

Abstract
Background
Clonal hematopoiesis of indeterminate potential (CHIP) and clonal cytopenia of undetermined significance (CCUS) are defined by somatic mutations in genes associated with myeloid neoplasms (MN) at a variant allele fraction (VAF) ≥ 0.02, in the absence and presence of cytopenia, respectively. CHIP/CCUS is highly prevalent in adults and defining predictors of MN risk would aid clinical management and research.
Methods
We analyzed sequenced exomes of healthy UK Biobank (UKB) participants (n = 438,890) in separate derivation and validation cohorts. Genetic mutations, laboratory values, and MN outcomes were used in conditional probability-based recursive partitioning and Cox regression to determine predictors of incident MN. Combined statistical weights defined a clonal hematopoiesis risk score (CHRS). Independent CHIP/CCUS patient cohorts were used to test prognostic capability of the CHRS in the clinical setting.
Results
Recursive partitioning distinguished CHIP/CCUS cases with 10-year probabilities of MN ranging from 0.0078 - 0.85. Multivariable analysis validated partitioning variables as predictors of MN. Key features, including single DNMT3A mutations, high risk mutations, ≥ 2 mutations, VAF ≥ 0.2, age ≥ 65 years, CCUS vs CHIP and red blood cell indices, influenced MN risk in variable direction. The CHRS defined low risk (n = 10018, 88.4%), intermediate risk (n = 1196, 10.5%), and high risk (n = 123, 1.1%) groups. In clinical cohorts, most MN events occurred in high risk CHIP/CCUS patients.
Conclusions
The CHRS provides simple prognostic framework for CHIP/CCUS, distinguishing a high risk minority from the majority of CHIP/CCUS which has minimal risk for progression to MN.
Introduction
A critical goal of cancer early detection is to identify individuals with pre-malignant states at greatest risk of progression. Clonal hematopoiesis (CH), a pre-malignant expansion of a population of blood cells derived from a single hematopoietic stem cell,1 is often caused by somatic mutations in leukemia driver genes.2–4 Under the broader category of CH, two conditions are formally defined. Clonal hematopoiesis of indeterminate potential (CHIP) is categorized by CH with somatic mutations detectable at a variant allele fraction (VAF) of ≥ 2% in the absence of a diagnosed blood disorder or cytopenia. Clonal cytopenia of undetermined significance (CCUS) describes CHIP in the presence of unexplained, persistent cytopenias.5,6
More than 10% of individuals over age 60 have CHIP or CCUS (CHIP/CCUS), and diagnosis rates are increasing, partially due to use of next generation sequencing (NGS) to evaluate unexplained cytopenias and “liquid biopsies” to evaluate solid malignancies.7,8 The overall rate of transformation for CH is ~0.5-1% per year. Similar to many premalignant states, most individuals with CHIP/CCUS do not progress to MN, though progression risks as high as 90% have been reported in certain populations.9,10 Risk stratification aids clinicians by identifying high risk patients in whom early intervention may be appropriate, while avoiding the toxicities11 associated with overdiagnosis, unnecessary monitoring and treatment in low risk patients.
Several studies have identified features associated with evolution to MN, including mutations in certain high-risk genes, specific patterns of co-mutation, larger clone size as determined by variant allele fraction (VAF) and having CCUS instead of CHIP.9,10,12–15 However, systematic risk prognostication tools do not exist for CHIP/CCUS. We leveraged analysis of genetic, laboratory and MN outcomes data from 438,890 UK Biobank (UKB) participants to definitively identify features of CHIP/CCUS which predict risk of MN. Statistically weighted features combined to yield the clonal hematopoiesis risk score (CHRS), a simple prognostic model which distinguishes high risk CHIP/CCUS from low risk CHIP/CCUS in population and patient cohorts.
Methods
UK Biobank cohorts and molecular annotation:
UK Biobank (UKB)16 data were extracted under application 50834 from a cohort of 502,490 participants aged 40-70 years recruited between 2006-2010. Detection of somatic variants in whole exome sequencing (WES) was as previously described15 and pathogenic somatic variants in at least 1 gene associated with CH or myeloid malignancy were used to define CH.2,17,18 A list of included genes and average coverage per gene has been previously published.19 Individuals with low abundance clones (defined by VAF < 0.02), missing laboratory values, and myeloid malignancy preceding or within 6 months of study enrollment were excluded from analysis. Of the 438,890 individuals eligible for study, 193,743 were used for model derivation and 245,147 were used for validation (Figure S1). Returned SNP-array data (“Return 3094”)20,21 was used to independently annotate mosaic chromosomal abnormalities (mCAs) using estimated break-points and relevance to hematologic malignancies based on cBioPortal for cancer genomics22 and the Atlas of Genetics and Cytogenetics in Oncology and Haematology.23 For this study, mCA refers to myeloid mCA (m-mCA) and “ambiguous” mCA (A-mCA) that were common to both myeloid and lymphoid malignancy.15 Lymphoid-specific mCA were not analyzed. Additional details on molecular analyses are available in Supplemental Methods.
CHIP and CCUS designations
CHIP and CCUS were defined by the presence of somatic mutations at VAF ≥ 0.02. CH in the absence of cytopenia was classified as clonal hematopoiesis of indeterminate potential (CHIP) and CH in persons with at least 1 cytopenia was classified as clonal cytopenia of undetermined significance (CCUS). Cytopenias were defined using World Health Organization5,24 (WHO) criteria (anemia = hemoglobin concentration (Hgb) < 13.0 g/dL in men and < 12.0 g/dL in women; thrombocytopenia = platelet counts (Plt) < 150 x 109 cells/L; and neutropenia = absolute neutrophil count (ANC) < 1.8 x 109 cells/L). Bone marrow analysis was unavailable for UKB participants.
Variables and outcomes of interest
Participants were followed from the time of study enrollment until death or 12/31/2021, whichever was earliest. Extracted variables included age, sex, laboratory values, self-reported smoking history, and prior cancer history. Prior cancer history was defined as solid or lymphoid malignancies occurring prior to initial study assessment. Table S1 shows the distribution of prior cancers.
The primary outcome of interest included incident myeloid neoplasia (MN) where MN was defined by diagnosis with myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML) or Philadelphia chromosome negative myeloproliferative neoplasms (Ph-MPN) any time after month 6 of study enrollment. Diagnoses were assessed by self-report and International Classification of Diseases version 10 (ICD-10) codes in linked national health record hospital data. Table S2 lists ICD-10 codes used.
