

Abstract
We have previously identified two distinct NADH oxidases corresponding to H2O2-forming oxidase (Nox-1) and H2O-forming oxidase (Nox-2) induced in Streptococcus mutans. Sequence analyses indicated a strong similarity between Nox-1 and AhpF, the flavoprotein component of Salmonella typhimurium alkyl hydroperoxide reductase; an open reading frame upstream of nox-1 also showed homology to AhpC, the direct peroxide-reducing component of S. typhimurium alkyl hydroperoxide reductase. To determine their physiological functions in S. mutans, we constructed knockout mutants of Nox-1, Nox-2, and/or the AhpC homologue; we verified that Nox-2 plays an important role in energy metabolism through the regeneration of NAD+ but Nox-1 contributes negligibly. The Nox-2 mutant exhibited greatly reduced aerobic growth on mannitol, whereas there was no significant effect of aerobiosis on the growth on mannitol of the other strains or growth on glucose of any of the strains. Although the Nox-2 mutants grew well on glucose aerobically, the end products of glucose fermentation by the Nox-2 mutant were substantially shifted to higher ratios of lactic acid to acetic acid compared with wild-type cells. The resistance to cumene hydroperoxide of Escherichia coli TA4315 (ahpCF-defective mutant) transformed with pAN119 containing both nox-1 and ahpC genes was not only restored but enhanced relative to that of E. coli K-12 (parent strain), indicating a clear function for Nox-1 as part of an alkyl hydroperoxide reductase system in vivo in combination with AhpC. Surprisingly, the Nox-1 and/or AhpC deficiency had no effect on the sensitivity of S. mutans to cumene hydroperoxide and H2O2, implying that the existence of some other antioxidant system(s) independent of Nox-1 in S. mutans compensates for the deficiency.
Streptococcus mutans, one of the principal causative agents of human dental caries, is considered to be a facultative anaerobe, and its energy metabolism depends strictly on glycolysis (16). One important characteristic distinguishing this organism from other oral streptococci is its ability to ferment mannitol and sorbitol (8). Although streptococci have a preference for anaerobiosis, O2 affected the growth on mannitol with a variation dependent on strains (10). The growth response to O2 was correlated with the ability of strains to induce NADH oxidase and superoxide dismutase (SOD) under aerobic conditions (10, 11). These findings suggested that NADH oxidase plays an important role in the regulation of the aerobic metabolism of mannitol.
Interestingly, two types of NADH oxidase were induced in O2-tolerant strains of S. mutans, including NCIB11723, NCTC10449, and Ingbritt. The two NADH oxidases purified from S. mutans NCIB11723 were identified as two distinct NADH oxidases corresponding to H2O2-forming oxidase (Nox-1) and H2O-forming oxidase (Nox-2) (12). Characteristics of these two enzymes differed remarkably (12):
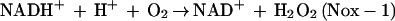 |
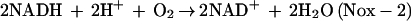 |
Nox-1 catalyzed the two-electron reduction of O2 by NADH, whereas Nox-2 catalyzed the four-electron reduction of O2 by NADH. The oxidase activity of Nox-1 was stimulated on addition of free flavin adenine dinucleotide (FAD), but that of Nox-2 was independent of free FAD. The subunit molecular masses were 55 kDa for Nox-1 and 50 kDa for Nox-2, estimated initially on the basis of mobility in sodium dodecyl sulfate-polyacrylamide gels and later on the basis of the deduced amino acid sequence of each structural gene (12, 13, 19). Moreover, antibodies raised against Nox-1 or Nox-2 reacted with their corresponding antigens but did not cross-react (12). Analysis of each structural gene, nox-1 and nox-2, also showed little homology of the deduced amino acid sequence between these enzymes and their separate positions on genomic DNA (13, 19).
S. mutans, like other types of lactic acid bacteria, lacks cytochromes and heme-containing proteins including catalase or heme peroxidases. Thus, it was contradictory to defense against O2 toxicity that an O2-tolerant S. mutans possesses Nox-1 generating a reactive oxygen species such as H2O2. However, located directly upstream of the nox-1 gene on the S. mutans chromosome was an ahpC gene encoding a peroxidase enzyme (AhpC) homologous with the structural gene of the nonflavoprotein component (AhpC) of Salmonella typhimurium alkyl hydroperoxide reductase, a defense system against oxidative stress (14). This finding implies that H2O2 produced by Nox-1 can be reduced to H2O by the AhpC, as follows: 2NADH + 2H+ + O2 → 2NAD+ + 2H2O (Nox-1 plus AhpC).
