

Abstract
Background:
Cardiomyopathy is characterized by the pathologic accumulation of resident cardiac fibroblasts that deposit extracellular matrix (ECM) and generate a fibrotic scar. However, the mechanisms that control the timing and extent of cardiac fibroblast proliferation and ECM production are not known, hampering the development of anti-fibrotic strategies to prevent heart failure.
Methods:
We used the Tcf21MerCreMer mouse line for fibroblast-specific lineage tracing and p53 gene deletion (called p53-CF KO). We characterized cardiac physiology, and used single-cell RNA-sequencing and in vitro studies to investigate the p53-dependent mechanisms regulating cardiac fibroblast cell cycle and fibrosis in left ventricle (LV) pressure overload induced by trans aortic constriction (TAC).
Results:
Cardiac fibroblast proliferation occurs primarily between days 7 and 14 following TAC in mice, correlating with alterations in p53 dependent gene expression. P53 deletion in fibroblasts led to a striking accumulation of Tcf21-lineage CF within the normal proliferative window, and precipitated a robust fibrotic response to LV pressure overload. However, excessive interstitial and perivascular fibrosis does not develop in p53-CF KO mice until after CF exit the cell cycle. Single cell RNA-sequencing revealed p53 null fibroblasts unexpectedly express lower levels of genes encoding important ECM proteins while they exhibit an inappropriately proliferative phenotype. In vitro studies establish a role for p53 in suppressing the proliferative fibroblast phenotype, which facilitates the expression and secretion of ECM proteins. Importantly, Cdkn2a expression and the p16Ink4a-Retinoblastoma cell cycle control pathway is induced in p53 null cardiac fibroblasts, which may eventually contribute to cell cycle exit and fulminant scar formation.
Conclusions:
This study reveals a mechanism regulating cardiac fibroblast accumulation and ECM secretion, orchestrated in part by p53-dependent cell cycle control that governs the timing and extent of fibrosis in LV pressure overload.
Keywords: Fibrosis, fibroblast, mouse, proliferation, heart
Subject Codes: Animal Models of Human Disease, Basic Science Research, Fibrosis, Translational Studies
Graphical Abstract
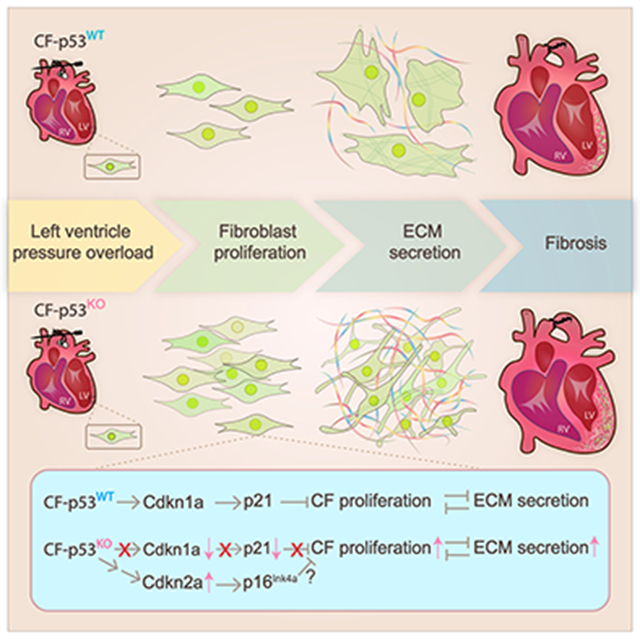
Resident cardiac fibroblasts respond to cardiac insult by progressing through phases defined by a pro-inflammatory phenotype, a proliferative phenotype, and finally a reparative phenotype, characterized by the deposition of extracellular matrix that generates a fibrotic scar. The cellular and molecular mechanisms governing the temporal control of the fibroblast phenotype are not well-understood, hampering the development of therapeutic interventions that allow for an appropriate healing response while preventing the development of pathological fibrosis. Here, we have found that fibroblast proliferation is in part controlled by the tumor suppressor protein, P53. P53 induces the expression of Cdkn1a (p21), which limits the extent of proliferation. P53 deletion releases an important break on the cell cycle, leading to the excessive accumulation of fibroblasts that generate a robust fibrotic response. Interestingly, p53 null fibroblasts still exit the cell cycle at the appropriate time, perhaps due to a compensatory induction of Cdkn2a / p14Ink4a; and the development of fibrosis occurs during the normal reparative window. Taken together, this study reveals a novel mechanism that governs the extent of fibroblast proliferation and the timing of scar formation, through the mutual antagonism between the proliferative and ECM-secretory fibroblast phenotypes.
Introduction
Chronic heart disease is invariably associated with an overabundance of extracellular matrix (ECM) within the myocardium. Thought to be an adaptive response to maintain cardiac integrity, sustained ECM deposition ultimately leads to formation of a fibrotic scar that impairs systolic and diastolic function and can initiate lethal arrhythmias1-3. The primary cellular source of ECM in the heart is resident cardiac fibroblasts (CF), which accumulate in disease and acquire a secretory phenotype, sometimes called an activated fibroblast or myofibroblast4, 5. Restraining CF accumulation and/or activation may prevent pathologic cardiac fibrosis and heart failure.
Animal studies have described the timecourse of cardiac remodeling, which is characterized by an initial proliferative phase of CF accumulation, followed by a reparative phase of ECM deposition by myofibroblasts, terminating in scar maturation and stabilization by CF with a post-mitotic matrifibrocyte phenotype6, 7. The various proliferative, reparative and pathologic CF phenotypes that contribute to the progression of fibrotic cardiac remodeling are beginning to be defined by scRNA-sequencing8-12. While the mechanisms that control myofibroblast activation and ECM deposition largely converge on biomechanical tension, canonical and non-canonical transforming growth factor (TGF)-β1 signaling, and Rho-Rho kinase-dependent pathways5, 13-17, the mechanisms that regulate the timing and extent of fibroblast accumulation and activation in heart disease remain elusive.
We recently found that p53 transcriptional targets are among the most dysregulated gene programs in CF during acute left LV pressure overload-induced remodeling 18. We subsequently found small proline rich (Sprr) protein-2b is induced in heart failure and stimulates MDM2-dependent p53 degradation in vitro, relieving constraints on CF cell cycle progression19. p53 is an important component of the DNA damage response and transcriptional regulator of cell cycle arrest that is often mutated in cancerous tumors 20. p53-dependent cellular senescence has also been linked to cardiac remodeling in mouse models of cardiac fibrosis21, 22, and regeneration23, 24. Indeed, overexpression of the p53 target gene, cyclin dependent kinase inhibitor 1a (Cdkn1a or p21), in transgenic mice prevents CF accumulation and reduces scar formation after myocardial infarction25. We hypothesized that p53 may control CF proliferation and fibrosis in LV pressure overload.
