

Abstract
Despite recent progress, it remains challenging to program biomacromolecules to assemble into discrete nanostructures with pre-determined sizes and topologies. We report here a novel strategy to address this challenge. By using two orthogonal pairs of heterodimeric coiled coils as the building blocks, we constructed six discrete supramolecular assemblies, each composed of a prescribed number of coiled coil components. Within these assemblies, different coiled coils were connected via end-to-side covalent linkages strategically pre-installed between the non-complementary pairs. The overall topological features of two highly complex assemblies, a “barbell” and a “quadrilateral” form, were characterized experimentally and were in good agreement to the designs. This work expands the design paradigms for peptide-based discrete supramolecular assemblies and will provide a route for de novo fabrication of functional protein materials.
Keywords: α-Helices, Coiled coils, Heterodimer, Nanostructures, Peptides, Supramolecular assemblies
Graphical Abstract
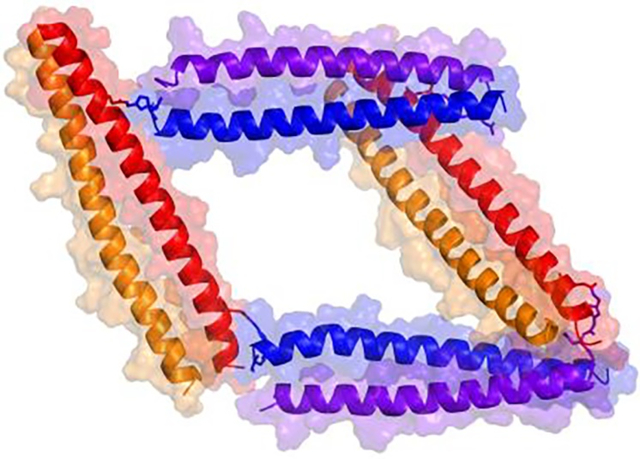
We report a novel strategy for constructing peptide-based supramolecular assemblies with pre-determined oligomeric states and topologies. By using four polypeptide chains that form two orthogonal heterodimeric coiled coils and pre-installing covalent linkages between the non-complementary pairs in strategic arrangements, several discrete supramolecular assemblies with pre-determined molar masses and topologies were constructed.
Introduction
Supramolecular assembly of peptides is a bio-inspired and powerful bottom-up approach that has been widely used to create a range of biomaterials with tunable functions for a variety of applications.[1] By implementing rational design strategies along with computational algorithms, nanostructured materials with different geometries and dimensions, such as nanofibers[2], nanotubes[3], nanosheets[2c, 4] and metal-coordinated assemblies,[5] can be fabricated via self-assembly of an individual peptide component or by co-assembly from multiple peptide entities. Although the attractive molecular interactions are always carefully designed to ensure the successful assembly, the termination of these cohesive forces are often not considered thoroughly, leading to difficulties in precisely controlling the size of resultant supramolecular assemblies. Nanostructured materials with monodisperse sizes are crucial for better quality control and an in-depth understanding of structure-function relationships.
A capping strategy that has emerged recently shows potential as a versatile method to alleviate the challenges of size control.[6] Physically mixing the capping component and non-capping building blocks with different molar ratios has been applied to successfully control and narrow the length distribution of peptide nanofibers[6a] and nanotubes[6b]. In addition to optimizing the molar ratio, it is also important to fine tune the relative strength of three different types of associations. “Heteromeric” associations between non-capping building blocks and capping components are desired, as are “homomeric” associations between the non-capping building blocks. The balance between these two types of association strongly influences the size distribution of the resultant assemblies. A third type of association, homomeric interactions between capping components, are undesired and should be curtailed or eliminated. Precise control of these three types of associations would ideally lead to the formation of discrete molecular assemblies with monodisperse and predictable size. Heterodimeric coiled coils formed by the intermolecular assembly of two complementary polypeptide chains are versatile building blocks and can be considered the simplest case of discrete peptide-based supramolecular assemblies. We considered whether the aforementioned precise control of three different types of associations could be achieved by linking multiple pairs of heterodimeric coiled coils together in strategically designed arrangements, leading to the predictable formation of complex assemblies.
The capabilities for precisely constructing peptide-based supramolecular structures assembled from a defined and pre-determined number of basic building blocks are still largely unrealized to date. There are, however, a few precedents demonstrating some successes for achieving such constructions using heterodimeric coiled coils. The Ghosh group has constructed four-arm dendrimers with monodisperse and pre-determined sizes, in which four identical heterodimeric coiled coils were tethered to a zero-generation poly(amidoamine) dendrimer core.[7] The Woolfson group has conjoined two or three heterodimeric coiled coils, thus fashioning putatively rod-like nanostructures with predicted molar masses.[8] Utilizing the co-assembly of three different polypeptide chains, the Keating group has designed triangular shaped nanostructures which are composed of three different covalently-connected heterodimeric coiled coils.[9] From the topological point of view, these precedent works used the termini of heterodimeric coiled coils as the linking points. Such a linking strategy allowed the relatively easy preparation of the linear polypeptide chains using either chemical synthetic methods or biological expression approaches. However, it may cause undesired intramolecular associations which could result in the formation of dead-end monomeric species and also limit the topological space that the final assembled products can access.[10]
We envisioned that installing covalent linkages between two or more different heterodimeric coiled coils in an end-to-side (termini-to-side chains) arrangement would be an effective strategy for embellishing peptide-based discrete supramolecular assemblies. In order to achieve the end-to-side linkages, we introduced branch points in the side chains of selected peptide components. The amino acid sequences of a peptide chain that can form coiled coils are often based on tandem seven-residue repeats, denoted (abcdefg)n. The residues located at the “f” positions of the heptad repeats are exposed to the solvent and have negligible impact on the coiled coil tertiary structure.[11] Thus, these positions were selected as the branch points in order to minimize perturbations to the folding propensities of the original peptide sequences. The undesired intramolecular associations may be largely obviated when two peptides are linked together via a covalent bond between the terminus of one peptide and the side chain of another peptide. This is presumably due to the mis-alignment of the peptide hydrophobic faces that could otherwise drive the formation of the dead-end monomeric species. In addition, the undesired side chain-side chain interactions that may be caused by side-to-side linking would also be largely avoided. The end-to-side linkage strategy would also assist in defining the relative orientation of the heterodimeric coiled coils that will be present within the resultant supramolecular assemblies. We expected that peptide-based discrete supramolecular assemblies with diverse topologies and predicted sizes could be obtained via a set of end-to-side linking strategies. Herein, we report the implementation of these strategic linkages, leading to the successful construction of several discrete supramolecular assemblies composed of a predicted number of dimeric coiled coil components, with molar masses ranging from ~20 kDa to ~42 kDa.
Results and Discussion
Our strategy of constructing discrete supramolecular assemblies relies on a set of peptide sequences that can form orthogonal pairs of heterodimeric coiled coils. Upon searching the literature for a set of such sequences with solved crystal structures, we identified four suitable SYNZIP peptides developed by the Keating group: SYNZIP-1, −2, −5 and −6.[12] These four peptides can orthogonally associate with their complementary sequences to form two hetero-specific parallel coiled coil dimers, the SYNZIP-1/SYNZIP-2 dimer (pdb id: 3HE5) and the SYNZIP-5/SYNZIP-6 dimer (pdb id: 3HE4). In this work, we used truncated versions of these four peptides (Scheme 1), which retained their folding abilities and specificities for orthogonal interactions.
Scheme 1.
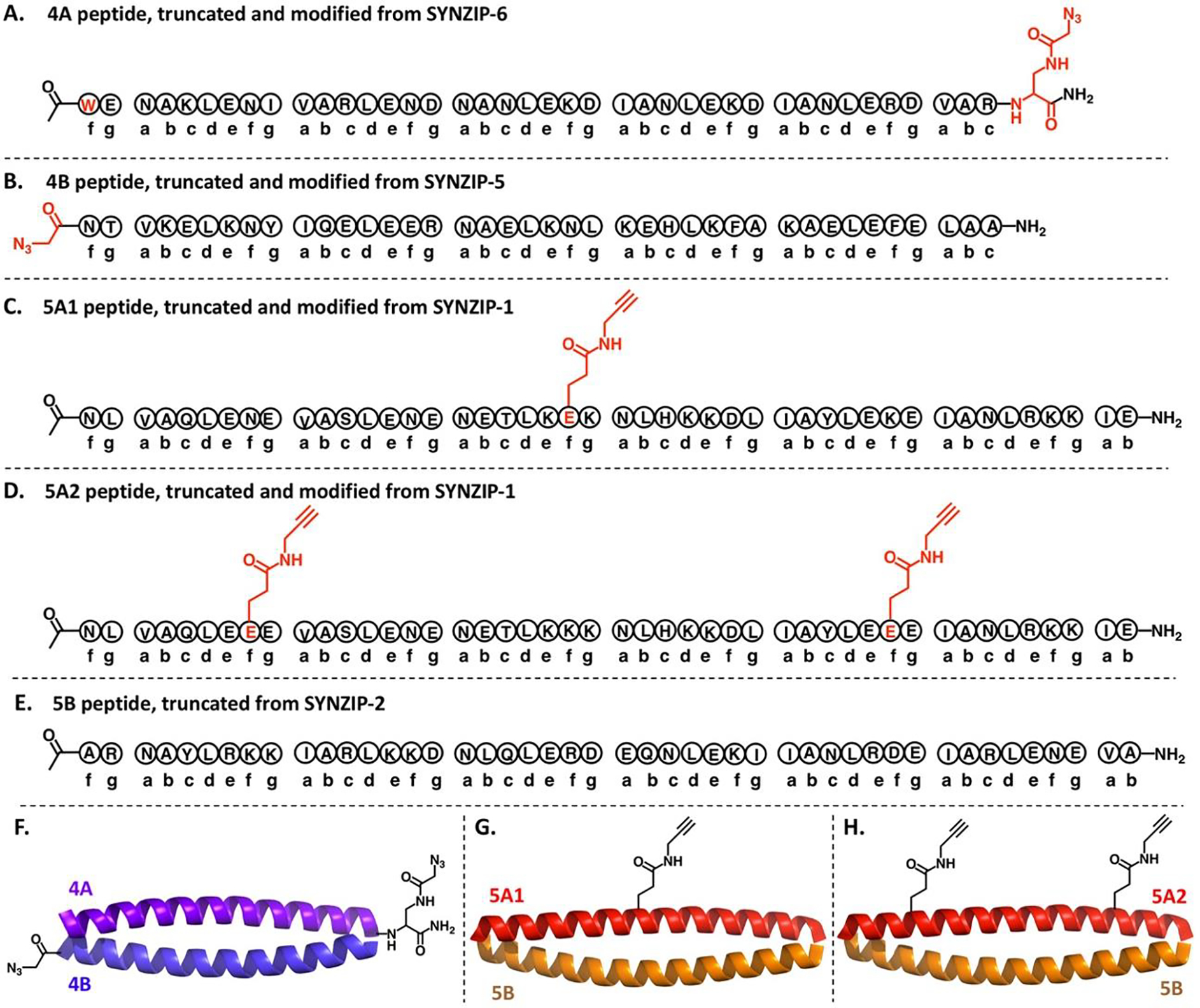
Amino acid sequences of each single chain peptide used in this work: (A) 4A, (B) 4B, (C) 5A1, (D) 5A2 and (E) 5B. Heptad repeat patterns are shown below the corresponding sequence. Mutations and side chain modifications are highlighted in red. Cartoon representations of three expected heterodimeric coiled coils: (F) 4A/4B dimer, (G) 5A1/5B dimer and (H) 5A2/5B dimer. Terminal and side chain modifications are highlighted by the corresponding chemical structures.