In a previous paper, we demonstrated in vitro that Nox-1, the H2O2-forming oxidase, functions as an NADH-dependent peroxidase in combination with the S. mutans AhpC (24). Consequently, we attempted to elucidate the physiological functions of Nox-1 and Nox-2 in S. mutans by constructing knockout mutants of Nox-1, Nox-2, and/or AhpC. We report here that Nox-2 plays an important role in regenerating NAD+, whereas Nox-1 contributes negligibly.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The bacterial strains and plasmids used in this study are described in Table 1. For transformation of S. mutans, GS-5 was routinely used instead of NCIB11723, which is the original source of the nox-1, nox-2, and ahpC genes described previously (13, 19). Strain GS-5 exhibits a high transformation efficiency compared with NCIB11723; the nucleotide sequences of these genes from GS-5 were confirmed to be almost 100% identical with those of nox-1, nox-2, and ahpC from strain NCIB11723. S. mutans cells were grown at 37°C in Todd-Hewitt broth (THB; Difco Laboratories, Detroit, Mich.), TY medium containing 1% glucose (TYG) or 1% mannitol (TYM) (10), or THB supplemented with 5% horse serum for generating competent cells. For anaerobic growth, 10 ml of fresh medium was inoculated with 0.1 ml of the late-log-phase anaerobic subculture and incubated without shaking in an anaerobic glove box (Hirasawa Works, Tokyo, Japan) under an atmosphere of 80% nitrogen, 10% hydrogen, and 10% carbon dioxide. For growth on agar plates, a portion (1 ml) of overnight anaerobic culture was diluted and spread onto the agar surface of appropriate medium. Then the plates were incubated for 60 h under anaerobic or aerobic conditions. Cultures were routinely incubated at 37°C. Escherichia coli cells were grown in L broth (18). Solid media were supplemented with 1.5% agar. When present in selective plates, antibiotics were used at the following concentrations: for S. mutans, erythromycin at 10 μg/ml and spectinomycin at 250 μg/ml; for E. coli, ampicillin at 100 μg/ml, erythromycin at 300 μg/ml, and spectinomycin at 50 μg/ml.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference |
|---|---|---|
| Strains | ||
| S. mutans | ||
| GS-5 | Wild type, serotype c | 31 |
| E22 | Δnox-1 Emr | This study |
| N2E | Δnox-2 Emr | This study |
| N2S | Δnox-2 Spcr | This study |
| B1 | ΔahpC Emr | This study |
| BEE | ΔahpC Δnox-1 Emr | This study |
| BES | ΔahpC Δnox-1 Spcr | This study |
| N2S-B1 | Δnox-2 ΔahpC Emr Spcr | This study |
| N2S-E22 | Δnox-1 Δnox-2 Emr Spcr | This study |
| N2E-BES | ΔahpC Δnox-1 Δnox-2 Emr Spcr | This study |
| E. coli | ||
| DH5α | supE44 ΔlacU169 (φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 9 |
| JM109 | recA1 supE44 endA1 hsdR17 gyrA96 relA1 thi Δ(lac-proAB)/F′ [traD36 proAB+ lacIqlacZΔM15] | 30 |
| K12 | ahpC+ ahpF+ (parent of TA4315) | 27 |
| TA4315 | ΔahpC ΔahpF | 27 |
| Plasmids | ||
| pUC18 | AprlacPOZ′ | 30 |
| pUC119 | AprlacPOZ′ | 30 |
| pTSE | Emr 0.9-kb BamHI | 2 |
| pSPC1 | Spcr 1.1-kb BamHI | 15 |
| pMS1 | 1.9-kb EcoRI fraction from λHS-1 containing nox-1 in pUC119 | This study |
| pNOX1-H | 1.6-kb HindIII fragment containing nox-1 in pKK223-3 | This study |
| pAN119 | 2.5-kb EcoRI-HindIII fragment from λHS-1 containing ahpC and nox-1 in pUC119 | This study |
| pSSW61 | 5.6-kb SacI fragment containing nox-2 in pMW118 | 19 |
| pB1 | ahpC::Emr | This study |
| pE22 | nox1::Emr | This study |
| pN2E | nox2::Emr | This study |
| pN2S | nox2::Spcr | This study |
| pBEE | ahpC::Emr::nox-1 | This study |
| pBES | ahpC::Spcr::nox-1 | This study |
Construction of plasmids for knockout of the target genes.
DNA manipulations were performed as described by Maniatis et al. (18). Plasmid pMS1 was obtained by subcloning the 1.9-kb EcoRI-EcoRI fragment from the original λHS-1 clone containing ahpC and nox-1 (13) into pUC119. Plasmid pNox-1H was constructed by subcloning a 1.6-kb HindIII-HindIII fragment engineered by PCR from the original pHS19 (13) into pKK223-3. Plasmid pAN119 was obtained by subcloning the 1.9-kb EcoRI-EcoRI fragment from pMS1 into pUC119 containing the 0.6-kb EcoRI-HindIII fragment derived from pNox-1H. Plasmid pN2EH was constructed by subcloning the 2.3-kb EcoRI-HindIII fragment from pSSW61 (19) into pUC18. Plasmids containing nox-1, nox-2, or ahpC inactivated by insertion of the Emr or Spcr gene were constructed. Plasmid pB1 was constructed by digesting pMS1, lacking the BamHI site in the multicloning site, with BamHI and ligating it to the BamHI Emr DNA fragment. Plasmid pE22 was constructed by digesting pMS1 with EcoT22 and ligating it to the BamHI Emr DNA fragment after generating blunt ends by treatment with deoxynucleoside triphosphates and the Klenow fragment of E. coli DNA polymerase I. Plasmids pBEE and pBES were constructed by digesting pMS1 lacking a BamHI site with BamHI and EcoT22, blunting as described for the pE22, and ligation to either the Emr (pBEE) or the Spcr (pBES) DNA fragment. Plasmids pN2E and pN2S were constructed by digesting pN2EH with XbaI and then blunting and ligating it to either the Emr (pN2E) or Spcr (pN2S) DNA fragment. All plasmids were transformed into E. coli DH5α, and the mutants were selected on Luria-Bertani medium (LB) plates containing either erythromycin or spectinomycin.
Transformation of S. mutans and homologous recombination.
Genetic transformation of DNA fragments into S. mutans was performed as described by Perry and Kuramitsu (22), with some modifications. S. mutans GS-5 was transformed to Emr with the 2.6-kb KpnI fragment of pE22, the 2.6-kb SacI-HindIII fragment of pN2E, the 2.6-kb KpnI fragment of pB1, or the 1.9-kb KpnI fragment of pBEE and to Spcr with the 2.5-kb EcoRI fragment of pBES or the 3.5-kb SacI-HincII fragment of pN2S. Transformants were selected on THB agar containing erythromycin or spectinomycin.
Screening of knockout mutants by direct PCR.