Statistical analysis
Statistical analyses were performed using R statistical software. Figures were made with R and GraphPad Prism. All statistical tests were two-sided with statistical significance determined by p-value of < 0.05. Categorical variables were compared using Fisher’s exact test and Wilcoxon rank-sum test was used to compare continuous variables. Cumulative incidence of MN was estimated using a competing risks approach with p-values determined by Gray’s test. Cumulative incidences are reported at 5- and 10-years. Overall survival was estimated using the Kaplan-Maier method with p-values determined by log-rank test.
Prognostic model derivation was performed using a two-staged approach. First, for CHIP/CCUS cases in the UKB with at least 10 years of follow-up (n=10,559), conditional-probability based recursive partitioning (RP) analysis was performed using the r-part package (https://cran.r-rproject.org/web/packages/rpart/index.html) and minimal complexity pruning with incident MN within 10 years as the single binary outcome. Additional information on RP method, including all variables used is available in Supplemental Methods. Variables identified by RP analysis were then used in multivariable Cox models to generate statistical weights. Regression models were adjusted for smoking status and prior cancer history, both independent risk factors for MN.25,26 Summed variable weights determined clonal hematopoiesis risk score (CHRS) values which were used define low, intermediate and high risk groups. Model performance was evaluated using receiver operator characteristics (ROC) analysis and model concordance (c-index).
Hematology patient cohorts
The Dana-Farber/Brigham and Women’s Hospital (DFCI/BWH) CHIP/CCUS cohort included all patients diagnosed in hematology clinics with CHIP/CCUS between 2014-2019 with follow-up until 12/1/2021. The Pavia CCUS cohort, an independent cohort of 99 patients with bone marrow biopsy confirmed CCUS, was followed between 2003-2019 at the Department of Hematology, Policlinico San Matteo and University of Pavia Italy. Patients with a prior MN history and those missing more than 1 CHRS variable were excluded. Remaining missing values were handled by stochastic regression imputation.
Results
Baseline characteristics of UK Biobank derivation cohort
We analyzed whole exome sequencing data from 193,743 study-eligible UKB participants and identified 11,337 individuals who met criteria for having CHIP or CCUS.5 Median follow-up time was 11.7 years [IQR 10.9, 12.6]. Compared to CHIP (n = 10,479), a greater proportion of CCUS (n = 858) was male (51.4% vs. 44.5%, p = 9.87x10−5) and CCUS was more commonly associated with prior cancer history (10.5% vs. 7.8%, p = 0.0072). No difference in age or smoking history was noted between CHIP and CCUS (Table 1 and Table S3). Anemia, thrombocytopenia and neutropenia were mostly mild and detected in 58.3%, 34.6% and 14.7% of CCUS, respectively (Table S4). Red cell distribution width (RDW) and mean platelet volume (MPV) were higher in CCUS compared to CHIP (Table S4).
Table 1.
Characteristics of UK Biobank Derivation Cohort
| No CHIP/CCUS (N = 182406) |
All CHIP/CCUS (N = 11337) |
CHIP (N = 10479) |
CCUS1 (N = 858) |
|
|---|---|---|---|---|
|
| ||||
| Demographics, Duration of Follow-up, Prior Cancer History and Smoking Status | ||||
|
| ||||
| Sex | ||||
|
| ||||
| Female | 100200 (54.9%) | 6235 (55.0%) | 5818 (55.5%) | 417 (48.6%) |
| Male | 82206 (45.1%) | 5102 (45.0%) | 4661 (44.5%) | 441 (51.4%) |
|
| ||||
| Age (years) | ||||
|
| ||||
| Median [IQR] | 57.0 [50, 63] | 62.0 [57, 66] | 62.0 [57, 66] | 62.0 [56, 66] |
|
| ||||
| Follow-up time2 (years) | ||||
|
| ||||
| Median [IQR] | 11.7 [10.8, 12.5] | 11.6 [10.8, 12.4] | 11.6 [10.8, 12.4] | 11.2 [10.6, 12.1] |
|
| ||||
| Any Smoking History | ||||
|
| ||||
| Yes | 102085 (56.0%) | 5703 (50.3%) | 5272 (50.3%) | 431 (50.2%) |
| No | 80322 (44.0%) | 5634 (49.7%) | 5207 (49.7%) | 427 (49.8%) |
|
| ||||
| Prior Cancer History3 | ||||
|
| ||||
| Prior Malignancy | 9844 (5.4%) | 908 (8.0%) | 818 (7.8%) | 90 (10.5%) |
| No Prior Malignancy | 172562 (94.6%) | 10429 (92.0%) | 9804 (92.2%) | 768 (89.5%) |
|
| ||||
| Laboratory Values and Cytopenias 4 | ||||
|
| ||||
| White Blood Cell Count (x109 cells/L) | ||||
|
| ||||
| Median [IQR] | 6.68 [5.69, 7.85] | 6.84 [5.77, 8.10] | 6.90 [5.82, 8.10] | 6.28 [4.87, 7.64] |
|
| ||||
| Hemoglobin (g/dL) | ||||
|
| ||||
| Median [IQR] | 14.1 [13.3, 15.0] | 14.1 [13.4, 15.0] | 14.2 [13.5, 15.1] | 12.6 [11.7, 14.0] |
|
| ||||
| Platelets (x109 cells/L) | ||||
|
| ||||
| Median [IQR] | 247 [213, 286] | 249 [213, 289] | 250 [216, 290] | 216 [142, 284] |
|
| ||||
| Neutrophil Count (x109 cells/L) | ||||
|
| ||||
| Median [IQR] | 4.03 [3.30, 4.95] | 4.16 [3.36, 5.13] | 4.20 [3.40, 5.15] | 3.67 [2.59, 4.83] |
|
| ||||
| Number of Cytopenias | ||||
|
| ||||
| 0 | 169801 (93.1%) | 10479 (92.4%) | 10479 (100%) | 0 (0%) |
| 1 | 12106 (6.6%) | 797 (7.0%) | 0 (0%) | 797 (92.9%) |
| 2 | 464 (0.3%) | 57 (0.5%) | 0 (0%) | 57 (6.6%) |
| 3 | 35 (0.0%) | 4 (0.04%) | 0 (0%) | 4 (0.5%) |
|
| ||||
| Anemia | ||||
|
| ||||
| No | 174878 (95.9%) | 10837 (95.6%) | 10479 (100%) | 358 (41.7%) |
| Yes | 7528 (4.1%) | 500 (4.4%) | 0 (0%) | 500 (58.3%) |
|
| ||||
| Thrombocytopenia | ||||
|
| ||||
| No | 178330 (97.8%) | 11040 (97.4%) | 10479 (100%) | 561 (65.4%) |
| Yes | 4076 (2.2%) | 297 (2.6%) | 0 (0%) | 297 (34.6%) |
|
| ||||
| Neutropenia | ||||
|
| ||||
| No | 180871 (99.2%) | 11211 (98.9%) | 10479 (100%) | 732 (85.3%) |
| Yes | 1535 (0.8%) | 126 (1.1%) | 0 (0%) | 126 (14.7%) |
|
| ||||
| Mean Corpuscular Volume (fL) | ||||
|
| ||||
| Median [IQR] | 91.3 [88.6, 93.9] | 91.4 [88.8, 94.1] | 91.4 [88.9, 94.1] | 90.6 [86.7, 94.1] |
|
| ||||
| Mean Platelet Volume (fL) | ||||
|
| ||||
| Median [IQR] | 9.20 [8.58, 9.94] | 9.18 [8.51, 9.92] | 9.15 [8.50, 9.90] | 9.47 [8.70, 10.60] |
|
| ||||
| Red Cell Distribution Width (%) | ||||
|
| ||||
| Median [IQR] | 13.3 [12.9, 13.9] | 13.4 [13.0, 14.0] | 13.4 [12.9, 13.9] | 14.0 [13.3, 15.1] |
A derivation cohort of 193,744 UK Biobank participants were evaluated by whole exome sequencing for presence or absence of clonal hematopoiesis (CH) and further classified as having clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenia of undetermined significance (CCUS) based on the absence or presence of cytopenia.
