

Summary
Inter-organelle membrane contacts are highly dynamic and act as central hubs for many biological processes, but the protein compositions remain largely unknown due to the lack of efficient tools. Here, we developed BiFCPL to analyze the contact proteome in living cells by a bimolecular fluorescence complementation (BiFC)-based proximity labeling (PL) strategy. BiFCPL was applied to study mitochondria-endoplasmic reticulum contacts (MERCs) and mitochondria-lipid droplet (LD) contacts. We identified 403 highly confident MERC proteins, including many transiently resident proteins and potential tethers. Moreover, we demonstrated that mitochondria-LD contacts are sensitive to nutrient status. A comparative proteomic analysis revealed that 60 proteins are up- or downregulated at contact sites under metabolic challenge. We verified that SQLE, an enzyme for cholesterol synthesis, accumulates at mitochondria-LD contact sites probably to utilize local ATP for cholesterol synthesis. This work provides an efficient method to identify key proteins at inter-organelle membrane contacts in living cells.
Subject areas: Membranes, Biophysics, Proteomics
Graphical abstract
Highlights
-
•
BiFCPL enables analysis of organelle membrane contact proteomes in living cells
-
•
403 mitochondria-endoplasmic reticulum contact proteins are identified
-
•
60 mitochondria-LD contact proteins are sensitive to metabolic challenge
-
•
SQLE accumulates at mitochondria-LD contacts under metabolic challenge
Membranes; Biophysics; Proteomics
Introduction
Mammalian cells are compartmentalized into membrane-bound organelles that maintain unique and efficient functions in different biological processes. Organelles physically interact with each other via membrane contact sites to carry out critical biological functions, such as organelle biogenesis, signal transduction, and the exchange of signals and metabolites.1 Inter-organelles contacts are dynamic and transient in response to changing conditions. Alterations in organelle interactions are closely related to neurodegenerative diseases, such as Alzheimer disease, and metabolic disorders, such as type 2 diabetes.1,2 However, the molecular machineries underlying inter-organelles contacts remain largely unknown.
There has been tremendous interest in identifying the protein compositions of inter-organelles contacts. Subcellular fraction analysis has been widely used and over 1000 proteins have been identified as components of mitochondria-endoplasmic reticulum contacts (MERCs, also known as mitochondria-associated ER membrane) in mammalian cells as well as animal tissues.3,4 However, a significant number of irrelevant proteins are included due to the complex procedures and contamination from other organelles. Moreover, MERC is highly dynamic, and proteins that are temporarily resident at MERC are easily missed during the centrifugation processes.5 Hence, a reliable spatial proteomic approach is required for the analysis of contact proteomes.
In recent years, proximity labeling strategies based on biotinylating enzymes, such as APEX2, BioID, and TurboID, have emerged as powerful tools for spatial proteomics. These enzymes are capable of labeling proximal proteins with biotin in living cells within 20 nm, which is comparable to the range of inter-organelle membrane contacts.6 As no specific contact marker protein has been reported to date,7,8 two recent studies utilized split forms of proximity labeling enzymes to map the MERC proteome in HEK293T cells. The two studies split the enzyme TurboID or BioID, the separate parts of which were targeted to mitochondrial outer membrane or ER membrane, respectively. Both studies identified over 100 proteins, but only a few of them were found in both studies. Many well-characterized MERC proteins, such as MFN2, RMDN3, GRP75, and ITPR3, were absent.8,9 Therefore, there is still a lack of reliable tools to probe the repertoire of the inter-organelle contact proteome.
Here, we described BiFCPL, a bimolecular fluorescence complementation-based proximity labeling strategy, which enables accurate and efficient analysis of inter-organelle contact proteomes in living mammalian cells. By using this method, we identified 403 MERC proteins, including most of the well-known proteins as well as many novel MERC residents. Moreover, we studied mitochondria-lipid droplet (LD) contacts in hepatocytes under various conditions and found that nutrient status significantly impacts the dynamics and proteomes of mitochondria-LD contacts. We further verified that squalene epoxidase (SQLE), which is the second rate-limiting enzyme in cholesterol biosynthesis that catalyzes the conversion of squalene to 2,3-epoxysqualene,10 accumulates at mitochondria-LD contact sites under metabolic challenge, probably to utilize mitochondria-derived ATP for cholesterol synthesis.
Results
BiFCPL enables the labeling of proteins at mitochondria-ER contacts
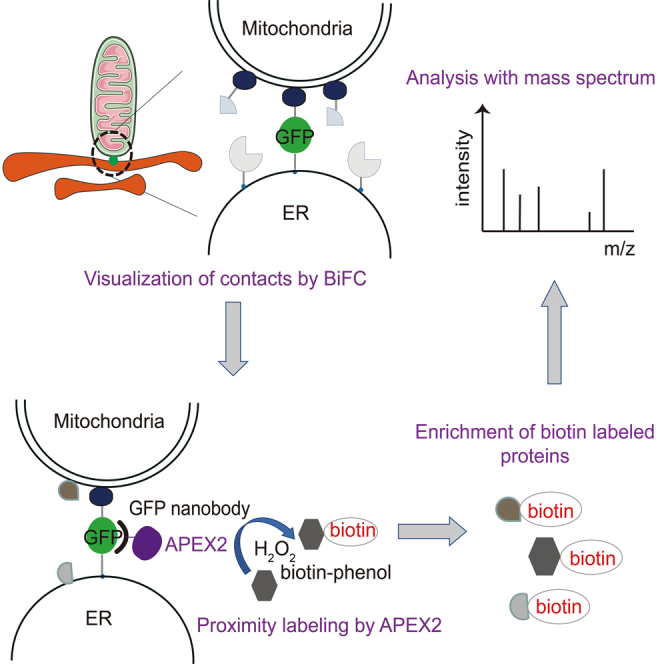
Bimolecular fluorescence complementation (BiFC) based on the split-GFP system has been used to detect inter-organelle membrane contacts in yeast and mammalian cells.11,12 The separate parts of superfolder GFP (spGFP), spGFP1-10 and spGFP11, were targeted to different organelle membranes and reconstituted into a complete fluorescent GFP when one organelle was juxtaposed to the other.13 The proximity labeling enzyme, APEX2, was recruited to the contact sites via the interaction between GFP and a nanobody against GFP. The addition of biotin-phenol and hydrogen peroxide subsequently initiates the biotinylation of proteins at the contact sites. The biotin-labeled proteins were then enriched with streptavidin-coated beads and analyzed using mass spectrometry. The BiFCPL enables the proteomic analysis of inter-organelle membrane contacts (Figure 1A).
Figure 1.

BiFCPL enables the labeling of proteins at mitochondria-ER contacts
(A) Designing principle of the method.
(B) Fluorescent images of biotinylated proteins. U2OS or MERC cells were transiently transfected with GNAP2. After biotin labeling, the cells were fixed and subjected to fluorescence labeling. Alexa 555-conjugated streptavidin was used to detect biotin-labeled proteins. Alexa 647-conjugated Tom20 antibody was used to label mitochondria. Scale bar, 5 μm. See also Figure S1. Contact, green; streptavidin, magenta; blue, mitochondria.
(C) Western blotting analysis of the enriched proteins. U2OS or MERC cells were transiently transfected with GNAP2, NLS-GNAP2, or CS-GNAP2. The cells were incubated with or without biotin-phenol for biotin labeling. The cells were then harvested, and biotin-labeled proteins were enriched by streptavidin magnet beads, separated by SDS-PAGE and detected with HRP-conjugated streptavidin (SA-HRP).
(D) Western blotting analysis of the enriched proteins. Enriched proteins were subjected to SDS-PAGE and immunoblotted with antibodies against MFN2, RMDN3, GFP, and ACTB.
It has been established that the GFP nanobody binds to reconstituted GFP rather than individual fragments.14,15 To further verify the interaction between individual split-GFP fragments and the GFP nanobody, we transiently expressed GFP nanobody-tagged APEX2 (referred to as GNAP2 below) along with mito-myc-2xspGFP11 or spGFP1-10-v5-ER in U2OS cells. Fluorescent streptavidin was used to detect biotin-labeled proteins. The individual GFP fragments localized in mitochondria or ER, respectively, but biotin-labeled proteins spread diffusely (Figure S1), suggesting that the GFP nanobody exhibits a weak binding affinity to the individual fragment.
Recently, a reporter was established by fusing spGFP1-10 to the ER-anchoring sequence of UBE2J2 and spGFP11 to the mitochondrial targeting sequence of TOMM70, respectively. This reporter enabled the imaging of dynamic MERC structure in living cells, and was used to study the distribution and dynamics of MERC during the cell cycle and under various stressful conditions.7 To characterize our method, we transiently expressed GNAP2 in wild-type U2OS cells or MERC reporter cells (referred to as MERC cells below) to minimize the artificial interference with native mitochondria-ER contacts caused by the binding between GFP and the GFP nanobody. In empty U2OS cells, a diffusive biotinylation pattern was observed. In MERC cells, the biotin-labeled proteins localized right at the contact sites and the fluorescent signal colocalized well with GFP (Figure 1B). These results indicated that BiFCPL enables protein biotinylation at MERC.
