

Abstract
Electroporation of the Cas9 ribonucleoprotein (RNP) complex offers the advantage of preventing off-target cleavages and potential immune responses produced by long-term expression of the nuclease. Nevertheless, the majority of engineered high-fidelity Streptococcus pyogenes Cas9 (SpCas9) variants are less active than the wild-type enzyme and are not compatible with RNP delivery. Building on our previous studies on evoCas9, we developed a high-fidelity SpCas9 variant suitable for RNP delivery. The editing efficacy and precision of the recombinant high-fidelity Cas9 (rCas9HF), characterized by the K526D substitution, was compared with the R691A mutant (HiFi Cas9), which is currently the only available high-fidelity Cas9 that can be used as an RNP. The comparative analysis was extended to gene substitution experiments where the two high fidelities were used in combination with a DNA donor template, generating different ratios of non-homologous end joining (NHEJ) versus homology-directed repair (HDR) for precise editing. The analyses revealed a heterogeneous efficacy and precision indicating different targeting capabilities between the two variants throughout the genome. The development of rCas9HF, characterized by an editing profile diverse from the currently used HiFi Cas9 in RNP electroporation, increases the genome editing solutions for the highest precision and efficient applications.
Keywords: CRISPR-Cas, protein delivery, RNP, HDR, hematopoietic stem cells, high-fidelity Cas9, specificity, genome editing
Graphical abstract
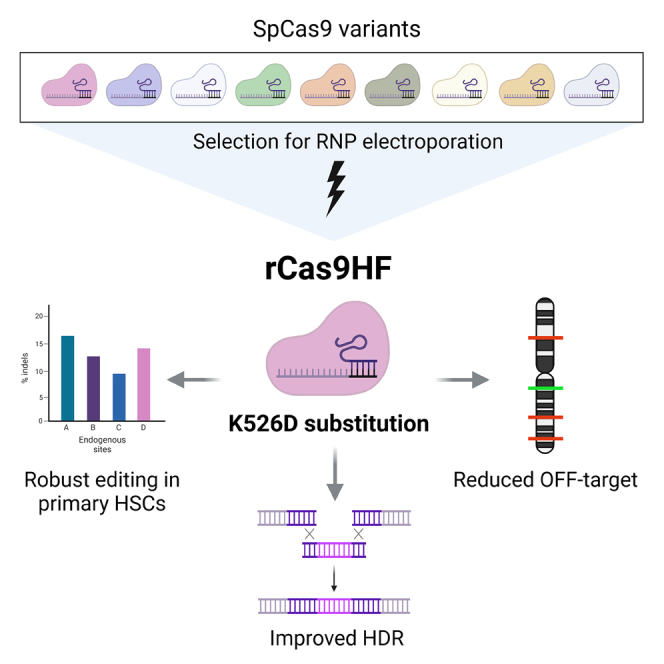
Cereseto, Casini, and colleagues have developed a high-fidelity variant of Cas9, rCas9HF, which allows efficient editing as a ribonucleoprotein. This variant showed editing efficacy similar to wild-type SpCas9 but higher precision in cell lines and in CD34+ primary hematopoietic stem cells; the rCas9HF variant improved homology-directed repair through ribonucleoprotein delivery.
Introduction
Encouraging results from experimental clinics demonstrate the high potential of the CRISPR technology to treat genetic diseases and cancer.1,2 Ribonucleoprotein (RNP) delivery is emerging as the method of choice for gene substitution through homology-directed repair3,4 or targeted sequence disruption as in γ-globin reactivation for the ex vivo treatment of β-thalassemia and sickle cell diseases.1,5,6,7
The rapid clearance of RNPs via cellular proteolysis is instrumental in limiting the off-target accumulation produced by Cas9 long-term residency.8,9 Nonetheless, off-target cleavages are best controlled by using high-fidelity Cas9 variants derived from Streptococcus pyogenes Cas9 (SpCas9). High-fidelity Cas9s were generated by either rational protein engineering10,11,12,13 or directed evolution approaches, as for evoCas9,14 resulting in limited tolerance to mismatches between the guide RNA and DNA substrates.14,15,16 A comparative analysis among the high-fidelity variants and the original wild-type SpCas9 (WT SpCas9) demonstrated a general trade-off between Cas9 activity and specificity resulting in an inverse correlation between efficiency and precision.17,18 The reduced editing efficacy of the high-fidelity SpCas9s can be compensated by sustained protein levels through long-term transgene expression via plasmid or viral vector delivery.19 Consequently, the fast protein-RNA decay following RNP electroporation is not compatible with the protein levels needed to obtain efficient editing with high-fidelity SpCas9s. In fact, even though a large number of high-fidelity variants have been produced, so far a single mutant, R691A (known as HiFi Cas9), is used in ex vivo RNP electroporation.13 Nonetheless, the editing efficacy and precision varies along the genome,14 and thus diverse variants responding to diverse editing requirements are needed to reach the highest number of genomic sites through CRISPR editing. To enlarge the spectrum of high-fidelity variants compatible with RNP delivery, we built on the protein engineering work that led to the identification of evoCas9.14 evoCas9 is characterized by four amino acid substitutions (M495V, Y515N, K526E, R661Q) and shows the highest specificity among all the high-fidelity SpCas9 variants so far developed.17,18 Nevertheless, its increased specificity is paralleled by a significant loss of activity on several target sites due to a trade-off between activity and specificity.17,18 Here, we identified the K526D mutant, which we named rCas9HF (recombinant high-fidelity Cas9), showing a favorable specificity profile, while preserving near-WT levels of activity when delivered as an RNP through electroporation. An in-depth comparative analysis between HiFi Cas9 and rCas9HF in several loci by targeted deep sequencing analysis demonstrated that the two variants have different efficiency and precision depending on the target genomic locus, thus providing alternative editing solutions and enriching the CRISPR toolbox. As a proof of concept, we challenged the two high-fidelity variants by editing a set of loci in primary human CD34+ blood stem cells including the γ-globin promoter, which is currently used as therapeutic target for sickle cell disease and is associated with a known off-target site generated with RNP editing.7 Finally, we comparatively evaluated their efficacy through RNP delivery in gene substitution experiments using donor DNAs to elucidate the advantages of high-fidelity variants in gene editing involving HDR repair.