The antibiotic-resistant colonies on plates were isolated as single colonies and analyzed for the insertion of the antibiotic resistance markers by direct PCR of the genomic DNA, using primers 1 (5′-AAGCTTCTTTCGTGTGTCCTACTGAG-3′) and 2 (5′-AAGCTTTGAATAGACTTAGCACGCGG-3′) for pB1, pE22, pBEE, and pBES and using primers 3 (5′-TGCGAGCTCGATTC TTGTATTA GCAGTCTTC-3′) and 4 (5′-ATAGAGCTCACTTTCAGACAGCAATA TACC-3′) for pN2E and pN2S.
Enzyme induction and preparation of cell extracts.
For enzyme induction, each culture was grown in five 50-ml Falcon tubes containing 50 ml of TYG or TYM until early log phase (A660 = 0.3) under strictly anaerobic conditions; then four of them were transferred to 500-ml flasks and incubated at 37°C with vigorous shaking under aerobic conditions. One flask at each time point (60, 120, 240, and 480 min) after exposure to air was cooled with ice water; then the bacteria were harvested by centrifugation at 12,000 × g for 10 min, washed twice with 50 mM potassium phosphate buffer containing 0.2 mM EDTA (pH 7.0), and stored at −80°C until use. To analyze enzyme induction at time zero, a chloramphenicol solution (250 μg/ml) was added to the remaining anaerobic culture grown to an A660 of 0.3, growth was stopped by cooling with ice water, and the cells were harvested, washed, and stored as described above. The frozen cells were thawed, suspended in 2 ml of the same buffer, and disrupted by sonication for 3 min with cooling intervals. After cell debris was removed by centrifugation at 25,000 × g for 30 min, the clear lysates were either used immediately or stored at −80°C. Protein concentration was measured by the dye-binding method (3).
Western blot analysis.
For analysis of enzyme induction by Western blotting, 5 μg of protein from the various extracts was separated on a sodium dodecyl sulfate-polyacrylamide gel and electrotransferred to a polyvinylidene difluoride membrane (Millipore, Tokyo, Japan). The membrane was blocked with 5% nonfat milk and reacted with either anti-S. mutans Nox-1 antibody, anti-S. mutans Nox-2 antibody, or anti-Amphibacillus xylanus AhpC antibody (a gift from Y. Niimura) and subsequently developed with a goat anti-rabbit antibody conjugated to alkaline phosphatase.
NADH oxidase and alkyl hydroperoxide reductase assays.
NADH oxidase activity in extracts was determined at 25°C by monitoring the oxidation of NADH in the reaction mixture (3 ml) at 340 nm as described previously (12). Nox-1- and AhpC-dependent peroxidase assays were carried out with cell extracts that were first subjected to ultrafiltration with CM-30 Centricon units (Amicon) to concentrate the samples and lower the concentrations of nonprotein and small protein components; activities were measured essentially as described previously (7, 23) in the presence of 200 μM NADH and 1 mM cumene hydroperoxide, except that the assays were carried out anaerobically in an Applied Photophysics DX.17MV stopped-flow spectrophotometer by mixing substrates in one syringe with the extracts and/or pure proteins in the other. For assays of AhpC in crude extracts, pure Nox-1 was included at a final concentration of 5 μM and a standard curve was generated with 0.1 to 0.5 μM pure S. mutans AhpC. For assays of Nox-1 in crude extracts, pure AhpC was included at a final concentration of 20 μM and a standard curve was generated with 0.005 to 0.06 μM pure Nox-1. Under these conditions, pure AhpC and Nox-1 exhibited specific activities of 59 and 67 U/mg, respectively, where 1 U of activity equals 1 μM NADH oxidized per min.
Analysis of growth and fermentation products.
Bacterial growth was monitored by measuring the increase in A660. Acetate, formate, lactate and pyruvate were measured with a carboxylic acid analyzer (Tokyo Rikakikai Ltd. model S-14) as described elsewhere (28).
Determination of glycolytic intermediates.
A portion (5 or 10 ml) of each culture after 60, 120, and 240 min of aeration in TYG (or TYM) medium was applied to a membrane filter (0.5-μm pore size, 47-mm polytetrafluoroethylene polymer; Toyo Roshi, Tokyo, Japan) under vacuum. The cells collected on the filters were used for the determination of intracellular levels of glycolytic intermediates of the Embden-Meyerhof pathway. Glycolytic intermediates in the cells washed with 0.9% NaCl were extracted with cold 0.6 M perchloric acid, and the extract was neutralized with 5 M K2CO3 at 0°C. Quantification of the intermediates in the neutralized extract was performed enzymatically by the method of Minakami et al. (20) with a double-wavelength spectrophotometer (model 557; Hitachi, Tokyo, Japan).
Sensitivity to killing by oxidants.
Disk inhibition assays were performed as described by Storz et al. (27) except that LB plates were used. For measurement of the sensitivity of TA4315 containing pNox1-H, 1 mM isopropyl-β-d-thiogalactopyranoside was added to soft agar just before mixing with the culture.
Survival assay after H2O2 and cumene hydroperoxide challenge.
All procedures were performed under anaerobic conditions. Overnight cultures were inoculated into 10 ml of fresh THB, grown to late log phase, used to inoculate two new cultures with 10 ml of THB each, and grown to an A660 of 0.18 to 0.2, after which an adaptive dose of H2O2 (10 μM) or cumene hydroperoxide (30 μM) was added to one of the two cultures. After 60 min, a lethal dose of H2O2 (100 μM) or cumene hydroperoxide (300 μM) was added for 15 or 30 min to both cultures, followed by dilution to measure viable cell counts. Diluted cells were plated on THB plates and incubated 48 h at 37°C to count CFU.
RESULTS
Construction of nox-1, nox-2, and/or ahpC mutants of S. mutans.