As shown in Scheme 1A, 4A is a 41-residue long peptide, initially synthesized to contain L-2,3-diaminopropanoic acid (Dap) as the C-terminal residue. An azido group was then installed in the side chain of the Dap residue through amide coupling between the side chain amino group and 2-azido acetic acid to complete the synthesis of 4A (Scheme S1). 4B is a 40-residue long peptide whose N-terminus was capped by an azidoacetyl group (Scheme 1B). 5A and 5B peptides both contain 46 residues. We opted to introduce branch points in the 5A peptide. 5A1 peptide was obtained by introducing one branch point in 5A by mutating Lys22 (the “f” position in the third heptad repeat) to Glu22. This subsequently enabled incorporation of an alkyne group through amide coupling between side chain carboxylic acid group of Glu22 and propargylamine (Scheme 1C and S2). We similarly introduced two branch points in 5A to obtain the 5A2 peptide. In this case, both Asn8 and Lys36 (“f” positions of the first and fifth heptad repeats) of 5A were mutated to glutamic acids, followed by installing the two side chain alkyne groups in the same way as for 5A1 (Scheme 1D). No mutation or side chain modification was made for 5B (Scheme 1E). The original sequences of 4B, 5A1, 5A2 and 5B peptides all contain tyrosine residues for accurate concentration determination by measurement of UV absorbance at 280 nm. Similarly, the Lys1 residue originally located at the N-terminus of 4A peptide was mutated to tryptophan (Scheme 1A). Each single chain peptide was synthesized using standard Fmoc-based solid phase peptide synthesis protocols. The purities were evaluated by analytical HPLC and the chemical identities were confirmed by mass spectrometry. (See supporting information for synthetic details and analytical data.)
Based on the design principles, 4A is complementary with 4B, while both 5A1 and 5A2 are complementary with 5B (Scheme 1F–1H). Stable dimeric coiled coils can be formed as a single species when the registered pairs are mixed at a 1:1 molar ratio. The hetero-association specificities of each single-chain peptide were confirmed by circular dichroism (CD) spectroscopy. Characteristic CD spectra of well-defined α-helical secondary structures (minima at 208nm and 222nm, respectively) were only observed for the mixtures of registered pairs with 1:1 molar ratios (Figure 1A). Isolated single chain peptides or equimolar mixtures of non-registered pairs showed CD spectra characteristic of weak helical secondary structures with very low thermal stabilities or non-helical secondary structures (Figure 1A, S18 and S19). Based on the temperature-dependent CD studies, the melting temperatures of the coiled coil tertiary structures were estimated at 47 °C for the 4A/4B mixture, 61°C for the 5A1/5B mixture and 63 °C for the 5A2/5B mixture (Figure 1B). The molar mass estimated by size exclusion chromatography in-line with multi-angle laser scattering (SEC-MALS) verified that the desired dimeric coiled coils were formed from the equimolar mixtures of registered pairs, 4A/4B, 5A1/5B and 5A2/5B (Figure 1C). Computer models of the three dimers were constructed based on the crystal structures of the parent coiled coil dimers.[12] Excellent agreements were found between the experimental small angle X-ray scattering (SAXS) data and the theoretical SAXS profiles modeled by the FoXS[13] computational platform (Figure 1D), suggesting that the overall solution-state structures of each dimeric coiled coil are well-represented by the corresponding model. These results indicate that the terminal and sidechain modifications we introduced have negligible perturbations to the heterodimeric coiled coil tertiary structures formed by the complementary pairs.
Figure 1.
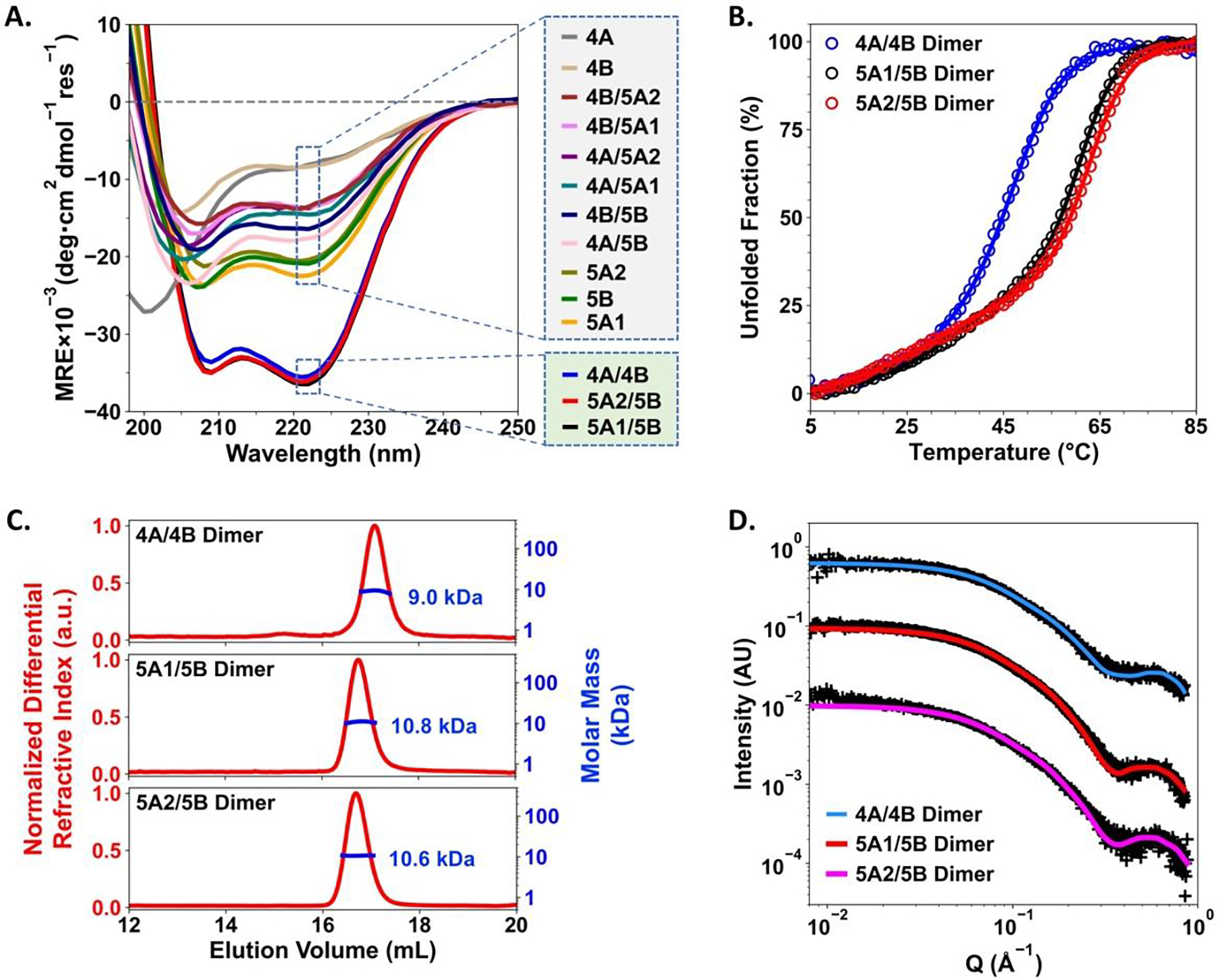
(A) Circular Dichroism spectra of single chain peptides in isolation, equimolar mixtures of non-registered pairs and equimolar mixtures of registered pairs. The “/” signs indicate equimolar mixtures. (B) Thermal denaturation profiles of heterodimeric coiled coils formed by the registered pairs, as monitored by CD at 222 nm. (C) SEC-MALS results of the three dimers. Expected molar mass, 4A/4B dimer: 9.7 kDa; 5A1/5B dimer: 10.7 kDa; and 5A2/5B dimer: 10.6 kDa. (D) Small angle X-ray scattering profiles of the 4A/4B, 5A1/5B and 5A2/5B dimers. Experimental data is represented by “+” symbols and theoretical scattering curves of computer models are shown in solid lines. Goodness of fit, χ2(4A/4B): 1.04; χ2(5A1/5B): 1.12; χ2(5A2/5B): 0.96.