Results
Identification of SpCas9 mutants suitable for RNP delivery
Among the SpCas9 high-fidelity variants so far developed, evoCas9 is the most precise, even though this is paralleled by low editing efficiency, especially on specific target sites.14,17,18 Since evoCas9 has been tested through endogenous expression via plasmid or viral vector delivery, we evaluated its efficacy following delivery as an RNP. We used a lipofection protocol reported before9 to evaluate the on- and off-target activity of the evoCas9 RNP by targeting EGFP expressed in HEK293T reporter cells.14 For the on target, we used a single guide RNA (sgRNA) fully matching the EGFP coding sequence, while the off targets were measured with two sgRNAs containing single mismatches at different positions along the same spacer. Despite observing high precision with the off-target sgRNAs (less than 5% off-target cleavages with evoCas9 as opposed to more than 40% with both WT SpCas9 and HiFi Cas9), we detected a very low activity with the on-target sgRNA (on average, 6–8 times less than HiFi Cas9 and WT SpCas9, respectively) (Figure 1A). These results confirmed the inverse correlation between efficiency and precision obtained with the high-fidelity SpCas9 variants,17,18 which turned to be more obvious by using the RNP delivery.13 Therefore, starting from the protein engineering work that produced evoCas9, we further refined our findings to finely tune the activity and specificity of the mutants for transient expression, aiming at enlarging the repertoire of SpCas9 variants for RNP delivery. Starting from the pool of most promising mutations identified during the development of evoCas9 (M495V, Y515N, K526E, and R661Q/S),14 we set to identify alternative combinations of amino acid substitutions that could increase the activity of the RNPs while maintaining the high-fidelity properties. Among these selected mutations, K526E was tested individually given the high specificity previously demonstrated.14 We thus produced recombinant proteins, including a combination of two or three substitutions, and tested them side by side with WT SpCas9 and HiFi Cas9 using our EGFP reporter cells and surrogate off-target models. As shown in Figure 1B, despite recovering part of the cleavage activity, all triple and most double mutants showed lower on-target editing efficacy compared with both HiFi Cas9 and WT SpCas9. Therefore, we picked the single K526E mutant as the best-performing mutant for further comparative analysis. The experiments were performed using a panel of six endogenous genomic targets and showed that only in the FANCF locus did K526E have similar efficiency to the WT and HiFi Cas9, while it was less active in the remaining sites (Figure 1C).
Figure 1.

Editing activity and precision of RNP from SpCas9 high-fidelity variants
(A and B) HEK293T cells stably expressing EGFP were lipofected with the indicated RNPs together with sgRNA perfectly matching the target (sgGFPon) or containing a single mismatch at different positions along the spacer as indicated (sgGFP12 and sgGFP18: mismatched nucleotides in positions 12 and 18 counting from the PAM, respectively). Loss of fluorescence was measured by cytofluorimetry 7 days post-lipofection. In (B), statistical significance was assessed using one-way ANOVA, comparing each mutant with WT SpCas9 separately for sgGFPon, sgGFP12, and sgGFP18. (C) Editing activity (percentage of indels) measured by sequencing (tracking indels by deconvolution [TIDE]) at the indicated endogenous genomic loci in U2OS cells 3 days after electroporation of WT SpCas9, HiFi Cas9, or K526E RNPs. Statistical significance was assessed using one-way ANOVA, comparing HiFi Cas9 and K526E with WT SpCas9. Data reported as mean ± SEM for n ≥ 2 biologically independent replicates.