To determine the physiological functions of the two distinct NADH oxidases in S. mutans, nox-1, nox-2, and/or ahpC mutants of S. mutans were constructed by homologous recombination between S. mutans genomic DNA and linearized DNA fragments from plasmids containing target genes interrupted by Emr or Spcr genes. Using this method, we constructed four Δnox-1, Δnox-2 (disrupted by Emr or Spcr), and ΔahpC single mutants, four ΔahpC Δnox-1 (disrupted by Emr or Spcr), ΔahpC Δnox-2, and Δnox-1 Δnox-2 double mutants, and one ΔahpC Δnox-1 Δnox-2 triple mutant (Table 1). Analysis of the chromosomal DNA of these mutants by direct PCR verified that the antibiotic resistance genes were introduced on the chromosomal DNA of these mutants (data not shown). Western blot analysis indicated that no products corresponding to AhpC, Nox-1, or Nox-2 were detected in cells of each knockout mutant as intended, either before or after induction by O2 (data not shown).
Aerobic induction of AhpC, Nox-1, and Nox-2.
The expression levels of AhpC, Nox-1, and Nox-2 in cell extracts from S. mutans wild-type strain GS-5 during aeration were analyzed by immunoblotting. The results shown in Fig. 1 indicated that AhpC, Nox-1, and Nox-2 proteins were induced by exposure to air and also revealed that a small amount of Nox-1 protein appeared in anaerobically grown cells, whereas no AhpC and Nox-2 proteins were observed in anaerobically grown cells.
FIG. 1.

Expression of the AhpC, Nox-1, and Nox-2 proteins in S. mutans wild-type strain GS-5 before and 60, 120, and 240 min after exposure to air on TYG medium. Each protein was analyzed by immunoblotting as described in Materials and Methods. Each lane was loaded with 5 μg of protein from the corresponding extract. The leftmost lane shows the purified enzymes applied as controls (from top to bottom, 50 ng of AhpC, 100 ng of Nox-1, and 100 ng of Nox-2).
Aerobic growth of nox-1 and nox-2 mutants.
S. mutans wild-type strain GS-5 and the Δnox-2, ΔahpC Δnox-1, and ΔahpC Δnox-1 Δnox-2 mutants grew well on glucose or mannitol under anaerobic conditions (data not shown). However, under aerobic conditions, the Δnox-2 and ΔahpC Δnox-1 Δnox-2 mutants were severely hampered in the ability to grow on mannitol. These mutants formed tiny colonies on mannitol agar plates even after 60 h of incubation at 37°C under air (Fig. 2). The aerobic conditions had no significant effect on the mannitol growth of the ΔahpC Δnox-1 mutant or on the glucose growth of any strain (Fig. 2). Under aerobic conditions, the Δnox-2 and ΔahpC Δnox-1 Δnox-2 mutants also demonstrated poor growth on sorbitol and formed tiny colonies on sorbitol agar plates but grew well on sorbitol anaerobically (data not shown).
FIG. 2.

Aerobic growth of S. mutans wild-type strain GS-5 and the ΔahpC Δnox-1, Δnox-2, and ΔahpC Δnox-1 Δnox-2 mutants on glucose and on mannitol agar plates. (A) Colonial morphologies of wild-type (a), ΔahpC Δnox-1 (b), Δnox-2 (c), and ΔahpC Δnox-1 Δnox-2 (d) cells aerobically grown on TYG plates, incubated at 37°C for 60 h; (B) colonial morphologies of wild-type (e), ΔahpC Δnox-1 (f), Δnox-2 (g), and ΔahpC Δnox-1 Δnox-2 (h) cells aerobically grown on TYM plates, incubated at 37°C for 60 h.
Aerobic induction of NADH oxidase activity.
We then explored the induction of NADH oxidase activities by exposure to air in wild-type strain GS-5 and the ΔahpC Δnox-1, and Δnox-2 mutants. During exposure to air for 4 h, wild-type strain GS-5 and the ΔahpC Δnox-1 mutant increased the level of NADH oxidizing activities 35-fold on glucose and 52-fold on mannitol (wild type) and 82-fold on glucose and 112-fold on mannitol (mutant) (Fig. 3). The enzyme activity of the ΔahpC Δnox-1 mutant was found to be over twofold higher than that of GS-5 in either medium. In contrast, the Δnox-2 mutant possessed low NADH oxidizing activity (less than 1 to 5% of that of the ΔahpC Δnox-1 mutant), and the activity was increased only 2.6-fold on glucose and 3.8-fold on mannitol during 4 h (Fig. 3). The low activity of NADH oxidase in the Δnox-2 mutant was consistent with the poor growth of this mutant either on agar plates (Fig. 2B) or in liquid medium (Fig. 3B) containing mannitol. In contrast, the Δnox-2 mutant grew well on glucose, to about the same level as wild-type strain GS-5 and the ΔahpC Δnox-1 mutant did, as mentioned above (Fig. 2A and 3A).
FIG. 3.

Aerobic induction of NADH oxidase activity in S. mutans wild-type strain GS-5 and the ΔahpC Δnox-1, Δnox-2, and ΔahpC Δnox-1 Δnox-2 mutants on TYG (A) and TYM (B) media. Anaerobically grown cultures in early log phase were exposed to air and induced at 37°C by shaking under air. Growth was monitored by measuring the optical density at 660 nm of cultures of wild-type (open squares), ΔahpC Δnox-1 (closed triangles), and Δnox-2 (gray circles) cells. At the time course before and after exposure to air, the cells were harvested and NADH oxidase activity in cell extracts was assayed for wild-type (open bars), ΔahpC Δnox-1 (black bars), and Δnox-2 (shaded bars) cells. Results shown are representative of three repeated experiments.