Cytopenias were defined such that individuals classified as CCUS had one or more of the following: anemia (hemoglobin <13 g/dL for male participants and hemoglobin <12 g/dL for female participants), thrombocytopenia (platelet count of <150K/uL) or neutropenia (absolute neutrophil count <1.8 K/uL).
Follow-up time is the number of years from sequencing to death or last follow-up (January 2021), whichever is earliest.
Prior Cancer History is defined as history of solid or lymphoid malignancy in the years prior to enrollment in study based on aggregated self-report and hospital records by International Classification of Disease (ICD-10) code (codes listed in Supplemental Table 1). Individuals with prior histories of myeloid malignancy were excluded. Cancer types contributing to prior cancer history in this population are indicated in Supplemental Table 3.
Hematologic parameters obtained from complete blood count and differential obtained at the time of sequencing.
Categorical variables are summarized by the number of events, N, and proportion (%). Continuous variables are summarized using median and interquartile range (IQR).
Consistent with prior reports,15,27 DNMT3A, TET2 and ASXL1 mutations were the most commonly mutated genes in CHIP/CCUS (Figure S2a). Individuals with CCUS had a higher VAF compared to CHIP (0.128 [IQR 0.076, 0.237] vs 0.111 [ 0.071, 0.189], p = 1.28x10−6, Figure S2b). Greater clonal complexity, defined by the presence of more than one mutation was higher in CCUS vs CHIP (15.0% vs 8.42%, p = 1.56 x 10−9) and CHIP/CCUS with a single mutation in DNMT3A (single DNMT3A) was the most common genotype (Figure S2c–e).
Genotype-specific risk of incident MN in UKB derivation cohort
Among the 11,337 individuals with CHIP/CCUS, there were 269 (2.37%) incident MN events (Figure 1a & Figure 1b). The cumulative incidence of MN was higher in CCUS compared to CHIP (Figure 1a & Table S5). Cox proportional hazards models performed with time to incident MN were performed for each gene and adjusted for sex, prior cancer history and smoking history. Hazards ratios > 5 were observed for mutations in SRSF2, SF3B1, ZRSR2, JAK2, RUNX1, and IDH2 (Figure 1c), and we classified these mutations as high risk. Mutations in splicing factor genes (SRSF2, SF3B1, and ZRSR2), AML-like genes (IDH1, IDH2, FLT3, and RUNX1), TP53-related genes (TP53 and PPM1D) were also evaluated as grouped variables in regression models (Figure 1d). Mutations in splicing factors and AML-like genes were associated with 9.26-fold (IQR 5.29, 16.2) and 13.8-fold (IQR 10.3, 18.4) increased risk of incident MN relative to other genotypes of CHIP/CCUS. We also classified these lesions as high risk. While no association was observed between TP53-related mutations and incident MN, we empirically added TP53 mutations to our final list of high risk mutations given prior data showing high penetrance for AML evolution,12,13 poor outcomes in TP53 mutant MDS/AML28–31, and the potential hazards of underestimating risk in TP53 mutant CHIP/CCUS. Compared to individuals without CHIP/CCCUS (No CHIP/CCUS), DNMT3A mutant had a 4.25-fold increased risk of MN (Figure S3). However, MN risk was markedly lower in DNMT3A mutant CHIP/CCUS compared to other genotypes (co-mutated DNMT3A: HR 0.273, 95% CI 0.209 – 0.355, p < 2 x 10−16, single DNMT3A: 0.188, 95% CI 0.138, 0.236, p < 2x10−16, Figure 1c & 1e–f).
Figure 1 – Features influencing risk of myeloid neoplasia (MN) in UKB participants with CHIP/CCUS.
