize evaporation and cover the reaction vessel with foil. Continue stirring while allowing the reaction to proceed for approximately 24 hr.
-
2.1.4.
After 24 hours, chill 200 proof ethanol (approximately 10x the reaction volume). On a stir plate, transfer the reaction dropwise to the chilled ethanol to precipitate the HA-NB and continue stirring for 20 min.
-
2.1.5.
Transfer the solution to 50 mL conical tubes then centrifuge at 5,000 x g for 10 min. Pour off the excess ethanol to dispose as waste. At this point, the HA-NB product should be white pellets in the conical tubes. Pull vacuum on the HA-NB in a dessicator to dry overnight.
-
2.1.6.
Purify the HA-NB using 12–14 kDa molecular weight cut-off dialysis tubing. Dissolve HA-NB in 2 M NaCl solution and transfer to dialysis tubing. Tie the tubing and secure with clamps if needed. Transfer the filled dialysis tubing to a bucket with 5 L of Milli-Q water. Dialyze the HA-NB against water overnight.
-
2.1.7.
The next day, remove the water and replace with 1 M NaCl solution for 30 min. Remove the NaCl solution then dialyze against Milli-Q water for 3 days, replacing the water daily.
NOTE: Liquid nitrogen is a hazardous substance. Wear the appropriate personal protective equipment when working with liquid nitrogen.
-
2.1.8.
Filter the dialyzed product using 0.2 μm vacuum-driven filter then transfer the filtered product to 50 mL conical tubes. Add liquid nitrogen to a cryogenic container then add the conical tubes of HA-NB to the liquid nitrogen to flash-freeze. After approximately 10 min, remove the conical tubes with forceps. Quickly remove the cap and cover with a lab-grade tissue. Secure the tissue with a rubber band and transfer to a lyophilization container or chamber and lyophilize. Stored the lyophilized product at −20 °C.
-
2.1.9.
Quantify norbornene modification by dissolving the HA-NB at 10 mg/mL in D2O and analyze via proton NMR (Figure 1A).16 To determine the amount of functionalization, first calibrate the D2O solvent peak to 4.8 PPM. Integrate the peak for the HA methyl protons (δ2.05) and calibrate the integration to 3.0. Next, integrate the peaks for the pendant norbornene groups at δ6.33 and δ6.02 (vinyl protons, endo). Normalize the integration of these peaks to the corresponding number of protons to determine the average degree of modification.3
Figure 1.
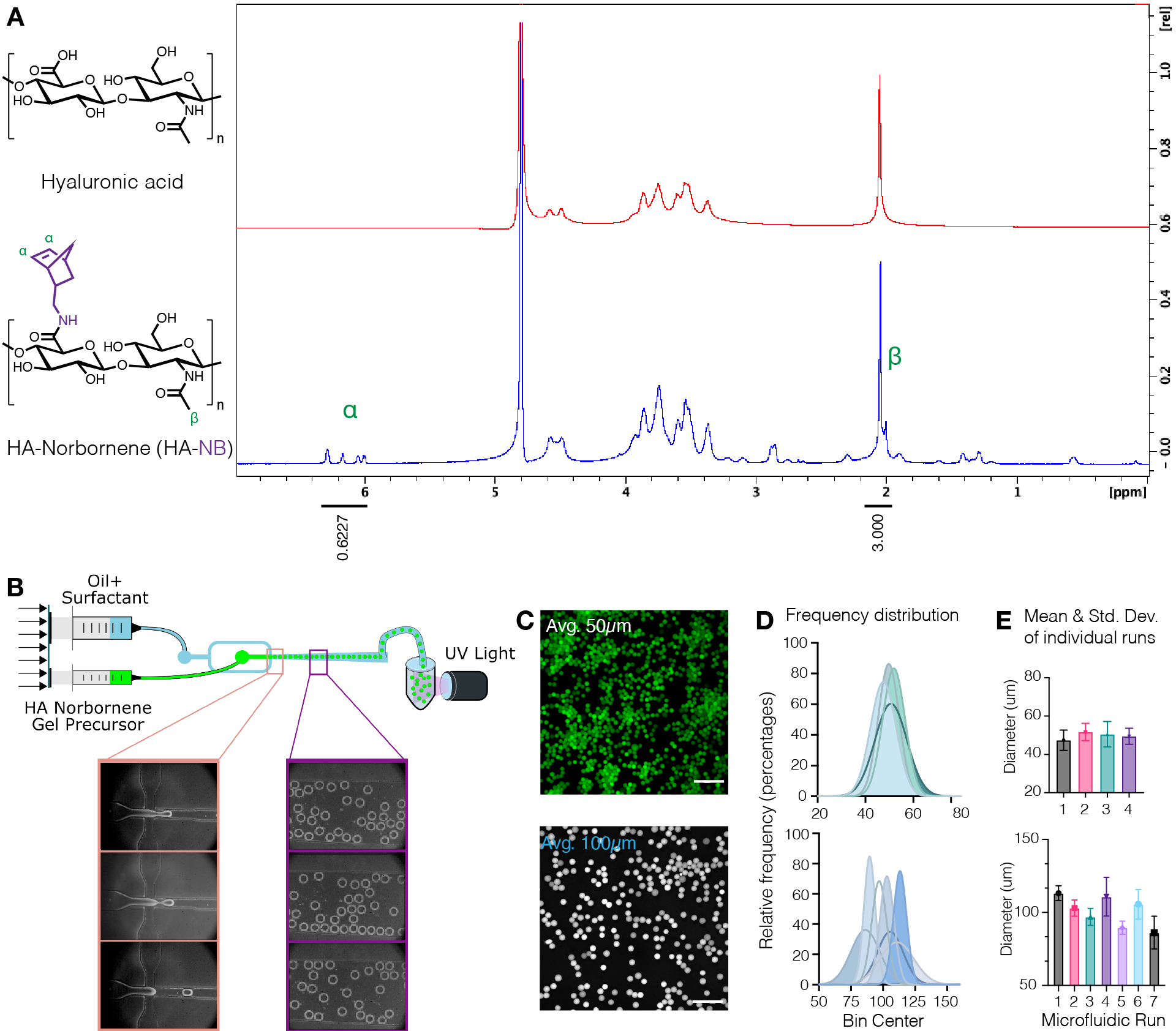
A) Approximately 31% of HA repeat units were successfully modified with NB, as determined by proton NMR analysis performed in deuterium oxide. 1H NMR shifts of pendant norbornenes at δ6.33 and δ6.02 (vinyl protons, endo), and δ6.26 and δ6.23 ppm (vinyl protons, exo) were compared to the HA methyl group δ2.05 ppm to determine functionalization. Reprinted from Acta Biomaterialia, Volume 150, Alexa R. Anderson, Ethan Nicklow, Tatiana Segura, Particle fraction is a bioactive cue in granular scaffolds, Copyright 2022, with permission from Elsevier.12 B) Schematic of the flow-focusing microfluidic device used to generate HA-NB μgels. C) Maximum intensity projections from confocal microscopy were used to visualize fluorescently labelled μgels (scale bar = 500 μm). D) Frequency distributions of microgel diameter from independent runs on the microfluidic setup demonstrate control over microgel size ~50 μm or ~100 μm depending on the device used. E) Microgel diameter is reported as the mean and standard deviation for each independent run. Reprinted from Advanced Materials, Volume 34, Issue 33, Wilson, et al., Stoichiometric post-modification of hydrogel microparticles dictates neural stem cell fate in microporous annealed particle scaffolds, Copyright 2022, with permission from Wiley.9
2.2. HA-NB Microgel Precursor
-
2.2.1.
Prepare 50 mM HEPES buffer (pH 7.5). Sterile filter the buffer using a 0.2 μm vacuum-driven filter. Using HEPES buffer, prepare respective 50 mM stocks of lithium phenyl(2,4,6,-trimethylbenzoyl)phosphinate (LAP) photo-initiator and tris(2-carboxyethyl)phosphine (TCEP) reducing agent. Keep the LAP solution away from light.
-
2.2.2.
Prepare the other microgel precursor components by preparing respective 50 mM stocks of di-thiol linker and RGD peptide in sterile DiH2O. Weigh out HA-NB and dissolve in HEPES buffer to prepare a 10 mg/mL stock.
-
2.2.3.
Combine the precursor components with final concentrations of 9.9 mM LAP, 0.9375 mM TCEP (4 thiol/TCEP), 3.75 mM di-thiol linker, 1 mM RGD, and 3.5 wt% (w/v) HA-NB by adding extra HEPES buffer to reach the desired final volume. Mix the precursor well with a positive displacement pipette.
-
2.2.4.