Interestingly, despite of the low NADH oxidase activity, Western blot analyses indicated that the expression levels of Nox-1 protein from extracts of both mannitol- and glucose-grown Δnox-2 cells were comparable to those of Nox-2 protein from the ΔahpC Δnox-1 mutant and wild-type strain GS-5 (data not shown).
Aerobic induction of alkyl hydroperoxide reductase activity.
The induction of alkyl hydroperoxide reductase activities in GS-5 and the Δnox-2 and ΔahpC Δnox-1 mutants by exposure to air for 4 h was explored. Alkyl hydroperoxide reductase activity of AhpC measured in the presence of added Nox-1 was relatively high in GS-5 grown on either glucose or mannitol (about 0.146 or 0.118 U/mg, respectively), with a low level of induction in the Δnox-2 mutant grown on glucose (to 1.4- to 1.7-fold over the wild-type level). No AhpC activity was detected in the ΔahpC Δnox-1 mutant grown on either glucose or mannitol. Alkyl hydroperoxide reductase activities of Nox-1 measured in the presence of added AhpC, on the other hand, were much lower and highly sensitive to conditions, varying from 0.0048 to 0.0865 U/mg. Mannitol-grown wild-type strain GS-5 exhibited approximately 5-fold higher Nox-1 activity than glucose-grown GS-5, while the absence of Nox-2 in the Δnox-2 mutant led to an 18-fold increase in this activity.
Fermentation end products from glucose and mannitol.
Although no significant difference in the aerobic growth on glucose was demonstrated between wild-type strain GS-5 and the Δnox-2 mutant, which exhibited a low level of NADH oxidase activity, it was conceivable that the high level of induced NADH oxidase activity in GS-5 and the ΔahpC Δnox-1 mutant affected the fermentation end products through a change in the ratio of NADH to NAD+. Thus, we examined whether the extremely low level of induced NADH oxidase activity in the Δnox-2 mutant affected the end products of aerobic fermentation of glucose. The end products of glucose or mannitol fermentation by the ΔahpC Δnox-1, Δnox-2, and wild-type strains after exposure to air for 4 h were analyzed. As shown in Fig. 4, the Δnox-2 mutant produced a large amount of lactate (39.4 mM) and less acetate (2.2 mM) in glucose media, whereas the ΔahpC Δnox-1 mutant and GS-5 produced less lactate (28.8 and 32.2 mM, respectively) and more acetate (7.5 and 7.2 mM, respectively), with small amounts of pyruvate (2.3 and 1.0 mM, respectively). In mannitol media, the ΔahpC Δnox-1 mutant and GS-5 produced markedly less lactate (14.7 and 15.7 mM, respectively) and a high amount of acetate (9.6 and 10.2 mM, respectively). The end products in each culture contained a small amount of formate derived from the anaerobic cultures used for enzyme induction by exposure to air at time zero. These results indicated that the deficiency of Nox-2 brought about the increased amount of lactate and decreased acetate in the end products of glucose fermentation under aerobic conditions.
FIG. 4.

Fermentation end products of S. mutans wild-type strain GS-5 and the ΔahpC, Δnox-1, and Δnox-2 mutants after 4 h of aeration in TYG (A) and TYM (B) media except that Δnox-2 was omitted. After 4 h of exposure to air, the cells were harvested and the cell-free culture media were assayed (see Materials and Methods).
Functions of Nox-1 and AhpC as alkyl hydroperoxide reductase in oxidative stress.
The alkyl hydroperoxide reductase, which is composed of AhpC and AhpF in S. typhimurium, has been identified as an antioxidant enzyme system capable of reducing organic hydroperoxides and hydrogen peroxide (14, 23). The ahpCF-defective mutants obtained in S. typhimurium and E. coli were hypersensitive to killing by cumene hydroperoxide (27). To determine whether Nox-1 functions as AhpF in vivo in combination with AhpC, we analyzed the sensitivity to killing by cumene hydroperoxide of E. coli TA4315 (ΔahpCF) transformed with either or both nox-1 and ahpC genes. Figure 5 shows that compared with the zone of inhibition for E. coli TA4315 induced by cumene hydroperoxide, this strain harboring pAN119 containing both nox-1 and ahpC demonstrated a striking reduction in diameter, to a size even less than that for E. coli K-12 (parent strain). Furthermore, TA4315 harboring pMS1 containing only ahpC also exhibited a reduction in diameter equal to that of K-12. In contrast, TA4315 harboring only the vector, pUC119, or pNox-1H containing nox-1 did not exhibit augmented resistance to cumene hydroperoxide.
FIG. 5.

Complementation of an ahpCF-deficient E. coli strain by S. mutans alkyl hydroperoxide reductase proteins. The zone of inhibition by 3% H2O2 or 3% cumene hydroperoxide (CHP) for E. coli TA4315 (ΔahpC ΔahpF) transformed with vector plasmid pUC119, pNox-1H containing nox-1, pMS1 containing ahpC, and pAN119 containing nox-1 and ahpC were compared with those of E. coli TA4315 as the sensitive control and E. coli K-12 (parent strain).
To identify the in vivo function of S. mutans alkyl hydroperoxide reductase in defense against peroxide stress, the same sensitivity assays of the ΔahpC, Δnox-1, ΔahpC Δnox-1 mutants and wild-type strain GS-5 were performed as described above except for the use of THB medium. Unexpectedly, the levels of sensitivity of these three mutants to killing by H2O2 and cumene hydroperoxide were almost the same as those of wild-type strains (data not shown). We also tested tert-butyl hydroperoxide and menadione under the same conditions but found no difference in sensitivities between the mutants and GS-5. These results indicated that defense systems of S. mutans for alkyl hydroperoxide differ from those of E. coli.
Adaptive responses and survival after H2O2 and cumene hydroperoxide challenge.