We then moved to the synthesis of the peptide conjugates using copper (I) catalyzed azide/alkyne cycloaddition (CuAAC) “click” chemistry. Four different conjugated peptides were successfully prepared through the formation of triazole bonds between the azido groups at the side chain branch points of 5A1 or 5A2 and terminal alkyne groups of 4A or 4B, Con_5A1–4A, Con_5A1–4B, Con_5A2-(4A)2 and Con_5A2-(4B)2 (Figure 2 and S11–S17). None of these four conjugated peptides showed CD spectra with mean residue ellipticity higher than 20,000 at 222nm (Figure 2C), indicating that they were all only partially helical and did not form coiled coils. In addition, sigmoidal transitions were not observed by CD for any of these conjugated peptides during the thermal unfolding experiments (Figure S20), further evidencing the absence, as desired, of intramolecular associations which would compete with the designed intermolecular interactions in the presence of the complementary folding partners.
Figure 2.
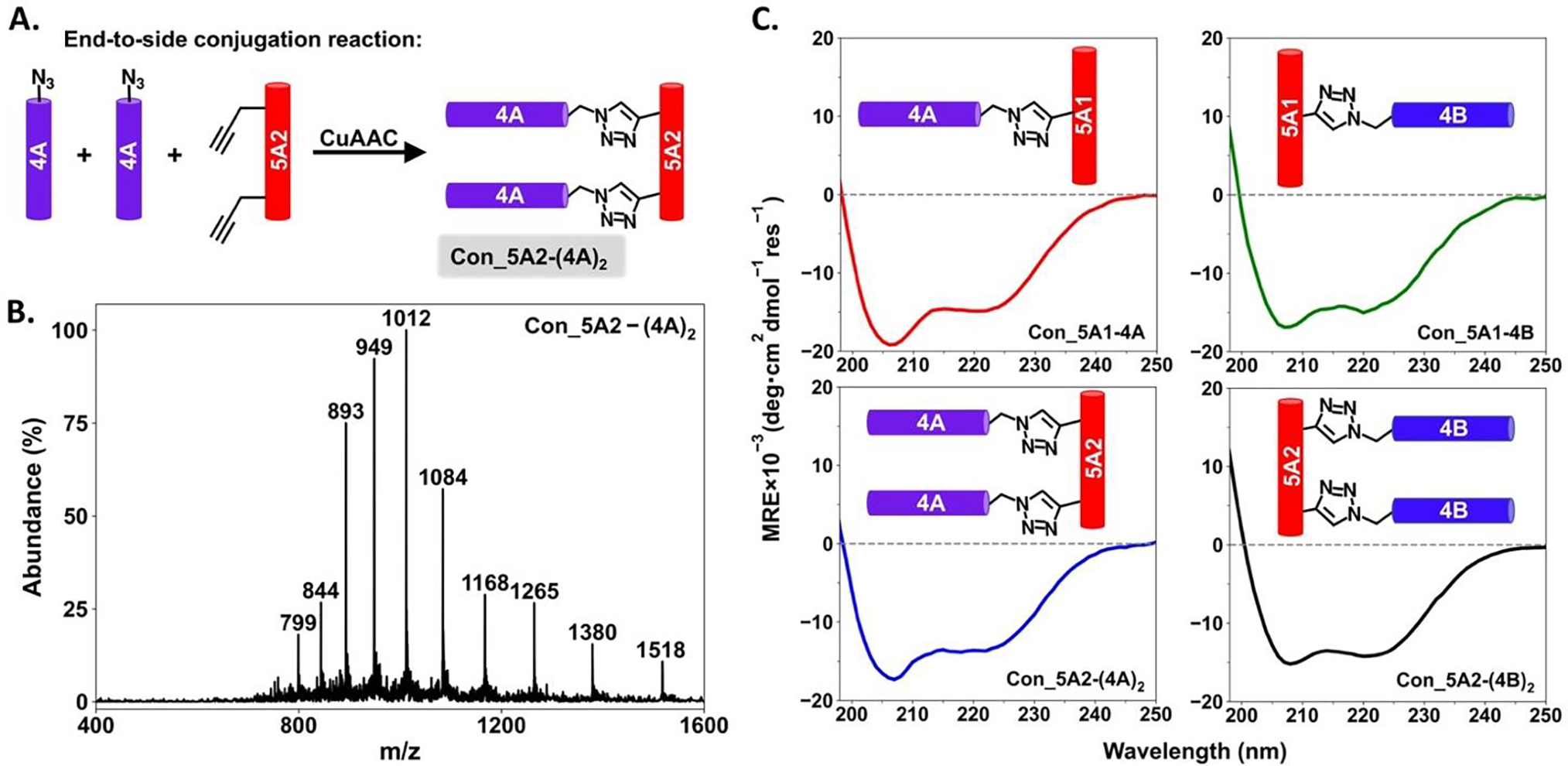
(A) Synthetic route for conjugating multiple peptide chains together at the branch points of 5A1 or 5A2 via CuAAC click chemistry, as exemplified for Con_5A2-(4A)2. (B) Mass spectrometry data for Con_5A2-(4A)2. Expected mass: [M+H]+/1=15171, [M+10H]10+/10=1518, [M+11H]11+/11=1380, [M+12H]12+/12=1265, [M+13H]13+/13=1168, [M+14H]14+/14=1085, [M+15H]15+/15=1012, [M+16H]16+/16=949, [M+17H]17+/17=893, [M+18H]18+/18=844, [M+19H]19+/19=799. (C) CD spectra of the four different conjugated peptides, Con_5A1–4A (upper left), Con_5A1–4B (upper right), Con_5A2-(4A)2 (lower left) and Con_5A2-(4B)2 (lower right). The subscripts indicate the number of peptide units. Schematic representation of each conjugated peptide is inserted to the corresponding CD spectrum. The weak signals at 208 nm and 222nm indicate the expected absence of coiled coil assemblies.
We conceived that barbell shaped supramolecular assembly composed of three dimeric coiled coil components could be constructed by mixing Con_5A1–4A, Con_5A1–4B and 5B with a 1:1:2 molar ratio (Figure 3C). Prior to constructing the barbell shaped assembly, two intermediate assemblies were constructed, pre-barbell_A and pre-barbell_B (Figure 3A and 3B). Pre-barbell_A assembly were obtained by preparing a mixture of Con_5A1–4A, 4B and 5B peptides with a 1:1:1 molar ratio. Two dimeric coiled coils were expected to be formed within this structure. Essentially, one 4A/4B dimer is covalently linked to one 5A1/5B dimer through a triazole bond formed between the C-terminus of 4A peptide and the side chain of 5A1 peptide. The CD spectra of this ternary mixture indicated that well-defined α-helical secondary structures were adopted by each peptide domain within the coiled coil context (Figure 3D). The coiled coil tertiary structures present within the pre-barbell_A assemblies were more resistant to thermal unfolding than that of isolated 4A/4B dimer but less resistant than isolated dimeric coiled coils of 5A1/5B (Figure 3E). This difference in thermal stabilities suggests that each dimeric coiled coil retained its native folding propensity in the context of pre-barbell_A assembly. The molar mass of pre-barbell_A assembly was estimated at ~21 kDa by SEC-MALS (Figure 3F top), in good agreement to the expected value of 20.6 kDa. Pre-barbell_B assembly were also successfully constructed by obtaining a mixture of Con_5A1–4B, 4A and 5B with 1:1:1 molar ratio. Pre-barbell_B showed near identical CD spectrum and thermal denaturation profile to that of pre-barbell_A (Figure 3D and 3E). The expected molar mass of pre-barbell_B (also 20.6 kDa) was similarly observed in the SEC-MALS analysis (Figure 3F middle). These results validated our hypothesis that these two intermediate assemblies are composed of one 4A/4B and one 5A1/5B dimeric coiled coil. They differ from each other by having the covalent linkage at different positions with respect to the 4A/4B dimer: at the C-terminus for pre-barbell_A and at the N-terminus for pre-barbell_B.
Figure 3.