To test whether the loss of editing activity could be recovered while preserving the specificity provided by the modification of the K526 residue, we tested all possible amino acid substitutions in this position. In this set of experiments, we induced higher editing through transient plasmid transfection instead of RNP delivery to enhance the sensitivity for both on- and off-target insertions or deletions (indels), as previously performed.13 Moreover, in addition to the matched sgRNA for on-target analysis, we used two sgRNAs, each containing two mismatched nucleotides (13-14 and 18-19) in the spacer sequence with increasing distance from the PAM nucleotides, to test the frequency of diverse off-target events. While the on-target activity did not differ significantly among the variants and WT SpCas9, the off-target cleavages varied, showing that the K526D variant was similarly precise as the K526E (Figures 2A and S1). Notably, the editing profile (higher on/off ratios) of both variants was better than the HiFi Cas9 mutant by inducing lower off-target activities (Figures 2A and S1). Given these results, we selected the K526D mutant for further characterization, and we named it rCas9HF.
Figure 2.

Identification and characterization of an optimized high-fidelity SpCas9 variant
(A) On-/off-target ratios obtained with fully matching sgRNA (on target) over two surrogate off targets (sgGFP13-14 and sgGFP18-19 with double mismatches in position 13-14 or 18-19 of the spacer, respectively) with K526 variants with the indicated amino acid substitutions. The editing was measured following transient plasmid transfection in HEK293-GFP reporter cells. The ratios are obtained using raw data from Figure S1. Statistical significance was assessed using one-way ANOVA, comparing each mutant with WT SpCas9 separately for on/off 13-14 and on/off 18-19. (B) Editing activities (percentage of indels) of WT SpCas9, HiFi Cas9, or rCas9HF RNPs were measured through tracking indels by TIDE analysis (n = 2 for K526D activity on HPRT locus). (C and D) Targeted deep sequencing analysis of the on targets (C) and previously validated off targets (D) after electroporation of WT SpCas9, HiFi Cas9, or rCas9HF RNPs in U2OS cells. The off-/on-target ratios calculated from the data in (C) and (D) are reported in Figure S2A. Statistical significance was assessed using paired t test to compare HiFiCas9 and rCas9HF; data reported as mean ± SEM for n ≥ 3 biologically independent replicates. (E) Percentage distribution of GUIDE-seq reads among the on-target and all off-target sites after electroporation of U2OS cells with the WT SpCas9, rCas9HF, and HiFi Cas9 RNPs; GUIDE-seq details are in Figure S3 and Tables S3–S11.
To investigate the editing performance of the rCas9HF RNP, we measured the indels produced in fifteen endogenous genomic loci. As shown in Figure 2B, rCas9HF had overall similar cleavage efficiency compared with the HiFi Cas9 mutant, thus suggesting that the glutamate-to-aspartate substitution at position K526 re-established near-WT activity. For a thorough comparative analysis among RNP delivery of WT SpCas9, rCas9HF, and HiFi Cas9, we selected the on-target sites from Figure 2B known to be associated with specific off-target sites7,14,20,21 and analyzed the on- and off-target activity by amplicon sequencing through next-generation sequencing (NGS) (Figures 2C and 2D). The analysis showed that WT SpCas9 is in general more active while producing higher off-target cleavages (Figures 2C and 2D). All the Cas9s had a similar editing efficacy in two sites (EMX1 and FANCF), while for most of the remaining sites (3 out of 5), HiFi Cas9 was more efficient than rCas9HF, except for CCR5, where our variant performed considerably better than HiFi Cas9 (Figures 2C and 2D). Nonetheless, the off-target analysis showed that rCas9HF produced less non-specific cleavages (Figures 2C and 2D). For a more direct interpretation of non-specific editing, we graphed the off/on ratios (Figure S2A). Overall, the results showed variable editing between the two high-fidelity variants, even though the off/on target profile is more favorable with the rCas9HF variant, given its higher specificity (Figures 2C, 2D, and S2A).
To further characterize the off-target events, we performed a genome-wide off-target analysis (GUIDE-seq method)22 following RNP delivery in three genomic loci (FANCF, VEGFAsite2, and VEGFAsite3) that are known to be associated with strong unspecific cleavages.14 Both the rCas9HF and the HiFi Cas9 variants generated significantly less off-target sites than the WT nuclease as absolute numbers (Figures S3A and S3B) as well as in percentages of on and off reads (Figure 2E). Furthermore, the comparison of the off-target sites captured by GUIDE-seq revealed that in addition to common sites, the engineered and WT variants showed either unique or differently shared off-target sites (Figure S4). Therefore, these results further indicate a more precise editing activity of both high-fidelity variants (slightly better for rCas9HF; Figures 2E, S3A, and S3B) and an off-target profile that is only partially overlapping among the diverse nucleases (Figure S4).
rCas9HF editing in CD34+ primary hematopoietic stem cells
Gene editing in CD34+ hematopoietic stem cells is of particular interest for the development of ex vivo gene therapies, with very encouraging recent results from the experimental clinic.1 We thus evaluated the activity and specificity of rCas9HF, HiFi Cas9, and WT SpCas9 on selected loci with therapeutic relevance in CD34+ stem cells (CCR5, HBB, HBG) through targeted deep sequencing analysis to increase sensitivity. After RNP electroporation in CD34+ cells, the editing produced by WT SpCas9 and the high-fidelity variants was measured at the on-target as well at the associated off-target (OT) sites.7,14,23 The three nucleases showed similar on-target efficiency (Figure 3A), while the OT cleavages were overall less frequent with the high-fidelity variants (Figure 3B). Additionally, the off/on values calculated from these editing data clearly showed the diverse editing and specificity of the rCas9HF and HiFi Cas9 variants, which might be relevant at targeted therapeutic sites, soliciting a careful evaluation of the SpCas9 variant to use for each specific locus (Figure S2B).