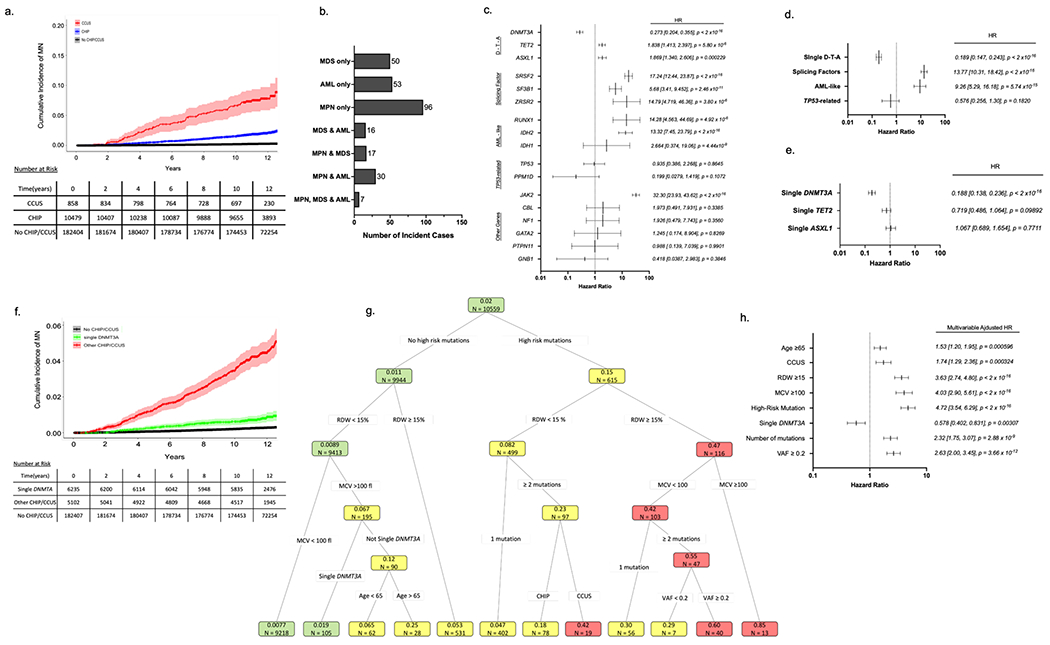
(a) Cumulative incidence of myeloid neoplasia (MN) in individuals with CHIP/CCUS compared to those without CHIP/CCUS. (b) Subtypes of MN among CHIP/CCUS patients who develop MN. (c) Univariate cox proportional hazard regression analysis for the 14 most commonly mutated genes in CH and (d) for groups of mutations including splicing factor mutations (SRSF2, SF3B1, ZRSR2) and AML-like mutations (IDH1, IDH2, FLT3, and RUNX1).) (e) Univariate analysis of single-DNMT3A, TET2 and ASXL1. (f) Cumulative incidence of MN for CHIP/CCUS possessing a single-DNMT3A mutation (green) compared to the cumulative incidence for all other CHIP/CCUS genotypes (red) and individuals without CHIP/CCUS (black). (g) For UK Biobank participants with at least 10-years of follow-up (n=10,559), recursive partitioning (RP) analysis was performed based on conditional probability of incident MN within 10-years. Of these, 207 incident MN events were recorded. Each node is annotated with number of individuals (n =) and probability of incident MN. Nodes are color coded as follows: probability ≤ 0.02 is green, 0.02-0.4 is yellow and >0.4 is red. Partitioning variable are all binary (presence vs absence of feature) and include high risk mutation (mutations in SRSF2, SF3B1, ZRSR2, IDH1, IDH2, FLT3, RUNX1 and JAK2), single DNMT3A, having ≥2 mutations, variant allele fraction (VAF) ≥0.2, having CCUS instead of CHIP, red cell distribution width (RDW) ≥15%, mean corpuscular volume (MCV) ≥100fl, and age ≥ 65 years. (h) Multivariable cox regression adjusted for assigned sex at birth, prior history of cancer and any history of smoking as confounders was performed on the entire cohort (n = 11,337) using features selected in RP analysis. For all Cox regression models hazard ratios (HR) are shown with error bars representing 95% confidence interval and numerical values for HR [95% confidence interval] and p-value for each feature analyzed.
Clonal hematopoiesis risk score (CHRS)
We used conditional probability-based RP analysis to identify critical predictors of 10 year probability of incident MN. A list of candidate variables used in analysis is provided in the Supplemental Methods. High risk mutations and single DNMT3A mutations were genotypes of greatest importance for classification. Other partitioning variables included, ≥ 65 years of age, CCUS vs CHIP, 2 or more mutations, a maximum VAF ≥ 0.2 (for any CH variant), MCV ≥ 100 femtoliters (fl) and RDW ≥ 15% (Figure 1g). Within sample size limitations, severity of cytopenia was not identified as a key partitioning variable. Cumulative incidence of MN was higher for CHIP/CCUS possessing each feature compared to cases lacking that feature (Figure S4 & Table S6).
RP analysis distinguished groups of CHIP/CCUS cases with a probability of incident MN by year 10 ranging from 0.0077 – 0.85 (Figure 1g), highlighting marked variability in risk of MN. Features identified in RP retained statistical significance in multivariable Cox regression analysis (Figure 1h). Regression coefficients (Table S6) for each variable were rounded to the nearest 0.5 and increased by 1 providing weighted scores for each prognostic variable (Table 2). Clonal hematopoiesis risk score (CHRS) values were calculated as the sum of scores for each prognostic variable. Most CHIP/CCUS cases had low CHRS values (Figure 2a). Score boundaries for risk groups were selected to prioritize creation of a low risk group with 10-year probability of incident MN < 2%. Three risk groups were defined (Figure 2b and Table S7): high risk (CHRS ≥ 12.5, n = 123, 1.1%), intermediate risk (CHRS 10 - 12, n = 1196, 10.5%) and low risk (CHRS ≤ 9.5, n =10018, 88.4%). Sample CHRS calculations and provisional calculator are provided in Supplemental Methods. The 10-year cumulative incidence of MN was 52.2 ± 4.96%, 7.83 ± 0.807, and 0.669 ± 0.0827% in high, intermediate and low risk CHIP/CCUS, respectively (Figure 2b & 2c). ROC analysis for the CHRS at each year of observation indicated an overall model concordance (c-index) of 0.807 ± 0.016 in the UKB derivation cohort. Notably, empiric addition of TP53 mutations as high risk did not significantly impact model c-index (Figure S5). Relative to low risk CHIP/CCUS, risk of incident MN was 11.8-fold and 101.1-fold higher in intermediate risk and high risk CHIP/CCUS (p < 2x10−16, Figure 2d). When we compared risk of incident MN in CHRS risk groups to individuals without CHIP/CCUS, we observed risk increases of 3.32-fold, 37.1-fold, and 348-fold in low, intermediate and high risk CHIP/CCUS, respectively (p < 2x10−16, Figure 2e).