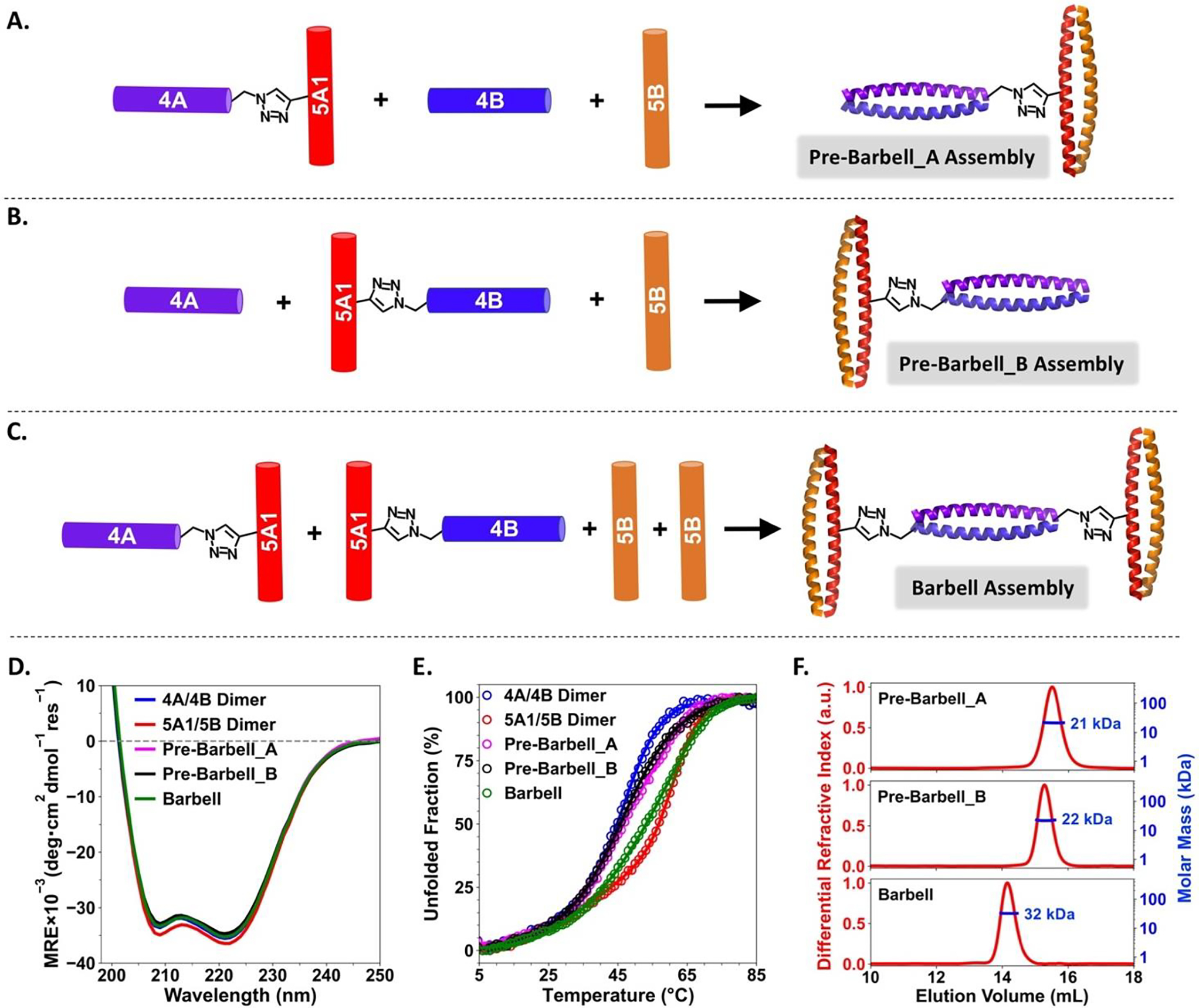
Preparation routes of pre-barbell_A (A), pre-barbell_B (B) and barbell shaped (C) assemblies. Comparison of CD spectra (D) and thermal denaturation profiles (E) between the dimeric building blocks (4A/4B and 5A1/5B dimers) and supramolecular assemblies in the barbell series. (F) SEC-MALS results of the three assemblies. Expected molar mass, pre-barbell_A assembly: 20.6 kDa; pre-barbell_B assembly: 20.6 kDa and barbell shaped assembly: 31.6 kDa.
The success of constructing both pre-barbell_A and pre-barbell_B assemblies indicated that the expected hetero-specific intermolecular associations are the dominant driving force in forming these two discrete supramolecular assemblies. The barbell shaped assembly were then constructed by preparing a mixture of Con_5A1–4A, Con_5A1–4B and 5B with a 1:1:2 molar ratio (Figure 3C). We expected one 4A/4B and two 5A1/5B dimeric coiled coils would form within this assembly. A CD spectrum with a strong α-helical signal was observed for this three-component mixture (Figure 3D). Due to the presence of one additional 5A1/5B dimer, the overall thermal stabilities of coiled coils tertiary structures presented within this assembly were stronger than that of the two pre-barbell assemblies, and somewhat weaker than that for the isolated 5A1/5B dimer (Figure 3E). SEC-MALS analysis indicated that a discrete supramolecular assembly with a molar mass ~32 kDa was obtained (Figure 3F bottom), in good agreement with the expected molar mass (31.6 kDa) of the barbell shaped assembly composed of two 5A1/5B dimers and one 4A/4B dimer.
We further conceived that replacing Con_5A1–4A and Con_5A1–4B in the barbell shaped assembly by Con_5A2-(4A)2 and Con_5A2-(4B)2, respectively, would lead to the formation of quadrilateral shaped structure in which each side is established by a dimeric coiled coil component (Figure 4C). Similar to the construction of the pre-barbell assemblies, two pre-quadrilateral assemblies were also constructed prior to attempting the construction of the quadrilateral shaped assembly. Pre-quadrilateral_A assembly was obtained by creating a mixture of Con_5A2-(4A)2, 4B and 5B with a 1:2:1 molar ratio (Figure 4A). Maintaining the same molar ratio but replacing the Con_5A2-(4A)2 and 4B with Con_5A2-(4B)2 and 4A, respectively, resulted in the formation of the pre-quadrilateral_B assembly (Figure 4B). The CD spectra of these two three-component mixtures generated strong characteristic α-helical signals, indicating well-defined α-helical secondary structures were observed for both pre-quadrilateral assemblies (Figure 4D). Upon heating, these two pre-quadrilateral assemblies unfolded less readily than isolated 4A/4B dimer but more readily than 5A2/5B dimer (Figure 4E). The expected molar mass determined by SEC-MALS analysis further confirmed that both of these two pre-quadrilateral assemblies contain two 4A/4B dimers and one 5A2/5B dimer (Figure 4F top and middle, expected mass: 31.6 kDa for both pre-quadrilateral assemblies, observed mass: ~30 kDa for pre-quadrilateral_A assembly and ~32 kDa for pre-quadrilateral_B assembly).
Figure 4.
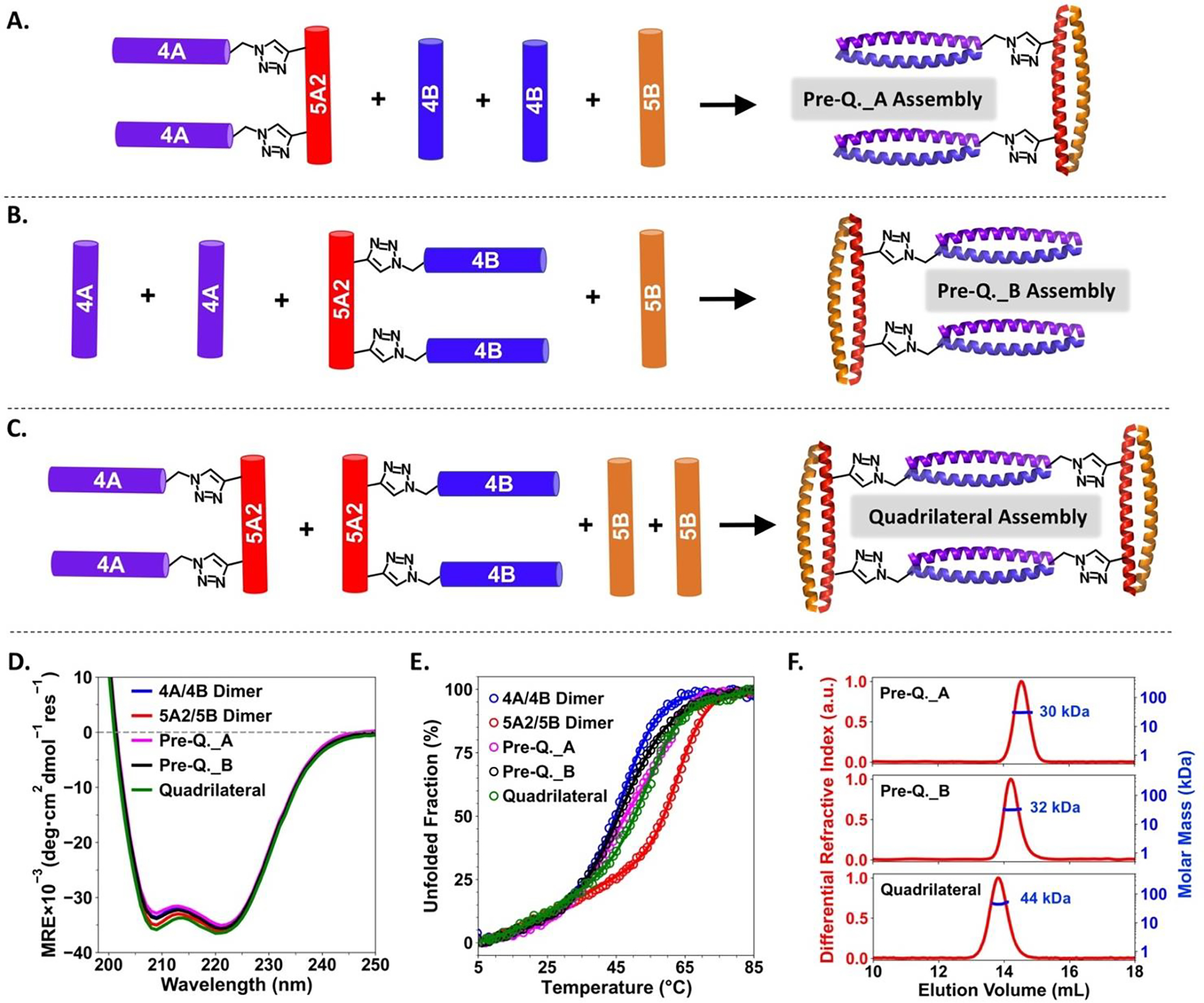
Preparation routes for pre-quadrilateral_A (A), pre-quadrilateral_B (B) and quadrilateral shaped (C) assemblies. Comparison of CD spectra (D) and thermal denaturation profiles (E) between the dimeric building blocks (4A/4B and 5A2/5B dimers) and the supramolecular assemblies in the quadrilateral series. (F) SEC-MALS results of the three assemblies in the quadrilateral series. Expected molar mass, pre-quadrilateral_A assembly: 31.6 kDa; pre-quadrilateral_B assembly: 31.6 kDa and quadrilateral shaped assembly: 42.6 kDa.