Figure 3.

Editing of clinically relevant genomic loci by RNP delivery of high-fidelity SpCas9 variants in CD34+ cells
Primary CD34+ cells were electroporated with WT SpCas9, HiFi Cas9, or K562D RNPs targeting the CCR5, HBG, and HBB loci. Targeted deep sequencing analyses were performed 6 days post-transfection at the on-target sites (A) or at reported identified off targets (B). The off-/on-target ratios for WT SpCas9 and the two SpCas9 high-fidelity variants (HiFi Cas9 and rCas9HF) calculated from (A) and (B) are reported in Figure S2B. Statistical significance was assessed using paired t test; data reported as mean ± SEM for n = 3 biologically independent replicates.
Improvement of editing through homology-directed repair by high-fidelity variants
High-fidelity variants are more efficient than WT SpCas9 in sequence substitution protocols through homology-directed repair (HDR) using donor templates. In fact, compared with WT SpCas9, high-fidelity variants offer the advantage of minimizing the re-editing of the repaired locus as a consequence of their lower tolerance to mismatches between the sgRNA and the integrated donor template carrying silenced mutations.7,14,23,24,25 We thus compared the performance of our best high-fidelity mutant, rCas9HF, with HiFi Cas9 and WT SpCas9 toward three benchmark genomic loci: EMX1, HBB, and BCR. In all tested loci, single-strand oligonucleotides (ssODNs) were used as donor DNAs containing a mismatched nucleotide within the range of the sgRNA target sequence (Table S1). Overall, the editing efficacy was similar among WT SpCas9 and the high fidelity variants, except for a lower editing by rCas9HF in one (the HBB site) out of three sites (Figure 4A). Nonetheless, the comparative analysis of non-homologous end joining (NHEJ)-generated indels compared with precise HDR showed that, in all cases, our variant produced more precise repairs (Figures 4A and 4B). This result is consistent with former reports demonstrating that increased precision correlate with higher HDR products.20
Figure 4.

Precise gene correction by HDR using high-fidelity RNPs
(A) U2OS cells were electroporated with WT SpCas9, HiFi Cas9, or rCas9HF RNPs targeting the EMX1, BCR, and HBB loci together with specific ssODN templates (Table S1). Indel formation (NHEJ) and HDR events were quantified at 3 days post-transfection. (B) HDR/indel ratios calculated for the EMX1, BCR, and HBB loci are from (A). (C) Quantification of HDR-mediated correction in the CFTR locus targeting two clinically relevant mutations causing cystic fibrosis. HEK293 cells stably expressing mutated CFTR minigenes were electroporated with WT SpCas9, HiFi Cas9, or rCas9HF RNP complexes and an ssODN donor. Indel frequencies and HDR levels were analyzed 3 days post-electroporation. (D) HDR/indel ratios calculated for the two CFTR loci are from (C). Data reported as mean ± SEM for n ≥ 2 biologically independent replicates; statistical significance was assessed using one-way ANOVA. Bars in (A) and (C) are superimposed.
Precise repair through HDR was measured in a therapeutically relevant locus, CFTR, mutations of which cause cystic fibrosis and which has been widely tested for the development of therapeutic genome editing approaches.26 In particular, we focused on two mutations: (1) the most common cystic fibrosis alteration consisting in a small deletion (CFTR-ΔF508) and (2) the CFTR 1717-1 G>A point mutation in intron 11 resulting in splicing defects.27 Cells treated with rCas9HF showed less indels than those produced by both WT SpCas9 and the HiFi Cas9 (Figure 4C), which correlated with higher HDR compared with WT SpCas9 or HiFi Cas9 (Figures 4C and 4D). As above (Figures 4A and 4B), increased editing precision through HDR is accompanied by lower NHEJ editing products generated by both high-fidelity variants (Figures 4A–4D).