Here, we deleted the p53 gene in fibroblasts using a tamoxifen-inducible Cre recombinase under the control of the Tcf21 locus (called p53-CF KO)26. p53 gene deletion leads to excessive CF accumulation in response to LV pressure overload; however, CF proliferation remains primarily restricted to the normal proliferative window. P53-CF KO mice eventually develop robust interstitial and perivascular fibrosis, but only after CF exit the cell cycle. scRNA-sequencing and in vitro assays reveal that CF proliferation suppresses the activated myofibroblast phenotype. Ultimately, Cdkn2a expression is induced in p53 deficient CF, likely promoting p16Ink4a – dependent cell cycle arrest and a robust fibrotic response. This study suggests p53 controls the timing of CF accumulation and the extent of cardiac fibrosis in LV pressure overload, and supports a paradigm of p53 – p16Ink4a crosstalk via the Cdkn2a locus in pathologic cardiac fibrosis.
Methods
A detailed description of all materials and methods can be found in the Expanded Materials and Methods and in the Major Resources Table in the Supplemental Materials. scRNA-sequencing data has been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE165455.
Results
Cardiac fibroblasts proliferate in the second week of LV pressure overload
Fibroblast proliferation in left ventricle pressure overload induced cardiac remodeling is reported to occur within a discrete time window, and we previously identified a profound disruption in p53-dependent transcription following TAC7 18. Upon re-analysis of this published RNA-seq data, we find that genes encoding mitotic cell cycle activators reached peak expression at 3 days, and waned by 10 days (Figure 1A). In contrast, genes associated with cell cycle arrest and fibroblast activation gradually increased through 10 days (Figure 1B, C). This gene expression data correlates with fibroblast proliferation - pulse chase BrdU incorporation in mice revealed that proliferation is restricted to days 7-14 post-TAC (Figure 1D, E). This proliferative window is shortly before we detect ECM deposition, LV hypertrophy, and functional decline, which gradually increase from 14-28 days post-TAC (Figure 1F-I). These data support temporally distinct peaks of cardiac fibroblast proliferation and the development of fibrosis in pressure overload, and suggest a correlation with p53-dependent cell cycle control (Figure 1J).
Figure 1. Acute transcriptional response of cardiac fibroblasts to left ventricle pressure overload.

A-C) Cardiac fibroblasts were isolated from C57BL/6J mice at baseline or at the indicated time after TAC surgery and subjected to RNA-sequencing18. Relative expression of select genes that indicate active cell cycle (A), cell cycle exit (B), and fibroblast activation (C). n=6 control; n=3 3-day TAC; n=3 10-day TAC. D) Mice were subjected to TAC surgery, and injected with BrdU for the indicated time periods prior to heart isolation at 28days post-TAC. Representative images of histological sections from hearts of indicated treatment stained with an antibody directed against BrdU (red), wheat germ agglutinin to mark cell membranes (WGA, green), and DAPI to mark nuclei (blue). E) Quantification of BrdU incorporation from (D) as a percentage of total nuclei. Data is represented as Mean +/− SEM. Data points indicate results from individual mice analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons. n=9 per timepoint. F) Hearts were isolated from mice at the indicated timepoints post TAC and stained with Picrosirius Red to visualize collagen. G-I) Serial echocardiographic assessment of left ventricle (LV) mass (G), LV ejection fraction (EF%) (H), and LV fractional shortening (FS%) (I). Violin plots depict data distribution of n=40 individual mice per timepoint analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons. J) Schematic describing the timing of various aspects of LV remodeling in pressure overload. Scale bar = 50μm (D), 100 μm (F).
p53 limits resident cardiac fibroblast expansion in left ventricle pressure overload
To examine the role of p53 in pressure overload-induced cardiac fibrosis in vivo, we deleted LoxP-targeted p53 in mice prior to TAC using a tamoxifen-inducible Cre-recombinase expressed at the fibroblast-specific Tcf21 gene locus (Figure 2A, B)26-28. Cre-mediated recombination was achieved in Tcf21MerCreMer;p53fl/fl;R26RmTmG mice by injecting tamoxifen (100 mg/kg for five consecutive days) to simultaneously delete p53 and indelibly label resident fibroblasts with EGFP (called p53-CF KO). TMX-injected Tcf21MerCreMer;p53+/+;R26RmTmG littermates were used as controls (called p53-CF WT). p53 immunoreactivity in perivascular fibroblasts was significantly reduced in p53-CF KO mice at 14 days post-TAC, compared to p53-CF WT mice (Figure 2C, D and Figure S1A).
Figure 2. p53 deletion stimulates cardiac fibroblast proliferation.

A, B) Schematic indicating genotype of mice used in study (A) and experimental timeline (B) of tamoxifen (TMX) injection, TAC surgery, BrdU injections, echocardiography, and tissue harvest. C) Heart sections were obtained from mice of the indicated genotype at 14 days post-TAC, and stained with an antibody directed against p53 (brown staining). D) Quantification of p53+ perivascular mesenchymal cells from (C) analyzed by 2-sided T-test. n= 4 (p53-CF WT) and 5 (p53-CF KO). E) Heart sections were obtained from mice of the indicated genotype at 28 days post-TAC. Representative images of sections that were stained with WGA and antibodies directed against PDGFRα and BrdU. F) Quantification of BrdU incorporation into PDGFRα+ cells from images in (E). n=9 (p53-CF WT), 21 (p53-CF HET), 10 (p53-CF KO); G) Heart sections were obtained from mice of indicated genotype at 28 days post-TAC. Representative images of sections that were incubated with antibodies directed against GFP (green fibroblasts), and stained with isolectin-B4 (IB4, red endothelial cells) and DAPI (blue, nuclei). H) Quantification of GFP+ cells identified in (G). n=9 per genotype. I) Heart sections were obtained from p53-CF WT or p53-CF KO mice at indicated times. Representative images of sections that were incubated with antibodies directed against Ki67 (red), PDGFRα (green), and DAPI (blue, nuclei). J) Quantification of Ki67+ nuclei identified in (I). All data points indicate results from individual mice, and data is represented as Mean +/− SEM. Data in F, H, J is analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons. Scale bar = 100μm (C); 50μm (E, G); 200μm (I).
To detect proliferating fibroblasts in BrdU injected mice, we immunostained heart sections with antibodies directed against BrdU and platelet-derived growth factor receptor (PDGFR)α. Following TAC, BrdU+ fibroblasts accumulated in all genotypes as expected, and BrdU incorporation in PDGFRα+ cells was further enhanced in both heterozygous and p53-CF KO mice by ~2- and 3-fold, respectively (Figure 2E, F). Importantly, the increase of BrdU-positivity in p53-CF KO mice after TAC corresponds to a significant expansion of Tcf21 lineage fibroblasts labeled by GFP (Figure 2G, H). We did not observe an overlap between GFP-labeled CF and isolectin-B4 (IB4)-labeled endothelial cells (ECs) (Figure 2G). Furthermore, there was no overlap between CFs and ECs as defined by co-staining for PDGFRα and the EC marker ERG (ETS-related gene)7, 29, 30, nor did we observe an increase in ERG+ EC in p53-CF KO mice (Figure S1B, C).
