

Summary
Alternative cleavage and polyadenylation (APA) is a widespread mechanism to generate mRNA isoforms with alternative 3′ untranslated regions. Here, we detail a protocol for detecting APA genome wide using direct RNA sequencing technology including computational analysis. We describe steps for RNA sample and library preparation, nanopore sequencing, and data analysis. Experiments and data analysis can be performed over a period of 6–8 days and require molecular biology and bioinformatics skills.
For complete details on the use and execution of this protocol, please refer to Polenkowski et al.1
Subject areas: Sequence Analysis, Gene Expression
Graphical abstract
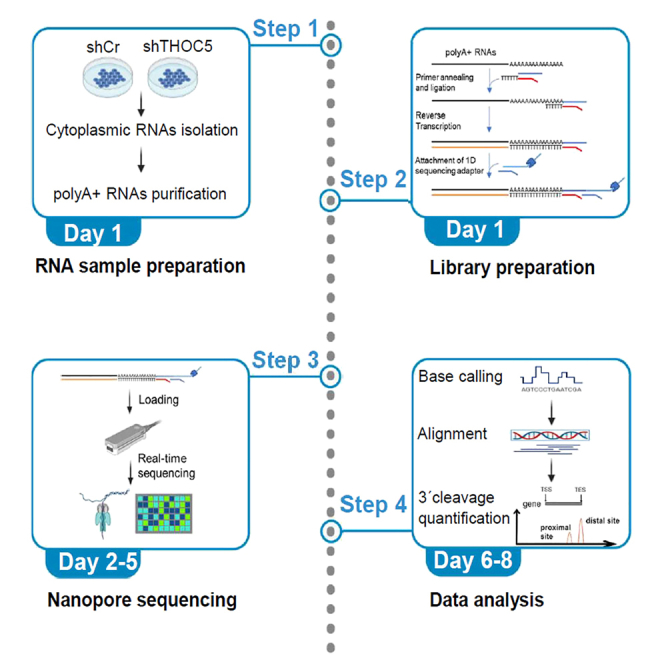
Highlights
-
•
Direct RNA sequencing using Oxford Nanopore sequencing technology
-
•
Mapping 3′ end cleavage sites using nanopore reads
-
•
Steps toward quantification of alternative polyadenylation in human cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Alternative cleavage and polyadenylation (APA) is a widespread mechanism to generate mRNA isoforms with alternative 3′ untranslated regions. Here, we detail a protocol for detecting APA genome wide using direct RNA sequencing technology including computational analysis. We describe steps for RNA sample and library preparation, nanopore sequencing, and data analysis. Experiments and data analysis can be performed over a period of 6–8 days and require molecular biology and bioinformatics skills.
Before you begin
This protocol allows the direct RNA sequencing from polyA-tail to the 5′end, thus it enables the precise mapping of 3′end cleavage sites. The first part of the protocol describes the lentivirus transduction of targeted shRNA and the isolation of cytoplasmic polyA-tailed RNAs. The second part of the protocol describes library preparation using the SQK-RNA002 kit for direct RNA sequencing. The third part describes the steps for direct RNA sequencing by Oxford Nanopore. The fourth part of the protocol describes the data analysis including quality control, basecalling, alignment and 3′end cleavage quantification.
Before beginning, buffer, media, and enzyme solutions must be prepared or purchased. Before nanopore sequencing, MinKNOW v21.06.0 must be installed to run the MinION flow cells. All software for base calling and alignment are run on Ubuntu 20.04 and need to be installed prior to starting the data analysis.
The detailed protocol below describes the specific steps for using HEK293 cells. However, in principle this method should also be applicable to other cell lines such as HeLa cells or NIH3T3 cells.
Preparing buffers
Timing: 0.5–1 h
Preparation of cells
All procedures must be performed in a biosafety level 2 (BSL2) laboratory.
-
1.
Maintain HEK293 cells in 10-cm tissue culture dishes in 10 mL of Dulbecco’s Modified Eagle Medium (DMEM) containing 25 mM glucose, 1 mM sodium pyruvate and supplemented with 2 mM L-glutamine and 10% fetal bovine serum (FBS) in the presence of antibiotics penicillin-streptomycin (P/S).
-
2.
Transduce HEK293 cells with control shRNA (shCr) and shRNA targeting THOC5 (shTHOC5-1 and shTHOC5-2). For the lentiviral infection, 5 × 105 cells (approximately 30%–40% confluency at the time of transduction) are plated in 10-cm dishes in complete DMEM. Lentiviral transduction was performed with a multiplicity of infections (MOI) of 10. Medium is replaced with infection mixture: 500 μL of the lentiviral particles (107 transducing unit (TU)/mL) in complete culture media (DMEM + 10% FBS) containing polybrene (final concentration of 8 μg/mL). Gently swirl the plate to mix. Incubate the cells for 48 h at 37°C and 5% CO2.
Note: Thaw the lentiviral particles (if frozen) on ice. Gently spin down before opening. Keep them on ice. Mix gently before use.
-
3.