Discussion
Two fundamental parameters in genome editing are efficiency and precision. CRISPR-Cas9 precision was highly improved through the generation of high-fidelity variants17 and by reducing intracellular levels of Cas9.20 One of the most successful methods for transient CRISPR-Cas9 expression is RNP electroporation, which results in the quickest degradation kinetics of the enzymatic complex in target cells (24–48 h persistence)8,9 compared with mRNA (48–72 h persistence) or plasmid delivery (>72 h persistence). Nevertheless, RNP delivery is hardly compatible with high-fidelity variants,13 resulting in low editing efficiency as also confirmed by our results (Figure 1). Very likely, the slow cleavage kinetics of the high-fidelity nucleases require longer target residency and higher enzymatic concentrations compared with the original WT SpCas9.8 In fact, so far, exclusively one high-fidelity mutant, the HiFi Cas9, has been identified to efficiently edit the genome through RNP electroporation and is thus unique for ex vivo editing.13,23
Nonetheless, the activity and precision of CRISPR-Cas nucleases throughout the genome is not homogeneous14 (as also demonstrated in this study; Figures 1C and 2B–2E), most likely due to different levels of chromatic compaction across different genomic loci.28,29,30 For this reason, an expansion of the editing toolbox is clearly needed to efficiently address all editing requirements. This can be obtained either by engineering CRISPR enzymes with improved properties, such as rCas9HF, or through the discovery of completely undescribed nucleases.31 Beyond editing through indels generated by NHEJ, another important use of high-fidelity variants through RNP delivery is gene substitution through HDR using DNA donor templates.4 The development of a high-fidelity SpCas9 nuclease working through RNP delivery, the rCas9HF variant, increases the opportunity for efficient and precise editing on a wider range of targeted loci and for more efficient HDR protocols.
The identification of the rCas9HF in this study was obtained by introducing a single point mutation in the WT nuclease. The amino acid interested by the substitution was originally identified during the development of evoCas9, which contained four mutations (M495V, Y515N, K526E, and R661Q/S) localized in the REC3 domain at protein sites where SpCas9 contacts the RNA:DNA duplex.14,32 The experimental work, which led to the identification of evoCas9, revealed that, similarly to other high-fidelity variants, the activity profile of the nuclease results from a trade-off between efficiency and specificity, with the highest precision obtained at the expense of nuclease activity.17,18
The best compromise to balance precision and efficiency for RNP delivery was obtained by installing a single mutation at position K526, which we found best works with an aspartate in place of the original glutamate present in evoCas9. Based on these results, we hypothesize that the increased activity observed with K526D, compared with K526E, stems from the aspartate’s shorter side chain: while both substitutions lead to a charge inversion, K526D is better accommodated within the context of neighboring residues, thus preserving the catalytic activity (see in silico analysis in Figure S5). Further structural studies are needed to strengthen the role of the K526E residue as a potential groundwork to further develop high-fidelity variants including other Cas9 orthologs for RNP delivery.
Finally, the immunogenicity of SpCas9 raises concern for its wider use in the clinic, as both humoral and cellular immune responses have been reported in the population.33 By cross checking the reported SpCas9 immunodominant epitopes34 with the amino acid region where the K526D substitution is located, we found no overlap, indicating that the mutation should not be able to modulate the immunological properties of the nuclease.
Here, we generated rCas9HF, a high-fidelity variant suitable for RNP delivery that is an alternative to the so-far unique HiFi Cas9. Despite the similar editing profile of the two high-fidelity nucleases, the in-depth analysis in primary CD34+ cells demonstrates that in particular for ex vivo clinical applications, the two variants can represent alternative tools to optimize editing approaches in terms of activity and precision. This represents a step forward in enriching the genome editing toolbox also by providing further insights on the generation of Cas9 mutants with enhanced targeting precision.
Materials and methods
Plasmids
A derivative of pX330 (Addgene #42230) where the sgRNA cassette has been removed by NdeI digestion was used to express SpCas9 in mammalian cells (pX-Cas9). pX-Cas9 plasmids encoding the different mutants were obtained by site-directed mutagenesis of the WT pX-Cas9 plasmid using the ODNs reported in Table S2. sgRNAs were expressed from a pUC19 plasmid containing a U6-driven sgRNA expression cassette. Desired spacer sequences were cloned as annealed ODNs (Table S1) into a double BbsI site immediately upstream of the sgRNA scaffold according to previously published cloning strategies.35
To express SpCas9 in bacterial cells for recombinant protein production, a modified version of the pET-28b-Cas9-His (Addgene #47327) was used. Briefly, an additional SV40 nuclear localization sequence (NLS) was added both at the N terminus and C terminus of the protein using standard cloning techniques, bringing the total number of NLSs to three (1 at the N terminus and 2 at the C terminus), generating pET-28b-NLS-Cas9-2xNLS-His. Corresponding plasmids to purify the different SpCas9 mutants were generated by site-directed mutagenesis using the ODNs reported in Table S2.
WT and 1717-1G>A-mutated CFTR minigenes were cloned into pcDNA3 (Invitrogen). The WT pMG1717-1WT minigene was obtained by PCR amplification and cloning of target regions from CFTR gene from HEK293T cells using the ODNs listed in Table S2. The pMG1717-1WT plasmid contains full exons 10, 11, 12, and 13, portions of intron 11, and full intron 12. The mutated pMG1717-1G>A minigene was obtained by site-directed mutagenesis of the WT minigene construct using the ODNs listed in Table S2.