Use a P1000 slowly pull up the entire mixture. Put the tip onto the end of a 1 mL syringe and eject the tip from the pipet. Pull the syringe plunger to load the mixture into the syringe. Add a 0.2 μm filter on the end of syringe and filter into a new Eppendorf tube. Centrifuge the filtered precursor solution to remove the bubbles produced during filtering.
-
2.2.5.
Again, use a P1000 slowly pull up the filtered precursor being careful not to create bubbles. If there are bubbles, gently tap the tip for them to dislodge and float to the top. Place the tip onto the end of a 1mL syringe and eject the tip from the pipet. Keep the syringe vertical and pull the syringe plunger slowly until the entire precursor solution is in the syringe. Add a blunt tip needle to the syringe and push the precursor through the tip of the needle. Wrap the syringe in foil to keep out of light.
2.3. Microgel Pinching Solution
-
2.3.1.
Prepare 5% v/v Span-80 in heavy white mineral oil and mix well. Desiccate to remove bubbles. Keep the surfactant/oil at room temperature wrapped in foil. Mix well and desiccate prior to each use.
-
2.3.2.
Use a 5 mL syringe, to draw up oil/surfactant mixture (minimize bubbles) until the distance between plunger and fingerhold is approximately equal to the distance of the precursor syringe. Add a blunt needle to the syringe and push the oil through the tip of the needle.
2.4. Microfluidic device setup
-
2.4.1.
Add a blunt needle to a 1 mL syringe and fill with synthetic hydrophobic treatment solution. Gently flow the solution through the microfluidic device until it pools at each inlet/outlet. Let solution dry in the device on the benchtop for approximately 30 minutes then pull vacuum on the outlet to remove excess solution. Secure the device with clamps on a tabletop microscope.
-
2.4.2.
Wrap a 15 mL conical tube with foil and place in a tube rack to serve as the microgel collection container. Use a ring stand with a clamp to place the UV light probe into the opening of the collection tube. Use a UV detector to measure the UV intensity, moving the probe until 20 mW/cm2 is achieved. Turn off the UV light until later.
-
2.4.3.
Cut tubing at a length that will reach from the microfluidic device to the collection container. On one end of the tubing, cut a 45° angle. Gently insert the angled end of the tubing into the outlet channel.
NOTE: Be gentle when inserting the tubing into the microfluidic device. Tears or rips around the inlet or outlet channels can cause leaks during microgel production.
-
2.4.4.
Secure both the precursor and oil phase syringes on dual-syringe pump. Cut 2 more pieces of tubing at a length that will reach from syringe tips to the microfluidic device. On one end of each tube, cut a 45° angle. Carefully secure the tubing (blunt end) on both syringe tips.
-
2.4.5.
Change the settings on the pump for the 1 mL syringe and include the approximate precursor volume. Slowly push the pump forward until enough pressure is applied to the syringe plungers to push both the oil and the precursor to the ends of the tubing, removing any air from the system. Let the pressure equalize 5–10 minutes prior to moving on to step 2.3.6.
-
2.4.6.
Gently insert the angled end of the tubing into the inlet channels of microfluidic device with the microgel precursor in the front inlet and the pinching oil in the back inlet. Move the pump forward in small increments until flow begins in the device and μgels begin to form at the flow-focusing region. Start the pump with a 0.4 μL/min flowrate and let the device run until it stabilizes. If needed, adjust the flow rate ± 0.1 μL/min in small increments to stabilize microgel production.
-
2.4.7.
Once microgel production stabilizes as shown in Figure 1B, replace your collection tube with a new tube, and turn on the UV light. Check the run periodically to ensure microgel production is stable over the duration of the run.
3. PURIFYING AND DRYING MICROGELS
3.1. Purification of μgels
-
3.1.1.
Prepare the microgel washing buffer (300 mM HEPES, 50 mM NaCl, 50 mM CaCl2) as well as the 2% (w/v) Pluronic F-127 surfactant solution in washing buffer. Filter the solutions using a 0.2 μm vacuum-driven filter for sterility.
-
3.1.2.
Centrifuge the microgel collection tube (5,000 x g) for 5 min. In a sterile hood, carefully aspirate the supernatant oil phase. Combine the μgels 1:1 with 2% Pluronic F-127 surfactant solution and vortex to mix well. Centrifuge (5,000 x g) for 5 min and aspirate the supernatant washing solution.
-
3.1.3.
Add washing buffer at 4x microgel volume and vortex to mix well. Centrifuge (5,000 x g) the mixture for 5 min and aspirate the washing solution. Complete 4–8 washes with washing buffer until the surfactant is removed from the system (i.e., no bubbles remain).
3.2. Fluorescent labelling of HA-NB μgels
-
3.2.1.
The in-house synthesis of a fluorescently labeled tetrazine relies on two base-catalyzed thiol-Michael addition reactions in series that has been well described and previously reported.3 For this work, an Alexa Fluor-488 was conjugated with tetrazine for the labeling of norbornene-modified μgels. The lyophilized product was dissolved in dimethylformamide at 1 mg/mL and stored at −20 °C.
-
3.2.2.
To fluorescently label the μgels, first prepare a working solution of Alexa Fluor 488-Tet by diluting the 1 mg/mL stock 1:14 in sterile 1x PBS. In a sterile hood, combine the μgels with the working solution of Alexa Fluor 488-Tet (2:1 by volume).
-
3.2.3.
Use a displacement pipette and mix well. Incubate the mixture for 1 hour at room temperature or overnight at 4 °C.
-
3.2.4.
Centrifuge (5,000 x g) and aspirate the staining solution. Wash the μgels twice with 1x PBS (1:1 by volume) to remove unreacted Alexa Fluor 488-Tet.
OPTIONAL: At this point, the fluorescently labeled μgels can be imaged on a confocal microscope to quantify the microgel size (Figure 1C–E).9 Methods for measuring microgel size have been thoroughly described by Roosa, et al.17
3.3. Drying HA-NB μgels
CAUTION: Ethanol is a highly flammable substance.
-
3.3.1.
Transfer purified μgels (Figure 2A) to a cryo-safe screw-cap tube using a positive displacement pipette. Add 70% ethanol to the purified μgels 50% (v/v) and mix well with a displacement pipette. Centrifuge for 5 minutes at 5,000 x g.
NOTE: The cryo-safe screw-cap tube can be weighed prior to adding μgels then weighed again after lyophilization to determine the mass of μgels. This is recommended to minimize error when using quantities less than 1 mg. Ensure that the scale is internally adjusted or calibrated prior to use.
-
3.3.2.
Aspirate the supernatant liquid and replace with 70% ethanol (50% v/v) (Figure 2B). Mix well with a displacement pipette. Incubate overnight 4 °C.
NOTE: Microgels can be stored in 70% ethanol at 4 °C prior to lyophilization for long-term storage if needed. Other lyophilization mediums can be used in this step if cryogel formation is desired (Figure 2D).
NOTE: Liquid nitrogen is a hazardous substance. Wear the appropriate personal protective equipment when working with liquid nitrogen.
-
3.3.3.
Briefly centrifuge to ensure the μgels are at the bottom of the screw-cap tube. Add liquid nitrogen to a cryogenic container then add the tube of μgels to flash-freeze.
-
3.3.4.
After 5–10 min, remove the tube of μgels with forceps. Quickly remove the cap and cover with a lab-grade tissue. Secure the tissue with a rubber band and transfer to a lyophilization container or chamber.
-
3.3.4.
Follow the manufacturer instructions for loading your sample on the lyophilizer. Lyophilize at 0.066 Torr and −63 °C. Store the lyophilized μgels (lyo-μgels) tightly sealed at room temperature.
NOTE: Lyophilization is complete when all liquid is removed from the tube and a dried product remains.
NOTE: Organic solvents can decrease the longevity of the rubber fixtures on common lyophilization systems.
Figure 2.