After 48 h, remove infection mixture and replace by the fresh medium (DMEM + 10% FBS + P/S). Incubate cells further for 2 days.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| shRNA control | Saran et al.2 | N/A |
| shRNA THOC5 | Saran et al.2 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM high-glucose medium | PAN Biotech | P04-05550 |
| Fetal bovine serum | Thermo Fisher | 10270098 |
| Penicillin-streptomycin-glutamine | Gibco | 10378016 |
| Polybrene | Sigma-Aldrich | TR-1003 |
| Tris hydrochloride pH8.0 (1 M) | Thermo Fisher | 15893661 |
| Sodium chloride (5 M) | Sigma-Aldrich | S6546 |
| NP40 (10% v/v) | Thermo Fisher | 13434269 |
| Diethylpyrocarbonat | Sigma-Aldrich | D5758 |
| Protease inhibitor cocktail | Sigma-Aldrich | P1860 |
| RNAse Inhibitor | NEB | M0314 |
| Isopropanol | Roth | 0733.1 |
| Nuclease-free water | Thermo Fisher | AM9937 |
| 10 mM dNTP solution | NEB | N0447 |
| Agencourt RNAClean XP beads | Beckman Coulter | A63987 |
| Ethanol | Roth | 5054.1 |
| PBS (phosphate buffered saline) | Thermo Fisher Scientific | 10010023 |
| RNAse- and DNAse-free Eppendorf tube | Thermo Fisher Scientific | AM12400 |
| Critical commercial assays | ||
| ReliaPrep™ miRNA Cell and Tissue Miniprep System | Promega | Z6210 |
| NEBNext® poly(A) mRNA magnetic isolation module | NEB | E7490 |
| Qubit dsDNA HS Assay-Kit | Thermo Fisher | Q32851 |
| Direct RNA sequencing kit | Oxford Nanopore Technologies | SQK-RNA002 |
| NEBNext Quick Ligation Module | NEB | E6056 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | 18080044 |
| Flow Cell Priming Kit | Oxford Nanopore Technologies | EXP-FLP002 |
| Qubit™ RNA High Sensitivity (HS) assay kit | Invitrogen | N/A |
| Deposited data | ||
| Nanopore-sequencing | N/A | Accession number GSE1733741 |
| Experimental models: cell lines | ||
| HEK293 cells | ATCC | Cat#CRL-1573 |
| Software and algorithms | ||
| MinKNOW software v21.06.0 | Oxford Nanopore Technologies | https://community.nanoporetech.com |
| Guppy v3.4.4 | N/A | https://nanoporetech.com/ |
| Guppy_on_SLURM | N/A | https://github.com/trandoanduyhai/guppy_on_slurm |
| Galaxy Bioinformatics Platform | As described by the Galaxy Community3 | Galaxy (usegalaxy.org) |
| Pycoqc | N/A |
https://github.com/a-slide/pycoQC Galaxy (usegalaxy.org) |
| Minimap2 | As described by Heng Li4 | https://github.com/lh3/minimap2 |
| Samtools | As described by Danecek et al.5 | https://github.com/samtools/samtools |
| NanoFilt | As described by De Coster et al.6 | https://github.com/wdecoster/nanofilt |
| Seqmonk v1.48.0 | Babraham institute | https://github.com/s-andrews/SeqMonk |
| Human polyA database | As described by Lee et al.7 | https://exon.apps.wistar.org/PolyA_DB/v3/ |
| Other | ||
| MinION Sequencing Device | Oxford Nanopore Technologies | MIN-101B |
| R9 version MinION SpotON Flow Cell | Oxford Nanopore Technologies | FLO-MIN106D |
| Magnetic separator | Invitrogen | 12321D |
| Thermal mixer | VWR | 75786-280 |
| Cooling microcentrifuge | VWR | 521-1647 |
| Vortex mixer | Heidolph | N/A |
| Ice bucket with ice | N/A | |
| Timer | N/A | |
| Thermal cycler | Biometra | N/A |
| Qubit fluorometer | Life Technologies | N/A |
| P1000 pipette and tips | Eppendorf | N/A |
| P200 pipette and tips | Eppendorf | N/A |
| P100 pipette and tips | Eppendorf | N/A |
| P20 pipette and tips | Eppendorf | N/A |
| P10 pipette and tips | Eppendorf | N/A |
| P2 pipette and tips | Eppendorf | N/A |
Materials and equipment
Cytoplasmic extraction buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris HCl pH = 8.0 (1 M) | 10 mM | 0.5 mL |
| NaCl (5 M) | 150 mM | 1.5 mL |
| NP40 (10% v/v) | 0.5% v/v | 2.5 mL |
| DEPC H2O | N/A | 45.5 mL |
| Total | 50 mL |
Note: The buffer can be stored at 4°C for 12 months. Add (1:100) Protease inhibitor cocktail for mammalian tissues (Sigma-Aldrich) and (1:100) RNAse Inhibitor (Biolabs) shortly before using.
Step-by-step method details
Isolation of cytoplasmic RNAs
Prior to the isolation step, prepare ice-cold PBS and pre-cool the centrifuge to 4°C.
Note: To ensure optimal cytoplasmic RNA yield (∼30 μg), prepare two plates of HEK293 in 10-cm tissue culture dishes at 80% cell confluency (9 × 106 cells). For other cell systems, 5–30 pg of total RNAs per cell are expected.
-
1.Nuclear-cytoplasm fractionation of HEK293 cells.
-
a.Discard the medium from the culture dishes. Place the culture dishes on ice.
-
b.Add 3 mL of cold PBS to each culture dish and carefully scrape the cells using a cell scraper (tilt the culture dish to maximize the collection of cells in the PBS solution).
-
c.Combine the cell suspension from both plates (2 × 3 mL) into a single 15 mL falcon tube and centrifuge at 1500 × g at 4°C for 2 min.
-
d.Remove the supernatant and resuspend the cell pellets with 800 μL cold cytoplasmic extraction buffer. Add 400 units of RNase Inhibitor (NEB) to the cell suspension and incubate on ice for 10 min.