Cells
U2OS cells were obtained from ATCC (HTB-96). HEK293 cells stably expressing multiple copies of EGFP (293multiEGFP cells) have been previously described.14 HEK293/CFTR-ΔF508 were obtained by transduction of a lentiviral vector expressing CFTR-ΔF50836 (a kind gift of Marianne Carlon, KU Leuven). HEK293/1717-1G>A stably expressing pMG1717-1G>A were produced by stable transfection of the BglII-linearized minigene plasmid in HEK293 cells. Cells were cultured in DMEM (Gibco) supplemented with fetal bovine serum (FBS; 10%, Gibco), glutamine (Gibco), and penicillin/streptomycin (Gibco) and maintained at 37°C in 5% CO2 humidified atmosphere. 293multiEGFP and HEK293CFTR-ΔF508 culture medium was additionally supplemented with 1 μg/mL puromycin. 500 μg/mL G418 (Thermo Fisher Scientific) was added to HEK293/1717-1G>A culture medium. Selection was removed during transfection experiments.
We obtained human peripheral blood CD34+ cells from patients with sickle cell disease. Samples eligible for research purposes were obtained from the “Hôpital Necker-Enfants malades” Hospital (Paris, France). Written informed consent was obtained from all adult subjects. All experiments were performed in accordance with the Declaration of Helsinki. The study was approved by the regional investigational review board (reference: DC 2014-2272, CPP Ile-de-France II “Hôpital Necker-Enfants malades”). CD34+ cells were purified by immunomagnetic selection with AutoMACS (Miltenyi Biotec) after immunostaining with the CD34 MicroBead Kit (Miltenyi Biotec). 48 h before transfection, CD34+ cells (5 × 105 cells/mL) were thawed and cultured at 37°C in 5% CO2 humidified atmosphere in a medium containing StemSpan (STEMCELL Technologies) supplemented with penicillin/streptomycin (Gibco), 250 nM StemRegenin1 (STEMCELL Technologies), and the following recombinant human cytokines (PeproTech): human stem cell factor (SCF) (300 ng/mL), Flt-3L (300 ng/mL), thrombopoietin (TPO) (100 ng/mL), and interleukin-3 (IL-3) (60 ng/mL).
Transfection of 293multiEGFP cells
RNP lipofection was performed 24 hours after seeding 1.5 × 105 293multiEGFP cells in a 24-well plate. Guide RNAs were obtained from IDT as separate chemically stabilized CRISPR RNAs (crRNAs) and trans-activating crRNA (tracrRNA), while WT SpCas9 and its mutants were purified as described (protein purification). RNPs were obtained assembling 6 pmol of previously annealed crRNA:tracrRNA and 3 pmol of SpCas9 protein of interest, then cells were lipofected using TransIT-X2 (Mirus Bio), according to the manufacturer’s protocol.
Plasmid transfections were performed by seeding 105 293multiEGFP cells in a 24-well plate the day before transfection. 400 ng of each pX-Cas9 plasmid (WT and mutants) together with 200 ng pUC19-sgRNA plasmids were transfected using the TransIT-LT1 reagent (Mirus Bio) according to the manufacturer’s protocol. Cells were collected 7 days post-transfection and analyzed by flow cytometry using FACSCanto (BD Biosciences) to evaluate EGFP knockout.
Protein purification
Recombinant nucleases were purified according to a previously published protocol with minor modifications.37 Briefly, each pET-28b-NLS-Cas9-2xNLS-His was transformed in Rosetta (DE3) pLysS E. coli competent cells (Novagen), and single colonies were inoculated and grown overnight, shaking at 37°C to obtain starter cultures, which were then diluted 1:100 in 1–2 L LB medium. Mass cultures were grown at 37°C until OD600 reached 0.6, they were then transferred at 18°C, and after 30 min, IPTG (final concentration 400 mM) was added to the medium to induce SpCas9 expression. Cultures were left shaking at 18°C overnight. Bacterial pellets were then re-suspended in lysis buffer (20 mM Tris [pH 8], 500 mM NaCl, and 5 mM imidazole) supplemented with lysozyme (0.5 mg/mL), incubated for 20 min at 4°C while shaking and lysed by sonication. After clarification by centrifugation (30,000 × g, 20 min, 4°C), the supernatants were mixed with NiNTA agarose beads (Protino, Macherey-Nagel) for initial protein isolation. According to protocol, two further steps of purification were performed: ion-exchange (IEX) chromatography (HiTrap SP FF column, GE Healthcare) followed by size-exclusion chromatography (HiLoad 16/600 Superdex 200 PG, GE Healthcare) using an AKTA Pure 25 FPLC system (GE Healthcare). After the final elution, fractions containing SpCas9 were pooled, concentrated using a centrifugal concentrator (10,000 MWCO) to reach at least 10 mg/mL, and stored in aliquots at −80°C until use.
Electroporation of U2OS cells
RNP electroporation experiments in U2OS and HEK293 minigene-expressing cells were performed using the Lonza 4D-Nucleofector (Lonza). RNP complexes were assembled in a final volume of 5 μL using 120 pmol previously annealed crRNA-tracrRNA (IDT) duplex and 100 pmol purified SpCas9. Complexes were mixed with 2 × 105 U2OS cells re-suspended in 20 μL SE buffer and electroporated using the CM-104 program. Genomic DNA was extracted 48 h post-electroporation to assess editing efficiency.