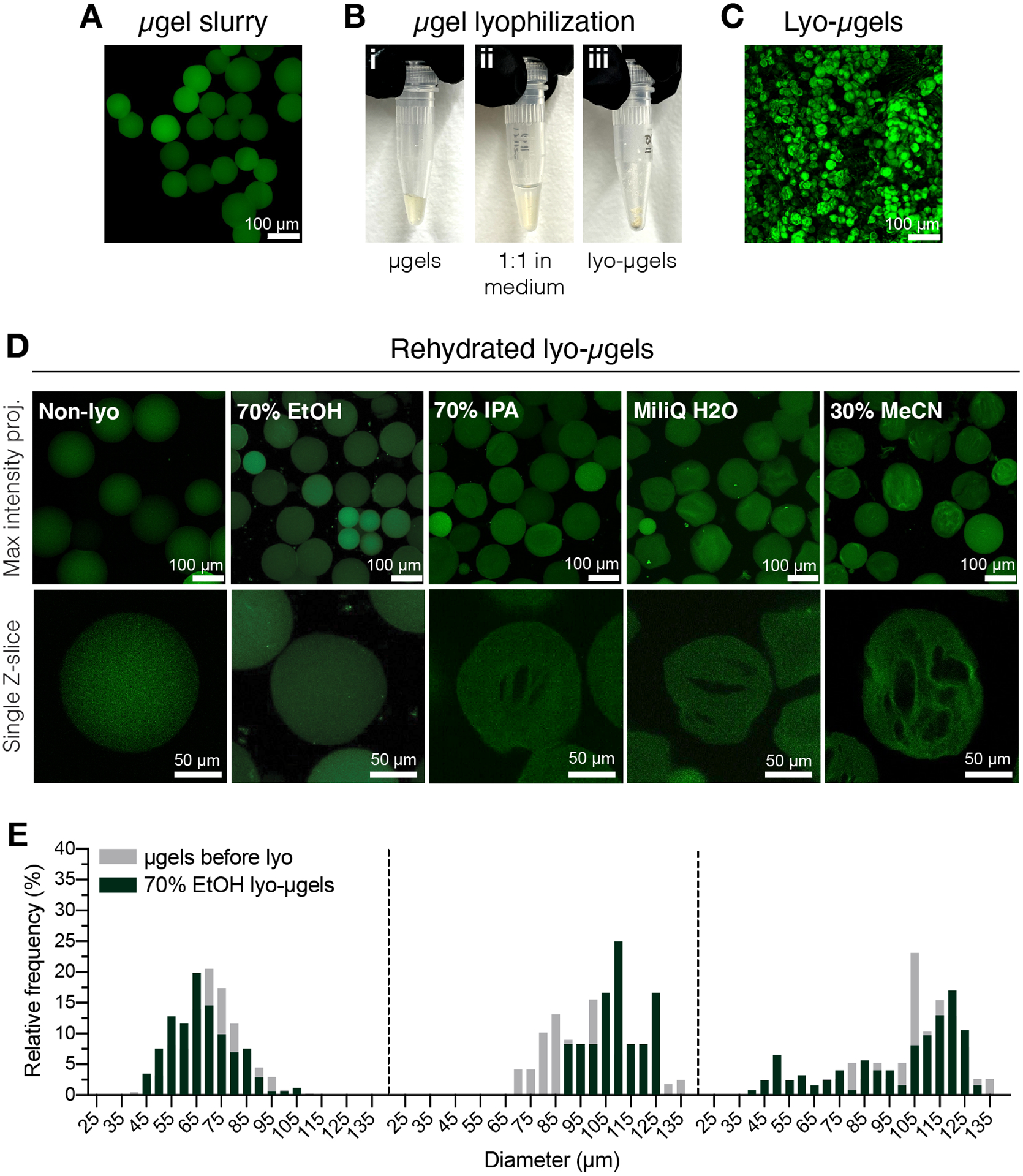
A) Maximum intensity projection of μgels in aqueous solution (scale bar = 100 μm). B) Purified μgels can be incubated 1:1 by volume in the lyophilization medium of choice and lyophilized. C) Maximum intensity projection of dried lyo-μgels (scale bar = 100 μm). D) Microgels are resuspended after lyophilization. 70% EtOH is recommended for retaining the μgels original properties throughout the lyophilization process; however, other mediums such as isopropyl alcohol (IPA), water, and acetonitrile (MeCN) can be used interchangeably to facilitate cryogel formation (scale bar = 100 or 50 μm as noted). E) Measurement of HA-NB microgel diameter before (gray) and after lyophilization (green) in 70% EtOH shown as frequency distributions for three microgel populations. Reprinted from Acta Biomaterialia, Volume 150, Alexa R. Anderson, Ethan Nicklow, Tatiana Segura, Particle fraction is a bioactive cue in granular scaffolds, Copyright 2022, with permission from Elsevier.12
4. MAP SCAFFOLD FABRICATION
4.1. Tetrazine linker synthesis
NOTE: Tetrazine linkers can be used to interlink μgels bearing free norbornene groups (Figure 3A). HA-tetrazine (HA-Tet) synthesis was adapted from Zhang, et al.18 using 79 kDa sodium HA with molar equivalents of 1:1:0.25 of HA-repeat units to DMTMM to tetrazine-amine (Figure 3B).12
Figure 3.
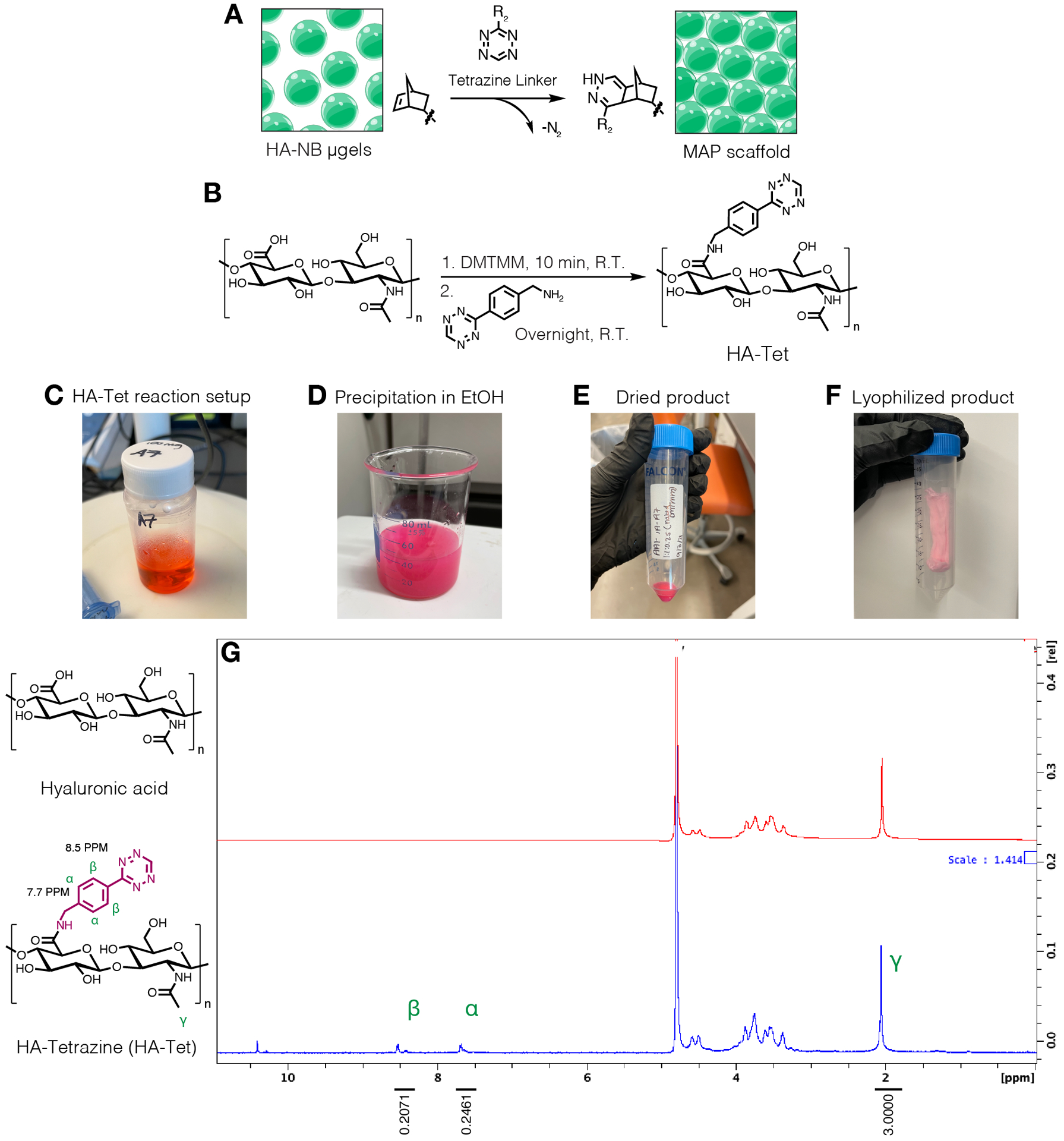
A) Schematic of HA-NB μgels being interlinked with a tetrazine linker to form MAP scaffolds. B) Reaction scheme for HA-Tet synthesis. C) The HA-Tet reaction was setup and allowed to react overnight followed by D) precipitation of HA-Tet in ethanol. E) Once purified and dried, the HA-Tet was rehydrated and lyophilized to yield F) a dried, light pink product. G) Proton NMR analysis shows successful modification of 11% of HA repeat units. Reprinted from Acta Biomaterialia, Volume 150, Alexa R. Anderson, Ethan Nicklow, Tatiana Segura, Particle fraction is a bioactive cue in granular scaffolds, Copyright 2022, with permission from Elsevier.12
-
4.1.1.Weigh the reactants. Dissolve the HA at 20 mg/mL in 200 mM MES buffer (pH ~6) by stirring in a beaker or flask on a stir plate. Once dissolved, add the DMTMM to the HA solution and allow to react for approximately 20 min at room temperature.