-
e.Centrifuge at 1500 × g and 4°C for 2 min.
-
f.Transfer and divide 800 μL of the supernatant (cytoplasmic fraction) into four new RNAse-free Eppendorf tubes (200 μL each). Nuclear pellets are discarded.
-
a.
CRITICAL: Dividing 800 μL of the cytoplasmic fraction into 4 × 200 μL is crucial to ensure efficient column throughput and optimal RNA yield.
-
2.Isolation of cytoplasmic RNAs.Note: We used the ReliaPrep™ miRNA Cell and Tissue Miniprep System for isolation for RNA from cytoplasmic fraction. However, it is not necessary to use the miRNA kit, because the length of most of polyA-tailed RNAs (mRNAs and lncRNAs) is longer than 200nt. Thus, they can be enriched with other RNA extraction column such as RNeasy Kit (Qiagen) or High Pure RNA Isolation Kit (Roche).Note: All four cytoplasmic fractions should be used to isolate the RNA material from step 1f. Separate column should be used for each 200 μL fraction.Steps 2a-e describe the RNA isolation for one cytoplasmic fraction reaction (200 μL).
-
a.Gently mix 200 μL of cytoplasmic fraction with 200 μL Lysis Buffer (LBA+TG) buffer, 130 μL RNA Dilution Buffer (RDB) and 739 μL isopropanol.
-
b.Transfer the mixture to the column and centrifuge at 11,600 × g at 25°C for 1 min. Remove the liquid from the collection tube. Repeat this step for the rest of the mixture using the same column.
-
c.Add 500 μL RNA Wash Solution (RWA) to column and centrifuge at 11,600 × g for 1 min at 25°C. Discard the liquid from the collection tube.
-
d.Add another 500 μL RWA to column and centrifuge at 16,000 × g for 2 min. Discard liquid from the collection tube.
-
e.Transfer the column into a new 1.5 mL elution tube.
-
f.Elute the RNAs by adding 50 μL nuclease-free water to column and incubate for 1 min at 25°C then centrifuge at 11,600 × g for 1 min. Collect all eluted cytoplasmic RNA into one single 1.5 mL elution tube.Note:Check the quantity and quality of the cytoplasmic RNA using nanodrop spectrophotometer. A yield of at least 30 μg is expected at this point. If A260/A280 and/or A260/A230 ratios are low, they indicate a contamination by proteins or salt. If A260/A280 ratio < 1.8 and A260/A230 < 1.7, users can follow the troubleshooting (problem 4). In addition to RNA quality check, the RNA integrity should be tested on a Bioanalyzer or RNA gel, especially for RNAs isolated from primary tissues.The purified RNAs can be stored at −80°C for up to one week.
-
a.
Isolation of cytoplasmic polyA-tailed RNA
Note: Keep all reagents on ice for optimal results. The following protocol is modified from NEBNext® poly(A) mRNA magnetic isolation module (E7490).
-
3.
Dilute the cytoplasmic RNAs with nuclease-free water to a final volume of 100 μL in a 1.5 mL RNAse-free tube.
Note: Prepare the thermal cycler at 65°C for step 5b. The lid needs to be heated at 95°C.
-
4.Washing the magnetic beads.
-
a.Add 100 μL of well resuspended NEBNext magnetic oligo-dT beads to 1.5 mL RNAse-free tube.
-
b.Wash the beads by adding 100 μL RNA binding buffer (provided in NEB #E7490 kit). Gently pipette the solution up and down 6 times using 100 μL pipette.
-
c.Place the tube in a magnetic rack at 25°C for approx. 1 min until the solution becomes clear.
-
d.Discard the supernatant from the tube by pipetting using 100 μL pipette without removing the tube from the magnetic rack or disturbing the beads.
-
e.Remove the tube from the magnetic rack.
-
f.Repeat steps 4b-e.
-
a.
-
5.RNA binding to the beads.
-
a.Resuspend the washed beads in 100 μL RNA binding buffer and add the 100 μL of cytoplasmic RNA from step 3. Mix the solution gently by pipetting up and down 3 times using 100 μL pipette. The pipette is set for 100 μL.
-
b.Transfer the bead suspension into 0.5 mL tube. Place the tube in the thermal cycler and heat the sample at 65°C for 5 min and cool to 4°C with the heated lid set at ≥75°C to denature the RNA and facilitate binding of the mRNA to the beads.
-
c.Resuspend the beads by gently pipetting the solution up and down 6 times using 100 μL pipette. The pipette is set to 100 μL.
-
d.Incubate the tube at 25°C for 5 min to allow the binding of polyA-tailed RNA to the beads.
-
e.Resuspend the beads by gently pipetting the solution up and down 6 times using 100 μL pipette. The pipette is set to 100 μL.
-
f.Incubate the tube at 25°C for another 5 min to allow the RNA to bind to the beads.
-
g.Place the tube in a magnetic rack at 25°C for 30 s–1 min until the solution becomes clear.
-
h.Discard the supernatant from the tube by pipetting without removing the tube from the magnetic rack or disturbing the beads.
-
i.Remove the tube from the magnetic rack.
-
a.
Note: Prepare the thermal cycler at 80°C for steps 7b and 7o. The lid temperature is set at 95°C.
-
6.Washing the magnetic beads.
-
a.Wash the beads by adding 200 μL wash buffer (provided in NEB #E7490 kit) to remove unbound RNA. Gently pipette the solution up and down 6 times using 1000 μL pipette. The pipette is set to 200 μL.