The construction of the quadrilateral shaped assembly was then carried out by preparing a mixture containing Con_5A2-(4A)2, Con_5A2-(4B)2 and 5B with a 1:1:2 molar ratio (Figure 4C). As expected, CD spectra consistent with well-defined α-helical secondary structures were observed for this three-component mixture (Figure 4D). In comparison to the two pre-quadrilateral assemblies, the thermal stability of quadrilateral shaped assembly increased slightly but was still weaker than the isolated 5A2/5B dimer (Figure 4E). The expected molar mass was also observed in the SEC-MALS analysis for the quadrilateral shaped assembly (Figure 4F bottom, expected mass: 42.6 kDa and observed mass: ~44 kDa)
The results from CD and SEC-MALS studies clearly indicated that discrete supramolecular assemblies with a pre-determined number of coiled coil components were successfully constructed. We further evaluated the structural features of the barbell and quadrilateral shaped assemblies by using Small Angle X-ray Scattering (Figure 5A). Guinier analysis of the SAXS data shows that these two assemblies have a similar radius of gyration (Rg), ~31 Å for barbell shaped assembly and ~33 Å for quadrilateral shaped assembly (Figure S33B and S34B). Such similarity was also found for the Rg estimated from pair-distance distribution (P(r)) functions, ~34 Å for barbell shaped assembly and ~36 Å for quadrilateral shaped assembly (Figure 5B). The P(r) functions of these two assemblies showed similar asymmetric profiles, while the maximum dimension (dmax) of quadrilateral assembly was estimated to be ~137 Å, 12 Å longer than that for the barbell assembly (dmax: ~125 Å).
Figure 5.
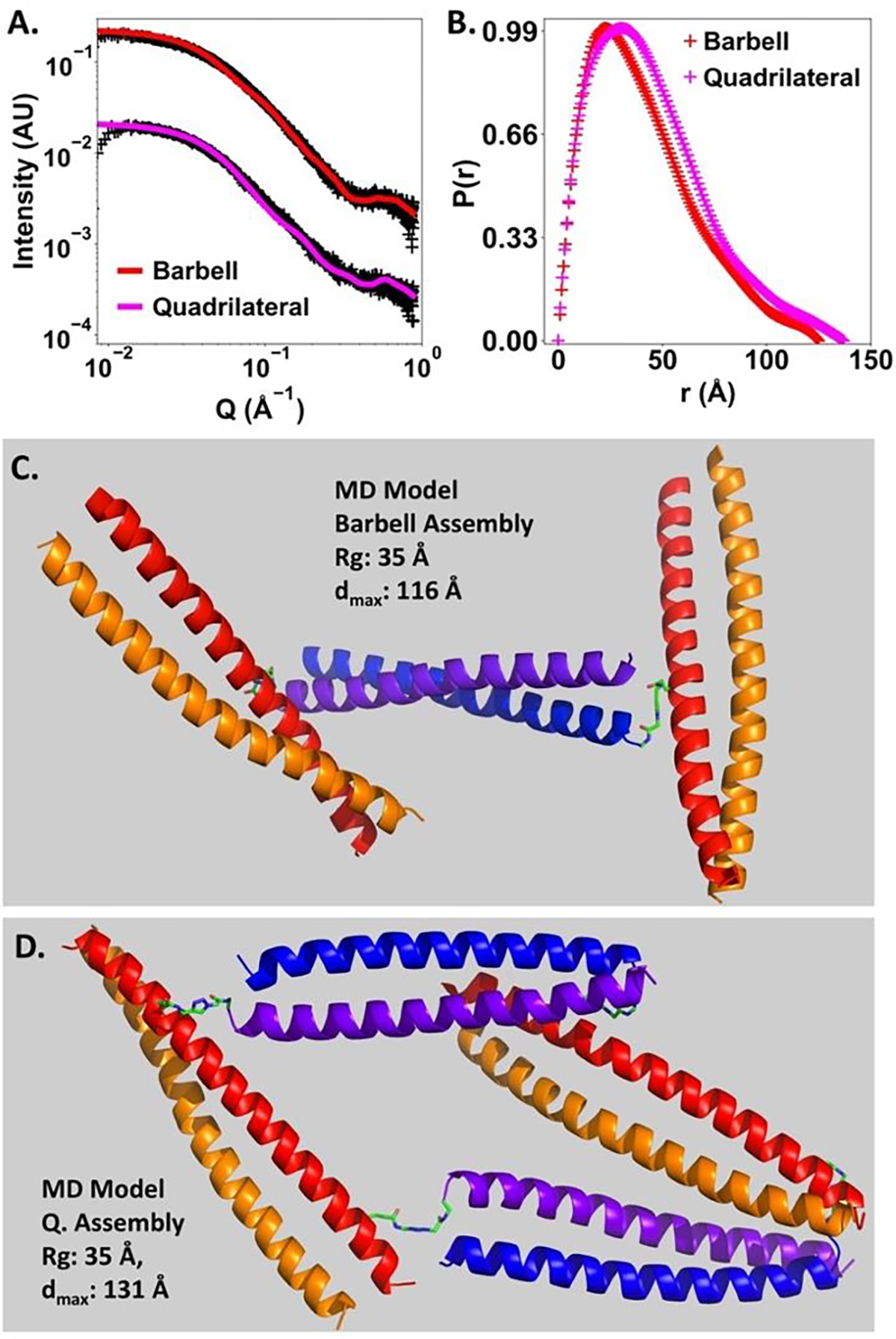
(A) Small angle X-ray Scattering (SAXS) analysis of the barbell and quadrilateral shaped assemblies. +: experimental data, solid line: theoretical curve calculated by FoXS for the representative MD models shown in (C) and (D). Goodness of fit, χ2(barbell): 0.93; χ2(quadrilateral): 0.87. (B) Pair distance distribution functions of the barbell and quadrilateral shaped assemblies. Structural parameters derived from P(r) functions, Rg(barbell): 33.7 Å, dmax(barbell): 125 Å. Rg(quadrilateral): 36.5 Å, dmax(quadrilateral): 137 Å.