To further characterize the alkyl hydroperoxide reductase of S. mutans, we studied the adaptive effect of H2O2 and cumene hydroperoxide treatment of the ΔahpC mutant and wild-type strain GS-5 under strictly anaerobic conditions. The results in Fig. 6 demonstrated that (i) both ΔahpC mutant and wild-type cells showed adaptive responses to both 10 μM H2O2 and 30 μM cumene hydroperoxide, leading to resistance to lethal doses of these oxidants (100 μM for H2O2 and 300 μM for cumene hydroperoxide) and (ii) there was no significant difference in sensitivities between the ΔahpC mutant and wild-type cells with or without adaptation.
FIG. 6.

Adaptive responses against H2O2 (A) and cumene hydroperoxide (B) killing in S. mutans wild-type strain GS-5 and the ΔahpC mutant. All procedures were performed under anaerobic conditions. (A) Uninduced wild type (open circles) and ΔahpC (open triangles); H2O2-induced (60 min of treatment with 10 μM H2O2) wild type (closed circular) and ΔahpC (closed triangle). (B) Uninduced wild type (open circles) and ΔahpC (open triangles); cumene hydroperoxide-induced (60 min of treatment with 30 μM cumene hydroperoxide wild type (closed circles) and ΔahpC (closed triangles). Experiments were repeated three times, and the data shown are the means of triplicates.
DISCUSSION
In sugar alcohol fermentation, S. mutans degrades 1 mol of mannitol or sorbitol to 2 mol of pyruvate with a concomitant generation of 3 mol of NADH by the metabolic steps of mannitol 1-phosphate (or sorbitol 6-phosphate) dehydrogenase and glyceraldehyde 3-phosphate dehydrogenase (Fig. 7). For smooth operation of glycolysis, NADH has to be oxidized to NAD+, but lactate dehydrogenase can oxidize only 2 mol of NADH (Fig. 7). Under strictly anaerobic conditions, part of the pyruvate can be converted to formate and acetyl coenzyme A by pyruvate formate-lyase (PFL) (1) and further degraded to ethanol along with the oxidation of the surplus NADH to NAD+. Thus, S. mutans PFL plays an important role in maintaining the intracellular balance of NADH and NAD+ in the anaerobic metabolism of sugar alcohol. Consequently, the PFL-defective mutant did not grow on sorbitol anaerobically and was detected as small colonies on sorbitol agar plates (29). On the other hand, under aerobic conditions S. mutans PFL is extremely sensitive to O2 and is inactivated (28); thus, NADH has to be oxidized by another pathway.
FIG. 7.

Aerobic pathway of glucose and mannitol metabolism in S. mutans. Solid arrows indicate the flow of electron in the catabolic pathway. FBP, fructose 1,6-bisphosphate; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase.
In the present study, we demonstrated that the Nox-2-defective mutant could not grow on mannitol under aerobic conditions. This finding clearly indicated that Nox-2, corresponding to the H2O-forming NADH oxidase, is an essential enzyme for the regeneration of NAD+ during aerobic mannitol metabolism in S. mutans. Furthermore, we demonstrated that the high level of Nox-2 activity dramatically affected the aerobic metabolism of not only mannitol but also glucose. Although no significant difference was observed in the aerobic growth on glucose between wild-type GS-5 and Nox-2-deficient mutant strains until stationary phase, the Nox-2-deficient mutant produced large amounts of lactate (more than 90%), in contrast to GS-5, which produced less lactate (78%) and more acetate (17%). This shift in the fermentation end products indicates that the Nox-2-deficient mutant could not convert pyruvate to acetate during aerobic metabolism. S. mutans has an additional branch in the pathway involving pyruvate dehydrogenase (PDH) (5), where pyruvate is oxidized to acetate along with the generation of NADH (Fig. 7). The NADH derived from PDH also has to be oxidized by NADH oxidase. That is, the Nox-2-deficient mutant cannot operate the PDH pathway.
Recently, it has been reported that NADH oxidase-overproducing Lactococcus lactis strains constructed by cloning the S. mutans nox-2 gene showed a shift from homo-lactic to mixed-acid fermentation along with a decreased NADH/NAD+ ratio during aerobic glucose catabolism (17). Although this metabolically engineered system in L. lactis is unnatural, these results supported our findings that in the presence of Nox-2 pyruvate was converted to acetate by PDH (5), whereas in the absence of Nox-2 pyruvate was converted mostly to lactate during aerobic glucose catabolism in S. mutans. PDH seems to function actively during mannitol fermentation, since more acetate was produced from mannitol than from glucose (Fig. 4). This is peculiar when more NADH generation from mannitol is considered. Moreover, levels of intracellular fructose 1,6-bisphosphate, an absolute activator for lactate dehydrogenase (4), were low during the first few hours of aerobic growth on mannitol (data not shown). This may explain the low production of lactate and the shift to the considerable production of acetate during mannitol metabolism.
Originally, Nox-1 was purified from an oxygen-tolerant strain of S. mutans and characterized as an H2O2-forming NADH oxidase (12). In vitro, Nox-1 was demonstrated to have NADH-dependent peroxidase activity in the presence of AhpC, resulting in catalysis of the full four-electron reduction of O2 to H2O, similar to the H2O-forming NADH oxidase, Nox-2 (24). Unexpectedly, the contribution of NADH oxidase activity by Nox-1 to mannitol growth seems negligible, since Nox-1 could not support aerobic mannitol growth in the absence of Nox-2 and the lack of Nox-1 enzyme had no effect on the mannitol growth (Fig. 2B and 3B). Thus, we suggest that Nox-1 is another NADH-oxidizing enzyme functionally distinct from Nox-2 and not important in energy metabolism.