-
b.Place the tube in a magnetic rack at 25°C until the solution becomes clear.
-
c.Discard the supernatant from the tube by pipetting without removing the tube from the magnetic rack or disturbing the beads.
-
d.Remove the tube from the magnetic rack.
-
e.Repeat steps 6a-d.
-
a.
-
7.Elution of the polyA-tailed RNA from the beads.
-
a.Add 50 μL of Tris buffer (provided in NEB #E7490 kit) to each tube and gently pipette the solution up and down 6 times using 100 μL pipette. The pipette is set to 50 μL.
-
b.Incubate the tubes at 80°C for 2 min, and then place them on the working bench until they reach 25°C.
-
c.Add 50 μL of RNA binding buffer to each sample and gently pipette the solution up and down 6 times to mix thoroughly using 100 μL pipette. The pipette is set to 50 μL.
-
d.Incubate the tubes at 25°C for 5 min.
-
e.Resuspend the beads by gently pipetting the solution up and down 6 times.
-
f.Incubate the tubes at 25°C for another 5 min to allow the RNA to bind to the beads.
-
g.Place the tube in a magnetic rack at 25°C until the solution becomes clear.
-
h.Discard the supernatant from the tube by pipetting without removing the tube from the magnetic rack or disturbing the beads.
-
i.Remove the tube from the magnetic rack.
-
j.Wash the beads with 200 μL wash buffer. Gently pipette the solution up and down 6 times.
-
k.Place the tube in a magnetic rack at 25°C until the solution becomes clear.
-
l.Discard the supernatant from the tube by pipetting without removing the tube from the magnetic rack or disturbing the beads.Note: To remove ethanol residue without disturbing the beads, use the 10 μL and 100 μL pipette tips as shown in the picture below (Figure 1).
-
m.Remove the tube from the magnetic rack.
-
n.Elute the RNA from the beads by adding 11 μL of Tris buffer. Gently pipette the solution up and down 6 times.
-
o.Incubate the tubes at 80°C for 2 min, and then place them on working bench until they reach 25°C.
-
p.Place the tube on magnetic rack at 25°C until the solution becomes clear.
-
q.Collect the purified polyA-tailed RNA by transferring the supernatant to a 1.5 mL DNA LoBind tube and then place the tube on ice.
-
r.Check the yield and the quality of the purified polyA-tailed RNA using Qubit fluorometer and Qubit RNA high sensitivity assay kit. Prepare the following component in a 0.2 mL tube and measure on a Qubit fluorometer:
Reagent Amount Qubit RNA buffer 198 μL Qubit reagent 1 μL Purified poly(A)-RNA 1 μL Total 200 μL Note: Expectation: approx. 500 ng of polyA-tailed RNA from 30 μg input RNAs. For isolation from polyA-tailed RNAs from primary cells or tissues we recommend to check the RNA integrity on a Bioanalyzer. A low yield of polyA-tailed RNA may be due to the RNA degradation from 3′end (troubleshooting: problem 5).
-
a.
Figure 1.

The use of the 10 μL and 100 μL pipette tips to remove ethanol residue
Library preparation
Note: The following protocol is modified from Direct RNA sequencing (SQK-RNA002 version: DRS_9080_V2_revO_14Aug2019) Nanopore protocol. Prepare a program at the thermal cycler for incubation at 50°C for 50 min, then 70°C for 10 min (step 9d). The lid temperature needs to be set at 95°C. Keep all reagents on ice for optimal results. For a quality library preparation 500 ng polyA-tailed RNA is used as a starting material. However 200–500 ng polyA-tailed RNA will also work. Lower than 200 ng polyA-tailed RNA may reduce the sequencing yield.
-
8.Ligation of a double stranded RT adapter (RTA) (provided by SQK-RNA002 kit) to polyA-tailed RNA.
-
a.Prepare in a 0.2 mL PCR tube and mix the reagents in the following order:
Reagent Amount NEBNext Quick Ligation Reaction Buffer 3 μL Purified polyA-tailed RNA 9 μL RNA CS (RCS), 110 nM(provided in SQK-RNA002 kit) 0.5 μL RT Adapter (RTA) 1.0 μL T4 DNA Ligase 1.5 μL Total 15 μL -
b.Gently mix by pipetting and incubate the reaction at 4°C for 10 min.Note: The incubation time could be extended up to 1 h. According to our experience extending of the incubation time does not affect the sequencing yields. Thus, having a break at this step is possible.
-
a.
-
9.Reverse transcription.
-
a.Mix the following reagents to make the reverse transcription master mix:
Reagent Amount Nuclease-free water 9 μL 10 mM dNTPs 2 μL 5× first-strand buffer 8 μL 0.1 M DTT 4 μL Total 23 μL -
b.Add the master mix into the 0.2 mL PCR tube from step 8b and mix gently by pipetting.
-
c.Add 2 μL of SuperScript III reverse transcriptase to reaction and mix gently by pipetting.
-
d.Incubate the tube in a thermal cycler at 50°C for 50 min, then 70°C for 10 min, and bring the sample to 4°C for 10 min.
-
e.Transfer the sample to a clean 1.5 mL Eppendorf DNA LoBind tube.
-
f.Resuspend the stock of Agencourt RNAClean XP beads by vortexing.
-
g.Add 72 μL of resuspended RNAClean XP beads to reverse transcription reaction from step 9e and mix it by pipetting.
-
h.Incubate the mixture on a Hula mixer (rotator mixer) for 5 min at 25°C.