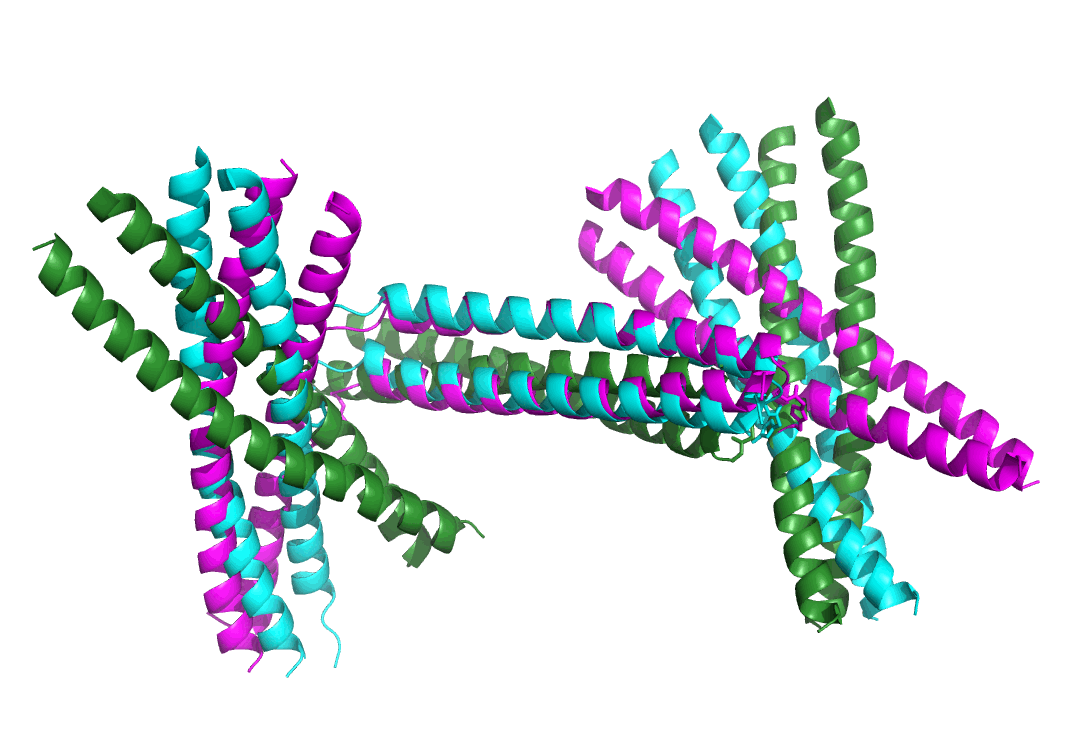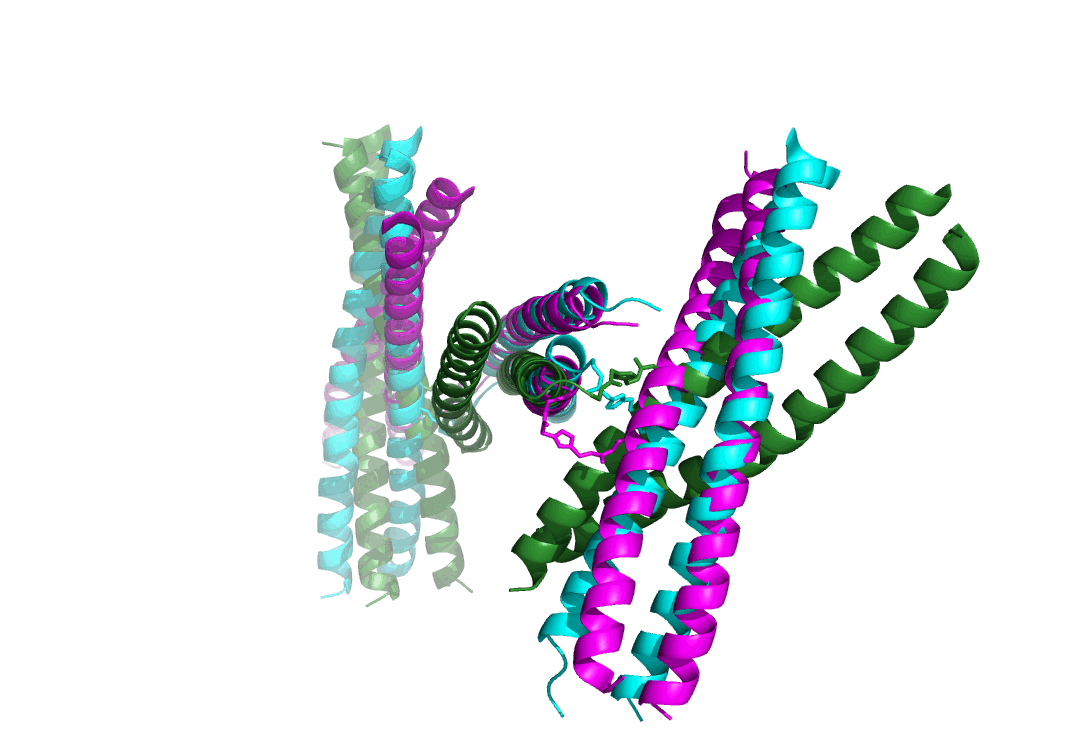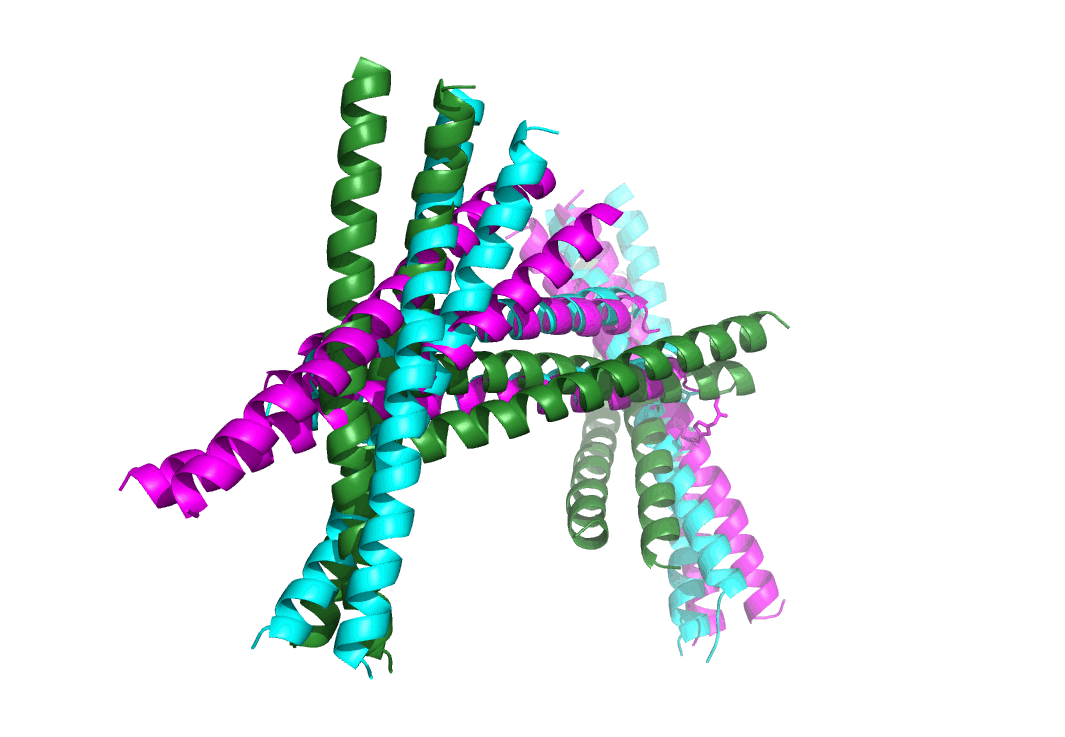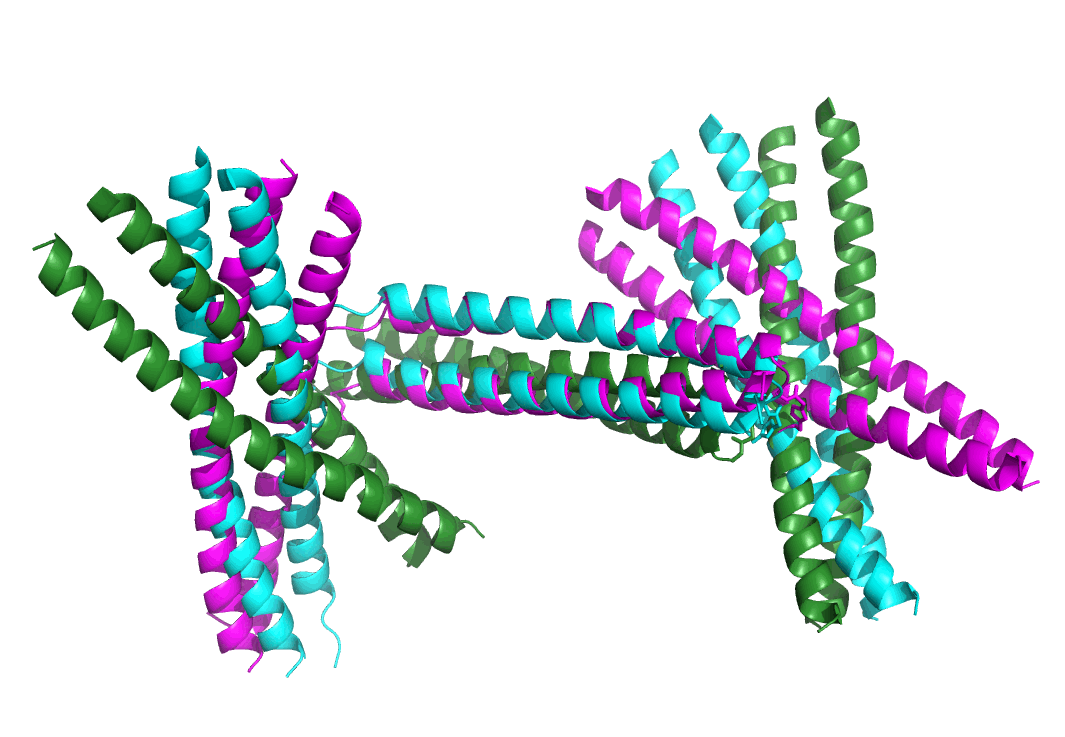
In order to extract more structural insights from the experimental SAXS data, molecular dynamics (MD) simulations were performed to generate computer models that could illuminate the experimental SAXS data. (See supporting information for the simulation details.) Good agreements were found between the experimental SAXS profile and those calculated from MD models by using the FoXS package[13] (Figure 5A, 5C and 5D). The overall structure of barbell shaped assembly resembles a distorted letter “H” form. The triazole linkers that connect one 4A/4B dimer and two 5A1/5B dimers in the barbell shaped assembly are not rigid by design. As a result, the extent of lateral distortion is somewhat variable. The experimental SAXS profile of barbell assembly could be well-fit by different MD models with different relative positions of one dimeric coiled coil component with respect to the other two (Figure 6A and 6B). The overall shapes of the MD models that properly correspond to the experimental SAXS data of quadrilateral assembly resemble parallelograms. Presumably due to the fact that the two 5A2/5B dimeric coiled coils were connected by two 4A/4B dimeric coiled coils, the overall structure of the quadrilateral shaped assembly is less flexible than that of barbell shaped assembly, for which two 5A1/5B dimers are connected by a single 4A/4B dimer (Figure 6B, 6C, S35–S38, Animation S1-S2 and supplementary pse files).
Figure 6.