Next, the biotin-labeled proteins were enriched and the labeling efficiency was assessed using Western blotting analysis. No biotinylation activities were observed in MERC cells when biotin-phenol or GNAP2 was omitted. To reduce the cytosolic background, different versions of GNAP2 were introduced, including a conditionally stable version of GNAP2 (referred to as CS-GNAP2 below) by adding a signal to target this protein to the proteasomal system,15 and a nucleus-localized GNAP2 (referred to as NLS-GNAP2 below) by adding a nucleus localization sequence in the N-terminus. These GNAP2 variants yielded strong biotinylation activities in MERC cells when biotin-phenol was included (Figure 1C).
To assess the specificity of BiFCPL, we checked the expression of certain proteins in the enriched samples. As shown in Figure 1D, two well-known MERC proteins, MFN2 and RMDN3 (also known as PTPIP51), were significantly enriched when biotin-phenol and GNAP2 variants were included. In comparison to GNAP2, the NLS-GNAP2 and CS-GNAP2 exhibited relatively weaker enrichment. ACTB was used to indicate cytosolic contamination. By comparing the signal (MFN2 and RMDN3) to noise (ACTB) in different conditions, we found GNAP2 exhibited significantly higher enrichment than NLS-GNAP2 or CS-GNAP2, so we chose GNAP2 for subsequent experiments. Taken together, these results suggested that BiFCPL enables the labeling of MERC proteins in living cells.
BiFCPL exhibits higher efficiency and accuracy than alternative approaches
To characterize the MERC proteome, we transiently expressed GNAP2 in empty U2OS or MERC cells. The experiments were performed with or without biotin-phenol, resulting in four groups: MERC, MERC-Ctrl, U2OS, and U2OS-Ctrl. After the cells were harvested and homogenized, crude mitochondria were purified to remove the cytosolic background. The biotin-labeled proteins were then enriched with streptavidin-coated beads and separated by SDS-PAGE. The whole band was cut and analyzed with liquid chromatography-tandem mass spectrometry (Figure S2A). The experiments were conducted reproducibly in three biological replicates.
Using label-free quantification analysis, we obtained 1220 quantifiable proteins. We then compared the MERC group with the other three groups, and the fold changes should be no less than four in at least two replicates. The U2OS group was used as diffuse cytosolic background. The MERC-Ctrl and U2OS-Ctrl groups were used to remove endogenous streptavidin-binding proteins. As a result, a total of 403 proteins met the above criteria and were categorized as MERC proteins (Figures S2B and S2C and Table S1).
Recently, split-TurboID and contact-ID utilized split proximity labeling strategies to map the MERC proteome in living HEK293T cells and revealed 101 and 115 MERC proteins, respectively.8,9 We then compared our data with those proteins obtained in the two studies. As shown in Figure 2A, although both split-TurboID and contact-ID utilized HEK293T cells as studying models, only 24 proteins were present in both studies, which implied MERC is rather dynamic and a repertoire of the MERC proteome is lacking. Only 5 proteins, OCIAD1, PGRMC1, FKBP8, ALDH3A2, and EMD, were included in both studies and our work. FKBP8 was validated by contact-ID to facilitate the formation of MERC and calcium transport at MERC.8 OCIAD1 was validated by split-TurboID to localize at MERC.9 PGRMC1 was shown to localize at mitochondria and ER and reside at MERC.16 EMD was shown to interact with FATE1 at MERC.17 More than 80% (363/403) of the proteins identified by BiFCPL were not included in either split-TurboID or contact-ID. We also compared our data with previous studies using subcellular fraction (Figure S3A).3,4,18 Most of the proteins (249/403) in our data were also identified by other studies despite the differences in organisms and cell types.
Figure 2.

BiFCPL exhibits higher efficiency and accuracy than alternative approaches
(A) UpSet plotting of the comparison of the MERC proteome identified by BiFCPL over proteomes identified by split-TurboID and contact-ID. The dot represented presence in the indicated proteome list and a line through the dots represented presence in the related proteome lists. See also Figures S2 and S3 and Table S1.
(B) A clustering analysis for biological processes of MERC proteins analyzed by WebGestalt. The length of the bar is correlated with the enrichment ratio of the indicated item.
(C) An interacting network between proteins located on the mitochondrial outer membrane and ER membrane. The yellow, blue, and red lines represent the interactions within ERM (ER membrane) proteins, interactions between ERM and MOM (mitochondria outer membrane) proteins, and interactions within MOM proteins, respectively. The circle indicates the value of log2(fold change).
Then, we checked the enrichment efficiency of well-characterized MERC proteins, including MFN1, MFN2, RMDN3, DNM1L, VAPA, VAPB, VDAC3, HSPA9, and ITPR3. These proteins have been proven by various studies to localize and conduct essential functions at MERC.19,20 As a result, these proteins were all highly enriched by BiFCPL (Figure S3B), but only two of them, VAPA and VAPB, were included in contact-ID, and only one of them, DNM1L, was included in split-TurboID.
Next, we submitted the MERC proteins to WebGetalt for enrichment analysis of Gene Ontology biological processes and the functional enrichment was displayed by EnrichmentMap.21 The resulting functional annotations of mitochondria- and ER-related processes were highlighted, including metabolic processes, mitochondria calcium ion homeostasis, mitochondria organization, and ER stress (Figure 2B), which are consistent with the functions of MERC demonstrated previously. A large proportion of proteins (196/403) were associated with mitochondria or ER, suggesting a high specificity. Together, these findings indicated that the MERC proteome revealed by BiFCPL is highly confident.
MERC is a specialized membrane structure organized by tethers. To date, several tethers, such as MFN2, IP3R–VDAC–GRP75, VAPB–PTPIP51, and the BAP31–FIS1 complex, have been demonstrated to directly bridge MERC formation.22 MERC tethers are typically dual-membrane localized proteins or a protein complex that drag mitochondria and the ER membrane together. Using BiFCPL, 30 mitochondrial outer membrane proteins and 66 ER membrane proteins were enriched. We then generated the interaction network between these proteins based on the STRING database (Figure 2C). The tethers, MFN2, IP3R–VDAC, and VAPB–PTPIP51(RMDN3) complex, were identified. The rest interacting protein pairs between mitochondrial outer membrane proteins and ER membrane proteins potentially function as tethers, which remains to be determined.
Validation of MERC proteins by fluorescence imaging
Afterward, we used live-cell imaging analysis for validation. We chose four proteins that spanned a wide range of enrichment ratios in our list. The candidates were tagged with EGFP, and MitoTracker and ER-DsRed were used to label mitochondria and ER in living U2OS cells, respectively.
As an ER protein, WFS1, ranked 43 in our list, formed puncta in the cytosol and was found transiently at contact sites (Figure 3A). WFS1 promotes calcium transfer between ER and mitochondria,23 so the time-dependent change of WFS1 location implied dynamic and transient calcium transfer at MERC. Ranking 100 in the list, some of the BAG2-EGFP puncta continuously colocalized with contacts, while in others, they occurred temporarily at contact sites (Figure 3B). BAG2 is a cochaperone24 and might help maintain protein-folding homeostasis at MERC. Ranking 235 in the list, SPTLC1-EGFP was an ER-resident protein and formed large puncta that frequently localized at MERC (Figure 3C). SPTLC1 may form a complex at MERC with SPTLC2,25 which was also included in our list. GLUD1 ranked 320 in our list and was quite dynamic. Some of the fluorescent signal temporarily localized at contact sites (Figure 3D). GLUD1 might coordinate the function of mitochondria and ER to reduce redox and maintain energy supply.26
Figure 3.

Validation of MERC proteins by fluorescence imaging in living cells
(A–D) Fluorescent images of living cells. U2OS cells were transiently transfected with ER-DsRed along with plasmids expressing WFS1-EGFP (A), BAG2-EGFP (B), SPTLC1-EGFP (C), or GLUD1-EGFP (D). The cells were then grown on coverslips to allow adherence and spreading. MitoTracker (deep red) was used to label mitochondria, and live cell images were taken at the indicated intervals with LSM 980 confocal microscopy equipped with a 63×/1.40 NA oil immersion objective (Zeiss). The smaller images were amplification of the boxed regions in the larger images. The upper images show overlap between mitochondria and ER. The lower images are merged images of the three channels. White arrows indicate mitochondria-ER contacts. Scale bar, 5 μm. See also Figure S4. Candidates, green; ER, magenta; blue, mitochondria.