-
4.1.2.
Dissolve the tetrazine-amine at 15 mg/mL in 200 mM MES buffer and add dropwise to the HA/DMTMM solution. Refer to Figure 3C for a photo of the HA-Tet reaction setup.
-
4.1.3.
Add parafilm to the opening of the reaction vessel to minimize evaporation and cover the reaction vessel with foil. Continue stirring while allowing the reaction to proceed for approximately 24 hr.
-
4.1.4.
After 24 hours, chill 200 proof ethanol (approximately 10x the reaction volume). On a stir plate, transfer the reaction dropwise to the chilled ethanol to precipitate the HA-Tet (Figure 3D) and continue stirring for 20 min.
-
4.1.5.
Transfer the solution to 50 mL conical tubes then centrifuge at 5,000 x g for 10 min. Pour off the excess ethanol to dispose as waste. Pull vacuum on the HA-Tet in a dessicator to dry overnight. An example of the dried product at this step in the protocol can be found in Figure 3E.
-
4.1.6.
Purify the HA-Tet using dialysis. Dissolve HA-Tet in 2 M NaCl solution and transfer to cellulose dialysis tubing with a 12–14 kDa molecular weight cut-off. Transfer the filled dialysis tubing to a bucket with 5 L of Milli-Q water. Dialyze the HA-Tet against water overnight.
-
4.1.7.
The next day, remove the water and replace with 1 M NaCl solution for 30 min. Remove the NaCl solution then dialyze against Milli-Q water for 3 days, replacing the water daily.
NOTE: Liquid nitrogen is a hazardous substance. Wear the appropriate personal protective equipment when working with liquid nitrogen.
-
4.1.8.
Filter the dialyzed product using 0.2 μm vacuum-driven filter then transfer the filtered product to 50 mL conical tubes. Add liquid nitrogen to a cryogenic container then add the conical tubes of HA-Tet to the liquid nitrogen to flash-freeze. After approximately 10 min, remove the conical tubes with forceps. Quickly remove the cap and cover with a lab-grade tissue. Secure the tissue with a rubber band and transfer to a lyophilization container or chamber and lyophilize. Stored the lyophilized product (Figure 3F) at −20 °C.
-
4.1.9.
Quantify tetrazine modification by dissolving the HA-Tet at 10 mg/mL in D2O and analyze via proton NMR (Figure 3G).16 To determine the amount of functionalization, first calibrate the D2O solvent peak to 4.8 PPM. Integrate the peak for the HA methyl protons (δ2.05) and calibrate the integration to 3.0. Next, integrate the peaks for the pendant tetrazine groups at δ8.5 (2H) and δ7.7 (2H) (aromatic protons). Normalize the integration of these peaks to the corresponding number of protons to determine the average degree of modification.12
4.2. Interlinking lyo-μgels to form MAP scaffolds for characterization
-
4.2.1.
Prepare the MAP scaffold components (i.e., μgels, HA-Tet, rehydration volume) based on the desired particle fraction (refer to Figure 4D–E). Weigh the lyo-μgels and reconstitute in 84% of the final MAP volume of 1x PBS based on the chosen wt % MAP.
-
4.2.2.
Dissolve the HA-Tet in 1x PBS at the chosen concentration (See NOTE below).
NOTE: Changing both the packing fraction (via wt % MAP) as well as the concentration of HA-Tet will alter bulk scaffold mechanical properties. For example, a 3.4 wt % MAP scaffold crosslinked with 0.02 mg/mL HA-Tet (annealing ratio of 2.6 mol Tet:mol HA-NB) generates MAP scaffolds with approximately 700 Pa shear storage modulus.12
-
4.2.3.
Use a displacement pipette to combine the HA-Tet and lyo-μgels and mix well. At this point, the mixture can be transferred via displacement pipette onto glass slides, well plates, or a container of the user’s choosing. Allow μgels to anneal at 37 °C for 25 minutes then use a spatula to transfer the MAP scaffolds to well plates filled with 1x PBS. Keep MAP scaffolds in 1x PBS until ready for characterization.
Figure 4.
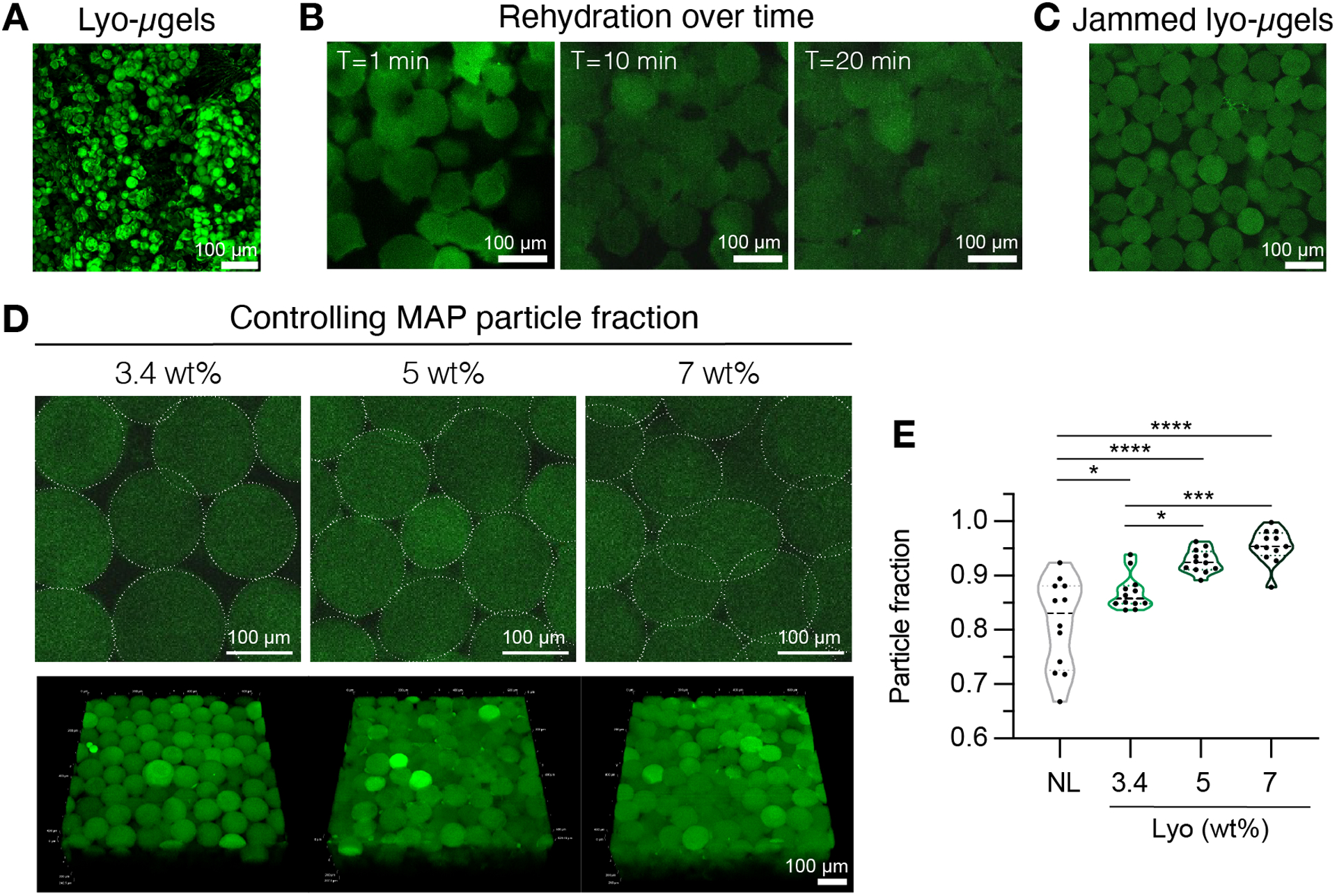
A) Maximum intensity projection of dried lyo-μgels (scale bar = 100 μm). B) After freeze-drying, rehydration of lyo-μgels is shown to take approximately 20 min (scale bar = 100 μm). C) Lyo-μgels can be rehydrated at varying wt % MAP to produce jammed μgels (scale bar = 100 μm). D) Increasing the wt % MAP when rehydrating lyo-μgels alters the particle fraction in MAP scaffolds, as shown by single Z-slices of MAP scaffolds and volume projections (scale bar = 100 μm). E) Using these user-defined wt % MAP scaffolds, unique particle fractions can be achieved (NL = non-lyophilized μgels). Reprinted from Acta Biomaterialia, Volume 150, Alexa R. Anderson, Ethan Nicklow, Tatiana Segura, Particle fraction is a bioactive cue in granular scaffolds, Copyright 2022, with permission from Elsevier.12
4.3. Calculating MAP scaffold particle fraction
-
4.3.1.