For RNP and ssODN delivery in HDR experiments, 120 pmol Cas9 protein and 150 pmol assembled crRNA-tracrRNA (IDT) were incubated at room temperature for 10–20 min, mixed with 120 pmol ssODN (IDT) and 120 pmol Alt-R Ca9 Electroporation Enhancer (IDT), and electroporated in a total of 2 × 105 U2OS or HEK293 stably expressing mutated CFTR minigenes in SE buffer (programs CM-104 and CM-130, respectively). ssODN donor sequences are reported in Table S1.
Electroporation of CD34+ cells
sgRNA containing both crRNA and tracrRNA sequences was obtained from Synthego (Table S1). RNP complexes were assembled in a final volume of 2.3–2.8 μL using 180 pmol sgRNA and 90 pmol purified SpCas9. CD34+ cells (2 × 105 cells/condition) were transfected in the presence of 180 pmol Alt-R Cas9 Electroporation Enhancer (IDT). We used the Lonza 4D-Nucleofector (Lonza), the P3 Primary Cell 4D-Nucleofector X Kit S (Lonza), and the CA137 program. After transfection, cells were kept in the same medium for 6 days. Genomic DNA was extracted 6 days post-electroporation, and the editing was assessed by deep-sequencing (Table S1).
Evaluation of genome editing by Sanger sequencing
Genomic DNA was extracted from cell pellets using the QuickExtract DNA extraction solution (Lucigen) according to the manufacturer’s instructions. Edited regions were amplified using ODNs listed in Table S1. Indels and HDR events were evaluated by deconvolution of chromatographic traces using the tracking indels by deconvolution (TIDE) software (http://shinyapps.datacurators.nl/tide/)38 or the Synthego ICE analysis tool (v.2) after Sanger sequencing the purified PCR products (EasyRun Service, Microsynth). Untreated cells were used as negative controls for calculating background modification frequencies.
Targeted deep-sequencing
Selected OT sites for CCR5, EMX1, FANCF, HBB, HBG, HEKsite4, and ZSCAN2 genomic loci, together with their relative on target, were amplified using the HOT FIREPol DNA Polymerase (Solis BioDyne) from genomic DNA of U2OS and CD34+ cells extracted 48 h after electroporation using WT SpCas9, HiFi Cas9, or rCas9HF. Amplicons were indexed by PCR using Nextera indexes (Illumina) and quantified with the Qubit dsDNA High Sensitivity Assay kit (Invitrogen). OT and on-target amplicons were pooled at a 10:1 M ratio and sequenced on an Illumina Miseq system using an Illumina Miseq Reagent kit v.3 for 300 cycles (2 × 150 bp paired end). The complete primer list used to generate the amplicons is reported in Table S1. Deep sequencing reads were analyzed using CRISPResso2 (v.2.0.45).39
OT evaluation
GUIDE-seq experiments were performed as previously described.22 Briefly, 2 × 105 U2OS cells were electroporated with the Lonza 4D-Nucleofector (Lonza) DN-100 program using the Cas9-gRNA RNP and adding 50 pmol dsODNs (crRNAs in Table S1); cells treated using WT SpCas9 and double-strand ODNs (dsODNs) were used as negative control. 3 days after transfection, cells were collected, and genomic DNA was extracted using NucleoSpin Tissue Kit (Macherey-Nagel) following the manufacturer’s instructions. Using the Focused Ultrasonicator (Covaris), genomic DNA was sheared to an average length of 500 bp. End-repair reaction was performed using NEBNext Ultra End Repair/dA Tailing Module and adaptor ligation using NEBNext Ultra Ligation Module as described by Nobles et al.40 Amplification steps were then performed following the GUIDE-seq original protocol.22 Following quantification of the libraries by Qubit dsDNA High Sensitivity Assay kit (Invitrogen), the MiSeq sequencing system (Illumina) was used with an Illumina Miseq Reagent kit (150 cycles, paired-end reads). Sequencing data were analyzed using the GUIDE-seq package (v.1.0.2).22 Visualization of aligned OT sites is available as a color-coded sequence grid (Figure S3B). GUIDE-seq data are listed in Tables S3–S11.
Statistical analysis
All statistics were calculated using GraphPad Prism. Ordinary one-way analysis of variance (ANOVA) was used in Figures 1, 2, and 4 as described in the captions. HiFi Cas9 and rCas9HF were compared using paired t test. Statistical significance was defined as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns, non-significant.
Acknowledgments
We thank lab members of Cereseto lab for helpful discussions. This work was supported by the European Union’s Horizon 2020 innovation program through the UPGRADE (Unlocking Precision Gene Therapy) project (grant agreement no. 825825), by the Horizon Europe EIC Pathfinder program under grant agreement no. 101071041 (AAVolution), and by the Italian Foundation for Cystic Fibrosis (FFC#3-2019). The authors are grateful to Gino Del Bon for supporting E.P. and A.U.’s fellowship through “progetto Sofia.”