To investigate the timing of CF proliferation in p53-CF KO mice, we stained sections of hearts obtained at baseline (2 weeks after TMX injection), 14 days post-TAC, or 28 days post-TAC with antibodies directed against Ki67 and PDGFRα. Although BrdU incorporation was observed in p53-CF KO fibroblasts over a two-week injection time-course even without TAC (Figure S1D, E), we did not observe increased Ki67 positivity at baseline (Figure 2I, J). In contrast, we observed a robust ~15-fold increase in Ki67 positive fibroblasts in p53-CF KO hearts at 14 days post-TAC, a timepoint coinciding with the later stages of the normal proliferative window when most control CF have already exited the cell cycle. Importantly, fibroblast proliferation in p53-CF KO hearts was nearly back to baseline, and identical to control hearts, at 28 days post-TAC. Taken together, these results indicate that p53 limits CF expansion in LV pressure overload primarily during the normal proliferative window, and that compensatory mechanisms may prevent CF proliferation in p53-CF KO hearts outside of the proliferative window.
Fibroblast-specific p53 deletion stimulates development of cardiac fibrosis in LV pressure overload
We hypothesized that accumulation of CF in p53-CF KO mice subjected to LV pressure overload might accelerate the time-course of ECM deposition. To test this, we stained heart sections obtained at baseline, 14- and 28-days post-TAC with PicroSirius Red to visualize collagen fibers. We did not detect excessive PicroSirius Red positivity in p53- CF KO mice prior to TAC, or 14 days post-TAC despite a significant accumulation of CF number (Figure 3A, B). However, p53 deletion subsequently precipitated a more robust fibrotic response; while all animals exhibit perivascular fibrosis, p53-CF KO mice developed fulminant interstitial fibrosis, leading to a significant increase in total cardiac fibrotic burden (Figure 3A, B). We further assessed ECM remodeling at 28 days post-TAC by staining for collagen hybridizing peptide (CHP), which labels denatured and single-stranded collagen. Large areas of CHP-positivity were present, primarily in the perivascular regions of p53-CF KO mice, suggesting ECM in p53-CF KO mice is more recently generated or is actively remodeling (Figure 3C, D).
Figure 3. p53 deletion in fibroblasts leads to an exaggerated fibrotic response in left ventricle pressure overload.

A) Heart sections were obtained from mice of indicated genotype at baseline, 14 days, or 28 days post-TAC, and stained with PicroSirius Red to visualize collagen fibrils. B) Quantification of PicroSirius Red staining from images in (A). For each genotype, n= 4 (day 0); n=5 (day 14); n=12 (day 28). C) Heart sections were obtained from mice of indicated genotype at 28 days post-TAC. Representative images of sections stained with Biotin-conjugated collagen hybridizing peptide (CHP, purple) to visualize denatured collagen as an indicator of active tissue remodeling, and wheat germ agglutinin (WGA, blue) to label cell membranes. D) Quantification of CHP staining from images in (C). n=9 (WT), 17 (HET), 8 (KO). Data is represented as Mean +/− SEM. Datapoints indicate results from individual mice. Data in is analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons. Scale bar = 150μm (A) or 50μm (C).
The excessive CF accumulation and fibrosis in p53-CF KO mice precipitated a subtle, but significant, decline in systolic cardiac function. Serial echocardiography demonstrated a decline in fractional shortening and ejection fraction, observable 28 days post-TAC (Figure 4A, B), but no observable diastolic dysfunction (Figure 4C). Hypertrophic remodeling was increased in both p53-CF KO and WT mice after TAC based on estimation of LV mass (Figure 4D). A further increase in cardiac mass in p53-CF KO mice was observed by visualizing CM cross-sectional area (CSA) in wheat germ agglutinin (WGA) stained sections (Figure 4E, F).
Figure 4. Left ventricle pressure overload-induced changes in cardiac physiology upon fibroblast specific p53 deletion.

A-D) Serial echocardiography was performed in mice of indicated genotype at baseline (0d) and 7, 14, 21, and 28 days (d) post-TAC to measure left ventricle (LV) ejection fraction (EF%) (A), LV fractional shortening (FS%) (B), diastolic function (E/E’ shown in C) and LV Mass (D). Data is represented as Mean +/− SEM. Datapoints indicate results from individual mice. Data is analyzed by two-way repeated measures ANOVA with Geisser-Greenhouse correction. E) Heart sections were obtained from mice of indicated genotype at baseline, 14 days post-TAC, or 28 days post-TAC. Representative images are shown of sections that were stained with wheat germ agglutinin (WGA) to visualize cardiomyocyte cell membranes. Scale bar = 40μm. F) Cardiomyocyte cross-sectional area (CSA) was quantified from images in (E). Results in (F) reflect at least ~300-400 cardiomyocytes per mouse. Data is represented as Mean +/− SEM. Data points indicate the average CSA across 3 images for an individual mouse. Data in (F) is analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons. n=3-5 mice per condition. Scale bar = 40μm
Single cell transcriptomics reveals a population of mitotic CF in LV pressure overload
Based upon the timecourse of CF proliferation and fibrosis observed in p53-CF KO mice, we speculated that fibroblast activation consists of an initially proliferative phenotype, followed shortly by the acquisition of a secretory phenotype responsible for ECM deposition, which are both impacted by p53 deletion. To address this hypothesis, we conducted scRNA-sequencing on fluorescently labeled CF isolated from p53-CF KO and p53-CF WT littermates at 14 days post-TAC, a timepoint coinciding with the end of the normal proliferative window (Figure 5A). Since WT fibroblasts have largely exited the cell cycle at 14 days, this approach allowed us to interrogate how p53 deletion affects the transition out of the proliferative fibroblast phenotype.
Figure 5. Single cell RNA-sequencing reveals highly proliferative population of cardiac fibroblasts.

A) Schematic indicating genotype of mice used in study, and experimental timeline of tamoxifen (TMX) injection, TAC surgery and Tcf21-lineage fibroblast isolation for single cell RNA-sequencing. B-D) Uniform Manifold Approximation and Projection (UMAP) of single Tcf21-lineage cell transcriptomes from p53-CF WT (1659 cells) and KO (3158 cells) mice obtained 14 days post-TAC, represented by genotype (B), cell identity (C) and cell cycle phase (D). Cluster 7 is defined by cells in G2/M phase of the cell cycle. E) Dot plot visualization of Gene ontology (GO) biological processes that are enriched in cluster 7, revealing p53-dependent neurological disorders, cellular senescence, and proliferation related processes. F) Expression of genes that define the highly proliferative cluster 7, depicted by violin plots. G) Dot plot visualization of gene expression for candidate p53 target genes sorted by genotype and cluster number. Color scale represents relative expression across all cells within a particular cluster, and size of dot represents the % of cells within cluster that express the indicated gene.