To further validate our data, we performed immunofluorescence staining in fixed cells. As shown in Figures S4A–S4H, MERC candidates were tagged with EGFP and mitochondria and ER were labeled with MitoTracker and an antibody against calnexin, respectively. Consistent with live-cell imaging analysis, all these proteins were concentrated at mitochondria-ER contact sites. Besides, we performed subcellular fraction analysis to purify mitochondria-associated ER membrane (referred to as MAM),27 which includes the MERC structure (Figure S4I). As shown in Figure S4J, the MAM fraction was enriched for TOMM70 (known MERC protein) but devoid of GAPDH (marker for cytosol) and cytochrome c (marker for mitochondrial matrix), indicating a high purity. The four MERC candidates were found to be enriched in MAMs. Taken together, these results suggested that BiFCPL captures both transiently and continuously resident MERC proteins, which infers that the MERC proteome is highly confident.
BiFCPL enables biotin labeling of proteins at mitochondria-LD contact sites
To extend the application of BiFCPL, we focused on mitochondria-lipid droplet (LD) contacts in hepatocytes. The mitochondria-LD interaction is crucial for maintaining the homeostasis of hepatic lipid metabolism,28 but the proteins involved in the mitochondria-LD interaction and their functional mechanism remain largely unknown, as it is difficult to purify the mitochondria-LD contact structure. In this study, a fluorescent reporter of mitochondria-LD contacts was established in HepG2 cells and APEX2 protein was recruited to the contact sites and enabled the biotinylation of contact proteins (Figure 4A).
Figure 4.

BiFCPL enables biotin labeling of proteins at mitochondria-LD contact sites
(A) Designing principle to characterize the spatial proteome at mitochondria-LD contacts.
(B) Representative single plane images of the reporter cell line (MLDCS) stably expressing mito-sfGFP11 and sfGFP1-10-Plin2. MitoTracker and LipidTox were used to label mitochondria and LDs, respectively. Scale bar, 5 μm. The lower images are a zoom-in version of the boxed region in the upper images. The white arrows mark contact sites between mitochondria and LDs. Scale bar, 1 μm. See also Figure S5. Contact, green; mitochondria, magenta; LDs, blue.
(C) Correlative light microscopy (LM) and electron microscopy (EM) analysis of the MLDCS cells. C1, superimposition of the LM image and EM image of the prepared lamella; C2-4, LM image (C2), EM image (C3), and correlation of LM and EM image (C4) of the boxed region in C1. Scale bar, 1 μm in C1 and 200 nm in C4. Contact, green; mitochondria, red.
(D and E) Representative fluorescence images of the wild-type HepG2 cells (D) or MLDCS cells. The cells were transiently transfected with GNAP2 and then subjected to fluorescence labeling after biotin labeling. Alexa 555-conjugated streptavidin was used to detect the biotin-labeled proteins. Scale bar, 5 μm. Contact, green; streptavidin, magenta; nucleus, blue.
To establish the mitochondria-LD contact reporter, we packaged lentiviruses and infected wild-type HepG2 cells to stably express mito-sfGFP11 and sfGFP1-10-Plin2. The appropriate cell clone was selected based on the following principles: 1. The GFP signal intensity should be modest in order to minimize the artificial interference to native mitochondria-LD interactions brought by bimolecular fluorescence complementation assay; 2. The GFP signal localized at the interface between mitochondria and LDs. The resulting cell clone was called the mitochondria-lipid droplet contact site reporter (MLDCS). As shown in Figure 4B, the LDs were either attached to or surrounded by mitochondria and GFP was present at the contact sites. GFP signals exhibited dot, semi-ring or ring-like structures, corresponding to different types of mitochondria-LD contacts (Figure S5A), which was consistent with a previous study.29 To further verify this reporter, we performed dual color correlative light and electron microscopy analysis. The GFP signal appeared right at the juxtaposition (Figure 4C), which suggested that this reporter is fairly specific.
To determine whether the expression of recombinant proteins affected the dynamics of mitochondria and LDs, we incubated the MLDCS cells with medium containing oleic acids (OA). As shown in Figure S5B, short time (4 h) OA treatment triggered the biogenesis of LDs, the majority of which were found to be free in the cytosol. GFP signals appeared when LDs were proximal to mitochondria.
Next, we transiently expressed GNAP2 in wild-type HepG2 cells or MLDCS cells. After biotin labeling, Alexa 555-conjugated streptavidin was used to detect biotinylated proteins. In wild-type HepG2 cells, biotinylated proteins diffusely spread throughout the cytosol (Figure 4D). However, in the MLDCS cells, a unique biotinylation pattern was observed at the contact sites (Figure 4E). Taken together, these results indicated that BiFCPL enables the labeling of proteins at mitochondria-LD contact sites in living cells.
Dynamics and proteomic analysis of mitochondria-LD contacts under metabolic challenges
To determine whether this reporter could detect the dynamics of mitochondria-LD contacts, we incubated MLDCS cells under various nutrient stresses and evaluated the changes in the fluorescent signals of GFP, mitochondria, and LDs (Figure S6A). The areas of GFP and mitochondria were calculated, as well as the number of LDs (Figures S6B and S6C). Compared to normal conditions, LD number and mitochondria-LD contact increased significantly when glucose was deprived, probably due to enhanced lipogenesis via autophagy.29 The cells were starved in HBSS for 16 h, but the LD number and mitochondria-LD contact remained unchanged. We then applied OA treatment to trigger LD biogenesis and found that LD number and mitochondria-LD contact increased significantly. Additionally, HBSS supplemented with OA, which mimicked starvation-induced hepatic lipid accumulation, also enhanced mitochondria-LD contact and increased LD number by approximately 3-folds, which was comparable to OA treatment, mimicking high-fat diet-induced hepatic lipid accumulation. These results provided direct imaging evidence that mitochondria-LD contact is sensitive to nutrient stresses.
Since mitochondria-LD contact was relatively low under normal conditions, which made it extremely difficult to enrich biotinylated proteins, we then incubated the cells for 16 h in full medium containing OA (referred to as OA condition below) or in HBSS containing OA (referred to as HBSS+OA condition below) to enhance the mitochondria-LD interaction. After biotin labeling, the cells were harvested and homogenized. The biotinylated proteins were then enriched with streptavidin-conjugated beads. When biotin-phenol (BP) was omitted, weak APEX2 activity was observed, whereas significantly more proteins were enriched under the other three conditions (Figures 5A and 5B).
Figure 5.

Dynamics and proteomic analysis of mitochondria-LD contacts under metabolic challenge
(A and B) The enriched biotin-labeled proteins were subjected to SDS-PAGE, followed by Western blotting analysis using streptavidin-HRP (A) or Coomassie blue staining (B). See also Figures S6 and S7, Tables S2 and S3 for details.
(C) A volcano plot showing biotinylated proteins under HBSS+OA conditions in comparison to OA conditions. The red dots represent 60 statistically significantly up- or downregulated proteins at mitochondria-LD contact sites upon starvation. See also Table S4 for details.
(D) The subcellular distribution of the 60 proteins with prior annotated localization information in the Uniprot database.
Next, the enriched samples were subjected to mass spectrometry analysis. A total of 3747 proteins were detected in at least two experiments of the three biological replicates. A principal component analysis of the three replicates indicated high reproducibility (Figure S7). MLDCS cells without BP were used to remove endogenous proteins that precipitated with streptavidin. Wild-type HepG2 cells were used as a control to eliminate cytosolic contamination. The ratio of the enriched samples over the controls was used to cut off diffuse cytosolic proteins. As a result, 136 proteins were enriched under OA conditions (Table S2), and 154 proteins were enriched under HBSS+OA conditions (Table S3).
It has been demonstrated that fatty acids induced by starvation are transferred into mitochondria for oxidation.29 We sought to identify metabolic challenge-sensitive proteins. These proteins may play critical roles in coordinating the functions of mitochondria and LDs. As a result, 60 proteins were significantly altered at mitochondria-LD contact sites (Figure 5C and Table S4). Twenty-eight proteins were more abundant at contact sites under OA conditions, whereas the rest 32 proteins were more abundant under HBSS+OA conditions. A subcellular localization Gene Ontology analysis revealed a high enrichment in the membrane and mitochondria (Figure 5D). Taken together, these results suggested that nutrient stresses had a significant impact on the dynamics and proteomes of mitochondria-LD contacts.
SQLE accumulates at mitochondria-LD contact sites under metabolic challenge
Next, we sought to verify our data. We selected SQLE for further validation because inter-organelles contacts play key roles in maintaining lipid homeostasis, and SQLE is a rate-limiting enzyme in cholesterol production. SQLE is an ER-resident protein and has been demonstrated as a component of LDs, and overexpression of SQLE leads to increased lipid accumulation in the mouse liver.30,31 However, its role in inter-organelle interactions has not been addressed.