-
i.Spin down the sample shortly, place it on magnetic rack at 25°C until the solution becomes clear and remove the supernatant by pipetting without disturbing the beads.
-
j.Wash the beads by adding 150 μL of freshly prepared 80% ethanol without disturbing the beads.CRITICAL: Freshly prepared 80% ethanol in nuclease free water should be used. Pipetting ethanol directly on the beads will decrease the output.
-
k.Keep the tubes on the magnetic rack, rotate the tubes 180°C and wait until the beads migrate toward the magnet and build a pellet.
-
l.Repeat the step 9k so that the beads migrate to the starting position.
-
m.Remove the 80% ethanol by pipetting without disturbing the beads.Note: To remove ethanol residue without disturbing the beads, use the 10 μL and 100 μL pipette tips shown in Figure 1.
-
n.Remove the tube from the magnetic rack and resuspend the pellet in 20 μL nuclease-free water. Incubate at 25°C for 5 min.
-
o.Place the tube in a magnetic rack at 25°C until the solution becomes clear.
-
p.Pipette 20 μL of eluate into a 1.5 mL DNA LoBind tube.
-
a.
-
10.Ligation of RNA sequencing adapter with motor protein (RMX) to the RNA molecule.
-
a.Prepare the reaction mixture in a 1.5 mL DNA LoBind tube in the following order:
Reagent Amount Reverse-transcribed RNA from step 9p 20 μL NEBNext Quick Ligation Reaction Buffer 8 μL RNA adapter (RMX) (provided in SQK-RNA002 kit) 6 μL Nuclease-free water 3 μL T4 DNA Ligase 3 μL Total 40 μL -
b.Mix the components by gently pipetting up and down using 100 μL pipette. The pipette is set for 50 μL.CRITICAL: Do not use a vortex mixer! The reaction mixture contains the motor protein (RMX).
-
c.Incubate the reaction mixture at 25°C for 10 min.
-
d.Resuspend the stock of Agencourt RNAClean XP beads by vortexing.
-
e.Add 40 μL of resuspended RNAClean XP beads to the adapter ligation reaction from step 10b and mix it gently by pipetting.
-
f.Incubate the mixture on a Hula mixer (rotator mixer) for 5 min at room temperature.
-
g.Spin down the sample shortly (centrifuge speed), place it in a magnetic rack at 25°C until the solution becomes clear, and remove the supernatant by pipetting without disturbing the beads.
-
h.Add 150 μL wash buffer (WSB, provided in SQK-RNA002 kit) to the beads. Close the tube lid and resuspend the beads by flicking the tube carefully.CRITICAL:Do not try to mix the beads by pipetting or vortexing; it will damage the motor protein annealed on the adapter.
CRITICAL:Gently flicking the tubes is more effective in removing unbound adapters than immediately aspirating after adding the wash buffer. A high number of unbound adapters will decrease the sequencing output.
-
i.Spin down the tube shortly and place the tube on a magnetic rack at 25°C until the solution becomes clear.
-
j.Discard the supernatant from the tube by pipetting without removing the tube from the magnetic rack or disturbing the beads.
-
k.Repeat steps 10h-j once.
-
l.Remove the tube from the magnetic rack and resuspend the beads in 21 μL Elution Buffer (ELB, provided in SQK-RNA002 kit) by gently pipetting.
-
m.Incubate for 10 min at 25°C.
-
n.Spin down the tube shortly and place the tube in a magnetic rack at 25°C until the solution becomes clear.
-
o.Transfer the 21 μL eluate into a clean 1.5 mL DNA LoBind tube.Optional: Quantity control of the purified sample from step 10o can be done using the Qubit fluorometer dsDNA high sensitivity assay kit. An approximate yield of 200 ng is expected. A lower yield of adapter annealed RNAs is due to the RNA degradation during library preparation or a lower than intended AMPure beads-to-sample ratio (troubleshooting: problem 6).
-
a.
Priming and loading the SpotOn flow cell
The reverse-transcribed and adapted RNA is now ready for loading into the flow cell.
-
11.
Thaw the RNA Running Buffer (RRB), Flush Tether (FLT) and one tube of Flush Buffer (FB) at room temperature.
-
12.
Mix the RNA Running Buffer (RRB), Flush Tether (FLT) and Flush Buffer (FB) tubes thoroughly by vortexing and spinning down at room temperature.
-
13.
Open the MinION Mk1B lid and slide the flow cell under the clip. Firmly press down the flow cell to ensure proper thermal and electrical contact.
-
14.
Slide the priming port cover clockwise to open the priming port (Figure 2).
-
15.Priming and loading the SpotON flow cell.
-
a.After opening the priming port, check for small air bubbles under the cover. Remove any bubbles by withdrawing 20–30 μL of buffer from the flow cell.
-
i.Set a 1000 μL Pipette to 200 μL.
-
ii.Insert the tip into the priming port.
-
iii.Turn the wheel until the dial shows 220–230 μL or until a small volume of a buffer can be seen entering the pipette tip.Visually check that there is a continuous buffer from the priming port across the sensor array.CRITICAL: Make sure that no bubble forms during pipetting. The bubble will induce unwanted pressure in the flow cell.
-
i.
-
b.Prepare the mixture for flow cell priming by adding 30 μL of thawed Flush Tether (FLT) directly into the Flush Buffer (FB) tube and mix by pipetting it gently at 25°C.