GFP+ fibroblasts were obtained from two p53-CF KO animals and two p53-CF WT animals by FACS (Figure S2A), and scRNA-sequencing was performed on individual cells captured using a 10x Genomics Chromium Controller. After exclusion of cell doublets based on unique molecular identifier counts, and filtering based on mitochondrial genes, we obtained high quality sequencing from 1659 wildtype and 3158 p53-CF KO cells (Figure S2B). Over-representation of p53-CF KO CF likely reflects their increased proliferation rate. To interrogate cellular heterogeneity within the Tcf21+ CF lineage, we used Seurat (v3.2.3) to generate uniform manifold approximation and projections (UMAP). CF from WT mice cluster into six unique populations, while CF from p53-CF KO mice are defined by seven unique populations (Figure S2C, D). Following canonical correlation analysis (CCA) and integration of p53-CF WT and p53-CF KO datasets, seven unique populations emerged across all cells (Figure 5B, C).
Cluster 7 (C7) is composed almost entirely of actively proliferating CF in G2/M phase (Figure 5D). In contrast, the vast majority of CF in other clusters reside in G1 or S phase, consistent with the proliferative window ending ~14 days post-TAC. Gene ontology (GO) analysis revealed C7 is enriched in pathways related to p53 signaling and function, senescence, mitosis, cell cycle, and nuclear processes (Figure 5E); comparative analysis of cell cycle progression genes revealed striking accumulation in C7 (Figure 5F). Importantly, canonical transcriptional targets of p53 involved in cell cycle arrest and apoptosis are uniformly down-regulated in p53-CF KO fibroblasts compared to control, regardless of cluster identity, confirming efficient deletion of p53 in Tcf21-lineage fibroblasts (Figure 5G). Of note, wildtype cells within the excessively mitotic C7 express high levels of p53 targets, suggesting that cell cycle exit may be initiating in this population, which becomes derailed by p53 deletion. Taken together, while p53 deletion leads to the accumulation of CF in LV pressure overload, most fibroblasts have exited the cell cycle at 14 days post-TAC even in the absence of p53.
Single cell transcriptomics defines cardiac fibroblast identities in LV pressure overload
To establish CF identities, we initially interrogated the expression of key marker genes across each cluster. The expression of Tcf21 and Pdgfra, which each define quiescent resident fibroblasts, is most prominent in C1, with intermediate expression in C2 and C3, and considerably lower expression in C4 (Figure 6A). In contrast, Postn and Acta2, well-established myofibroblast markers, predominate in C4, with intermediate levels in C2 and C3, and minimal expression in C1 (Figure 6B). Hierarchical clustering and identification of the most enriched genes in clusters 1 and 4 further establish their identity as quiescent and activated fibroblasts, respectively (Figure 6C). Here, we found quiescent fibroblasts express high levels of Inmt, and antioxidant genes such as glutathione-s-transferases (Gstt1, Gstm1) and flavin containing dimethylaniline monooxygenase 2 (Fmo2) (Figure 6C, D and Figure S3A). We also identified an enrichment of Igfbp3, Sfrp2, Fgl2, Cxcl1, and Cxcl12 in more quiescent fibroblasts (Figure 6C, D), which are of particular interest due to their roles in inflammation and angiogenesis31, 32.
Figure 6. Single cell RNA-sequencing defines cardiac fibroblast phenotypes and the transition from quiescent to activated myofibroblast.

A, B) The expression of markers of the quiescent (A) and activated (B) fibroblast phenotype is represented as violin plots across all fibroblast clusters. C) Hierarchical clustering reveals the genes that define the identity of cluster 1 (quiescent) and cluster 4 (myofibroblast). D, E) Violin plots display the expression of representative genes that define quiescent cluster 1 cells (D) and activated cluster 4 cells (E). F) Dot plot visualization of GO terms that are enriched based on comparison between indicated clusters. Data reveals a stepwise progression from quiescent to activated myofibroblast traversing clusters 1, 2, 3, and 4.
In contrast to C1, C4 is enriched in scleraxis (Scx), a transcriptional activator of the contractile myofibroblast phenotype33, and genes that encode ECM proteins and other myofibroblast markers such as Postn, Acta2, Tagln2, Fn1, Ltbp2, Cd200, Cilp, Cthrc1, Fmod, Thbs4, Tnmd, and Lox (Figure 6C, E and Figure S3B). We also identified an enrichment in Runx1, a transactivator of the cartilage phenotype, and cartilage oligomeric matrix protein (Comp), a component of the mature cardiac scar6. Finally, we observed genes that were not previously associated with the myofibroblast phenotype, such as Crlf2, Ddah1, and Mgp. Inter-cluster GO term comparisons established a stepwise increase in biological processes associated with ECM deposition progressing from C1 to C4, with C2 and C3 representing transitional / proto-myofibroblast phenotypes (Figure 6F). The remaining two clusters, C5 and C6, did not fit neatly within the progression from quiescence to activated CF. GO term analysis identified pathways related to angiogenesis and vasculature development within C5, which we hypothesize may represent a perivascular support cell (Figure S3C). C6 is a relatively rare population defined by GO terms related to inflammation and immune function, which we hypothesize might serve as a resident sentinel of tissue injury (Figure S3D). In summary, scRNA-sequencing defined 7 primary CF phenotypes, with C1-C4 representing the progression from quiescent to activated fibroblast and C7 representing proliferative CF.
Cardiac fibroblast developmental trajectory in LV pressure overload is altered by p53 deletion
In order to determine how p53 deletion impacts the CF response to pressure overload, we established developmental trajectory in pseudotime using Monocle, which revealed 11 cell states and five nodal points (Figure 7A, B). The start of pseudotime (state 1) is characterized by GO terms related to the cellular response to cytokines and negative regulation of motility, and predicted regulation by transcription factors such as Tcf21 (CF quiescence), Oct4 (self-renewal capacity), and Rela (inflammation) (Figure S4A, B). Integrating cell identities described by Seurat with the pseudotime trajectory revealed an enrichment of C1 cells in state 1, confirming the start of pseudotime is represented by a quiescent CF phenotype (Figure 7C). At the other extreme, the end of pseudotime (state 11) is characterized by GO terms such as ECM organization and cytokine-mediated signaling pathways, and predicted transcriptional regulation by Smad2 and Atf3, known regulators of LV pressure overload-induced cardiac remodeling (Figure S4C, D) 34. The end of pseudotime harbors an over-abundance of C4 activated myofibroblasts (Figure 7D). Transcriptional signatures that are enriched in State 11 (late pseudotime) vs. 1 (early pseudotime) include GO terms related to ECM, actin binding, cell adhesion, and TGF-β1 signaling, confirming State 11 cells exhibit a myofibroblast phenotype (Figure 7E).
Figure 7. p53 deletion in fibroblasts alters developmental trajectory in left ventricle pressure overload.