The intracellular localization of SQLE was verified by high-resolution fluorescence microscopy. As shown in Figure 6A, under normal conditions, SQLE localized in the ER. Its localization was further verified under metabolic challenges. Under OA conditions, SQLE formed ring-like structures on LDs (Figure 6B). Since LDs are derived from the ER,32 it is reasonable that in the presence of excessive lipids, SQLE translocates from the ER to LDs. Surprisingly, under HBSS+OA conditions, SQLE formed clusters on LDs, which concentrated at the contact sites between mitochondria and LDs (Figures 6C and 6D). The percentage of SQLE protein that colocalized with LDs or mitochondria was subsequently calculated. Since the number of LDs is low in hepatocytes under normal conditions, there was nearly no colocalization between SQLE and LDs. OA treatment significantly enhanced the colocalization between SQLE and LDs and the increase was even higher under HBSS+OA conditions (Figure S8A). Similarly, OA or HBSS+OA treatment also enhanced the association between SQLE and mitochondria (Figure 6F). These findings together suggested that SQLE localizes to the ER under normal conditions and translocates onto LDs under excessive lipid conditions.
Figure 6.

SQLE accumulates at mitochondria-LD contact sites under metabolic challenge
(A–C) Representative single-plane fluorescent images of HepG2 cells stably expressing SQLE-HA. The cells were incubated in normal conditions (A), OA (B), or HBSS+OA (C) conditions for 16 h and subjected to immunofluorescence labeling. An antibody against HA and CANX (calnexin) was used to detect the SQLE-HA protein and ER, respectively. MitoTracker and LipidTox were used to label mitochondria and LDs, respectively. Scale bar, 5 μm. The lower images are zoom-in views of the boxed regions in the upper images. Scale bar, 2 μm. See also Figures S8 and S9. SQLE-HA, green; mitochondria, magenta; LDs, blue, CANX, gray.
(D) A plot profile of the fluorescence intensity of the white line in (C).
(E and F) Quantification of the percentage of SQLE protein localized at mitochondria-LD contacts (E) or with LDs (F). N = 8 images were analyzed. The experiments shown are a representative of three replicates. Data are presented as the mean ± SEM. NS, no significance. ∗∗p < 0.01, ∗∗∗∗p < 0.0001 (two-tailed t-test).
Next, we compared the proportion of SQLE that localized at mitochondria-LD contact sites. We found that SQLE was significantly enriched at mitochondria-LD contact sites under OA treatment and the buildup was more significant under HBSS+OA treatment (Figure 6E). To further confirm this phenomenon, we isolated crude mitochondria and assessed protein expression. Consistent with microscopy analysis, a relatively small amount of SQLE protein was attached to mitochondria under normal conditions. OA treatment modestly increased SQLE protein levels in the mitochondrial fraction. Under HBSS+OA treatment, a dramatically higher level of SQLE protein was attached to mitochondria (Figure S8B). We also detected the expression of SQLE under these conditions and found that OA treatment and HBSS+OA treatment enhanced SQLE expression, but no significant difference was observed in between (Figure S8C). Taken together, these results suggested that SQLE translocates onto LDs under excessive fatty acids and accumulates at mitochondria-LD contact sites under HBSS+OA conditions.
Since SQLE is an essential enzyme for cholesterol synthesis and is enriched at mitochondria-LD contact sites under HBSS+OA conditions, we sought to verify whether cholesterol synthesis was affected. Filipin is a widely used fluorescent indicator for free cholesterol.33 The cells were exposed to the indicated conditions and then stained with filipin. As shown in Figure S9A, under either normal conditions or OA treatment conditions, free cholesterol was dominantly distributed on the plasma membrane, and very little cholesterol was found in the cytosol. However, under HBSS+OA conditions, a large proportion of free cholesterol was distributed in the cytosol. When comparing the filipin intensity among different conditions, we discovered a significant increase under HBSS+OA conditions (Figure S9B). These results suggested that metabolic challenge affects the synthesis of cholesterol.
Since mitochondria are major places to synthesize ATP, we speculated that SQLE might utilize local mitochondrial ATP to synthesize cholesterol. Oligomycin A suppresses the oxidative phosphorylation of ADP to ATP by blocking proton channels, thus inhibiting mitochondrial ATP synthase activity.34 We then incubated the cells under the indicated conditions with or without oligomycin A. After filipin staining, the fluorescence intensity was measured and compared. Oligomycin A treatment significantly decreased cholesterol synthesis especially under HBSS+OA conditions (Figure S9C). Collectively, these results indicated that SQLE probably utilizes local mitochondrial ATP to synthesize cholesterol under HBSS+OA conditions.
Discussion
Inter-organelle membrane contacts have been fascinating and challenging due to their important biological functions and a lack of reliable tools. Here, we developed BiFCPL to study the compositions and dynamics of contact proteomes in living mammalian cells. Using BiFCPL, we revealed 403 MERC proteins in living U2OS cells with high specificity and accuracy, including most of the well-known MERC proteins and many previously undiscovered proteins. Moreover, we investigated the protein compositions of mitochondria-LD contacts under metabolic challenges. We discovered 60 challenge-sensitive proteins and further validated that SQLE accumulates at mitochondria-LD contact sites probably to synthesize cholesterol under metabolic challenge.
BiFC assay based on split-GFP is easy to use and has been widely employed to visualize protein interactions.35 Several MERC reporters based on the split-GFP system have been established with different mammalian cell lines and successfully applied to study the dynamics of MERC under various conditions.7,12,36 Though split-GFP system is the most commonly used, BiFC based on other fluorescent proteins, such as VENUS, is also applicable to detect inter-organelle contacts.11 It would be noticeable that reconstitution of most of the split fluorescent proteins is irreversible, thus inducing artificial contacts. Considering there are currently no specific markers for inter-organelle membrane contacts,7,8 the BiFC assay could still be a useful tool to detect inter-organelle contacts as long as the individual fragments were expressed at a moderate rate, to minimize artificial interference with native organelle interactions.
Identifying the spatial proteomes of inter-organelle contacts has been challenging due to the lack of specific and efficient tools. Subcellular fraction analysis has been widely used but often produces confusing results, as a large number of irrelevant proteins were included. Split-TurboID and contact-ID based on split proximity labeling enzymes revealed a much smaller MERC proteome consisting of approximately 100 proteins.8,9 Though the authors emphasized the high specificity of these two methods, less than 30% of the proteins were present in both studies and many well-known MERC proteins were absent from both studies. In this study, BiFCPL takes advantage of BiFC to visualize inter-organelle contacts and a proximity labeling strategy to probe the local proteome in living cells. Multiple sample preparation and data filtering steps were used to minimize contamination by proteins from the cytosol and other organelles as much as possible. Moreover, the full-length APEX2 protein generates stronger biotinylation activity than split forms of proximity labeling enzymes.37,38 The lack of a need to introduce rapamycin also brings some advantage, as the addition of extra chemicals may interfere with MERC. Additionally, split-TurboID and contact-ID need a 4 and 16 h incubation time, respectively, while APEX2 needs a much shorter time (45 min), which gives BiFCPL more flexibility to compare time-dependent contact proteomes.
We found that GNAP2 is more suitable than NLS-GNAP2 or CS-GNAP2. The majority of the biotin-labeled proteins of NLS-GNAP2 occurred in the nucleus and would be discarded during the sample preparation processes. We also had the concern that the broken nuclei during the homogenization process would induce contamination of nucleus proteins. The biotinylation activity of CS-GNAP2 was weaker than GNAP2, which may result from less active APEX2 proteins in the cytosol. As a result, BiFCPL revealed a MERC proteome consisting of 403 proteins with high accuracy and efficiency. Most of the well-known MERC proteins and many transiently resident MERC proteins were identified. The applicability of our system in vivo is constrained by the need for exogenous biotin-phenol and hydrogen dioxide, but it could still be a reliable method to study spatial proteomes at inter-organelle contacts in vitro.
The liver plays a central role in lipid metabolism. Hepatocytes take up, synthesize, store, and secrete lipids in response to fasting and feeding to maintain whole-body lipid homeostasis.39 Abnormal triglyceride and cholesterol accumulation in the liver is the hallmark of non-alcoholic fatty liver disease (NAFLD), which affects approximately 30% of adults worldwide.40 Mitochondria-LD contacts have been closely related to metabolic stresses in the liver,41,42 but the protein composition remains unclear. In this study, we developed a reliable fluorescent reporter and provided direct imaging evidence linking mitochondria-LD contacts and nutrient stresses. We then utilized OA and HBSS+OA treatment to mimic high-fat diet-induced NAFLD and starvation-induced NAFLD, respectively. We found that over 100 proteins were enriched at mitochondria-LD contact sites under OA or HBSS+OA conditions. More importantly, 60 proteins, the majority of which were membrane proteins, were either up- or downregulated at mitochondria-LD contact sites under metabolic challenge. These proteins may be essential for the proper function of mitochondria-LD contacts.