For improved image quality, transfer MAP scaffold to a glass coverslip using a spatula. Image MAP scaffolds on a confocal microscope using the laser for FITC excitation and emission. Image MAP scaffolds on a 20X objective and obtain a Z-stack traversing 250–300 μm in the Z-direction with a step size of 2.5 μm. Make note of the μm/pixel calibration of the image.
-
4.3.2.
Import the Z-stack image into IMARIS software.
Select the “Add new Surfaces” button. Check the box to “Segment only a Region of Interest” then select the blue arrow button “Next: Region of Interest”.
-
4.3.3.
Define a region of interest, keeping track of the X-, Y-, and Z-dimensions of the volume being analyzed. Select the blue arrow button “Next: Source Channel”.
NOTE: A recommended Z-height for the region of interest should include a minimum of 2 μgels.
NOTE: X- and Y-dimensions are in units of pixels while the Z-dimension is number of steps.
-
4.3.4.
Use the Source Channel drop-down list to select the FITC channel. Check the box next to “Smooth” and input a surface detail of 2.50 μm. Under Thresholding, select “Absolute Intensity” then select the blue arrow button “Next: Threshold”.
-
4.3.5.
Use the suggested thresholding value for the FITC channel. Rotate the 3D projection to assess the rendering quality and adjust as needed. Select “Next: Classify Surfaces”.
NOTE: The “Back” button can be used to edit previous steps in the process, such as Z-dimension, as needed.
-
4.3.6.
Check that “Number of Voxels” is 10.0 then select the green double arrow button “Finish: Execute all creation steps and terminate the wizard”.
NOTE: Volume rendering parameters can be store for batch analysis so that the same settings are applied to analyze all scaffolds.
-
4.3.7.
To export the data, select the “Statistics” tab then the “Detailed” tab. Use the second drop-down box to select the variable “Volume”. Select the floppy disk button “Export Statistics on Tab Display to File” and save as a Microsoft Excel file (.xls) when prompted.
-
4.3.8.
Open the file and use the SUM function on Column A “Volume” to determine the total volume (μm3) of the μgels in the region of interest.
-
4.3.9.
Convert the dimensions of the region of interest that was analyzed from pixels to μm. Use the μm/pixel calibration of the image from step 4.3.1. to convert the X- and Y-dimensions. Multiply the Z-dimension (number of steps) by the step size for the image to convert the Z-dimension to μm. Calculate the volume of the region of interest (μm3) by multiplying the X-, Y-, and Z-dimensions.
-
4.3.10.
To determine the particle fraction of the scaffold, divide the total volume of the μgels in the region of interest (found in step 4.3.8.) by the volume of the region of interest (found in step 4.3.9.).
4. 3D CELL CULTURE IN MAP SCAFFOLDS
5.1. Prepare cell culture devices
-
5.1.1.
To create a custom cell culture device for these experiments (Figure 3A–C), use a 3D printer to print a negative mold using the CAD file found in Supplementary File 1.
NOTE: The dimensions of the cell culture device are as follows: 94.9 mm x 94.9 mm x 4.8 mm with the total well height being 2.6 mm. The diameter of the inner wells and outer wells is 4mm and 6 mm, respectively.
-
5.1.2.
Mix polydimethylsiloxane (PDMS) elastomer base to the curing agent at a 10:1 ratio by mass. Pour the PDMS mixture into a large plastic petri-dish and degas in a desiccator for approximately 30 minutes or until all bubbles have disappeared.
-
5.1.3.
Once all bubbles have disappeared, carefully place the 3D printed mold into the PDMS to minimize the formation of new bubbles. Place in the oven at 60 °C for at least 2 hours to cure the PDMS.
-
5.1.4.
Use a knife or razor blade to gently trace around the parameter of the culture device then carefully remove the mold. Use a 4 mm biopsy punch to remove any PDMS from the bottom of the wells. Cut the devices to size to fit on a glass coverslip.
NOTE: Cell culture devices can also be bonded to glass slides, but glass coverslips improve sample imaging.
-
5.1.5.
Use tape to remove dust from the bottom side of the culture devices. Place clean glass coverslips and culture devices (bottom side up) on a hot plate at 135 °C for at least 15 minutes to remove moisture.
-
5.1.6.
In a fume hood, use a corona plasma gun on high on both the glass coverslip and the bottom side of the device for 30 seconds, then quickly bond the treated surfaces together. Gently apply pressure to ensure a good seal between the culture device and glass coverslip.
-
5.1.7.
Repeat step 3.1.6. for all devices then place in a 60 °C oven overnight to secure the bond. Autoclave the devices to sterilize before use in vitro.
3.2. Cell Culture in MAP scaffolds
-
3.2.1.
Prepare the MAP scaffold components (i.e., μgels, HA-Tet, media volume) based on the desired particle fraction (refer to Figure 2E–G). Weigh lyo-μgels in a sterile hood and reconstitute in 84% of the final MAP volume of cell media based on the chosen wt % MAP.
NOTE: These methods require the user to weigh the lyo-microgel product for rehydration. For small masses (1 mg or less), it is suggested to first weigh the cryotube before adding and lyophilizing μgels, then reweigh the tube after lyophilization to determine the mass of the product to minimize error.
-
3.2.2.
Dissolve the HA-Tet in cell media in a final MAP volume of 16%.
NOTE: The following steps for preparing cells for seeding in MAP scaffolds can be altered depending on the cell type being used. This protocol describes the steps for seeding D1 mouse mesenchymal cells.
-
3.2.3.
Once D1 mouse mesenchymal cells have reach 70–80% confluency, aspirate the media and wash cells with 1x PBS. Lift the cells by adding enough volume of 1% trypsin-EDTA to cover to surface of the tissue culture vessel. Incubate at 37 °C for 1–3 minutes then quench the trypsinization by adding DMEM media supplemented with 1% pen/strep and 10% fetal bovine serum at 2x the volume of trypsin-EDTA.
-
3.2.4.
Centrifuge the cell suspension at 100 x g for 5 min at room temperature to pellet the cells. Aspirate the supernatant media and resuspend the cells in 1 mL DMEM media. Ensure the cell suspension is well mixed, then transfer 20 μL to a new microcentrifuge tube. Add 20 μL trypan blue solution and mix well. Use 20 μL of this mixture to count the cells using either a hemocytometer or automated cell counter.
-
3.2.5.
Transfer the number of cells needed for seeding 10,000 cells/μL MAP to a new microcentrifuge tube. Centrifuge at 100 x g for 5 min at room temperature to pellet the cells. Carefully aspirate the supernatant media from the cell pellet without aspirating the cells.
-
3.2.6.
Add the μgels and crosslinker to the cell pellet with a displacement pipette. Mix well with a displacement pipette then seed 10 μL of the mixture per well. When plating, pipette in a circular motion to evenly distribute the mixture in the well.
-
3.2.7.
Allow μgels to anneal at 37 °C in the cell incubator for 25 minutes before adding cell media to fill the wells (~50 μL of media per well). Maintain the 3D cultures at 37 °C and change media as needed. To avoid aspirating the scaffold when changing media, stabilize the pipette tip along the ridge of the upper well.
NOTE: When adding or removing liquid from the culture wells, rest the end of the pipet tip on the ledge above the MAP scaffold to minimize the chance of disrupting or aspirating the scaffold from the well.
-
3.2.8.
At the desired time points, fix samples by removing the media and adding 50 μL of 4% paraformaldehyde per well for 30 minutes at room temperature. Wash the samples three times with 50 μL 1x PBS or preferred buffer. At this point in the protocol, standard methods for immunofluorescence or fluorescence staining can be followed, using 50 μL per well as the working volume.
-
3.2.9.
Image cells in MAP scaffolds on a confocal microscope using a 20X objective and obtain a Z-stack traversing 200–250 μm in the Z-direction with a step size of 2.5 μm. An example of fluorescence staining with DAPI (nuclear stain diluted 1:1000 in the 0.15% triton-x in 1x PBS) and phalloidin-647 (F-actin stain diluted 1:40 in the 0.15% triton-x in 1x PBS) is shown in Figure 5 with confocal micrographs.
NOTE: These methods for fixation and cell staining specifically describe the use of fluorescent stains, but immunostaining with primary and/or secondary antibody conjugations can be performed in these scaffolds as well following manufacturer instructions using 50 μL as the working volume per well. The representative images in Figure 5 were fixed on day 3 of D1 cell culture in MAP scaffolds.