A, B) Monocle generated developmental trajectory (A) reveals the distribution of 11 cell states across pseudotime (B). C-D) The integration of cell identities within pseudotime reveals the association of cell state 1 with quiescent C1 fibroblasts (C), state 11 with activated C4 myofibroblasts (D). E) Dot plot representation of GO terms derived from genes that are enriched in State 11 compared to State 1. F) Highly proliferative cluster 7 fibroblasts are distributed bi-modally in pseudotime. G) Visualization of cluster 7 based on genotype reveals equal proportion of WT and p53 null fibroblasts in early pseudotime, while p53 null fibroblasts are over-represented in late pseudotime. H) Proportionality analysis reveals significant over-representation of WT or p53-CF KO cells in each cluster. I) Dot plot representation of GO terms that are derived from genes that are downregulated in p53 null fibroblasts in state 1. J) Dot plot representation of genes representative of the impact of p53 deletion on fibroblast activation in state 1 and 11.
Unexpectedly, we found that mitotic C7 cells localize bimodally, at the extreme poles of pseudotime (Figure 7F). Early pseudotime is composed of proliferative CF obtained from WT and p53-CF KO hearts at expected proportions (Figure 7G). In contrast, the proliferative CF found in late pseudotime includes an over-abundance of cells obtained from p53-CF KO hearts. Proportionality analysis across all cells revealed an enrichment in WT cells in C2 (quiescent / early transition fibroblasts). In contrast, p53 null cells are significantly over-represented in C3 (proto-myofibroblasts) and C4 (fully active myofibroblasts), as well as in C7 (mitotic fibroblasts) (Figure 7H).
Given the over-abundance of p53-CF KO myofibroblasts at 14 days post-TAC, it was curious we did not observe increased cardiac fibrosis at this timepoint. Surprisingly, p53-CF KO cells exhibit a reduction in GO terms related to ECM production and binding (Figure 7I), driven by a reduction in Col1a1, Col3a1, Col14a1 and Eln expression (Figure 7J and Figure S5). It is interesting to speculate that p53 deletion accelerates CF proliferation at the expense of the ECM secretory phenotype, leading to accumulation of proto-myofibroblasts poised to produce a robust, but delayed, fibrotic response after cell cycle exit.
Mutually antagonistic programs of CF proliferation and activation are governed by p53
To investigate whether the proliferative and ECM secretory CF phenotypes are functionally linked via p53, we isolated CF from neonatal p53fl/fl mice or WT mice, cultured them overnight in low serum (0.5% FBS), then infected them with adenovirus directing the expression of Cre recombinase to delete p53 (Ad-Cre) or a control adenovirus expressing β-galactosidase (Ad-β-gal). CF were then treated with TGF-β1/AngII to induce myofibroblast activation. qRT-PCR confirmed deletion of p53 in Ad/Cre-treated p53fl/fl CF (Figure 8A). TGF-β1/AngII stimulation led to an increase in expression of the p53-dependent cell cycle inhibitor Cdkn1a, which was prevented by p53 deletion (Figure 8B). p53 deletion in CF increased proliferation as measured by Cyquant assay (Figure 8C), and flow cytometric analysis of propidium iodide-stained cells (Figure 8D). While p53 deletion accelerated the cell cycle, TGF-β1/AngII-dependent fibroblast activation was ameliorated, as revealed by a significant reduction in the expression of Acta2, Col1a1, and Postn (Figure 8E-G), and decreased abundance of Acta2+ myofibroblasts (Figure 8H, I). Further, p53 null CF secrete less collagen, as revealed by hydroxyproline detection in conditioned media (Figure 8J).
Figure 8. p53 deletion accelerates cardiac fibroblast cell cycle at the expense of myofibroblast activation.

Cardiac fibroblasts were isolated from WT or p53-fl/fl mice and treated with adenovirus directing the expression of β-galactosidase (β-Gal) or Cre-recombinase (Cre) for 24 hrs. Cardiac fibroblasts of indicated genotype and transduction were treated with vehicle or TGF-β1(10ng/mL)/AngII (1μM) for an additional 24 hrs (for RNA) or 72 hrs (for protein) prior to indicated assay. A, B) qRT-PCR reveals the expression of Trp53 (A) and Cdkn1a (B). C) Cardiac fibroblast proliferation was quantified by Cyquant assay. D) Cell cycle phase was determined by flow cytometry of propidium iodide-stained cells. E-G) qRT-PCR reveals the expression of Acta2 (E) Col1a1 (F) and Postn (G). H) Immunocytochemistry using an antibody directed against Acta2 (red) reveals activated myofibroblasts in p53–fl/fl cultures treated with TGF-β1 (10ng/mL)/AngII (1μM) and Ad/β-Gal or Ad/Cre for the indicated time. Nuclei are labeled with DAPI (blue). Scale bar = 50 μm. I) Quantification of Acta2 staining as a percentage of total cells shown in (H). J) Hydroxyproline incorporation assay reveals the relative level of collagen (Pro-OH content) in the conditioned media. K - O) Cardiac fibroblasts were isolated from p53-fl/fl mice and treated +/− Ad/Cre +/− Ad/Cdkn1a, followed by stimulation with TGF-β1(10ng/mL)/AngII (1μM) for 72 hours to induce myofibroblast activation. qRT-PCR reveals the expression of Trp53 (K) and Cdkn1a (L). Immunocytochemistry revealed Acta2+ stress fibers (white), Ki67+ proliferating cells (red), and nuclei (DAPI, blue) (M). Ki67+ nuclei (%) was quantified in (N) and Acta2+ area/cell is quantified in (O). Scale bar in (M) = 100 μm. P) p53 was deleted by Ad/Cre treatment 72 hrs after myofibroblast activation with TGF-β1(10ng/mL)/AngII (1μM). 24 hrs later, RNA was isolated and qRT-PCR was performed to investigate expression of indicated genes. Q) Heart sections obtained from mice of indicated genotype 28 days post-TAC were incubated with antibodies directed against PDGFRα (fibroblasts, green) and p16Ink4a (red). DAPI labels nuclei (blue). Scale bar = 10 μm. R) Quantification of PDGFRα / p16Ink4a double positive cells in (Q). Datapoints indicate results of individual biological replicate cell culture well or individual mouse. Data is represented as Mean +/− SEM. Data is analyzed by Kruskal-Wallis test followed by Dunn’s test to calculate pairwise comparisons (C, K, L, M, O); or by two-way repeated measures ANOVA with Geisser-Greenhouse correction (A, B, D, E, F, G, I, J); or by unpaired, two-tailed t-test with Welch’s correction (P, R).