Based on a search of the sequence database, the S. mutans AhpC protein deduced from the partial sequence of the ahpC gene was identified as a member of AhpC/thiol-specific antioxidant family, a widely distributed class of antioxidant enzymes including the AhpC component of S. typhimurium (6). Furthermore, the Nox-1 protein as well as the AhpF component of S. typhimurium was also identified as a member of the AhpF/thioredoxine reductase family (6). The proposed catalytic mechanism for alkyl hydroperoxide reductase of S. typhimurium involves substrate peroxide reduction by the AhpC protein, with subsequent reduction of the AhpC by the AhpF coupled to either NADH or NADPH oxidation (23).
In vitro, Nox-1 functioned as an alkyl hydroperoxide reductase when combined with AhpC. Particularly for S. mutans lacking catalase and heme-containing peroxidases, the peroxidase activity of Nox-1 combined with AhpC should be important at least in defense against peroxide-mediated stress. In in vitro experiments, the purified proteins of alkyl hydroperoxide reductase, Nox-1 and AhpC, from S. mutans are mechanistically very similar to those from S. typhimurium, and can each interact with the S. typhimurium alkyl hydroperoxide reductase partner, AhpF or AhpC, for efficient catalysis of peroxide reduction (24). Nox-1 and AhpF also share the property that their oxidase activity is stimulated upon addition of free FAD (24). This apparent activation is the result of the ability of these enzymes to reduce free FAD and the subsequent nonenzymatic reaction of free reduced FAD with oxygen. S. mutans Nox-1 clearly plays a role similar to that of S. typhimurium AhpF.
In this report, we explored the properties of Nox-1 as an AhpF in S. mutans. The expression of Nox-1 is not highly correlated with alkyl hydroperoxide reductase activity, as shown by assays of cell extracts and by Western blot analysis of the proteins over the course of induction by aerobiosis. It may be that in vivo, the high levels of AhpC in these and other bacteria (26) allow for the maintenance of a large pool of activated (reduced) AhpC for rapid detoxification of any peroxides formed, while Nox-1 levels are modulated as necessary to efficiently maintain this pool of reduced AhpC. The antioxidant activity of Nox-1 in combination with AhpC in vivo was demonstrated by the increase in resistance to cumene hydroperoxide of an ahpCF-deficient E. coli mutant, TA4315, transformed with both structural genes, nox-1 and ahpC (Fig. 5). In these studies, the ability of pMS1 (encoding only AhpC) to itself impart this resistance is likely to be due to the demonstrated ability of S. mutans AhpC to be reduced by E. coli thioredoxin reductase and thioredoxin in the absence of Nox-1 or AhpF (25). However, the actual significance of the antioxidant activity of Nox-1 combined with AhpC in S. mutans was unclear, since no significant difference in resistance to cumene hydroperoxide and H2O2 between the Nox-1 and/or AhpC-deficient mutants and the wild-type strain was demonstrated.
Furthermore, even in the absence of AhpC, the mutant ΔahpC showed increased tolerance toward oxidants following treatment by sublethal doses of H2 O2 and cumene hydroperoxide to approximately the same level as that of the wild-type strain (Fig. 6). The finding that the AhpC-deficient mutant induced resistance to such oxidants despite the lack of catalase in streptococci implies that S. mutans has at least one other inducible organic hydroperoxide resistance gene in addition to ahpC; possibilities include a glutathione peroxidase gene (26) or a new organic hydroperoxide resistance gene like that from Xanthomonas campestris pv. phaseoli (21). The identification of antioxidants other than the alkyl hydroperoxide reductase system in S. mutans awaits further study.
In conclusion, the most striking finding in the present study is that Nox-2, identified as H2O-forming NADH oxidase, plays an important role in aerobic energy metabolism in O2-tolerant S. mutans. Nox-1, identified as H2O2-forming NADH oxidase, on the other hand, was shown to contribute negligibly in either function, as NADH-dependent oxidase or as NADH-dependent peroxidase in combination with AhpC. Presumably, the function of Nox-1 as alkyl hydroperoxide reductase is masked by overlapping effects of some other antioxidant system(s) in S. mutans. It is noteworthy that only Nox-1 among the three proteins was present in anaerobically grown cells and expressed during aerobic growth to amounts comparable to those of Nox-2 protein, despite the low NADH oxidase activity. These findings suggest that Nox-1 plays a role distinct from that of Nox-2 in S. mutans.
ACKNOWLEDGMENTS
We thank Yoichi Niimura for generously providing anti-A. xylanus AhpC sera and Al Claiborne for helpful discussions.
This work was supported by ISRP grant 09044200 to Y. Kamio from the Ministry of Education, Science, Sports, and Culture of Japan.