Overexpression or depletion of SQLE significantly up- or downregulates cholesterol synthesis, respectively.31,43 Here, we demonstrated that SQLE accumulates at mitochondria-LD contact sites under HBSS+OA conditions. The contact between mitochondria and ER has been shown to function as a platform for local ATP transfer to drive the activity of FABP4 to take up fatty acids in endothelial cells.34 Similarly, we demonstrated that SQLE probably utilizes mitochondria-derived ATP to synthesize cholesterol, which is consistent with the fact that peri-LD mitochondria provide ATP for triacylglyceride synthesis, hence promoting the growth of LDs.44 It has been demonstrated that starvation enhances lipolysis in adipose tissue and then increases cholesterol synthesis in the liver.45,46,47,48 Consistently, we discovered abnormal accumulation of free cholesterol under HBSS+OA conditions, which infers that SQLE may mediate starvation-induced NAFLD. In conclusion, our study suggested that SQLE probably coordinates the function of ER, mitochondria, and LDs to synthesize cholesterol during the pathogenesis process of NAFLD, which potentializes SQLE as a therapeutic target.
Inter-organelle membrane contact is widespread and affects a variety of biological processes. BiFCPL is highly adaptable to any other inter-organelle membrane contact by targeting individual GFP fragments to the opposing organellar membranes. This enables the investigation of both the dynamics and molecular architecture of distinct inter-organelle contacts under diverse physiological conditions, thereby enabling the identification of key proteins and more importantly, disease-related therapeutic targets.
Limitations of the study
Though BiFCPL has been successfully applied to study mitochondria-endoplasmic reticulum contacts and mitochondria-lipid droplet contacts in living mammalian cells, there are a few limitations. First, the split-GFP system is irreversible, which may induce artificial membrane contacts, so it would be careful when choosing the appropriate cell clones. Second, proximity strategy based on APEX2 easily generates background labeling activities, so it would be important to set appropriate controls. Last, the application of AEPX2 in vivo is constrained for the need of exogenous biotin-phenol and hydrogen dioxide, so alternative labeling strategies may be utilized to probe organelle contact proteomes in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MYC-tag Polyclonal antibody | Proteintech | Cat#16286-1-AP; RRID: AB_11182162 |
| V5 tag antibody | Abcam | Cat#ab27671; RRID: AB_471093 |
| HRP conjugated streptavidin | Pierce | Cat#21130 |
| HA Tag Monoclonal Antibody | Invitrogen | Cat#26183; RRID: AB_10978021 |
| SQLE Polyclonal antibody | Proteintech | Cat#12544-1-AP; RRID: AB_2195888 |
| COXIV Polyclonal antibody | Proteintech | Cat#11242-1-AP; RRID: AB_2085278 |
| Tom20 antibody | Santa Cruz | Cat#sc-17764; RRID: AB_628381 |
| MFN2 Polyclonal antibody | Proteintech | Cat#12186-1-AP; RRID: AB_2266320 |
| GFP tag Polyclonal antibody | Proteintech | Cat#50430-2-AP; RRID: AB_11042881 |
| Calnexin Polyclonal antibody | Proteintech | Cat#10427-2-AP; RRID: AB_2069033 |
| TOMM70 Polyclonal antibody | Proteintech | Cat#14528-1-AP; RRID: AB_2303727 |
| GAPDH Monoclonal antibody | Proteintech | Cat#60004-1-Ig; RRID: AB_2107436 |
| Cytochrome c Polyclonal antibody | Proteintech | Cat#10993-1-AP; RRID: AB_2090467 |
| WFS1 Polyclonal antibody | Proteintech | Cat#26995-1-AP; RRID: AB_2880717 |
| SPTLC1 Polyclonal antibody | Proteintech | Cat#15376-1-AP; RRID: AB_2286678 |
| BAG2 Polyclonal antibody | Proteintech | Cat#29820-1-AP; RRID: AB_2918348 |
| GLUD1 Polyclonal antibody | Proteintech | Cat#14299-1-AP; RRID: AB_2110515 |
| Streptavidin, Alexa Fluor 555 conjugate | Invitrogen | Cat#S21381; RRID: AB_2307336 |
| Goat anti-Mouse IgG, Alexa Fluor 488 | Invitrogen | Cat#A11001; RRID: AB_2534069 |
| Goat anti-Rabbit IgG, Alexa Fluor 488 | Invitrogen | Cat#A11008; RRID: AB_143165 |
| Donkey anti-Rabbit IgG, Alexa Fluor 568 | Invitrogen | Cat#A10042; RRID: AB_2534017 |
| Goat anti-Mouse IgG, Alexa Fluor 647 | Invitrogen | Cat#A21235; RRID: AB_2535804 |
| Donkey anti-Rabbit IgG, Alexa Fluor 546 | Invitrogen | Cat#A10040; RRID: AB_2534016 |
| Chemicals and reagents | ||
| MitoTracker Deep Red FM | Invitrogen | Cat#M22426 |
| HCS LipidTOX Red | Invitrogen | Cat#H34476 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#B2064 |
| Oleic acid | Sigma-Aldrich | Cat#O1008 |
| Hank’s Balanced Salt Solution (HBSS) | Gibco | Cat#14025076 |
| DMEM, basic medium | Gibco | Cat#C11995500BT |
| Biotin-phenol | Sigma-Aldrich | Cat#41994-02-9 |
| Streptavidin Magnetic Bead | Pierce | Cat#88817 |
| Filipin | Sigma-Aldrich | Cat#F9765 |
| Lipofectamine 3000 transfection kit | Invitrogen | Cat#L3000015 |
| 8% PFA | Novon | Cat#SS0308 |
| Percoll | Sigma-Aldrich | Cat#p4937 |
| Plasmids | ||
| plx304-spGFP1-10-ERt | Tong Chao | N/A |
| pLVX-Mitot-spGFP11 | Tong Chao | N/A |
| pCDH-mito-sfGFP11 | This paper | N/A |
| pCDH-sfGFP1-10-Plin2 | This paper | N/A |
| pApex2-GFPnanobody-C1 | This paper | N/A |
| pQCXIP-sqle-ha | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health, Bethesda, MD, USA. | https://ImageJ.nih.gov/ij/download.html |
| Graphpad Prism | Graphpad Prism | https://www.graphpad.com/scientificsoftware/prism/; RRID: SCR_002798 |
| Zen 3.0 Blue for line profile analysis | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html; RRID:SCR_010279 |
| FLUOVIEW FV1200 Viewer | Olympus | https://www.olympus-lifescience.com/en/ |
| Oligonucleotides | ||
| Primer name and sequence | Source | Identifier |
| Gfp1-10-v5 up: ATGTCCAAAGGA GAAGAACTGTTTAC |
This paper | N/A |
| Gfp1-10-v5 down: CGTAGAATCGAG ACCGAGGAGA |
This paper | N/A |
| Plin2 up: ATGGCATCCGTTG CAGTTGATCCACAA |
This paper | N/A |
| Plin2 down: TTAATGAGTTTTATG CTCAGATCGCTG |
This paper | N/A |
| pCDH-Gfp1-10-v5-plin2 up: AAGAATTCATGTCCAAAGGAGA AGAACTGTTTAC |
This paper | N/A |
| pCDH-Gfp1-10-v5-plin2 down: TTGCGGCCGCTTAATGAGTTTTAT GCTCAGATCGCTG |
This paper | N/A |
| pCDH-mito-sfGFP11 up: AAGCTAGCCCACCATGGTGGGT CGGAACAGCGCCAT |
This paper | N/A |
| TTGGATCCTTAAGTGATGCCTG CGGCGTTAA |
This paper | N/A |
| GNAP2-up: AAGCTAGCATGGGAA AGTCTTACCCAACTGT |
This paper | N/A |
| GNAP2-down: TTGGATCCTCAA GAGGACACGGTCACCTGGGTA |
This paper | N/A |
| NLS-GNAP2-up: ATGCCAAAAAAG AAAAGAAAAGTTGGAAAGTCTTAC CCAACTGT |
This paper | N/A |
| pQCXIP-ha-sqle up: AAACCGGTAT GTGGACTTTTCTGGGCATTGCCA |
This paper | N/A |
| pQCXIP-ha-sqle down: TTGGATCCTTAA GCGTAGTCTGGGACGTCGTATGGGTA ATGAACCATATACTTCATTTCTG |
This paper | N/A |
| SPTLC1 up: AAGCTAGCATGGCGACC GCCACGGAGCAGT |
This paper | N/A |
| SPTLC1 down: TTACCGGTGGGAGCA GGACGGCCTGGGCTAC |
This paper | N/A |
| BAG2 up: AAGCTAGCATGGCTCA GGCGAAGATCAACG |
This paper | N/A |
| BAG2 down: TTACCGGTGGATTGAA TCTGCTTTCAGCATTTTG |
This paper | N/A |
| GLUD1 up: AAGAATTCATGTACCG CTACCTGGGCGAAG |
This paper | N/A |
| GLUD1 down: TTACCGGTGGTGTGAA GGTCACACCAGCTTCAT |
This paper | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tao Xu (xutao@ibp.ac.cn).