-
c.Load 800 μL of the priming mix into the flow cell through the priming port (avoid introducing air bubbles). Wait for 5 min to equilibrate the flow cell.Note: Connect the SpotON flow cell loaded MinION device with the computer. Prior to use, the flow cell can be checked by performing a Flow Cell Check using MinKNOW (the MinION device software). The Flow Cell Check will report the number of nanopores available for sequencing (> 1,100 nanopores should be active). The Flow Cell Check will be performed as following:
-
i.Connect the device to a computer (a window of a flow cell appears and a note with “Flow cell not checked”).
-
ii.Click symbol ≡.
-
iii.Then “Flow cell check”.
-
iv.Then “Start”.
-
v.Note: After the device reaches 37°C, it will start scanning the number of active channels. If the temperature is fluctuated, the flow cell may lose the contact with the device (troubleshooting: problem 2 and 3).
-
i.
-
d.Prepare the library for loading by following these steps:
-
i.Mix the 20 μL of prepared RNA library with 17.5 μL nuclease-free water.
-
ii.In a new tube, prepare the library for loading as follows:
Reagent Amount RNA Running Buffer (RRB) 37.5 μL RNA library in nuclease-free water ( from step 15d i) 37.5 μL Total 75 μL
-
i.
-
e.Complete the flow cell priming:
-
i.Gently lift the SpotON sample port cover to make the SpotON sample port accessible.
-
ii.Load 200 μL of the priming mix into the flow cell via the priming port (not the SpotON sample port), avoiding the introduction of air bubbles.
-
i.
-
f.Mix the prepared library gently by pipetting prior to loading.
-
g.Add 75 μL of prepared library to the flow cell via SpotON sample port in dropwise fashion. Ensure each drop flows into the port before adding the next.
-
h.Gently replace the SpotON sample port cover, making sure the bung enters the SpotON port, close the priming port and close the MinION Mk1B lid.
-
a.
Figure 2.

SpotON flow cell map (Retrieved from streetscience.community/protocols/beer-dna-sequencing/)
Nanopore direct RNA sequencing
-
16.
Open the MinKNOW software (21.06.0).
-
17.Select “Start” then “Start sequencing” and fill in the information, as requested:
-
a.Selected positions:
-
i.Position: Select the MinION device.
-
ii.Choose an experiment number and sample ID.
-
iii.Verify the flow cell ID number. The flow cell model will be recognized by MinKNOW.
-
i.
-
b.Kit:
-
i.Selected kit: SQK-RNA002.
-
i.
-
c.Run options:
-
i.Run length: 72 h.
-
ii.Bias voltage: −180 mV.
-
i.
-
d.Base calling: Off.
-
e.Output: FAST5 (On).
-
a.
Note: After starting the sequencing run, it is recommendable to monitor the initial result after around 1 h to ensure that more than 90% of the pores are sequencing samples.
Data analysis
The complete data analysis pipeline is depicted in Figure 3. Here, we describe a step-by-step protocol, including the expected outcome.
Figure 3.

Strategy for data analysis
(A) Cytoplasmic polyA-tailed mRNAs were subjected to direct RNA-seq. The nanopore reads can be used for mapping 3′end cleavage, quantitating gene expression and identification of spliced isoforms. To map and quantitate 3′end cleavage sites, raw nanopore reads were trimmed to the last 200 nucleotides from the 3′end. The 3′end cleavage site usage at the distal and the proximal site were quantitated based on the human polyA database. Full-length reads are used for gene expression quantification and alternative splicing analysis.
(B) Due to the nature of long read sequencing, reads of transcripts cleaved at the distal site also cover the proximal site. Thus, to calculate the read coverage for 3′ cleavage site we trimmed the reads to the last 200 nucleotides from the 3′end.
(C) An overview of the data analysis pipeline (Steps 18–21).
Base calling and alignment
Note: Base calling is a process to convert the electrical signals (FAST5 data) into nucleotide sequences (fastq data) (Figure 3C).
-
18.Base calling:
-
a.Use Guppy, a data processing toolkit containing Oxford Nanopore Technologies’ base calling algorithms, to translate the raw electrical signal from the sequencer into a nucleotide sequence in fastq format. Guppy v6.4.8 for GPU mode is available for download at community.nanoporetech.com/downloads. Guppy is installed according to Oxford Nanopore’s instruction (community.nanoporetech.com/docs/prepare/library_prep_protocols/Guppy-protocol/v/gpb_2003_v1_revas_14dec2018/linux-guppy). All base calling operations are launched in GPU mode, using a Nvidia Tesla A100 40GB GPU. For base calling, we use the script runbatch_gpu_guppy.sh (available for download in github.com/trandoanduyhai/guppy_on_slurm).
-
b.For base calling RNA data the config file rna_r9.4.1_70bps_hac.cfg needs to be added to the “runbatch_gpu_guppy.sh” script. Alternatively, user can also provide flowcell model and sequencing kit as the guppy parameters. For example, guppy_basecaller --flowcell FLO-MIN106 --kit SQK-RNA002. Guppy can choose the config file automatically according to the provided flowcell model and sequencing kit.
-
a.
-
19.Alignment:
-
a.After base calling, data was aligned to the human reference genome GRCh37 using minimap2.4 Open a terminal, navigate to the run folder and run:
-
a.