To establish a causative relationship between p53 deletion, proliferation rate, and the myofibroblast phenotype we conducted a rescue experiment by transducing p53fl/fl CF with Ad-Cre +/− Ad-Cdkn1a, followed by TGF-β1/AngII treatment. Alteration of p53 and Cdkn1a expression was confirmed by qRT-PCR (Fig. 8K, L). p53 deletion increased cell cycle as demonstrated by ~ 2-fold increase in Ki67 positive CF nuclei, and proliferation rate was reduced to background levels by Cdkn1a overexpression (Figure 8M, N). p53 deletion also led to a reduction in cell spreading and Acta2+ stress fiber formation, indicators of the myofibroblast phenotype (Figure 8M, O). Importantly, the reduction in cell area and Acta2+ stress fiber formation in p53 null CF was partially reversed by overexpression of Cdkn1a. Of note, Cdkn1a overexpression in control cultures reduced proliferation below background, and led to a substantial increase in Acta2+ stress fiber formation and cell spreading. These results were supported by qRT-PCR detection of Acta2, Col1a2, Col3a1, Postn and Fn1, which all trended downwards when cell cycle activity was stimulated by p53 deletion, and recovered upon Cdkn1a overexpression (Figure S6A).
Next, we investigated the impact of p53 deletion on cell cycle and the secretory phenotype in activated myofibroblasts by stimulating p53fl/fl CF cultures with TGF-β1/AngII for 72 hours prior to transduction with Ad-β-gal or Ad-Cre. Although p53 was effectively deleted and Cdkn1a expression was suppressed (Figure 8P), we did not observe any alterations in fibroblast activation or proliferation as demonstrated by qRT-PCR (Figure S6B), immunostaining for Acta2 (Figure S6C, D), and hydroxyproline detection in conditioned media (Figure S6E). Of note, we observed a compensatory increase in the expression of the Cdkn2a gene in myofibroblast cultures that lacked p53 (Figure 8P). Cdkn2a encodes two proteins (p16Ink4a and p14Arf) involved in retinoblastoma (Rb1) and MDM2/p53-mediated cell cycle control, respectively35, 36. Based on the finding that p53 deletion stimulated Cdkn2a expression, and p53-CF KO fibroblasts ultimately exit the cell cycle and contribute to a robust scar following TAC, we asked whether p16Ink4a/Rb1 might compensate for p53 loss in vivo. While we observed minimal p16Ink4a immunoreactivity in control mice, p16Ink4a was robustly expressed in PDGFRα-positive CF of p53-CF KO mice at 28 days post-TAC (Figure 8Q, R). Taken together, these data suggest that p53 and Cdkn2a play a central role in regulating CF accumulation and the timing and extent of cardiac fibrosis in LV pressure overload.
Discussion
Here, we report the timing and extent of resident CF proliferation is regulated by p53 during LV pressure overload. Our data also suggest that the proliferative and secretory phenotypes are at least partially mutually antagonistic. Deletion of p53 in Tcf21-lineage resident CF accelerates cell cycle, ultimately leading to an exaggerated fibrotic response. However, stimulation of CF proliferation is associated with a transient reduction in ECM deposition, leading to a delay in cardiac fibrosis.
p53 is as master regulator of the DNA damage response, and induces cell cycle arrest and apoptosis. Genotoxic insult prior to S phase leads to cell cycle arrest in G1 via the DNA damage checkpoint. Ataxia telangiectasia mutated (ATM), a phosphoinositol 3-kinase-related family serine/threonine kinase, is recruited to double-strand DNA breaks, leading to the phosphorylation and activation of checkpoint kinase 2 (CHK2) and p5337, 38. Subsequently, p53 induces a transcriptional response that facilitates cell cycle arrest, cellular senescence, or apoptotic cell death, depending upon the duration and severity of damage39, 40. Previous reports implicate ATM in pathologic cardiac remodeling and regeneration through diverse mechanisms including the DNA damage response23, 24, 41-43. In the heart, Angiotensin II promotes reactive oxygen species (ROS) production via angiotensin II receptor (AT1R)-dependent activation of Nox144, and ROS induces DNA damage via single and double strand breaks that activate the ATM/p53 system45, 46. We previously found that p53 stability is reduced in CF treated with TGF-β1 / H2O219. Thus, our findings may link pressure overload and ROS production to p53-dependent control of CF proliferation.
The CF response to cardiac insult transitions through various temporally regulated phases of wound healing, including inflammatory, proliferative, secretory (ECM generation), and maturation (scar remodeling)6, 7. The proliferative phase generally occurs early in the response to cardiac insult, prior to overt scar formation. We have found that genes governing cell cycle exit become induced concomitant with myofibroblast genes18. This led us to question whether CF proliferation and ECM secretion are mutually antagonistic. The data presented here support this hypothesis, but also suggest the answer is not straightforward. While p53-deficient CF become excessively proliferative following TAC, p53-CF KO mice exhibit a delayed but exaggerated fibrotic response.
scRNA-sequencing analyses of CF obtained at 14 days post-TAC did not define a clear distinction between proliferative and secretory CF. In fact, highly proliferative p53 null CF can still acquire a myofibroblast phenotype. Thus, a potential weakness of this study is the limited animal numbers (n=2 WT and 2 KO) analyzed by scRNA-sequencing; while statistical power is provided by the large number of individual cells analyzed, increasing the number of individual animals may allow for identification of more subtle differences. Nevertheless, we found that a majority of p53 null fibroblasts accumulate as proto-myofibroblasts that are poised to contribute to fibrosis, and those that acquire the myofibroblast phenotype express lower levels of genes encoding ECM proteins than WT CF. Likewise, deletion of p53 in cultured CF stimulates the cell cycle while suppressing myofibroblast activation. It is interesting to speculate that p53 may directly target genes that encode both cell cycle inhibitors and ECM components, providing a single unifying transcriptional mechanism controlling the transition out of the proliferative phenotype. However, since overexpression of Cdkn1a in p53 null CF at least partially restores the myofibroblast phenotype, it is likely that p53 independent mechanisms are at least partially responsible for the mutual antagonism. Metabolic changes are reported to impact fibroblast plasticity47, suggesting reallocation of energy and resources from cell replication to ECM generation may be necessary for myofibroblast activation. Regardless of the transcriptional mechanism, our data indicate that cell cycle exit facilitates the transition to a fully secretory fibroblast phenotype.
The specific timing of CF proliferation following cardiac insult suggests tight control over cell cycle is essential for proper healing. As a master regulator of the cell cycle, cellular senescence and apoptosis, p53 activity is governed by post-translational modifications that include phosphorylation, ubiquitination and acetylation. The primary mode of p53 regulation is via the E3 ubiquitin ligase MDM2, which is responsible for ubiquitination and proteasomal degradation of p5348-51. We previously discovered a small proline rich protein (Sprr2b) that is induced in CF during LV pressure overload. Sprr2b promotes ubiquitination of p53 by MDM2, leading to its proteasomal degradation and relieving constraints on the cell cycle19, 48-51. Inhibition of p53 using a small molecule MDM2 antagonist is reported to block the senescence associated secretory phenotype (SASP), which induces various pathophysiological processes, including inflammation and angiogenesis52-54. Intercellular communication between CF and myocytes may therefore be partially responsible for the cardiac hypertrophy observed following TAC. It is also possible that increased matrix deposition, which occurs concomitant with hypertrophy, decreases ventricular compliance and exacerbates pathological cardiac remodeling. Future studies should investigate whether alterations in p53 activity in activated fibroblasts alters paracrine signaling programs to impact phenotypes including cardiac hypertrophy.