NOTE: Plasma treatment of glass surfaces results in increased hydrophilicity which has been shown to enhance cell adhesion. Cells will likely be observed spreading along the bottom of the cell culture wells but should not be included in cell counts or cell volume quantification for assessing cell response in MAP scaffolds.
Figure 5.
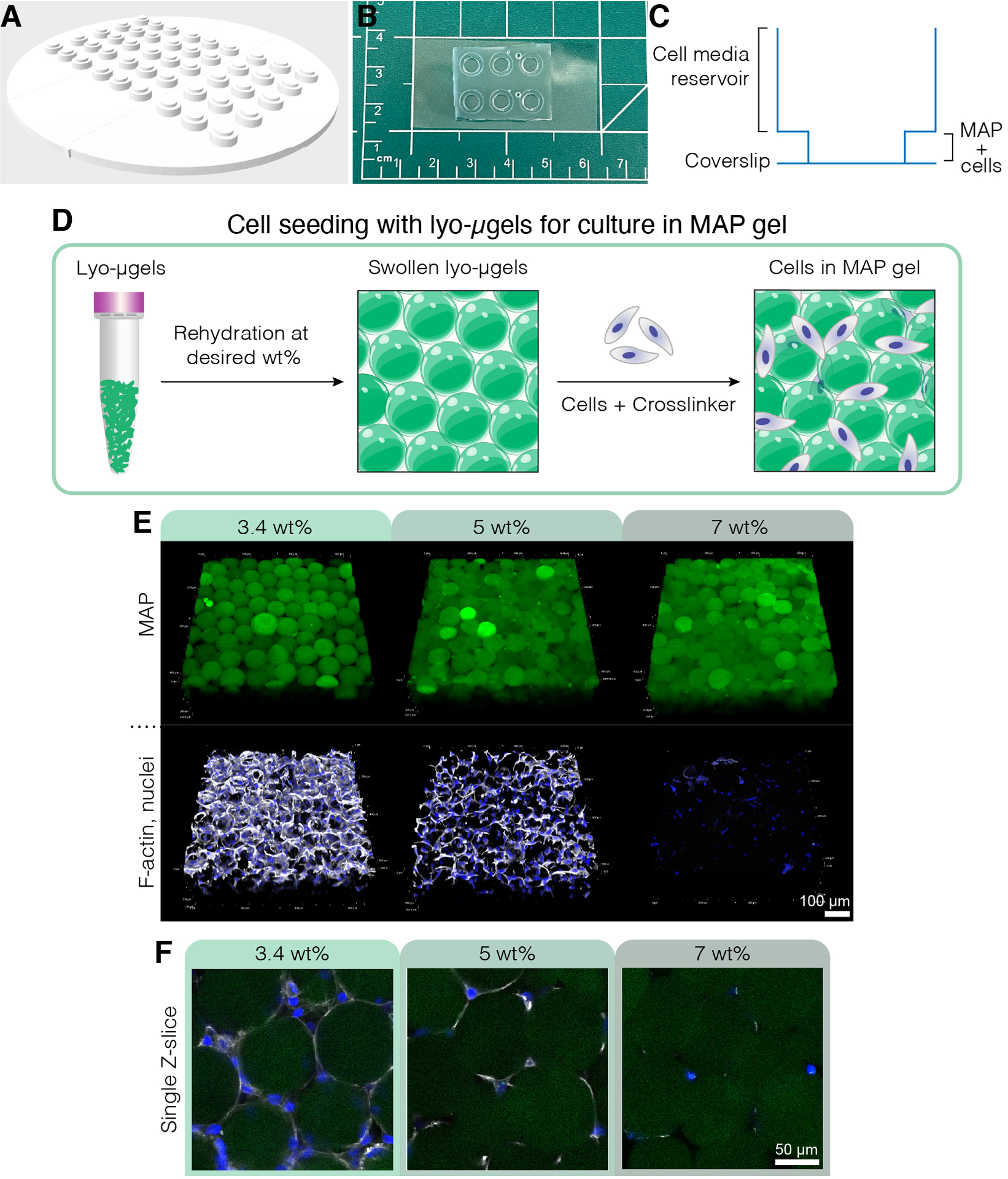
A) The mold for creating cell culture wells can be 3D printed and cast with PDMS. The entire mold is 95 mm in diameter, the large wells are 6 mm in diameter, and the small inner wells are 4 mm in diameter. B) Once cast with PDMS, the cell culture devices are plasma bonded to coverslips for improved microscopy capabilities. C) The cross section of a cell culture well depicts the reservoir for cell media (~50 μL) and a smaller reservoir for seeding MAP scaffold with cells (~10 μL). D) The process of seeding cells in MAP scaffolds first relies on the rehydration of lyo-μgels at the user’s desired wt %, followed by mixing with cells and the crosslinker for interlinking the μgels. E) Cells can be encapsulated in MAP scaffolds (green) with varied wt % MAP. Representative images are from day 5 of D1 cell culture in MAP scaffolds (scale bar = 100 μm). F) Single Z-slices show differences in cell growth in scaffolds comprised of different wt % MAP (scale bar = 50 μm). Reprinted from Acta Biomaterialia, Volume 150, Alexa R. Anderson, Ethan Nicklow, Tatiana Segura, Particle fraction is a bioactive cue in granular scaffolds, Copyright 2022, with permission from Elsevier.12
REPRESENTATIVE RESULTS:
The aim of these methods was to demonstrate the preparation of microporous annealed particle (MAP) scaffolds with a bio-orthogonal crosslinking scheme as well as controlled particle fractions for 3D cell culture. First, HA was modified with norbornene pendant groups to be used in both microgel formation and interlinking to form MAP scaffolds. Using these methods, approximately 31% of HA repeat units were successfully modified with a norbornene functional handle (Figure 1A). Microfluidic devices with a flow-focusing region (Figure 1B) were shown to produce HA-NB μgels of either ~50 μm or ~100 μm in diameter (Figure 2C–D). The μgels used throughout the rest of this work had a mean diameter of 92 μm (Q1 = 79 μm, Q3 = 103 μm).
To control the particle fraction, μgels were dried via lyophilization (Figure 2A–C) to produce a product that can be weighed by the user and rehydrated to achieve swollen μgels. The medium for lyophilizing the μgels was optimized to prevent cryogel formation (i.e., internal defects) by using 70% ethanol, but it has also been demonstrated that cryogels were achieved using other mediums for lyophilization if cryogels are desired by the user (Figure 2D). A comprehensive study of different lyophilization mediums for microgel cryogel formation can be found in the work by Anderson, et al.12 Using confocal microscopy, HA-NB microgel diameter was quantified both before and after lyophilization with 70% ethanol (Figure 2E), showing no significant difference in microgel size with this drying process.
To facilitate bio-orthogonal interlinking of HA-NB μgels, a linear HA-Tet crosslinker was synthesized. Proton NMR spectroscopy showed successful modification of 11% of HA repeat units a tetrazine pendant group using the steps detailed in this work (Figure 1E), and the reaction yield was 95%. Using the HA-Tet linker, the dried lyo-microgel product was rehydrated with specified volumes to achieve different wt % (w/v) formulations of μgels in MAP scaffolds. These user-defined wt % of μgels in MAP scaffolds corresponded to unique particle fractions in the scaffolds (Figure 4D–E).
These methods also detailed the process for seeding cells in MAP scaffolds for 3D culture (Figure 3D) using bio-orthogonal annealing schemes. The lyo-μgels were rehydrated at the desired weight percentages in the cell media then mixed with the cell pellet and HA-Tet for interlinking the μgels. This mixture was then plated in cell culture devices to yield cells encapsulated in the void space between μgels in MAP scaffolds. Cells in 3D culture in MAP scaffolds were fixed on day 5, stained, and imaged on a confocal microscope as shown in Figure 3E. An example of the cells in the void space of the MAP scaffolds for different wt % formulations is shown in Figure 3F.