Materials availability
This study did not generate new unique reagents. For the plasmids, please contact with the lead contact.
Experimental model and subject details
Cell culture and treatment
The reporter U2OS cell line, referred to as MERC cells, was a kind gift from Prof. Chao Tong’s group. HepG2 cells (1101HUM-PUMC000035, National Infrastructure of Cell Line Resource, China) and U2OS cells were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/L glucose supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin under 5% CO2. Transient transfection of the appropriate plasmids into the cells at ∼60% confluence was performed using Lipofectamine 3000 according to the manufacturer’s instructions. The oleic acids were dissolved in 0.1 M Tris, pH 8.0 to make a stock solution, which was then diluted to a final concentration of 100 μM in culture medium or Hank’s Balanced Salt Solution (HBSS) to make a working solution. All the reagents are available in the key resources table.
Generation of stable cell lines
The stable MLDCS cell line was generated by lentiviral infection. The lentiviruses stably expressing mito-sfGFP11 and sfGFP1-10-Plin2 were packaged and concentrated by the Obio Inc. (Shanghai, China). Briefly, the plasmids were transfected into 293T cells along with packaging plasmids (pLP1, pLP2, and pLP/VSVG). After 72 h, the lentivirus containing supernatant were collected and concentrated. HepG2 cells were then infected with these two viruses supplemented with 10 μg/mL polybrene and then selected with 2 μg/mL puromycin. Single GFP-positive cells were then sorted by flow cytometry and maintained in DMEM supplemented with puromycin. The appropriate cell clone, termed MLDCS cell, was selected with a confocal microscope equipped with a 63×/1.40 NA oil immersion objective (LSM980, Zeiss, Germany).
For the generation of stable cell lines expressing SQLE-HA, the retroviruses were packaged by transfecting PlatE cells with pQCXIP-SQLE-HA and two packaging plasmids (VSVG and Phit). The virus-containing supernatant was collected after 48 h and filtered to infect HepG2 cells with 10 μg/mL polybrene. The cells were then selected and maintained with 2 μg/mL puromycin to generate stable cell lines.
Method details
Plasmids
To construct pCDH-sfGFP1-10-v5-Plin2, sfGFP1-10-v5 was amplified from plx304-sfGFP1-10-ERt.7 The overlapping products of sfGFP1-10-v5-plin2 were then inserted into pCDH-CMV-MCS-EF1-Puro vector. Mito-myc-sfGFP11 was synthesized by the GENEWIZ Biotechnology Co. Ltd. (Suzhou, China) and then inserted into the pCDH-CMV-MCS-EF1-Puro vector. For the construction of pApex2-GFP nanobody-C1 to express GFP nanobody tagged APEX2 protein (referred to as GNAP2), the coding sequence of APEX2 was amplified from pCDNA3-flag-APEX2.37 The GFP nanobody sequence was synthesized and ligated into the C-terminus of APEX2. The resulting Apex2-GFP nanobody sequence was then inserted into the pEGFP-C1 vector in replacement of the EGFP sequence. NLS-GNAP2 was constructed by introducing a nucleus localized sequence (CCAAAAAAGAAAAGAAAAGTT) into the N terminal of GNAP2. CS-GNAP2 was constructed by adding a proteasomal system targeting sequence as described previously.15 The coding sequence of human SQLE was amplified from the cDNA of HepG2 cells and an HA tag was inserted at the C-terminus. The resulting SQLE-HA was then inserted into the PQCXIP vector. For biological validation, plasmids expressing WFS1-EGFP and ER-DsRed were described previously.49 The coding sequences of BAG2, SPTLC1 and GLUD1 were introduced into the pEGFP-N1 vector through restriction enzymes. The sequences of all the primers used in this study are available in the key resources table.
Correlative light and electron microscopy
The CLEM analysis was performed with a novel cryogenic corelated light, ion and electron microscope.50 Briefly, the MLDCS cells were grown on lacey 200 mesh gold EM grids (T10012Au, Beijing XXBR Technology Co., Ltd.) overnight to allow adherence and spreading. After staining with MitoTracker, the grid was plunge-frozen in liquid ethane using an automatic plunge-freezer (EM GP2, Leica, Germany). Light microscopy images were acquired using integrated confocal microscope. Then the grids were coated with Pt and focused ion beam (FIB) milling was conducted. The cryo-electron tomography data were acquired on a 300 kV Titan Krios cryo-transmission electron microscope equipped with a BioQuantum energy filter and a K2 Summit Direct electron detector (Gatan, USA).
Mitochondria and LD staining
The mitochondria were stained with 1 μM MitoTracker deep red (M22426, Thermo Fisher Scientific, USA). The LDs were stained with HCS LipidTox red (H34476, Thermo Fisher Scientific, USA) at a dilution of 1/1000. HepG2 cells were grown on coverslips and stimulated with different treatments. The cells were then incubated with the indicated media containing MitoTracker and LipidTox for 30 min at 37°C. The fluorescent images were captured using a confocal microscope equipped with a 63×/1.40 NA oil immersion objective (LSM980, Zeiss, Germany).
Biotin-phenol labeling
HepG2, MLDCS, U2OS or MERC cells were transiently transfected with GNAP2 and then incubated under the indicated conditions. Biotin-phenol (BP) labeling was performed as previously described.51 Briefly, biotin-phenol (BP) was added to the medium at a concentration of 0.5 μM for 45 min at 37°C H2O2 was then added at a final concentration of 1 mM to initiate biotin-phenol labeling. After 1 min, the reaction was stopped by switching the medium into a quenching solution containing 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox. After washing with phosphate buffered saline (PBS) three times, the cells were either fixed for immunofluorescence labeling or harvested for enrichment analysis.
Immunofluorescence labeling
The cells grown on coverslips were fixed with 4% paraformaldehyde (PFA) at room temperature for 20 min, washed with PBS and permeabilized for 3 min with PBS containing 0.1% Triton X-100. The cells were then blocked with 5% goat serum in PBS for 1 h and incubated with appropriate primary antibodies. After washing with PBS three times, the cells were incubated with fluorescent secondary antibodies and mounted with DAPI for nucleus staining. The fluorescent images were acquired using a confocal microscope equipped with a 63×/1.40 NA oil immersion objective (LSM980, Zeiss, Germany) or a 60×/1.40 NA oil immersion objective (FV1200, Olympus, Japan). The images were then analyzed with ZEN 2 blue edition or ImageJ.
Enrichment of biotinylated proteins
After biotin-phenol labeling, the cells were harvested, washed with PBS and then resuspended in a buffer containing 225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA and 30 mM Tris–HCl, pH 7.4 supplemented with proteinase inhibitor.27 The cells were then homogenized using glass tissue grinders. The cell integrity was monitored under a light microscope to ensure more than 80% efficiency of cell damage. The homogenized cells were centrifuged at 500 g for 5 min at 4°C to remove unbroken cells and nuclei and the supernatant was collected and centrifuged again under the same conditions. The supernatant was then centrifuged at 9,000 g for 10 min at 4°C and the pellet containing crude mitochondria was resuspended in 1 mL RIPA buffer (Beyotime Biotechnology, Shanghai, China) containing 50 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS, pH 7.4 supplemented with proteinase inhibitor. The lysate was then clarified on ice by ultrasonication. The supernatant was collected after centrifugation at 12,000 g for 15 min at 4°C and incubated overnight with 100 μL streptavidin-coated magnetic beads with rotation at 4°C. The beads were then washed with RIPA buffer twice, and then washed sequentially with 1 M KCl, 0.1 M Na2CO3 and 2 M urea, and RIPA buffer twice. The biotinylated proteins were eluted by boiling the beads with sodium dodecyl sulfate (SDS) loading buffer at 100°C for 10 min. The eluted proteins were analyzed by Western blotting or mass spectrometry (MS).
Subcellular fraction
The cells were harvested and homogenized as above. Mitochondria and MAM (mitochondria associated ER membrane) were separated from the resuspended crude mitochondria using percoll medium (225 mM mannitol, 25 mM HEPES, 1 mM EGTA, 30% Percoll (v/v) (pH 7.4)) by ultracentrifugation at 100,000g for 60 min. To isolate ER and cytosol, the supernatant from crude mitochondria were collected and centrifugated at 100,000g for 60 min.
Western blotting
The enriched biotinylated proteins were separated by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and then transferred onto nitrocellulose membranes. After blocking with 5% milk in TBST (Tris-buffered saline, 0.1% Tween 20), the membrane was incubated with appropriate primary antibodies, followed by appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Sungene Biotech, Tianjin, China). The reactive bands were revealed by chemiluminescence (GE Healthcare, Piscataway, NJ, USA) and imaged with the ChemiScope series (Clinx, Shanghai, China).