In the present study, we observe a compensatory induction of Cdkn2a gene expression in p53 null CF. The Cdkn2a locus generates alternatively transcribed mRNAs encoding p14Arf and P16Ink4a, which control MDM2 and Rb1 activity, respectively. p14Arf and P16Ink4a thus represent complementary means to regulate cell cycle exit and cellular senescence from a common genetic locus55,56. However, the antagonistic role of p14Arf on MDM2 activity is futile in the absence of p53. We observe an enrichment of p16Ink4a levels upon p53 deletion at 28 days post-TAC. p16Ink4a dependent Rb1 activation induces cell cycle arrest and is a well-established marker of cellular senescence57, 58. TGF-β1 induces p16Ink4a expression, connecting myofibroblast activation to cellular senescence programs59; indeed, myofibroblasts acquire a senescent-like phenotype in the mature scar that is thought to facilitate structural stability6. Since p53 deletion primarily accelerates proliferation in the second week post-TAC, we hypothesize that p53 controls the magnitude of CF accumulation within the normal time-window. Other factors, such as P16Ink4a / Rb1, may solidify cell cycle exit at later timepoints. It is important to note that scRNA-seq did not detect induction of Cdkn2a in p53 null fibroblasts in vivo; however, this experiment was performed at 14 days post-TAC, when the proliferative window is just beginning to wane. Future studies are required to investigate whether Cdkn2a and p16Ink4a become active later in LV-pressure overload to ensure cell cycle exit.
Overall, these results suggest inhibition of CF proliferation may provide therapeutic benefit in fibrotic cardiac remodeling. Indeed, overexpression of Cdkn1a in transgenic mice suppressed ischemia-induced CF proliferation and was proposed as an anti-fibrotic strategy25. MDM2 inhibitors that stabilize the p53 protein are currently undergoing clinical trials with the goal of combating various neoplasias60, 61, and may be considered for blocking CF accumulation in heart failure. Our data suggests cell cycle inhibition strategies would be most effective early in the disease progression, but might not provide benefit if used in established heart failure. Our data also imply that cell cycle arrest may have the unintended consequence of stimulating ECM deposition by existing fibroblasts. Future studies are certainly warranted to investigate the potential that pharmacological inhibition of CF proliferation, via p53-dependent cell cycle exit or other means, can prevent the development of fibrosis and cardiac functional decline in heart disease.
Supplementary Material
Novelty and Significance.
What Is Known?
Fibroblasts secrete excessive amounts of extracellular matrix (ECM) in response to pathologic cardiac insults such as left ventricle (LV) pressure overload and myocardial infarction.
Cardiac fibroblasts proliferate during a precise time-window following cardiac insult, leading to the accumulation ECM-secreting cells.
Strategies that prevent fibroblast accumulation in mouse heart disease models have been shown to attenuate the development of pathological cardiac fibrosis.
What New Information Does This Article Contribute?
Cardiac fibroblast proliferation in a mouse model of LV pressure overload is limited in part by the tumor suppressor protein, P53, and its transcriptional target Cdkn1a.
Deletion of P53 specifically in fibroblasts leads to their excessive proliferation in LV pressure overload and the development of pathological fibrosis.
The proliferative and reparative phases of the cardiac injury response remain temporally distinct even upon P53 deletion, at least partially due to mutually antagonistic control of the proliferative and ECM-secretory gene programs in fibroblasts.
Sources of Funding:
This work was supported by grants from the National Institutes of Health to EMS (R01HL133761, R01HL144867, R01HL136179, and UL1-TR002001), RMB (F32HL136066 and T32HL007937-15), JKL (T32HL066988-15), and a grant from the American Heart Association to JKL (15POST25550114).
Non-standard abbreviations and Acronyms:
- Acta2
Smooth muscle alpha actin 2
- AngII
Angiotensin II
- Atf3
Activating transcription factor 3
- ATM
Ataxia telangiectasia mutated kinase
- BrdU
Bromodeoxyuridine
- CCA
Canonical correlation analysis
- Cdkn1a
Cyclin dependent kinase inhibitor 1a
- Cdkn2a
Cyclin-dependent kinase inhibitor 2a
- CF
Cardiac fibroblast
- CHK2
Checkpoint kinase 2
- CHP
Collagen hybridizing peptide
- CSA
Cross-sectional area
- EC
Endothelial cell
- ECM
Extracellular matrix
- EGFP
Enhanced green fluorescent protein
- ERG
ETS-related gene
- FACS
Fluorescence-activated cell sorting
- FN
Fibronectin
- LV
Left ventricle
- MDM2
Mouse double minute 2 homolog
- P53
Tumor protein p53
- PDGFR α
Platelet-derived growth factor receptor a
- Postn
Periostin
- ROS
Reactive oxygen species
- SASP
Senescence associated secretory phenotype
- TAC
Trans aortic constriction
- TCF21
Transcription factor 21
- TGF-β1
Transforming growth factor beta 1
- TMX
Tamoxifen
- UMAP
Uniform manifold approximation and projections
- WGA
Wheat germ agglutinin
Footnotes
Disclosures: None
References
- 1.Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde AV, Conway SJ, Graff J and Frangogiannis NG. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ Res. 2019;124:1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JA and Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. [DOI] [PubMed] [Google Scholar]
- 3.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. [DOI] [PubMed] [Google Scholar]
- 4.Moore-Morris T, Tallquist MD and Evans SM. Sorting out where fibroblasts come from. Circ Res. 2014;115:602–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z and Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol. 2018;114:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, Kurmangaliyev YZ, Nasiri R, Ahadian S, Nguyen T, et al. Type V Collagen in Scar Tissue Regulates the Size of Scar after Heart Injury. Cell. 2020;182:545–562e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE and Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation. 2020;142:1448–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J and Martin JF. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garvin AM, De Both MD, Talboom JS, Lindsey ML, Huentelman MJ and Hale TM. Transient ACE (Angiotensin-Converting Enzyme) Inhibition Suppresses Future Fibrogenic Capacity and Heterogeneity of Cardiac Fibroblast Subpopulations. Hypertension. 2021;77:904–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lighthouse JK and Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol. 2016;91:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K and Davis J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crider BJ, Risinger GM Jr., Haaksma CJ, Howard EW and Tomasek JJ. Myocardin-related transcription factors A and B are key regulators of TGF-beta1-induced fibroblast to myofibroblast differentiation. J Invest Dermatol. 2011;131:2378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexanian M, Przytycki PF, Micheletti R, Padmanabhan A, Ye L, Travers JG, Gonzalez-Teran B, Silva AC, Duan Q, Ranade SS, et al. A transcriptional switch governs fibroblast activation in heart disease. Nature. 2021;595:438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janbandhu V, Tallapragada V, Patrick R, Li Y, Abeygunawardena D, Humphreys DT, Martin E, Ward AO, Contreras O, Farbehi N, et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell. 2022;29:281–297e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lighthouse JK, Burke RM, Velasquez LS, Dirkx RA Jr., Aiezza A 2nd, Moravec CS, Alexis JD, Rosenberg A and Small EM. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke RM, Lighthouse JK, Quijada P, Dirkx RA Jr., Rosenberg A, Moravec CS, Alexis JD and Small EM. Small proline-rich protein 2B drives stress-dependent p53 degradation and fibroblast proliferation in heart failure. Proc Natl Acad Sci U S A. 2018;115:E3436–E3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vousden KH and Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. [DOI] [PubMed] [Google Scholar]