Table 2:
Clonal hematopoiesis risk score (CHRS) values
| Prognostic Variable | 0.5 | 1 | 1.5 | 2 | 2.5 |
|---|---|---|---|---|---|
| Single DNMT3A | present | absent | – | – | – |
| High Risk Mutation | – | absent | – | – | present |
| Mutation Number | – | 1 | – | ≥ 2 | – |
| Variant Allele Fraction | – | < 0.2 | – | > 0.2 | – |
| Red Cell Distribution Width | – | < 15 | – | – | ≥ 15 |
| Mean Corpuscular Volume | – | < 100 | – | – | > 100 |
| Cytopenia | – | CHIP | CCUS | – | – |
| Age | – | < 65y | ≥ 65y | – | – |
Figure 2 – Clonal hematopoiesis risk score (CHRS) distribution and risk stratification in UKB Derivation Cohort.
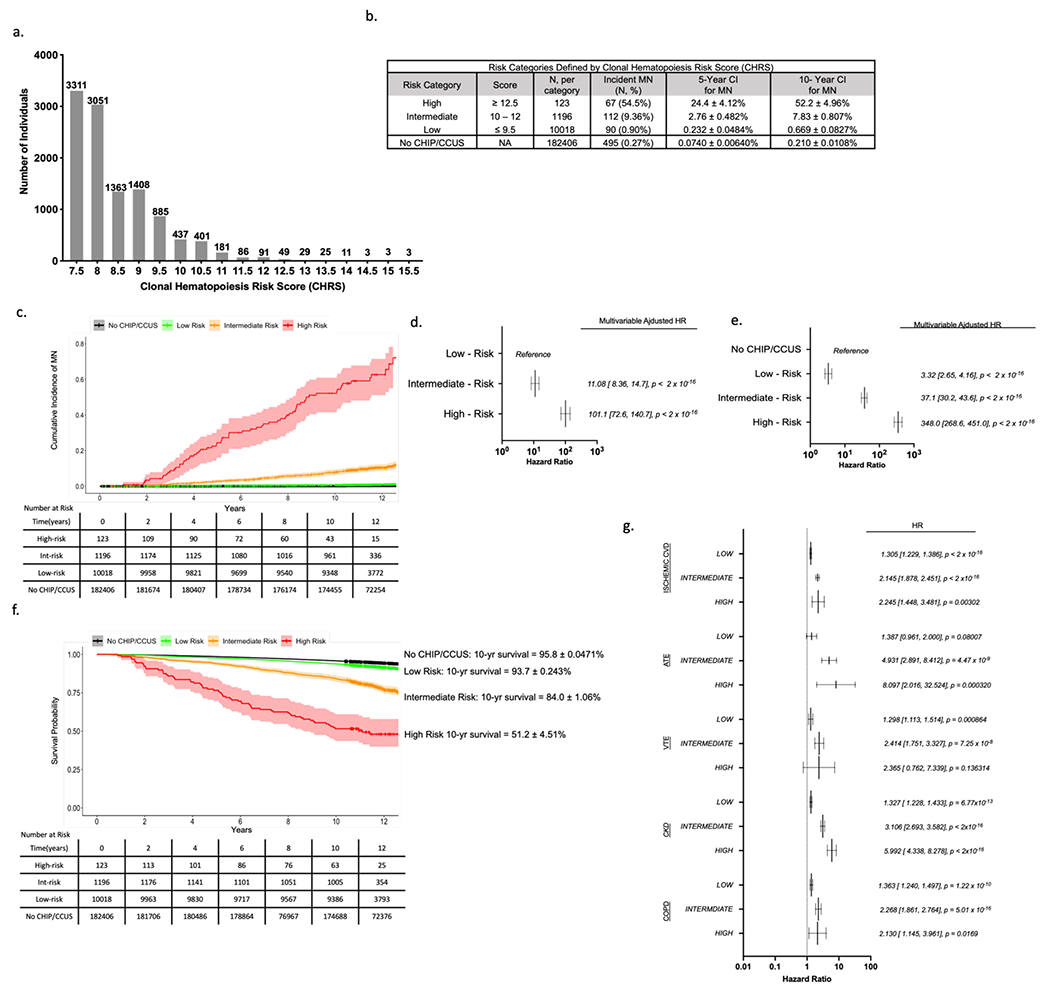
(a) Number of individuals with each possible CHRS value (number of individuals with each score is indicated above the bar). (b) Risk-Categories were defined by CHRS value with cutoffs chosen to minimize risk in low-risk strata. For each category, the number individuals in the risk group, number of myeloid neoplasia (MN) events and crude event rate (N, %), as well as the 5- and 10-year cumulative incidences (± standard deviation) is shown. Cumulative incidence of MN for individuals without CHIP or CCUS (No CHIP/CCUS) in the derivation cohort is included for reference. (c) Cumulative incidence curves of MN by CHRS risk category. Curves correspond to cumulative incidence analysis used to derive figures in panel (b). Hazard ratios for incident MN were determined for CHRS risk strata using Cox proportional hazards models adjusted for sex, smoking history and history of prior cancer. Hazard ratios were calculated in models with low risk strata as the reference population (d) and using the population of 182406 UKB participants in the No CHIP/CCUS group as the reference population (e). (f) Survival by CHRS risk category is shown, with 10-year survival is annotated to the right of the graph for each category. For both cumulative incidence and survival curves, Black = No CHIP/CCUS, Green = Low Risk, Orange = Intermediate Risk, and Red = High Risk. The ribbon about each curve indicates the 95% confidence interval. Tables show number at risk. (g) results of Cox regression analyses for non-malignant outcomes by CHRS risk group. For all outcomes, NO CHIP/CCUS is the reference population. Outcomes shown include ischemic cardiovascular disease (CVD) which is a composite of atherosclerosis, ischemic heart failure, myocardial infarction and stroke; arterial thromboembolic events (ATE); venous thromboembolic events (VTE); chronic kidney disease (CKD); and chronic obstructive pulmonary disease (COPD). For panels (d), (e) and (g), forest plots indicate HR (95% confidence interval) and p-values for main effects.
CHRS groups showed clear survival differences. In high risk CHIP/CCUS, 10-year survival was 51.2 ± 4.51% compared to 84.0 ± 1.06% in intermediate, 93.7 ± 0.243% in low risk CHIP/CCUS and 95.8 ± 0.0471% in individuals without CHIP/CCUS (Figure 2f). Higher risks for CH-related comorbidities, including ischemic cardiovascular disease (CVD), arterial and venous thromboembolic disease (ATE/VTE), chronic kidney disease (CKD) and chronic obstructive pulmonary disease (COPD) were observed in high risk CHIP/CCUS compared to intermediate and low risk CHIP/CCUS (Figure 2g & Table S8).