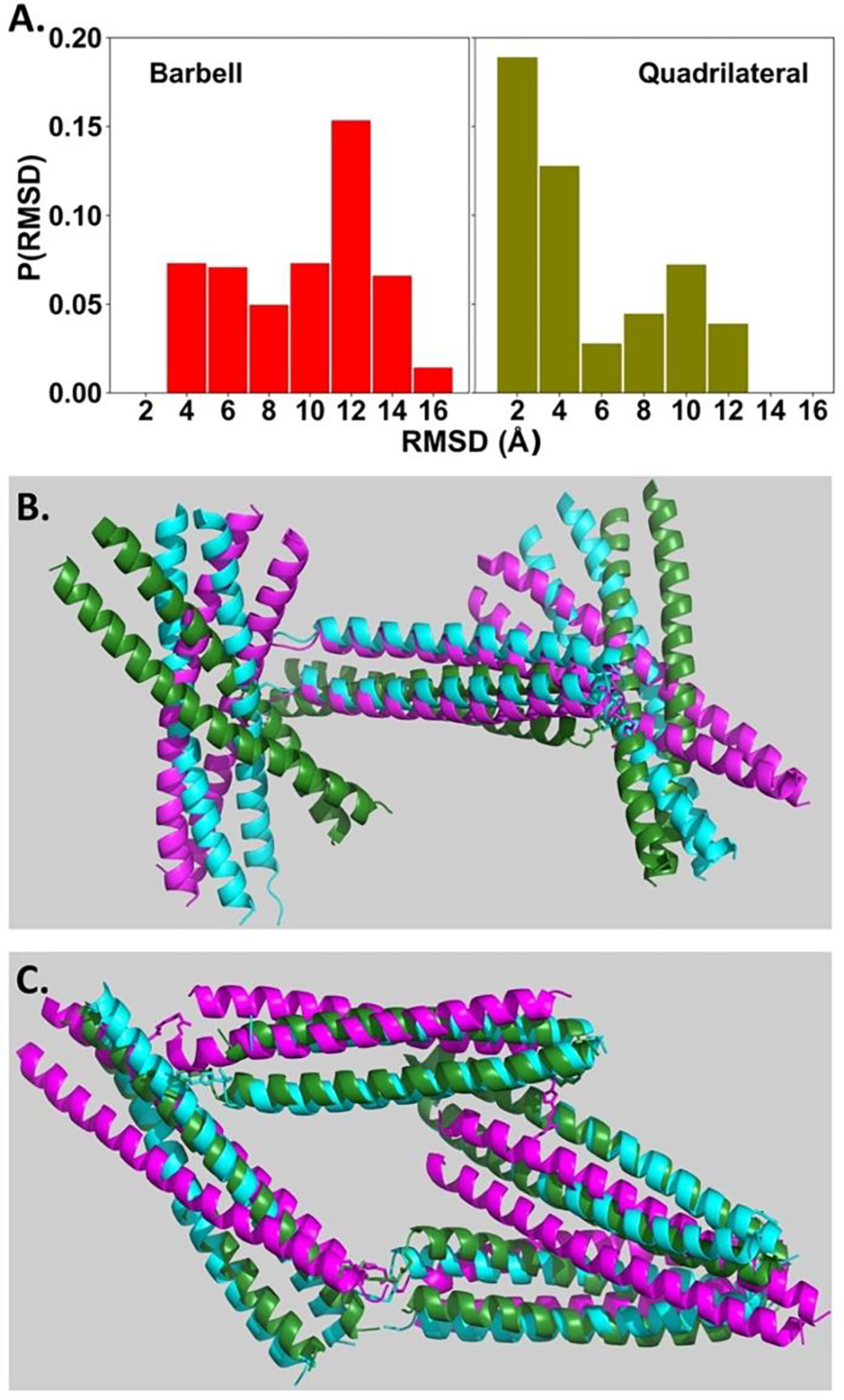
(A) Histogram plots of Cα-RMSD values of all Molecular Dynamics models that could fit the experimental SAXS data of barbell (left) and quadrilateral (right) shaped assemblies. Three different models of barbell (B) and quadrilateral (C) shaped assemblies are overlaid. The cyan-coloured model was used as the reference for calculating Cα-RMSD values. The green- and magenta-coloured representative models show the median and largest Cα-RMSD values, respectively. (See Figure S35–S38, Amination S1-S2 and supplementary pse files for more details of analysis.)
The structural features of the quadrilateral assembly were further investigated by transmission electron microscopy (TEM) using the negative staining method. We observed protein particles with consistent apparent diameters ~13 nm (Figure 7 and S39). In addition, the expected pore structure exhibited by the quadrilateral shaped assembly were also observed for several particles suitably oriented on the TEM grid (red circular highlights in Figure 7).
Figure 7.

TEM image of the quadrilateral shaped assembly. Red circles highlight five single particles with visible pore structures. See Figure S39 for other TEM images
In comparison to prior studies in which discrete supramolecular assemblies were obtained after empirical trial and error,[14] the strategy reported in this work allows us to pre-determine the size and approximate topologies of the resultant supramolecular assemblies. Notably, supramolecular assemblies composed of four dimeric coiled coil components have also been constructed by the Woolfson group.[14f] Only one pair of peptides that form heterodimeric coiled coils were used in their study. The two complementary peptide sequences were spaced by a (glycine-asparagine)n linker within one linear peptide chain. Discrete supramolecular assemblies with an oligomeric state of four were obtained only when n equaled to three and after an annealing process. Larger sized aggregates were obtained when n was less than three. Smaller nanoscale objects composed of three or two dimeric coiled coil components were formed when n was larger than three. Prior to the empirical experimentation, it may not have been feasible to anticipate reliably the relationships between the linker length and the size/heterogeneities of the resultant supramolecular assemblies.
Among the six discrete supramolecular assemblies constructed in this work, the quadrilateral shaped assembly is the only one that has the geometry of an enclosed shape. For this reason, when we have a ternary mixture of Con_5A2-(4A)2, Con_5A2-(4B)2 and 5B with a 1:1:2 molar ratio, it is possible that larger sized species could form in competition with the desired formation of discrete quadrilateral shaped assembly. Assuming the two 4A/4B dimeric coiled coils of the quadrilateral shaped assembly formed sequentially, after one 4A/4B dimeric coiled coil was formed between one Con_5A2-(4A)2 and one Con_5A2-(4B)2, there were mainly two possible pathways for the remaining 4A and 4B components to assemble with the complementary sequences. First, the remaining 4A and 4B chains would pair together to form another 4A/4B dimeric coiled coil which would constitute the other edge of the quadrilateral shaped assembly (Figure 8A). Second, it is also possible for the remaining 4A and 4B chains to pair with complementary sequences from other Con_5A2-(4A)2 and Con_5A2-(4B)2 (Figure 8B). Fibrillar assemblies with uncontrolled length could result from the second pathway. We considered that the probability of the first pathway would be much higher because the effective local concentrations of 4A and 4B components are much higher than the overall concentrations of conjugated peptides in our experiments. Due to the end-to-side conjugation strategy, the relative orientation of the remaining 4A and 4B were also pre-aligned in a form close to parallel alignment. This alignment would further facilitate their hetero association and lead to the formation of quadrilateral shaped assembly. In addition, the hetero-specific associations between 4A and 4B components are orthogonal to that between 5A2 and 5B components which constitute the other two sides of the quadrilateral assemblies. As a consequence, the predicted quadrilateral shaped assemblies are likely favored relative to other possible assemblies.
Figure 8.
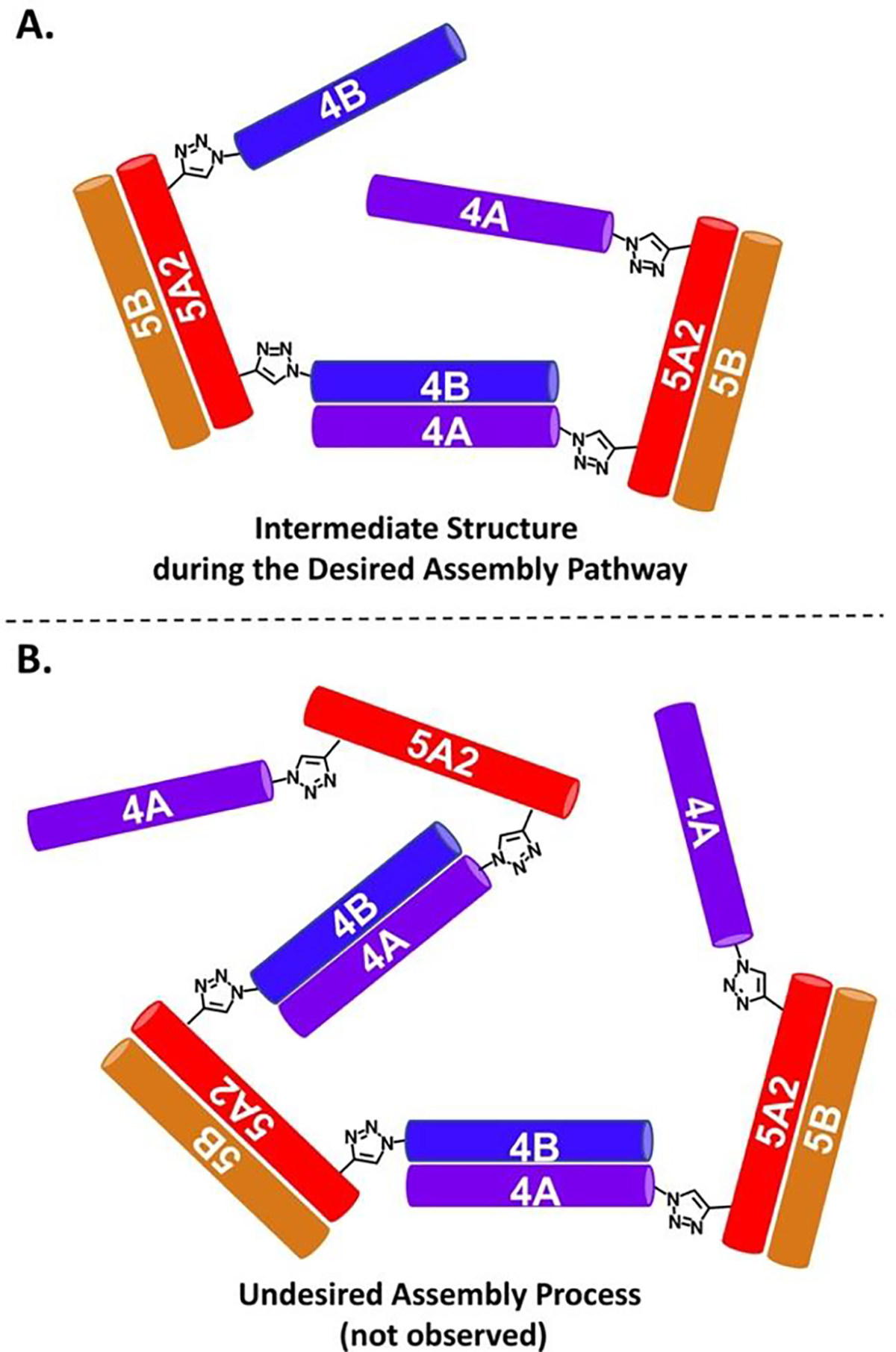
(A) Plausible intermediate state during formation of the desired quadrilateral shaped assembly. (B) Hypothetical intermediate state with low probability.