DISCUSSION:
Microfluidic production of HA-NB μgels has been shown to generate μgels with a narrower range of size distribution than emulsion batch production.3, 9 The μgels described in this protocol were formulated using an MMP-cleavable crosslinker (Ac-GCRDGPQGIWGQDRCG-NH2) to support material degradation. However, HA-NB μgels can also be crosslinked using an alternative di-thiol linker such as dithiothreitol (DTT) which is non-degradable. Similarly, other photo-initiators, such as Irgacure 2959 2-hydroxy-4-(2-hydroxyethoxy)-2-methylpropiophenone, can be used in the HA-NB precursor instead of LAP to facilitate thiol-ene crosslinking.10 These μgels were composed of 3.5 wt% HA which was based on previous work which demonstrated the viscosity of a 3.5 wt% precursor solution was amenable for pinching into droplets on a microfluidic platform.6 Others have described HA-NB microgel fabrication on flow-focusing microfluidic devices using lower wt% formulations (3 wt% HA-NB) as well.8, 10, 11
The microgel lyophilization process relies on freezing crosslinked polymer networks. Defects can occur during the freezing process when crystal structures in the lyophilization medium form and act as porogens, and the resulting cryogels exhibit enhanced porosity and different mechanical properties compared to the original material.19, 20 To date, there have been methods described for freeze-drying μgels that prevent cryogel formation for polyethylene glycol (PEG)13, gelatin methacryloyl (GelMa)14, and HA formulations12 using different lyophilization mediums. While cryogels can be advantageous for some applications, 70% ethanol was highlighted in this protocol as the lyophilization medium because it allowed the HA μgels to retain their original size, shape, and stiffness while also minimizing internal defects once rehydrated.12 70% ethanol is a common lab reagent that incorporates an additional benefit of sterilization in the process of MAP fabrication. If cryogels are desired, the user could interchange the 70% ethanol with another lyophilization medium of their choosing for this protocol.
The methods described in this work for drying HA-NB μgels were introduced to circumvent the variability in particle fraction and produce consistent MAP scaffolds with user-defined wt% formulations. The study of controlled particle fraction at different wt% rehydration was limited to the study of spherical μgels. Other microgel shapes, such as rods8, 21 or irregular shapes10, 22, have not been investigated to assess the relationship between wt% MAP and particle fraction. Particle fraction has been assessed in MAP scaffolds using consistent proportions of rehydrated lyo-μgels (84%) and HA-Tet crosslinker (16%); however, these fractions could be altered at the user’s discretion as long as the HA-Tet volume is sufficient for dissolving HA-Tet at the desired crosslinking ratio. The norbornene-tetrazine click chemistry reaction has been effective in annealing μgels to form MAP scaffolds using bi-functional23, multi-arm3, as well as the linear12 tetrazine linker described in these methods. If desired, the molar ratio of Tet:HA-NB can be varied to achieve different stiffnesses of the bulk MAP scaffolds.12
3D cell culture in MAP scaffolds has been described previously with numerous cell types, such as endothelial cells8, fibroblasts1, 3, 4, 24, neural progenitor cells9, and mesenchymal stem cells25, 26. Residual ethanol was observed via proton NMR after HA-NB μgels were lyophilized in 70% ethanol; however, it was also confirmed that the trace amounts of ethanol did not impact cell viability.12 Culture in MAP scaffolds comprised of lyo-μgels has only been demonstrated thus far with mouse mesenchymal stem cells12; however, this protocol for seeding cells in MAP scaffolds with lyo-μgels could be interchanged with other cell types and their corresponding cell media. For D1 cell culture, F-actin intensity and total cell volume has been shown to decrease as the wt% MAP increases.12
This work described methods for freeze-drying that allows μgels to either retain their original properties or produce microgel cryogels. Rehydrating the lyo-μgels at defined weight percentages leads to consistent particle fractions in MAP scaffolds which allows the user to assess cell responses within these user-defined microarchitectures. Creating MAP scaffolds with consistent particle fractions improves the reproducibility of experimental results for MAP users, extending beyond cell responses to mass transport and mechanical properties as well.12 Additionally, the use of a lyophilized product is beneficial for improving the material shelf-life and will be advantageous for future translation of MAP scaffolds to a clinical setting.
Supplementary Material
Supplementary File 1: CAD file used to 3D print the mold for cell culture devices.
Table-Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| 1 mL Luer-Lok™ syringe sterile, single use, polycarbonate | BD | 309628 | |
| 5 mL Luer-Lok™ syringe sterile, single use, polycarbonate | BD | 309646 | |
| Alexa Fluor 488 C5 maleimide | Invitrogen | A10254 | For synthesis of fluorescently-labeled tetrazine |
| Alexa Fluor 647 Phalloidin | Invitrogen | A22287 | For staining cell culture samples |
| Aluminum foil | VWR | 89107–726 | |
| Biopsy punch with plunger, 1.0 mm | Integra Miltex | 69031–01 | |
| Biopsy punch, 4 mm | Integra Miltex | 33–34 | |
| Blunt needle, 23G 0.5”, Non-Sterile, Capped | SAI Infusion Technologies | B23-50 | |
| Bottle-top vacuum filter, 0.22 μm | Corning | CLS430521 | |
| Calcium chloride | VWR | 1B1110 | For microgel washing buffer |
| Capillary-piston assemblies for positive-displacement pipettes, 1000 μL max. volume | Rainin | 17008609 | |
| Capillary-piston assemblies for positive-displacement pipettes, 25 μL max. volume | Rainin | 17008605 | |
| Capillary-piston assemblies for positive-displacement pipettes, 250 μL max. volume | Rainin | 17008608 | |
| Centrifuge tube, 15 mL | CELLTREAT | 667015B | |
| Centrifuge tube, 50 mL | CELLTREAT | 229421 | |
| Chloroform, ACS grade, Glass Bottle | Stellar Scientific | CP-C7304 | For synthesis of fluorescently-labeled tetrazine |
| Corona plasma gun, BD-10A High Frequency Generator | ETP | 11011 | |
| CryoTube Vials, Polypropylene, Internal Thread with Screw Cap | Nunc | 368632 | |
| D1 mouse mesenchymal cells | ATCC | CRL-12424 | Example cell line for culture in MAP gels |
| DAPI | Sigma-Aldrich | D9542 | For staining cell culture samples |
| Deuterium oxide, 99.9 atom % D | Sigma-Aldrich | 151882 | For NMR spectroscopy |
| Dialysis tubing, regenerated cellulose membrane, 12-14 kDa molecular weight cut-off | Spectra/Por | 132703 | For purifying HA-NB and HA-Tet |
| Diethyl ether | VWR | BDH1121-4LPC | For synthesis of fluorescently-labeled tetrazine |
| Dimethylformamide | Sigma-Aldrich | 277056 | For synthesis of fluorescently-labeled tetrazine |
| 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) | TCI-Chemicals | D2919 | For modifying HA |
| Dithiothreitol (DTT) | Thermo Scientific | R0861 | Non-degradable dithiol linker (substitute for MMP-cleavable peptide) |
| Dulbecco’s Modified Eagle’s Medium - high glucose, w/ 4500 mg/L glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | D6429–500ML | For D1 cell culture |
| EMS Paraformaldehyde, Granular | VWR | 100504–162 | For making 4% PFA |
| Ethanol absolute (200 proof) | KOPTEC | 89234–850 | |
| Heating Plate | Kopf Instruments | HP-4M | |
| 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) | Sigma-Aldrich | H3375 | |
| Sodium hyaluronate, 79 kDa average molecular weight, produced in bacteria Streptococcus zooepidemicus, pharmaceutical grade, microbial contamination <100 CFU/g, bacterial endotoxins <0.050 IU/mg | Contipro | N/A | 79 kDa average molecular weight was used for HA-Tet synthesis, but these methods but could be adapted for other molecular weights. |
| IMARIS Essentials software package | Oxford Instruments | N/A | Microscopy image analysis software |
| Infusion pump, dual syringe | Chemyx | N/A | |
| Kimwipe | Kimberly-Clark | 34120 | |
| Laboratory stand with support lab clamp | Geyer | 212100 | |
| Liquid nitrogen | Airgas | NI 180LT22 | |
| Lithium Phenyl(2,4,6-trimethylbenzoyl)phosphinate | TCI-Chemicals | L0290 | |
| Methyltetrazine-PEG4-maleimide | Kerafast | FCC210 | For synthesis of fluorescently-labeled tetrazine |
| 2-(4-Morpholino)ethane Sulfonic Acid (MES) | Fisher Scientific | BP300–100 | For modifying HA |
| Micro cover glass, 24×60mm No. 1 | VWR | 48393–106 | |
| Mineral oil, heavy | Sigma-Aldrich | 330760 | |
| MMP-cleavable dithiol crosslinker peptide (Ac-GCRDGPQGIWGQDRCG-NH2) | GenScript | N/A | |
| 5-Norbornene-2-methylamine | TCI-Chemicals | 95–10–3 | For HA-NB synthesis |
| Packing tape | Scotch | 3M 1426 | |
| Parafilm | Bemis | PM996 | |
| PEG(thiol)2 | JenKem Technology USA | A4001-1 | For synthesis of fluorescently-labeled tetrazine |
| Petri dish, polystyrene, disposable, Dia. x H=150 × 15 mm | Corning | 351058 | |
| Pluronic F-127 | Sigma-Aldrich | P2443 | For washing HMPs |
| Phosphate buffered saline (PBS) 1x | Gibco | 10010023 | |
| RainX water repellent glass treatment | Grainger | 465D20 | |
| RGD peptide (Ac-RGDSPGERCG-NH2) | GenScript | N/A | |
| Rubber bands | Staples | 112417 | |
| Sodium chloride | Chem-Impex | 30070 | For dialysis |
| Span® 80 for synthesis | Sigma-Aldrich | 1338–43–8 | |
| Sylgard 184 Silicone Elastomer | Electron Microscopy Science | 4019862 | |
| Syringe filter, Whatman™ Uniflo, 0.2 μm PES, 13 mm diameter | Cytvia | 09–928–066 | |
| Tetraview LCD digital microscope | Celestron | 44347 | |
| Tetrazine-amine HCl salt | Chem-Impex | 35098 | For HA-Tet synthesis |
| Triethylamine | Sigma-Aldrich | 471283 | For synthesis of fluorescently-labeled tetrazine |
| Tris(2-carboxyethyl)phosphine (TCEP) | Millipore Sigma | 51805–45–9 | |
| Triton X-100 | VWR | 97063–864 | |
| Trypan blue solution, 0.4% | Thermo Fisher Scientific | 15250061 | |
| Trypsin EDTA (0.25%), Phenol red | Fisher Scientific | 25–200–056 | For lifting adherent cells to seed in MAP gels |
| Tygon® formula E-3603 laboratory tubing | Millipore Sigma | Z765244 | |
| UV curing system controller, LX500 LED | OmniCure | 010–00369R | |
| UV curing head, LED spot UV | OmniCure | N/A | |
| UV light meter, Traceable | VWR | 61161–386 | |
| Vacuum dessicator | Bel-Art | 08–594–15C | |
| X-Acto Z Series Precision Utility Knife | Elmer’s | XZ3601W |
ACKNOWLEDGMENTS:
The authors would like to thank the National Institutes of Health, the National Institutes of Neurological Disorders and Stroke (1R01NS112940, 1R01NS079691, R01NS094599), and the National Institute of Allergy and Infectious Disease (1R01AI152568). This work was performed in part at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (award number ECCS-2025064) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). The authors would like to thank the lab’s former post-doc Dr. Lucas Schirmer as well as Ethan Nicklow for their assistance in generating the 3D printed device for cell culture experiments.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/64554.