> minimap2 –ax splice –uf –k14 $referenceGenome.fa $sample.fastq | samtools sort -@ 8 -O BAM -o $outfile.bam - && samtools index $outfile.bam
Note: The alignment of full-length reads can be used to quantitate the cytoplasmic expression level. To install minimap2, user can acquire precompiled binaries with: curl -L https://github.com/lh3/minimap2/releases/download/v2.26/minimap2-2.26_x64-linux.tar.bz2 | tar-jxvf-/minimap2-2.26_x64-linux/minimap2
Trimming of nanopore reads
-
20.To map and quantify the 3′end cleavage sites, raw nanopore reads are trimmed from the 5′ end to a uniform length so that the last 200 nucleotides before the polyA tail are retained. The tool NanoFilt6 is applied for the read trimming. The python script is freely available at (https://github.com/wdecoster/nanofilt/blob/master/scripts/get_read_ends.py).Note: Users do not need to install NanoFilt the whole tool. In order to run get_read_ends.py script, users only need python with biopython dependency installed.
-
a.Open a terminal, navigate to the run folder and run:> python get_read_ends.py --bases_from_end 200 reads.fastq.gz | gzip > last_200_bp.fastq.gz
-
b.For downstream analysis trimmed reads are then aligned to the GRCh37 human genome reference using minimap2:> minimap2 –ax splice –uf –k14 –t16 $referenceGenome.fa last_200_bp.fastq.gz | samtools sort -@ 8 -O BAM -o $outfile.bam - && samtools index $outfile.bam
-
a.
Expected outcomes
In this experiment, we sequenced shCr and shTHOC5 samples separately on two R9.4 flow cells for approx. 48 h. To create a base calling report the “sequencing_summary.txt” file generated by guppy (step 18) was loaded into pycoqc module of galaxy (usegalaxy.org). The number of sequencing pores decreased over time (Figure 4A). Generally, we stop the sequencing if the number of sequencing pores is less than 30 out of 512. Please note that we did not do any flow cell wash and re-sequence.
Figure 4.

Expected outcome of nanopore direct RNA-seq experiment generated by pycoqc
(A) Number of RNAs were sequenced within 50 h.
(B) Base call summary.
(C) Read length distribution.
(D) The read quality over sequencing time.
(E) The read quality distribution.
We expected an outcome of 2–3 million reads for each flowcell (Figure 4B) using RNAs isolated from HEK293 cells with the read length ranging from 100 bp to 5 kb (Figure 4C). The sequencing depth depends on the amount of initial RNA, RNA complexity from different species8,9 (troubleshooting: problem 7).
We observed a slight decrease of read quality over the time (Figure 4D). Only reads having a quality ≥ 7 were base called by Guppy (Figure 4E). Please note that low read quality may be due to the quality of flow cells or contaminants that are present in the library (troubleshooting: problem 8).
Quantification and statistical analysis
-
1.The quantification of 3′ cleavage is performed with Seqmonk v1.48.0 using the human polyA database.10 SeqMonk is a program that enables the visualization and analysis of mapped sequence data and is freely available at (Babraham Bioinformatics - SeqMonk Mapped Sequence Analysis Tool).
-
a.To run Seqmonk on Windows, download the seqmonk zip file, and unzip it on your machine → double click seqmonk.exe to run the program.
-
b.Open Seqmonk and start a new project.
-
c.Choose “Import Data” to import the mapped sequence data (bam file).
-
d.Download the human polyA database (hg19.apadb_v2_final.bed), freely available at http://exon.umdnj.edu/polya_db/.Note: Seqmonk does not accept annotation file in bed format; it needs to be converted into GTF or GFF format.
-
e.Choose “Import Annotation” to import the human polyA database.
-
f.To quantify the 3′end cleavage at the genome-wide scale: Go to “Data” → “Define Probes” → “Feature Probe Generator” → “Features to design around” → “PAS sites”. Seqmonk will calculate the read coverage of proximal and distal cleavage sites.
-
g.Go to “Data” → “Quantitate existing probes” –> “Read count quantitation” → To normalize the read counts.
-
h.Go to “Reports” → “Annotated Probe Report” to export the normalized read counts overlapped with annotated polyA sites.
-
i.Genes that contain more than two annotated polyadenylation sites (PAS) and more than 5 reads at distal PAS sites are selected for downstream analysis. The ratios between the proximal- and distal cleavage sites will be calculated. Comparing the ratios of shCr and shTHOC5 will reflex the changes of polyA site usage. The biological replicates will be subjected to statistical tests (student’s t-test, two-sided). In Figure 5, we show C4ORF46 (A) and NUDT21 (B) genes as an example.
-
a.
Figure 5.

Examples for 3′end cleavage quantification
(A and B) 3′end cleavage quantification of C4ORF46 (A) and NUDT21 (B) genes. The number of reads overlapped with proximal- and distal cleavage sites were calculated and normalized to the largest dataset.
Limitations
The 3′end cleavage quantification described in this protocol is based on the annotated human polyA database. Thus, the unannotated polyA sites will not be quantitated. We observed a number of unannotated polyA sites were detected in our cell system. Figure 6 shows two genes as examples: NUDT21 (A, one unannotated site) and EEF2 (B, nine unannotated sites).
Figure 6.

Examples of genes having unannotated polyA sites
(A and B) (A) NUDT21, (B) EEF2. Unannotated polyA sites are the read clusters that are not overlapped with any annotated polyA sites located in 3′UTR-, 5′UTR-, Exon- and Intron regions.
Troubleshooting
Problem 1
Low disk space
The sequencing reaction (Step 17) will stop if there is not enough space on the local drive (less than 30 GB).
Potential solution
-
•
Release the disk space by transferring the raw sequencing reads (FAST5 files) to another disk drive.
-
•
Restart the sequencing reaction (Steps 16–17)
-
•
After the sequencing is completed, copy all FAST5 files into one folder for base calling (Step 18).