In gel digestion
The enriched biotinylated proteins were separated by SDS-PAGE and stained with Coomassie G-250. In-gel protein digestion was performed as previously described.37 In brief, manually cut protein lanes were de-stained with 40% acetonitrile in 50 mM NH4HCO3, followed by dehydration with 100% acetonitrile. The disulfide bonds were reduced with 10 mM dithiothreitol (DTT) at 56°C for 45 min and then alkylated with 55 mM iodoacetamide at 25°C for 60 min in the dark. The gel pieces were sequentially washed with 50 mM NH4HCO3 and 50% acetonitrile in 50 mM NH4HCO3 and then dehydrated with 100% acetonitrile. After drying using a SpeedVac, the gels were rehydrated using 100 ng/μL trypsin on ice for 30 min. The proteins were digested overnight at 37°C and the reaction was quenched with 1.0% trifluoroacetic acid (TFA). The tryptic peptides were extracted twice with 60% acetonitrile containing 0.1% TFA and then dried using a SpeedVac.
Mass spectrometry
All nano-LC-MS/MS experiments were conducted on an Orbitrap Exploris 480 (Thermo Scientific, USA) equipped with an Easy n-LC 1200 HPLC system (Thermo Scientific, USA). The peptides were loaded onto a 100 μm id×2 cm fused silica trap column packed in-house with reversed-phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch GmbH, Germany) and then separated on an a 75 μm id×20 cm C18 column packed with reversed-phase silica (Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch GmbH, Germany). The peptides bound on the column were eluted with a 73-min linear gradient. Solvent A consisted of 0.1% formic acid in water solution and solvent B consisted of 80% acetonitrile and 0.1% formic acid. The segmented gradient was 4–9% B, 3 min; 9–20% B, 22 min; 20–30% B, 20 min; 30–40% B, 15 min; 40–95% B, 3 min; 95% B, 10 min at a flow rate of 300 nL/min.
MS analysis was conducted with an Orbitrap Exploris 480 mass spectrometer (Thermo Scientific, USA). With the data-dependent acquisition mode, the MS data were acquired at a high resolution of 60,000 (m/z 200) across the mass range of 350–1500 m/z. The target value was 3.00E+06 with a maximum injection time of 22 ms. The data dependent mode was selected as the cycle time mode with a duration of 2 s. The precursor ions were selected from each MS full scan with an isolation width of 1.6 m/z for fragmentation in the ion routing multipole at a normalized collision energy of 28%. Subsequently, the MS/MS spectra at a resolution of 15,000 at m/z 200 were acquired. The target value was 7.50E+04 with a maximum injection time of 22 ms. The dynamic exclusion time was 40 s. For the nano electrospray ion source setting, the spray voltage was 2.0 kV. There was no sheath gas flow and the heated capillary temperature was 320°C.
Proteomic analysis
The raw data from the Orbitrap Exploris 480 were analyzed with Proteome Discovery version 2.4.1.15. The Sequest HT search engine was used for protein identification according to the Uniprot database of human proteins (updated on 09/2018). The following parameters were used to search the database: 10 ppm precursor mass tolerance; 0.02 Da fragment ion tolerance; up to two missed cleavages; carbamidomethyl cysteine and oxidized methionine as a variable modification. FDR analysis was performed with Percolator, and FDR <1% was set for protein identification. The proteins used for the quantitative analysis were further filtered according to the following criteria: 1) total spectral counts of proteins should be at least 3; 2) at least two unique peptides were required; and 3) no razor peptides were allowed. After normalizing protein abundances using RUVg,52 differential expression analysis of proteins was carried out by edgeR,53 and proteins with p value less than 0.01 and fold change above 2 were considered as high confidence candidates.
Filipin staining
HepG2 cells were cultured under the indicated conditions and then fixed with 4% PFA for 20 min at room temperature. After washing with PBS, the cells were incubated with 0.05 mg/mL filipin in PBS for 1 h at room temperature. After washing with PBS, the cells were imaged with a confocal microscope equipped with a 60×/1.40 NA oil immersion objective (FV1200, Olympus, Japan) under the same parameters. For each group, n = 11–18 fields were imaged and the filipin intensity was calculated.
Acknowledgments
We thank Prof. Chao Tong from Zhejiang University for providing the MERC cells and plasmids. We thank Prof. Hong Zhang, Dr. Liqing Liu, and Ce Jia from the Institute of Biophysics for their suggestions on manuscript organization. We thank the staff from the Institute of Biophysics, particularly, Jifeng Wang for the support in mass spectrometry analysis, and Yan Teng for the support in fluorescence imaging. We thank the editors and reviewers for their constructive suggestions. This work was supported by the National Natural Science Foundation of China (grant no. 32100536, 62105356, 82200884, 92254306), the National Science Foundation for Distinguished Young Scholars of China (grant no. T2225020), and the Scientific Instrument Development Project of the CAS (grant no. GJJSTD20210001). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author contributions
Conceptualization, M.Z. and T.X.; Methodology & Investigation, M.Z., B.K., K.X., J.L., W.L., and M.L.; Methodology & Software, X.Z. and J.H.; Writing – Original Draft, M.Z. and B.K.; Writing – Review & Editing, J.L., Z.L., W.J., J.H., and T.X.; Project Administration, W.J., J.H., and T.X.; Supervision, T.X.
Declaration of interests
The authors declare no competing interests.
Published: June 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107159.
Contributor Information
Wei Ji, Email: jiwei@ibp.ac.cn.
Junjie Hou, Email: houjj_pro@163.com.
Tao Xu, Email: xutao@ibp.ac.cn.
Supplemental information
Data and code availability
The raw mass spectrometry data in this study are available via ProteomeXchange with the identifiers PXD035813 (for mitochondria-LD contact) and PXD035801 (for MERC). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The proteomic data of MERC are available in Table S1. The proteomic data of mitochondria-LD contact are available in Tables S2, S3 and S4. Mendeley Data: https://doi.org/10.17632/r2xsfj7hb8.1.
References
- 1.Prinz W.A., Toulmay A., Balla T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020;21:7–24. doi: 10.1038/s41580-019-0180-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rossini M., Pizzo P., Filadi R. Better to keep in touch: investigating inter-organelle cross-talk. FEBS J. 2021;288:740–755. doi: 10.1111/febs.15451. [DOI] [PubMed] [Google Scholar]
- 3.Poston C.N., Krishnan S.C., Bazemore-Walker C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM) J. Proteonomics. 2013;79:219–230. doi: 10.1016/j.jprot.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 4.Ma J.H., Shen S., Wang J.J., He Z., Poon A., Li J., Qu J., Zhang S.X. Comparative Proteomic Analysis of the Mitochondria-associated ER Membrane (MAM) in a Long-term Type 2 Diabetic Rodent Model. Sci. Rep. 2017;7:2062. doi: 10.1038/s41598-017-02213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X., Jiang C., Yu L., Yang A. Current and Emerging Approaches for Studying Inter-Organelle Membrane Contact Sites. Front. Cell Dev. Biol. 2020;8:195. doi: 10.3389/fcell.2020.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi C.R., Rhee H.W. Proximity labeling: an enzymatic tool for spatial biology. Trends Biotechnol. 2022;40:145–148. doi: 10.1016/j.tibtech.2021.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Yang Z., Zhao X., Xu J., Shang W., Tong C. A novel fluorescent reporter detects plastic remodeling of mitochondria-ER contact sites. J. Cell Sci. 2018;131 doi: 10.1242/jcs.208686. [DOI] [PubMed] [Google Scholar]
- 8.Kwak C., Shin S., Park J.S., Jung M., Nhung T.T.M., Kang M.G., Lee C., Kwon T.H., Park S.K., Mun J.Y., et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA. 2020;117:12109–12120. doi: 10.1073/pnas.1916584117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho K.F., Branon T.C., Rajeev S., Svinkina T., Udeshi N.D., Thoudam T., Kwak C., Rhee H.W., Lee I.K., Carr S.A., Ting A.Y. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA. 2020;117:12143–12154. doi: 10.1073/pnas.1919528117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill S., Stevenson J., Kristiana I., Brown A.J. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metabol. 2011;13:260–273. doi: 10.1016/j.cmet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Shai N., Yifrach E., van Roermund C.W.T., Cohen N., Bibi C., IJlst L., Cavellini L., Meurisse J., Schuster R., Zada L., et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018;9:1761. doi: 10.1038/s41467-018-03957-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cieri D., Vicario M., Giacomello M., Vallese F., Filadi R., Wagner T., Pozzan T., Pizzo P., Scorrano L., Brini M., Calì T. SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018;25:1131–1145. doi: 10.1038/s41418-017-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calì T., Brini M. Quantification of organelle contact sites by split-GFP-based contact site sensors (SPLICS) in living cells. Nat. Protoc. 2021;16:5287–5308. doi: 10.1038/s41596-021-00614-1. [DOI] [PubMed] [Google Scholar]