Validation of CHRS Model
We used a distinct subset of 245,147 UKB participants as a validation cohort. Validation and derivation cohort characteristics were similar (Table S9). Of the 16,274 individuals with CHIP/CCUS, 14,755 (90.6%) were low risk for incident MN (Figure S6 & Table S10), similar to the rate of low risk CHIP/CCUS in the derivation cohort. The c-index for the CHRS in the validation cohort was 0.799 ± 0.015 indicating the model performed equally well in both validation and derivation datasets.
Patients with CHIP/CCUS referred to hematology clinics within tertiary referral centers have a higher risk of developing MN relative to individuals with incidentally detected CH in population cohorts like UKB. We applied the CHRS model to two independent patient cohorts for validation in the clinical setting. CH variants for the 646 patients in the DFCI/BWH CHIP/CCUS cohort were detected by clinical NGS32 on peripheral blood or bone marrow (30.2%) in patients evaluated for unexplained cytopenia or for suspected CH identified on solid tumor or hereditary cancer sequencing panels. The Pavia CCUS cohort includes 99 patients with bone marrow biopsy proven CCUS diagnosed in hematology clinics at the University of Pavia. Cohort characteristics are summarized in Table S11. Rates of mutations in DNMT3A, TET2, ASXL1 and high risk genes are shown in Figure 3a & 3c.
Figure 3 – External validation of CHRS in Hematology Patient Cohorts.
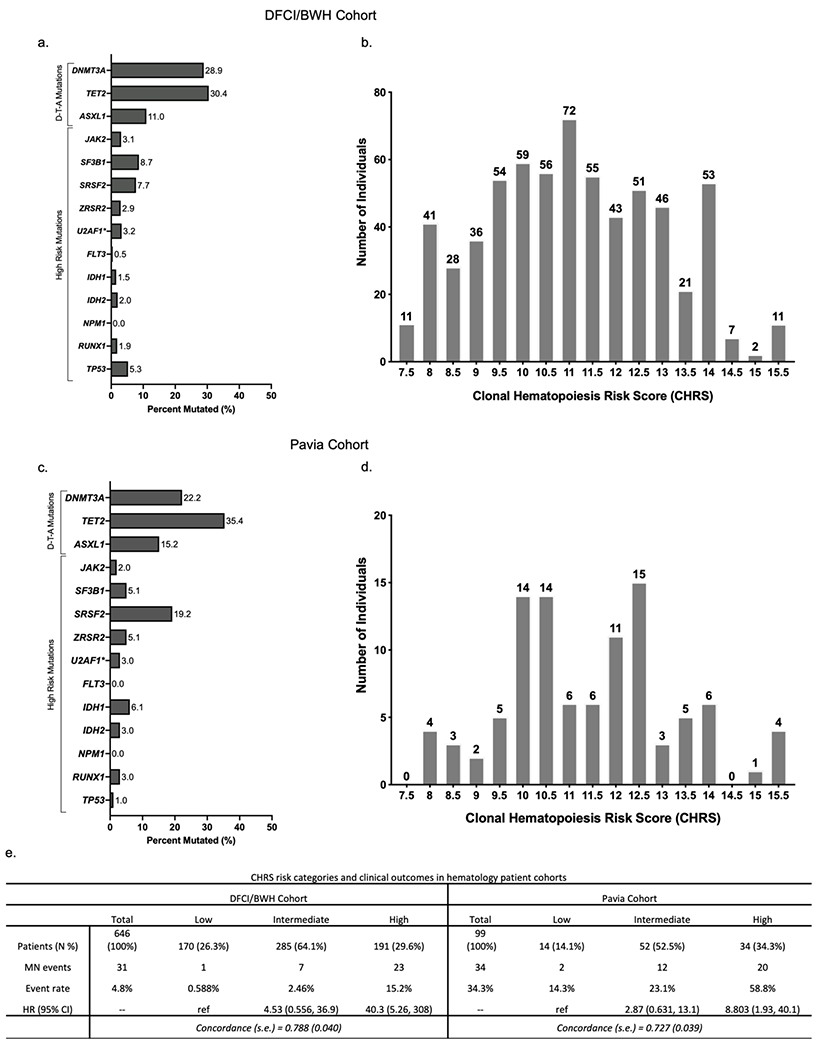
Distribution of mutations (a & c) and CHRS values in (b & d) in the DFCI/BWH CH cohort and Pavia CCUS cohorts. (e) CHRS risk categories and outcomes (incident MN) in DFCI/BWH cohort (left) and Pavia cohort (right). Number of patients with in each category is shown with percentage (N %). The number of incident myeloid neoplasia (MN) cases (MN events) and the event rate (MN events relative to number of individuals in that category, expressed as percentage). Cox proportional hazard models were used to obtain hazard ratios (95% CI) for each CHRS risk strata and performance of the CHRS model is estimated by the concordance statistic (c-index ± standard error) when applied to each cohort.
Both hematology patient cohorts had higher CHRS values relative to UKB cohorts (Figure 3b & d), yet the CHRS predicted MN outcomes well. Crude event rates for MN were 4.8% (n = 31) and 31.3% (n = 31) in the DFCI/BWH and Pavia cohorts, respectively. Most MN events occurred in high risk patients and the HR for MN was 40.3 (95% CI 5.26, 308) in the DFCI/BWH cohort and 8.80 (95% CI 1.93, 40.1) in the Pavia cohort (Figure 3e). Despite brief follow-up, ROC analysis indicated a c-index of 0.788 (± 0.040) for the DFCI/BWH cohort and 0.727 (± 0.039) for the Pavia cohort (Figure S7).
Influence of mosaic chromosomal abnormalities (mCA) on risk of MN
Chromosomal mosaicism is not yet routinely assessed in patients with cytopenia or CHIP/CCUS, so mCAs were not used to derive the CHRS. However, 11.1% of low risk CHIP/CCUS that progressed to MN had co-occurring mCA (Table S12). We analyzed the association of myeloid mCA with risk of incident MN. Cumulative incidence of MN was substantially higher in CHIP/CCUS with ≥ 1 co-occurring mCA (Figure S8), consistent with prior reports of mCA as independent risk factors for MN.15,20,21,33–36 Among CHRS risk groups, MN risk was higher when mCA were present. In high risk CHIP/CCUS, the 10-year cumulative incidence of MN was 83.3 ± 8.20% with mCA and 43.1 ± 5.61% without mCA (Figure S8g). When mCA are treated as “high risk” somatic alterations in the CHRS, 1.49% of CHIP/CCUS cases are reclassified resulting in a modest increase in c-index to 0.826 ± 0.15 (Figure S9).