Problem 2
Temperature fluctuation
Causes:
-
•
If the cold flow cell is inserted directly into the device, condensate can form between the metal plate and the back of the flow cell that will also cause temperature fluctuation.
-
•
The flow cell has lost contact with the device (Step 17).
Potential solution
-
•
The flow cell should be equilibrated to room temperature before being inserted into the device.
-
•
Re-insert the flow cell and press it down to make sure the connector pins are firmly in contact with the device.
Problem 3
MinKNOW shows "Failed to reach target temperature"
It may happen when the instrument is placed in a location that is colder than 25°C or with poor ventilation (which leads to the flow cells overheating) (Step 17).
Potential solution
-
•
Try moving the instrument to a location with better temperature control and ventilation.
Problem 4
Poor A260/A280 or A260/230 ratios
A low A260/A280 can indicate contamination by proteins, while a low A260/A230 can indicate salt contamination.
Potential solution
-
•
Repeat the RNA extraction procedure or use an RNA clean-up kit to remove proteins and salts from the RNA sample.
Problem 5
Low yields of polyA-tailed RNAs
Low yields of polyA-tailed RNAs generally result from degraded RNAs from 3′end.
Potential solution
To improve the quality of RNAs.
-
•
Reduce exposure to environmental RNases.
-
•
Cells for isolating RNAs need to be in a good condition.
-
•
Store the RNA properly after isolation.
-
•
For isolating RNAs from tissue, the problem could be that the homogenization was too hard and the sample was heated for too long. Try to optimize the homogenization.
Problem 6
Low recovery of adapter annealed RNAs (step 10o)
Causes:
-
•
PolyA-tailed RNAs are degraded during library preparation.
-
•
Due to a lower than intended AMPure beads-to-sample ratio.
Potential solution
-
•
Using RNAse- and DNAse-free tubes and pipette tips to prevent RNAse- and DNAse-contamination.
-
•
AMPure beads settle quickly, so ensure they are well resuspended before adding them to the sample. When the AMPure beads-to-sample ratio is lower than 0.4:1, DNA fragments of any size will be lost during the clean-up.
Problem 7
Low sequencing output
Causes:
-
•
If the pore occupancy is lower than 40%, the library may not be enough loaded on the flow cell.
-
•
If the pore occupancy is close to 0, no tether is on the flow cell.
Potential solution
-
•
5–50 fmol of good quality library can be loaded on to a MinION Mk1B/GridION flow cell. Please quantify the library before loading and calculate mols using tools like the Promega Biomath Calculator, choosing "dsDNA: μg to pmol".
-
•
Tethers are adding during flow cell priming (FLT tube). Make sure FLT was added to FB before priming.
Problem 8
Low read quality
Causes:
-
•
Quality of the flow cells.
-
•
Contaminants are present in the library.
Potential solution
-
•
Since the flow cell contains biological pores which have a definite half-life, the sequencing should be performed as soon as possible after the delivery of flow cells.
-
•
Using HAC (High ACcuracy) mode instead of FAST mode for base calling with guppy will help to reduce the error rates.8
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Doan Duy Hai Tran (Tran.Doan@mh-hannover.de).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (Ta 111/13-3 to D.D.H.T) and Junge Akademie 2.0 (Medizinische Hochschule Hannover) to D.D.H.T.
Author contributions
D.D.H.T. conceived the study. M.P. and A.B.A. performed experiments. M.P., D.D.H.T., S.B.d.L., M.S., and G.K. analyzed data. D.D.H.T., M.P., and A.B.A. drafted the manuscript with input from all authors.
Declaration of interests
The authors declare no competing interests.
Data and code availability
-
•
Nanopore data have been deposited at GEO and are publicly available. Accession number is listed in the key resources table.
-
•
This study did not generate code beyond the described assembly and quality control command-line interface commands.
References
- 1.Polenkowski M., Allister A.B., Burbano de Lara S., Pierce A., Geary B., El Bounkari O., Wiehlmann L., Hoffmann A., Whetton A.D., Tamura T., Tran D.D.H. THOC5 complexes with DDX5, DDX17, and CDK12 to regulate R loop structures and transcription elongation rate. iScience. 2023;26:105784. doi: 10.1016/j.isci.2022.105784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saran S., Tran D.D.H., Ewald F., Koch A., Hoffmann A., Koch M., Nashan B., Tamura T. Depletion of three combined THOC5 mRNA export protein target genes synergistically induces human hepatocellular carcinoma cell death. Oncogene. 2016;35:3872–3879. doi: 10.1038/onc.2015.433. [DOI] [PubMed] [Google Scholar]
- 3.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Coster W., D'Hert S., Schultz D.T., Cruts M., Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J.Y., Yeh I., Park J.Y., Tian B. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res. 2007;35:D165–D168. doi: 10.1093/nar/gkl870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delahaye C., Nicolas J. Sequencing DNA with nanopores: troubles and biases. PLoS One. 2021;16:e0257521. doi: 10.1371/journal.pone.0257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leger A., Amaral P.P., Pandolfini L., Capitanchik C., Capraro F., Miano V., Migliori V., Toolan-Kerr P., Sideri T., Enright A.J., et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021;12:7198. doi: 10.1038/s41467-021-27393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang R., Nambiar R., Zheng D., Tian B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46:D315–D319. doi: 10.1093/nar/gkx1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
Nanopore data have been deposited at GEO and are publicly available. Accession number is listed in the key resources table.
-
•
This study did not generate code beyond the described assembly and quality control command-line interface commands.