REFERENCES
- 1.Abbe K, Takahashi S, Yamada T. Involvement of oxygen-sensitive pyruvate formate-lyase in mixed-acid fermentation by Streptococcus mutans under strictly anaerobic conditions. J Bacteriol. 1982;152:175–182. doi: 10.1128/jb.152.1.175-182.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki H, Shiroza T, Hayakawa M, Sato S, Kuramitsu H K. Cloning of a Streptococcus mutans glucosyltransferase gene coding for insoluble glucan synthesis. Infect Immun. 1986;53:587–594. doi: 10.1128/iai.53.3.587-594.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradford M M. A rapid and sensitive methods for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Brown A T, Wittenberger C T. Fructose 1,6-diphosphate-dependent lactate dehydrogenase from a cariogenic streptococcus: purification and regulatory properties. J Bacteriol. 1972;122:1126–1135. doi: 10.1128/jb.110.2.604-615.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlsson J, Kujala U, Edlund M B K. Pyruvate dehydrogenase activity in Streptococcus mutans. Infect Immun. 1985;49:674–678. doi: 10.1128/iai.49.3.674-678.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chae H Z, Robison K, Poolr L B, Church G, Storz G, Rhee S G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis H R, Poole L B. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 8.Hamada S, Slade H D. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol Rev. 1980;44:331–384. doi: 10.1128/mr.44.2.331-384.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 10.Higuchi M. Effect of oxygen on the growth and mannitol metabolism of Streptococcus mutans. J Gen Microbiol. 1984;130:1819–1826. doi: 10.1099/00221287-130-7-1819. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi M. Reduced nicotinamide adenine dinucleotide oxidase involvement in defense against oxygen toxicity of Streptococcus mutans. Oral Microbiol Immunol. 1992;7:309–314. doi: 10.1111/j.1399-302x.1992.tb00594.x. [DOI] [PubMed] [Google Scholar]
- 12.Higuchi M, Shimada M, Yamamoto Y, Hayashi T, Kamio Y. Identification of two distinct NADH oxidases corresponding to H2O2-forming oxidase and H2O-forming oxidase induced in Streptococcus mutans. J Gen Microbiol. 1993;139:2343–2351. doi: 10.1099/00221287-139-10-2343. [DOI] [PubMed] [Google Scholar]
- 13.Higuchi M, Shimada M, Matsumoto J, Yamamoto Y, Rhaman A, Kamio Y. Molecular cloning and sequence analysis of the gene encoding the H2O2-forming NADH oxidase from Streptococcus mutans. Biosci Biotechnol Biochem. 1994;58:1603–1607. doi: 10.1271/bbb.58.1603. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson F S, Morgan R W, Christman M F, Ames B N. An alkyl hydroperoxide reductase involved in the defense of DNA against oxidative damage: purification and properties. J Biol Chem. 1989;264:1488–1496. [PubMed] [Google Scholar]
- 15.LeBlanc D J, Lee L N, Inamine J M. Cloning and nucleotide base sequence analysis of a spectinomycin adenyltransferase AAD(9) determinant from Enterococcus faecalis. Antimicrob Agents Chemother. 1991;35:1804–1810. doi: 10.1128/aac.35.9.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loesche W J. Role of Streptococcus mutans in human dental decay. Microbiol Rev. 1986;50:353–380. doi: 10.1128/mr.50.4.353-380.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez de Felipe F, Kleerebezem M, de Vos W M, Hugenholtz J. Cofactor engineering: a novel approach to metabolic engineering in Lactococcus lactis by controlled expression of NADH oxidase. J Bacteriol. 1998;180:3804–3808. doi: 10.1128/jb.180.15.3804-3808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maniatis T, Fritsch E F, Sambrook J. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 19.Matsumoto J, Higuchi M, Shimada M, Yamamoto Y, Kamio Y. Molecular cloning and sequence analysis of the gene encoding the H2O-forming NADH oxidase from Streptococcus mutans. Biosci Biotechnol Biochem. 1996;60:39–43. doi: 10.1271/bbb.60.39. [DOI] [PubMed] [Google Scholar]
- 20.Minakami S, Suzuki C, Saito T, Yoshikawa H. Studies on erythrocyte glycolysis. I. Determination of the glycolytic intermediates in human erythrocytes. J Biochem. 1965;58:543–550. doi: 10.1093/oxfordjournals.jbchem.a128240. [DOI] [PubMed] [Google Scholar]
- 21.Mongkolsuk S, Praituan W, Loprasert S, Fuangthong M, Chamnongpol S. Identification and characterization of a new organic hydroperoxide resistance (ohr) gene with a novel pattern of oxidative stress regulation from Xanthomonas campestris pv. phaseoli. J Bacteriol. 1998;180:2636–2643. doi: 10.1128/jb.180.10.2636-2643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry D, Kuramitsu H K. Genetic transformation of Streptococcus mutans. Infect Immun. 1981;32:1295–1297. doi: 10.1128/iai.32.3.1295-1297.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poole L B, Ellis H R. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 24.Poole L B, Shimada M, Higuchi M. NADH oxidase-1 and a second component encoded upstream of nox-1 comprise an alkyl hydroperoxide reductase system in Streptococcus mutans. In: Stevenson K J, Massey V, Williams C H Jr, editors. Flavins and flavoproteins 1996. Calgary, Alberta, Canada: University of Calgary Press; 1997. pp. 769–772. [Google Scholar]
- 25.Poole, L. B. Unpublished observations.
- 26.Roe, B. A., S. Clifton, M. McShan, and J. Ferretti. Streptococcal genome sequencing project. University of Oklahoma, Norman, Okla.
- 27.Storz G, Jacobson F S, Tartaglia L A, Morgan R W, Silveira L A, Ames B N. An alkyl hydroperoxide reductase induced by oxidative stress in Salmonella typhimurium and Escherichia coli: genetic characterization and cloning of ahp. J Bacteriol. 1989;171:2049–2055. doi: 10.1128/jb.171.4.2049-2055.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi N, Abbe K, Takahashi-Abbe S, Yamada T. Oxygen sensitivity of sugar metabolism and interconversion of pyruvate formate-lyase in intact cells of Streptococcus mutans and Streptococcus sanguis. Infect Immun. 1987;55:652–656. doi: 10.1128/iai.55.3.652-656.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto Y, Sato Y, Takahashi-Abbe S, Abbe K, Yamada T, Kizaki H. Cloning and sequence analysis of the pfl gene encoding pyruvate formate-lyase from Streptococcus mutans. Infect Immun. 1996;64:385–391. doi: 10.1128/iai.64.2.385-391.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 31.Wetherell J R, Jr, Bleiweis A S. Antigens of Streptococcus mutans: characterization of a polysaccharide antigen from walls of strain GS-5. Infect Immun. 1975;12:1341–1348. doi: 10.1128/iai.12.6.1341-1348.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]