Discussion
Here, we report genetic and laboratory features required to predict incident MN in CHIP/CCUS and incorporate them into the CHRS, a novel prognostic model which predicts myeloid malignancy outcomes. The CHRS robustly defines three distinct CHIP/CCUS risk groups and demonstrates the low absolute risk of progression to overt MN in the vast majority of CHIP and CCUS.
This large cohort analysis enabled estimates of absolute risk of incident MN, unlike prior case-control studies in CH.9,10,14 Among UKB participants, individuals with CHRS-defined low risk CHIP/CCUS (~90% of CHIP/CCUS cases), like those without CHIP/CCUS, have a < 1% 10-year cumulative incidence of MN, whereas high risk CHIP/CCUS (1.1% of CHIP/CCUS cases) has a > 50% 10-year cumulative incidence of MN.
CHRS risk groups correlated well with survival and select CH-related non-malignant comorbidities such as ischemic cardiovascular disease.37,38 Given the prevalence of CHIP/CCUS, this finding has broad implications. Shared predictors allude to common pathophysiology and the potential for early intervention strategies that improve overall survival via prevention of both malignant and non-malignant outcomes in CHIP/CCUS. Dedicated risk models for non-malignant outcomes would clarify this possibility.
We definitively demonstrate marked genotype specificity for risk of progression in CHIP/CCUS, as observed in MDS.39–42 Specifically, our data highlight a more benign risk profile for DNMT3A mutant CHIP/CCUS, particularly single DNMT3A cases which have an 80% lower risk of MN relative to other genotypes. These findings are consistent with lower growth rates for DNMT3A mutant HSC clones compared to other CH mutations43 and with low rates of progression to donor-derived MDS/AML in stem cell transplant recipients with DNMT3A mutant donor-derived CH.44 We also established a category of high risk mutations which includes mutations in splicing factor genes (SRSF2, SF3B1, and ZRSR2), AML-like genes (IDH1, IDH2, FLT3 and RUNX1), JAK2 and TP53. Notably, the splicing factor U2AF1 is absent from our analysis due to an erroneous duplication on chromosome 21 in the hg38 reference genome45 which precluded reliable calls of U2AF1 variants. Recent data have demonstrated the rapid clonal expansion and high rate of transformation to myeloid malignancy46 for U2AF1 mutant CH. Datasets with reliable U2AF1 variant calls are necessary to confirm that mutations in U2AF1 like other splicing factor genes are high risk.
CHRS performance was not significantly altered by the empiric inclusion of TP53 as a high risk mutation. Heterogeneity of MN risk among TP53-mutant CHIP/CCUS is probable, with outcomes influenced by clonal selection pressures such as chemotherapay,47 clone size,48 allelic state,49 and clonal complexity.50 While assessment of the aforementioned factors and variant-specific risk may enhance precision of CH risk algorithms, we were constrained by lack of serial samples, limited information about timing and duration of selection pressures, and small sample sizes for specific variants. The importance of clonal complexity and allelic state enhance the case for assessment of mCA in patients with CHIP/CCUS and our analysis indicates these are best incorporated into the CHRS as one would high risk mutations.
Compared to prior published models predicting risk of MN in adults,51,52 the CHRS permits risk assessment using objective data obtained at a single time point from two peripheral blood tests: a next generation sequencing (NGS) panel and a complete blood count with differential. Despite limited duration of follow-up, the CHRS defines high and low risk in typical CHIP/CCUS patients and c-index >0.7 in two independent cohorts validates the CHRS in clinical settings. To facilitate clinical uptake, the CHRS calculator derived in this article is available in the Supplementary Appendix at https://evidence.nejm.org/ and at https://www.chrsapp.com.
CHIP/CCUS provides a substrate for developing early cancer detection programs which center identification of somatic mutations in cancer driver genes. However, because somatic mosaicism is ubiquitous and CHIP/CCUS is highly prevalent53 with substantial heterogeneity in outcomes, health systems would become overwhelmed if all individuals with CHIP/CCUS were referred for extensive hematologic evaluation. The CHRS provides an intuitive and adoptable framework for prognostication in CHIP/CCUS. In so doing, the CHRS aids clinical decision and research, allowing prioritization of intensive surveillance and therapeutic intervention in the minority of CHIP/CCUS patients who are most likely to progress to overt MN.
Supplementary Material
Acknowledgements
This work was supported by grants from the NIH (R01HL082945, P01CA066996, P50CA206963 and R35CA253125), Adelson Medical Research Foundation, Howard Hughes Medical Institute and Fondation Leducq awarded to BLE. LDW was supported by grants from the Edward P Evans foundation for MDS, the ASH/RWJF Harold Amos Medical Faculty Development Program and the Wood Foundation. AN was supported by funds from Knut and Alice Wallenberg Foundation (KAW2017.0436). PN was supported by grants from the NIH (R01HL142711, R01HL148050, R01HL151283, R01HL148565), Fondation Leducq, and Massachusetts General Hospital (Hassenfeld Research Scholar). SJ is supported by the Burroughs Wellcome Foundation Career Award for Medical Scientists, Foundation Leducq, Ludwig Center for Cancer Stem Cell Research, the Leukemia and Lymphoma Society, and the NIH Director’s New Innovator Award (DP2-HL157540). RCL was supported by a Scholar award from The Leukemia & Lymphoma Society. The authors thank Anne Charles and the data management team of the Dana-Farber Hematologic Malignancy Data Repository for facilitating access to CH patient data and thank the patients for their invaluable contribution to this effort.