Annealing is a commonly used technique during supramolecular assembly processes to circumvent paths leading to undesired thermodynamic states. For instance, the Keating group applied a rational annealing process composed of heating, slow cooling and fast cooling to generate the desired protein nanotriangles and avoid the formation of undesired side products.[9] Annealing was not required to obtain the six discrete supramolecular assemblies constructed in this work, indicating the absence of strong competing molecular interactions that would otherwise interfere with the formation of the desired discrete supramolecular assemblies.
Conclusion
By using two pairs of orthogonally interacting peptides and installing covalent triazole linkages between the non-complementary pairs in a strategic end-to-side arrangement, we have successfully constructed six discrete supramolecular assemblies with a pre-determined number of coiled coil components. All coiled coil components retained their native folding abilities and orthogonal dimer-interaction specificity in the context of the supramolecular structures. Hetero specific interactions between complementary peptide chains are the foundation of this work. The molar mass and approximate topologies of the resultant supramolecular assemblies are directed by the number and the location of the branch points we introduced to the parent 5A peptide, and by the stoichiometry of the multi-component mixture.
From the design point of view, our strategy is highly expandable and customizable. One-dimensional assemblies with pre-determined length could be constructed when branch points are also introduced to 5B peptide. We only used dimeric coiled coils in this work, while peptides that can form coiled coils with a higher order of oligomeric states are also candidates to be exploited. In this fashion, replacing 5A1/5B or 5A2/5B dimers may allow construction of other two- or three-dimensional structures with pre-determined sizes and tunable structural properties. The interior pores of the quadrilateral assemblies may be particularly suitable for functionalization and/or sequestration of ligands in future efforts to develop applications of these assemblies.
Our design paradigm is not limited to coiled coil peptides -- collagen mimetic peptides that form homo or hetero collagen triple helices could also be utilized as alternatives to 5A1/5B or 5A2/5B dimers. In addition, the N- and C-termini of the 5A1/5B dimer, 5A2/5B dimer and their potential replacements are amenable to alternative functionalization. Thus, we believe the strategy reported in this work will have broad potential in constructing customized supramolecular assemblies for a wide range of envisioned applications.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This study was supported by award CHE-2002890 from the National Science Foundation (NSF). L.J. would like to gratefully acknowledge the New York University Chemistry Department for the Margaret and Herman Sokol Fellowship. N.J.T. acknowledges support from NIH R01AI108889. The authors would like to thank Dr. Shahid Khan for the assistance on SEC purifications. Dr. Jia Ma from the Chemistry Department of Columbia University is greatly appreciated for the assistance on performing the SEC-MALS analysis. This work was supported in part through the NYU IT High Performance Computing resources, services, and staff expertise. Joseph Sall, Dr. Feng-Xia (Alice) Liang and Dr. William J. Rice from NYU Grossman School of Medicine are greatly acknowledged for assistance with TEM imaging. The authors acknowledge the contribution of James Eastwood for assistance with this manuscript. The Circular Dichroism Spectropolarimeter used in this work was acquired through the support of New York University. SAXS measurements were performed at Beamline 12-ID-B of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
References
- [1].a) De Santis E, Ryadnov MG, Chem. Soc. Rev. 2015, 44, 8288–8300 [DOI] [PubMed] [Google Scholar]; b) Makam P, Gazit E, Chem. Soc. Rev. 2018, 47, 3406–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim BJ, Yang D, Xu B, Trends Chem. 2020, 2, 71–83 [Google Scholar]; d) Boyle AL, Woolfson DN, Chem. Soc. Rev. 2011, 40, 4295–4306 [DOI] [PubMed] [Google Scholar]; e) Raymond DM, Nilsson BL, Chem. Soc. Rev. 2018, 47, 3659–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Moore AN, Hartgerink JD, Acc. Chem. Res. 2017, 50, 714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hauser CA, Zhang S, Chem. Soc. Rev. 2010, 39, 2780–2790 [DOI] [PubMed] [Google Scholar]; c) Parmar AS, James JK, Grisham DR, Pike DH, Nanda V, J Am Chem Soc 2016, 138, 4362–4367. [DOI] [PubMed] [Google Scholar]
- [3].a) Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N, Nature 1993, 366, 324–327 [DOI] [PubMed] [Google Scholar]; b) Gao X, Matsui H, Adv. Mater. 2005, 17, 2037–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Valéry C, Artzner F, Paternostre M, Soft Matter 2011, 7, 9583–9594 [Google Scholar]; d) Rho JY, Cox H, Mansfield EDH, Ellacott SH, Peltier R, Brendel JC, Hartlieb M, Waigh TA, Perrier S, Nat. Commun. 2019, 10, 4708. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Burgess NC, Sharp TH, Thomas F, Wood CW, Thomson AR, Zaccai NR, Brady RL, Serpell LC, Woolfson DN, J. Am. Chem. Soc. 2015, 137, 10554–10562. [DOI] [PubMed] [Google Scholar]
- [4].a) Jiang T, Xu C, Zuo X, Conticello VP, Angew. Chem. Int. Ed. Engl. 2014, 53, 8367–8371 [DOI] [PubMed] [Google Scholar]; b) Dai B, Li D, Xi W, Luo F, Zhang X, Zou M, Cao M, Hu J, Wang W, Wei G, Zhang Y, Liu C, Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2996–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Tavenor NA, Murnin MJ, Horne WS, J. Am. Chem. Soc. 2017, 139, 2212–2215; [DOI] [PubMed] [Google Scholar]; b) Scheib KA, Tavenor NA, Lawless MJ, Saxena S, Horne WS, Chem. Commun. 2019, 55, 7752–7755. [DOI] [PubMed] [Google Scholar]
- [6].a) Fries CN, Wu Y, Kelly SH, Wolf M, Votaw NL, Zauscher S, Collier JH, Adv. Mater. 2020, 32, 2003310; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Adler-Abramovich L, Marco P, Arnon ZA, Creasey RCG, Michaels TCT, Levin A, Scurr DJ, Roberts CJ, Knowles TPJ, Tendler SJB, Gazit E, ACS Nano 2016, 10, 7436–7442. [DOI] [PubMed] [Google Scholar]
- [7].Zhou M, Bentley D, Ghosh I, J. Am. Chem. Soc. 2004, 126, 734–735. [DOI] [PubMed] [Google Scholar]
- [8].Bromley EHC, Sessions RB, Thomson AR, Woolfson DN, J. Am. Chem. Soc. 2009, 131, 928–930. [DOI] [PubMed] [Google Scholar]
- [9].Park WM, Bedewy M, Berggren KK, Keating AE, Sci. Rep. 2017, 7, 10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou M, Ghosh I, Org. Lett. 2004, 6. [DOI] [PubMed] [Google Scholar]
- [11].a) Grigoryan G, Keating AE, Curr. Opin. Struct. Biol. 2008, 18, 477–483; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Woolfson DN, Adv. Prot. Chem. 2005, 70, 79–112. [DOI] [PubMed] [Google Scholar]
- [12].Reinke AW, Grant RA, Keating AE, J. Am. Chem. Soc. 2010, 132, 6025–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A, Biophys. J. 2013, 105, 962–974; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A, Nucleic Acids Res. 2016, 44, W424–W429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Jiang L, Xu D, Namitz KE, Cosgrove MS, Lund R, Dong H, Small 2016, 12, 5126–5131 [DOI] [PubMed] [Google Scholar]; b) Jiang L, Yang S, Lund R, Dong H, Biomater. Sci. 2018, 6, 272–279 [DOI] [PubMed] [Google Scholar]; c) Raman S, Machaidze G, Lustig A, Aebi U, Burkhard P, Nanomedicine 2006, 2, 95–102 [DOI] [PubMed] [Google Scholar]; d) Boato F, Thomas RM, Ghasparian A, Freund-Renard A, Moehle K, Robinson JA, Angew. Chem. Int. Ed. Engl. 2007, 46, 9015–9018 [DOI] [PubMed] [Google Scholar]; e) Noble JE, De Santis E, Ravi J, Lamarre B, Castelletto V, Mantell J, Ray S, Ryadnov MG, J. Am. Chem. Soc. 2016, 138, 12202–12210 [DOI] [PubMed] [Google Scholar]; f) Boyle AL, Bromley EH, Bartlett GJ, Sessions RB, Sharp TH, Williams CL, Curmi PM, Forde NR, Linke H, Woolfson DN, J. Am. Chem. Soc. 2012, 134, 15457–15467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.