- 21.Meyer K, Hodwin B, Ramanujam D, Engelhardt S and Sarikas A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J Am Coll Cardiol. 2016;67:2018–28. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh AK, Bhattacharyya S and Varga J. The tumor suppressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. J Biol Chem. 2004;279:47455–63. [DOI] [PubMed] [Google Scholar]
- 23.Sarig R, Rimmer R, Bassat E, Zhang L, Umansky KB, Lendengolts D, Perlmoter G, Yaniv K and Tzahor E. Transient p53-Mediated Regenerative Senescence in the Injured Heart. Circulation. 2019;139:2491–2494. [DOI] [PubMed] [Google Scholar]
- 24.Feng T, Meng J, Kou S, Jiang Z, Huang X, Lu Z, Zhao H, Lau LF, Zhou B and Zhang H. CCN1-Induced Cellular Senescence Promotes Heart Regeneration. Circulation. 2019;139:2495–2498. [DOI] [PubMed] [Google Scholar]
- 25.Pu W, Han X, He L, Li Y, Huang X, Zhang M, Lv Z, Yu W, Wang QD, Cai D, et al. A genetic system for tissue-specific inhibition of cell proliferation. Development. 2020;147. [DOI] [PubMed] [Google Scholar]
- 26.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino S, Vooijs M, van Der Gulden H, Jonkers J and Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 29.Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He L, Huang X, Kanisicak O, Li Y, Wang Y, Li Y, Pu W, Liu Q, Zhang H, Tian X, et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J Clin Invest. 2017;127:2968–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen B and Frangogiannis NG. Chemokines in Myocardial Infarction. J Cardiovasc Transl Res. 2020. [DOI] [PubMed] [Google Scholar]
- 32.Das S, Goldstone AB, Wang H, Farry J, D'Amato G, Paulsen MJ, Eskandari A, Hironaka CE, Phansalkar R, Sharma B, et al. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019;176:1128–1142e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagchi RA, Roche P, Aroutiounova N, Espira L, Abrenica B, Schweitzer R and Czubryt MP. The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype. BMC Biol. 2016;14:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dick FA and Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith J, Tho LM, Xu N and Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. [DOI] [PubMed] [Google Scholar]
- 38.Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y and Chen DJ. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7. [DOI] [PubMed] [Google Scholar]
- 39.Stracker TH and Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mochan TA, Venere M, DiTullio RA Jr., and Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–91. [PubMed] [Google Scholar]
- 41.Foster CR, Zha Q, Daniel LL, Singh M and Singh K. Lack of ataxia telangiectasia mutated kinase induces structural and functional changes in the heart: role in beta-adrenergic receptor-stimulated apoptosis. Exp Physiol. 2012;97:506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foster CR, Daniel LL, Daniels CR, Dalal S, Singh M and Singh K. Deficiency of ataxia telangiectasia mutated kinase modulates cardiac remodeling following myocardial infarction: involvement in fibrosis and apoptosis. PLoS One. 2013;8:e83513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia L, Zhang W, Ma Y, Chen B, Liu Y, Piao C, Wang Y, Yang M, Liu T, Zhang J, et al. Haplodeficiency of Ataxia Telangiectasia Mutated Accelerates Heart Failure After Myocardial Infarction. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–85. [DOI] [PubMed] [Google Scholar]
- 45.Imlay JA and Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–9. [DOI] [PubMed] [Google Scholar]
- 46.Krohn K, Maier J and Paschke R. Mechanisms of disease: hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat Clin Pract Endocrinol Metab. 2007;3:713–20. [DOI] [PubMed] [Google Scholar]
- 47.Lombardi AA, Gibb AA, Arif E, Kolmetzky DW, Tomar D, Luongo TS, Jadiya P, Murray EK, Lorkiewicz PK, Hajnoczky G, et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun. 2019;10:4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang S, Jensen JP, Ludwig RL, Vousden KH and Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–51. [DOI] [PubMed] [Google Scholar]
- 49.Honda R and Yasuda H. Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene. 2000;19:1473–6. [DOI] [PubMed] [Google Scholar]
- 50.Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW and Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18:565–76. [DOI] [PubMed] [Google Scholar]
- 51.Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, El-Deiry WS and Yang X. Critical role for Daxx in regulating Mdm2. Nat Cell Biol. 2006;8:855–62. [DOI] [PubMed] [Google Scholar]
- 52.Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, Aird KM, Iwasaki O, Kossenkov AV, Schultz D, et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 2019;21:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJ, et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med. 2016;8:362ra144. [DOI] [PubMed] [Google Scholar]
- 54.Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, Desprez PY, Benz C, Davalos AR and Campisi J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018;8:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rivandi M, Khorrami MS, Fiuji H, Shahidsales S, Hasanzadeh M, Jazayeri MH, Hassanian SM, Ferns GA, Saghafi N and Avan A. The 9p21 locus: A potential therapeutic target and prognostic marker in breast cancer. J Cell Physiol. 2018;233:5170–5179. [DOI] [PubMed] [Google Scholar]
- 56.Bostrom J, Meyer-Puttlitz B, Wolter M, Blaschke B, Weber RG, Lichter P, Ichimura K, Collins VP and Reifenberger G. Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C (p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol. 2001;159:661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simboeck E and Di Croce L. p16INK4a in cellular senescence. Aging (Albany NY). 2013;5:590–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campisi J and d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. [DOI] [PubMed] [Google Scholar]
- 59.Vijayachandra K, Higgins W, Lee J and Glick A. Induction of p16ink4a and p19ARF by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol Carcinog. 2009;48:181–186. [DOI] [PubMed] [Google Scholar]
- 60.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–40. [DOI] [PubMed] [Google Scholar]
- 61.Reis B, Jukofsky L, Chen G, Martinelli G, Zhong H, So WV, Dickinson MJ, Drummond M, Assouline S, Hashemyan M, et al. Acute myeloid leukemia patients' clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica. 2016;101:e185–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muzumdar MD, Tasic B, Miyamichi K, Li L and Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 63.Pachitariu M and Stringer C. Cellpose 2.0: how to train your own model. Nat Methods. 2022;19:1634–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.