DISCLOSURES:
ARA and TS have filed a provisional patent on this technology.
REFERENCES:
- 1.Griffin DR; Weaver WM; Scumpia PO; Di Carlo D; Segura T, Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nat Mater 2015, 14 (7), 737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daly AC; Riley L; Segura T; Burdick JA, Hydrogel microparticles for biomedical applications. Nature Reviews Materials 2020, 5 (1), 20–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darling NJ; Xi W; Sideris E; Anderson AR; Pong C; Carmichael Thomas, S.; Segura T, Click by Click Microporous Annealed Particle (MAP) Scaffolds. Advanced Healthcare Materials 2020, 9 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Truong NF; Kurt E; Tahmizyan N; Lesher-Pérez SC; Chen M; Darling NJ; Xi W; Segura T, Microporous annealed particle hydrogel stiffness, void space size, and adhesion properties impact cell proliferation, cell spreading, and gene transfer. Acta biomaterialia 2020, 94, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfaff BN; Pruett JL; Cornell NJ; de Rutte J; Di Carlo D; Highley CB; Griffin DR, Selective and Improved Photoannealing of Microporous Annealed Particle (MAP) Scaffolds. ACS Biomaterials Science & Engineering 2021, 7 (2), 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sideris E; Griffin DR; Ding Y; Li S; Weaver WM; Di Carlo D; Hsiai T; Segura T, Particle Hydrogels Based on Hyaluronic Acid Building Blocks. ACS Biomaterials Science & Engineering 2 2016, 2 (11), 2034–2041. [DOI] [PubMed] [Google Scholar]
- 7.Caldwell AS; Campbell GT; Shekiro KMT; Anseth KS, Clickable Microgel Scaffolds as Platforms for 3D Cell Encapsulation. Advanced Healthcare Materials 2017, 6 (15), 1700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qazi TH; Wu J; Muir VG; Weintraub S; Gullbrand SE; Lee D; Issadore D; Burdick JA, Anisotropic Rod-Shaped Particles Influence Injectable Granular Hydrogel Properties and Cell Invasion. Advanced Materials 2021, 2109194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson KL; Lesher-Pérez SC; Naffa MM; Kelly SH; Segura T, Stoichiometric Post Modification of Hydrogel Microparticles Dictates Neural Stem Cell Fate in Microporous Annealed Particle Scaffolds. Advanced Materials 2022, 2201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muir VG; Qazi TH; Shan J; Groll J r.; Burdick, J. A., Influence of Microgel Fabrication Technique on Granular Hydrogel Properties. ACS Biomaterials Science & Engineering 2021, 7 (9), 4269–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Highley CB; Song KH; Daly AC; Burdick JA, Jammed Microgel Inks for 3D Printing Applications. Advanced Science 2018, 6 (1), 1801076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson AR; Nicklow E; Segura T, Particle fraction as a bioactive cue in granular scaffolds. In Review 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pruett L; Ellis R; McDermott M; Roosa C; Griffin D, Spatially heterogeneous epidermal growth factor release from microporous annealed particle (MAP) hydrogel for improved wound closure. Journal of Materials Chemistry B 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheikhi A; Di Lisa D; de Rutte J; Akouissi O; Di Carlo D; Khademhosseini A, Microengineered Emulsion-to-Powder Technology for the High-Fidelity Preservation of Molecular, Colloidal, and Bulk Properties of Hydrogel Suspensions. ACS Applied Polymer Materials 2019, 1 (8), 1935–1941. [Google Scholar]
- 15.Brower K; White AK; Fordyce PM, Multi-step Variable Height Photolithography for Valved Multilayer Microfluidic Devices. Journal of Visualized Experiments 2017, 119, e55276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nuclear Magnetic Resonance (NMR) Spectroscopy. JoVE Science Education Database 2022, Organic Chemistry. [Google Scholar]
- 17.Roosa C; Pruett LJ; Trujillo J; Rodriguez A; Pfaff BN; Cornell NJ; Flanagan C; Griffin DR, Microfluidic Synthesis of Microgel Building Blocks for Microporous Annealed Particle Scaffold. Journal of Visualized Experiments 2022, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H; Dicker KT; Xu X; Jia X; Fox JM, Interfacial bioorthogonal crosslinking. ACS Macro Letters 2014, (8), 727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welzel PB; Friedrichs J; Grimmer M; Vogler S; Freudenberg U; Werner C, Cryogel Micromechanics Unraveled by Atomic Force Microscopy-Based Nanoindentation. Advanced Healthcare Materials 2014, 3 (11), 1849–1853. [DOI] [PubMed] [Google Scholar]
- 20.Plieva F; Huiting X; Galaev IY; Bergenståhl B; Mattiasson B, Macroporous elastic polyacrylamide gels prepared at subzero temperatures: control of porous structure. Journal of Materials Chemistry 2006, 16 (41), 4065–4073. [Google Scholar]
- 21.Rommel D; Mork M; Vedaraman S; Bastard C; B. GLP; Kittel Y; Vinokur R; Born N; Haraszti T; De Laporte L, Functionalized Microgel Rods Interlinked into Soft Macroporous Structures for 3D Cell Culture. Advanced Science 2022, 9 (10), 2103554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurt E; Segura T, Nucleic Acid Delivery from Granular Hydrogels. 2021, 11 (3), 2101867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isaac A; Jivan F; Xin S; Hardin J; Luan X; Pandya M; Diekwisch TG; Alge DL, Microporous Bio-orthogonally Annealed Particle Hydrogels for Tissue Engineering and Regenerative Medicine. ACS Biomaterials Science & Engineering 2019, 5 (12), 6395–6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Truong NF; Lesher-Pérez SC; Kurt E; Segura T, Pathways Governing Polyethylenimine Polyplex Transfection in Microporous Annealed Particle Scaffolds. Bioconjugate Chemistry 2019, 30 (2), 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh J; Griffin DR; Archang MM; Feng AC; Horn T; Margolis M; Zalazar D; Segura T; Scumpia PO; Di Carlo D, Enhanced in vivo delivery of stem cells using microporous annealed particle scaffolds. Small 2019, 15 (39), 1903147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F; Truong VX; Fisch P; Levinson C; Glattauer V; Zenobi-Wong M; Thissen H; Forsythe JS; Frith JE, Cartilage tissue formation through assembly of microgels containing mesenchymal stem cells. Acta biomaterialia 2018, 77, 48–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1: CAD file used to 3D print the mold for cell culture devices.