Conflicts of interest and disclosures
BLE has received research funding from Celgene, Deerfield, and Novartis and consulting fees from GRAIL. He serves on the scientific advisory boards for Skyhawk Therapeutics, Exo Therapeutics, and Neomorph Therapeutics and TenSixteen Bio, all unrelated to this work. DN is a current equity holder in Madrigal pharmaceuticals, unrelated to this work. ML has received research funding from Novartis and Abbvie and honoraria from Pfizer, all unrelated to this work. RCL has received consulting fees from bluebird bio, Takeda Pharmaceuticals, Qiagen, Nuprobe, and Thermo Fisher, all unrelated to this work. RMS reports grants from AbbVie, Agios, Arog, and Novartis and has received personal fees from AbbVie, Actinium, Agios, Argenx, Apteva, Astella, AstraZeneca, Biolinerx, Celgene, Daiichi-Sankyo, Elevate, Gemoab, Janssen, Jazz, Macrogenics, Novartis, Otsuka, Pfizer, Hoffman LaRoche, Stemline, Syndax, Syntrix, Syros, Takeda, and Trovagene, all unrelated to this work. DD has received research funding from Abbvie, Glycomimetics and Novartis as well as consulting fees from Blueprint Medicines, Incyte, Forty-Seven, Autolus, Agios, Amgen, Shire, Takeda, Novartis, Pfizer and Jazz, all unrelated to this work. RS is a member on the board of directors of Kladis, Be the Match/National Marrow Donor Program and Juno and has received personal fees from Alexion, Gilead, Rheos, Jazz and Vor Biopharma, all unrelated to this work. AGB is a current holder of stock options in TenSixteen Bio, unrelated to this work. SJ is a consultant to Novartis, AVRO Bio, Roche Genentech, and Foresite Labs, and is on the scientific advisory board and holds equity interest in TenSixteen Bio and Bitterroot Bio, all unrelated to this work. PN reports grant support from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Genentech, and Novartis, advisory board participation and equity interest in TenSixteen Bio and spousal employment at Vertex, all unrelated to this work. The remaining authors declare no competing financial interests.
References
- 1.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genovese G, Jaiswal S, Ebert BL, McCarroll SA. Clonal hematopoiesis and blood-cancer risk. N Engl J Med. 2015;372(11):1071–1072. [DOI] [PubMed] [Google Scholar]
- 4.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Integrating Morphological, Clinical, and Genomic Data. Blood. 2022. [Google Scholar]
- 7.Razavi P, Li BT, Brown DN, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25(12):1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Ulrich BC, Supplee J, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res. 2018;24(18):4437–4443. [DOI] [PubMed] [Google Scholar]
- 9.Malcovati L, Galì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galì A, Todisco G, Catamo E, et al. Relationship between clone metrics and clinical outcome in clonal cytopenia. Blood. 2021;138(11):965–976. [DOI] [PubMed] [Google Scholar]
- 11.Gondek LP, DeZern AE. Assessing clonal haematopoiesis: clinical burdens and benefits of diagnosing myelodysplastic syndrome precursor states. Lancet Haematol. 2020;7(1):e73–e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24(7):1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niroula A, Sekar A, Murakami MA, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. 2021;27(11):1921–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol. 2017;35(14):1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bick AG, Weinstock JS, Nandakumar SK, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586(7831):763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agrawal M, Niroula A, Cunin P, et al. TET2-mutant clonal hematopoiesis and risk of gout. Blood. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loh PR, Genovese G, Handsaker RE, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018;559(7714):350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh PR, Genovese G, McCarroll SA. Monogenic and polygenic inheritance become instruments for clonal selection. Nature. 2020;584(7819):136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huret JL, Ahmad M, Arsaban M, et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013;41(Database issue):D920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenberg PL, Tuechler H, Schanz J, et al. Cytopenia levels for aiding establishment of the diagnosis of myelodysplastic syndromes. Blood. 2016;128(16):2096–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroll ME, Murphy F, Pirie K, Reeves GK, Green J, Beral V. Alcohol drinking, tobacco smoking and subtypes of haematological malignancy in the UK Million Women Study. Br J Cancer. 2012;107(5):879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goel S, Hall J, Pradhan K, et al. High prevalence and allele burden-independent prognostic importance of p53 mutations in an inner-city MDS/AML cohort. Leukemia. 2016;30(8):1793–1795. [DOI] [PubMed] [Google Scholar]
- 29.Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. [DOI] [PubMed] [Google Scholar]
- 30.Bowen D, Groves MJ, Burnett AK, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23(1):203–206. [DOI] [PubMed] [Google Scholar]
- 31.Grob T, Al Hinai ASA, Sanders MA, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2022;139(15):2347–2354. [DOI] [PubMed] [Google Scholar]
- 32.Kluk MJ, Lindsley RC, Aster JC, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J Mol Diagn. 2016;18(4):507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao T, Ptashkin R, Bolton KL, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021;12(1):338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs KB, Yeager M, Zhou W, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terao C, Suzuki A, Momozawa Y, et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature. 2020;584(7819):130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weeks LD, Marinac CR, Redd R, et al. Age-related diseases of inflammation in myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2022;139(8):1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bersanelli M, Travaglino E, Meggendorfer M, et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J Clin Oncol. 2021;39(11):1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard E, Tuechler H, Greenberg Peter L, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence;0(0):EVIDoa2200008. [DOI] [PubMed] [Google Scholar]
- 43.Fabre MA, de Almeida JG, Fiorillo E, et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature. 2022;606(7913):335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibson CJ, Kim HT, Zhao L, et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J Clin Oncol. 2021:Jco2102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller CA, Walker JR, Jensen TL, et al. Failure to Detect Mutations in U2AF1 due to Changes in the GRCh38 Reference Sequence. The Journal of Molecular Diagnostics. 2022;24(3):219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pritzl SL, Gurney M, Badar T, et al. Clinical and Molecular Spectrum and Prognostic Outcomes of U2AF1 Mutant Clonal Hematopoiesis-Prospective Mayo Clinic Cohort Leukemia Research. 2022;In Press. [DOI] [PubMed] [Google Scholar]
- 47.Bolton KL, Ptashkin RN, Gao T, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sallman DA, Komrokji R, Vaupel C, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2016;30(3):666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haase D, Stevenson KE, Neuberg D, et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia. 2019;33(7):1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oster HS, Crouch S, Smith A, et al. A predictive algorithm using clinical and laboratory parameters may assist in ruling out and in diagnosing MDS. Blood Adv. 2021;5(16):3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Radhachandran A, Garikipati A, Iqbal Z, et al. A machine learning approach to predicting risk of myelodysplastic syndrome. Leuk Res. 2021;109:106639. [DOI] [PubMed] [Google Scholar]
- 53.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.