- 14.Kubala M.H., Kovtun O., Alexandrov K., Collins B.M. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 2010;19:2389–2401. doi: 10.1002/pro.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariotti N., Rae J., Giles N., Martel N., Sierecki E., Gambin Y., Hall T.E., Parton R.G. Ultrastructural localisation of protein interactions using conditionally stable nanobodies. PLoS Biol. 2018;16:e2005473. doi: 10.1371/journal.pbio.2005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGuire M.R., Mukhopadhyay D., Myers S.L., Mosher E.P., Brookheart R.T., Kammers K., Sehgal A., Selen E.S., Wolfgang M.J., Bumpus N.N., Espenshade P.J. Progesterone receptor membrane component 1 (PGRMC1) binds and stabilizes cytochromes P450 through a heme-independent mechanism. J. Biol. Chem. 2021;297 doi: 10.1016/j.jbc.2021.101316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doghman-Bouguerra M., Granatiero V., Sbiera S., Sbiera I., Lacas-Gervais S., Brau F., Fassnacht M., Rizzuto R., Lalli E. FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep. 2016;17:1264–1280. doi: 10.15252/embr.201541504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Wen Y., Dong J., Cao C., Yuan S. Systematic In-Depth Proteomic Analysis of Mitochondria-Associated Endoplasmic Reticulum Membranes in Mouse and Human Testes. Proteomics. 2018;18 doi: 10.1002/pmic.201700478. [DOI] [PubMed] [Google Scholar]
- 19.Lee S., Min K.T. The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cell. 2018;41:1000–1007. doi: 10.14348/molcells.2018.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aoyama-Ishiwatari S., Hirabayashi Y. Endoplasmic Reticulum-Mitochondria Contact Sites-Emerging Intracellular Signaling Hubs. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.653828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenberg-Bord M., Shai N., Schuldiner M., Bohnert M. A Tether Is a Tether Is a Tether: Tethering at Membrane Contact Sites. Dev. Cell. 2016;39:395–409. doi: 10.1016/j.devcel.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 23.Angebault C., Fauconnier J., Patergnani S., Rieusset J., Danese A., Affortit C.A., Jagodzinska J., Mégy C., Quiles M., Cazevieille C., et al. ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aaq1380. [DOI] [PubMed] [Google Scholar]
- 24.Qin L., Guo J., Zheng Q., Zhang H. BAG2 structure, function and involvement in disease. Cell. Mol. Biol. Lett. 2016;21:18. doi: 10.1186/s11658-016-0020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aaltonen M.J., Alecu I., König T., Bennett S.A., Shoubridge E.A. Serine palmitoyltransferase assembles at ER-mitochondria contact sites. Life Sci. Alliance. 2022;5 doi: 10.26508/lsa.202101278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boese A.C., Kang S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021;42 doi: 10.1016/j.redox.2021.101870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieckowski M.R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 28.Ma X., Qian H., Chen A., Ni H.M., Ding W.X. Perspectives on Mitochondria-ER and Mitochondria-Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells. 2021;10 doi: 10.3390/cells10092273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rambold A.S., Cohen S., Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zembroski A.S., Andolino C., Buhman K.K., Teegarden D. Proteomic Characterization of Cytoplasmic Lipid Droplets in Human Metastatic Breast Cancer Cells. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.576326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D., Wong C.C., Zhou Y., Li C., Chen H., Ji F., Go M.Y.Y., Wang F., Su H., Wei H., et al. Squalene Epoxidase Induces Nonalcoholic Steatohepatitis Via Binding to Carbonic Anhydrase III and is a Therapeutic Target. Gastroenterology. 2021;160:2467–2482.e3. doi: 10.1053/j.gastro.2021.02.051. [DOI] [PubMed] [Google Scholar]
- 32.Olzmann J.A., Carvalho P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019;20:137–155. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X., Cai B., Yang X., Sonubi O.O., Zheng Z., Ramakrishnan R., Shi H., Valenti L., Pajvani U.B., Sandhu J., et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metabol. 2020;31:969–986.e7. doi: 10.1016/j.cmet.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ibrahim A., Yucel N., Kim B., Arany Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metabol. 2020;32:309–319.e7. doi: 10.1016/j.cmet.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romei M.G., Boxer S.G. Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu. Rev. Biophys. 2019;48:19–44. doi: 10.1146/annurev-biophys-051013-022846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kakimoto Y., Tashiro S., Kojima R., Morozumi Y., Endo T., Tamura Y. Visualizing multiple inter-organelle contact sites using the organelle-targeted split-GFP system. Sci. Rep. 2018;8:6175. doi: 10.1038/s41598-018-24466-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue M., Hou J., Wang L., Cheng D., Lu J., Zheng L., Xu T. Optimizing the fragment complementation of APEX2 for detection of specific protein-protein interactions in live cells. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-12365-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Y., Branon T.C., Martell J.D., Boassa D., Shechner D., Ellisman M.H., Ting A. Directed Evolution of Split APEX2 Peroxidase. ACS Chem. Biol. 2019;14:619–635. doi: 10.1021/acschembio.8b00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rui L. Energy metabolism in the liver. Compr. Physiol. 2014;4:177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le M.H., Yeo Y.H., Li X., Li J., Zou B., Wu Y., Ye Q., Huang D.Q., Zhao C., Zhang J., et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022;20:2809–2817.e28. doi: 10.1016/j.cgh.2021.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Arruda A.P., Pers B.M., Parlakgül G., Güney E., Inouye K., Hotamisligil G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014;20:1427–1435. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krahmer N., Najafi B., Schueder F., Quagliarini F., Steger M., Seitz S., Kasper R., Salinas F., Cox J., Uhlenhaut N.H., et al. Organellar Proteomics and Phospho-Proteomics Reveal Subcellular Reorganization in Diet-Induced Hepatic Steatosis. Dev. Cell. 2018;47:205–221.e7. doi: 10.1016/j.devcel.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 43.Yang F., Kou J., Liu Z., Li W., Du W. MYC Enhances Cholesterol Biosynthesis and Supports Cell Proliferation Through SQLE. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.655889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benador I.Y., Veliova M., Mahdaviani K., Petcherski A., Wikstrom J.D., Assali E.A., Acín-Pérez R., Shum M., Oliveira M.F., Cinti S., et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metabol. 2018;27:869–885.e6. doi: 10.1016/j.cmet.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Ginneken V., Verhey E., Poelmann R., Ramakers R., van Dijk K.W., Ham L., Voshol P., Havekes L., Van Eck M., van der Greef J. Metabolomics (liver and blood profiling) in a mouse model in response to fasting: a study of hepatic steatosis. Biochim. Biophys. Acta. 2007;1771:1263–1270. doi: 10.1016/j.bbalip.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Sokolović M., Sokolović A., van Roomen C.P.A.A., Gruber A., Ottenhoff R., Scheij S., Hakvoort T.B.M., Lamers W.H., Groen A.K. Unexpected effects of fasting on murine lipid homeostasis--transcriptomic and lipid profiling. J. Hepatol. 2010;52:737–744. doi: 10.1016/j.jhep.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 47.Xu H., Jiang Y., Miao X.M., Tao Y.X., Xie L., Li Y. A Model Construction of Starvation Induces Hepatic Steatosis and Transcriptome Analysis in Zebrafish Larvae. Biology. 2021;10 doi: 10.3390/biology10020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaziri N.D., Liang K.H. Hepatic HMG-CoA reductase gene expression during the course of puromycin-induced nephrosis. Kidney Int. 1995;48:1979–1985. doi: 10.1038/ki.1995.500. [DOI] [PubMed] [Google Scholar]
- 49.Wang L., Liu H., Zhang X., Song E., Wang Y., Xu T., Li Z. WFS1 functions in ER export of vesicular cargo proteins in pancreatic beta-cells. Nat. Commun. 2021;12:6996. doi: 10.1038/s41467-021-27344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W., Lu J., Xiao K., Zhou M., Li Y., Zhang X., Li Z., Gu L., Xu X., Guo Q., et al. Integrated multimodality microscope for accurate and efficient target-guided cryo-lamellae preparation. Nat. Methods. 2023;20:268–275. doi: 10.1038/s41592-022-01749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhee H.W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Risso D., Ngai J., Speed T.P., Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lun A.T.L., Chen Y., Smyth G.K. It's DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol. Biol. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mass spectrometry data in this study are available via ProteomeXchange with the identifiers PXD035813 (for mitochondria-LD contact) and PXD035801 (for MERC). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. The proteomic data of MERC are available in Table S1. The proteomic data of mitochondria-LD contact are available in Tables S2, S3 and S4. Mendeley Data: https://doi.org/10.17632/r2xsfj7hb8.1.