Author contributions
A. Casini and A. Cereseto designed the research. E.P. and A.B. performed the protein engineering and genome editing analyses. D.A. analyzed the protein structure. G.F., M.B., and A.M. analyzed the RNP editing in primary cells from sickle cell disease patients. K.A.B. performed genome editing experiments. A.U. and S.A. performed HDR experiments. M.C. performed all the bioinformatic analyses. E.P., A.U., and S.A. performed GUIDE-seq experiments.
Declaration of interests
A. Casini and A. Cereseto are founders of and hold shares in Alia Therapeutics, a genome editing company. A. Casini is an employee of Alia Therapeutics, and A. Cereseto is a consultant for Alia Therapeutics. A patent application covering the technology disclosed in this manuscript has been filed, and A. Casini and A. Cereseto are listed as inventors.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.03.007.
Contributor Information
Anna Cereseto, Email: anna.cereseto@unitn.it.
Antonio Casini, Email: antonio.casini@aliatx.com.
Supplemental information
Data and materials availability
GUIDE-seq and targeted deep sequencing data have been deposited at BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under the accession number BioProject: PRJNA939956. All other relevant data are available from the authors upon request.
References
- 1.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 2.Hirakawa M.P., Krishnakumar R., Timlin J.A., Carney J.P., Butler K.S. Gene editing and CRISPR in the clinic: current and future perspectives. Biosci. Rep. 2020;40 doi: 10.1042/BSR20200127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghetti S., Burigotto M., Mattivi A., Magnani G., Casini A., Bianchi A., Cereseto A., Fava L.L. CRISPR/Cas9 ribonucleoprotein-mediated knockin generation in hTERT-RPE1 cells. STAR Protoc. 2021;2:100407. doi: 10.1016/j.xpro.2021.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavani G., Fabiano A., Laurent M., Amor F., Cantelli E., Chalumeau A., Maule G., Tachtsidi A., Concordet J.-P., Cereseto A., et al. Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in human hematopoietic stem cells. Blood Adv. 2021;5:1137–1153. doi: 10.1182/bloodadvances.2020001996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frati G., Miccio A. Genome editing for β-hemoglobinopathies: advances and challenges. J. Clin. Med. 2021;10:482. doi: 10.3390/jcm10030482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A., et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6:eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S., Kim D., Cho S.W., Kim J., Kim J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 10.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casini A., Olivieri M., Petris G., Montagna C., Reginato G., Maule G., Lorenzin F., Prandi D., Romanel A., Demichelis F., et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018;36:265–271. doi: 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu R., Liang L., Freed E.F., Gill R.T. Directed evolution of CRISPR/Cas systems for precise gene editing. Trends Biotechnol. 2021;39:262–273. doi: 10.1016/j.tibtech.2020.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Lee J.K., Jeong E., Lee J., Jung M., Shin E., Kim Y.-H., Lee K., Jung I., Kim D., Kim S., Kim J.S. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018;9:3048. doi: 10.1038/s41467-018-05477-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid-Burgk J.L., Gao L., Li D., Gardner Z., Strecker J., Lash B., Zhang F. Highly parallel profiling of Cas9 variant specificity. Mol. Cell. 2020;78:794–800.e8. doi: 10.1016/j.molcel.2020.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim N., Kim H.K., Lee S., Seo J.H., Choi J.W., Park J., Min S., Yoon S., Cho S.-R., Kim H.H. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020;38:1328–1336. doi: 10.1038/s41587-020-0537-9. [DOI] [PubMed] [Google Scholar]
- 19.Lino C.A., Harper J.C., Carney J.P., Timlin J.A. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018;25:1234–1257. doi: 10.1080/10717544.2018.1474964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petris G., Casini A., Montagna C., Lorenzin F., Prandi D., Romanel A., Zasso J., Conti L., Demichelis F., Cereseto A. Hit and go CAS9 delivered through a lentiviral based self-limiting circuit. Nat. Commun. 2017;8:15334–15339. doi: 10.1038/ncomms15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M., et al. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell. 2019;24:821–828.e5. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Idoko-Akoh A., Taylor L., Sang H.M., McGrew M.J. High fidelity CRISPR/Cas9 increases precise monoallelic and biallelic editing events in primordial germ cells. Sci. Rep. 2018;8:15126. doi: 10.1038/s41598-018-33244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato-Inui T., Takahashi G., Hsu S., Miyaoka Y. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 with improved proof-reading enhances homology-directed repair. Nucleic Acids Res. 2018;46:4677–4688. doi: 10.1093/nar/gky264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maule G., Arosio D., Cereseto A. Gene therapy for cystic fibrosis: progress and challenges of genome editing. Int. J. Mol. Sci. 2020;21:3903. doi: 10.3390/ijms21113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welcome to CFTR2 | CFTR2. https://www.cftr2.org/
- 28.Uusi-Mäkelä M.I.E., Barker H.R., Bäuerlein C.A., Häkkinen T., Nykter M., Rämet M. Chromatin accessibility is associated with CRISPR-Cas9 efficiency in the zebrafish (Danio rerio) PLoS One. 2018;13:e0196238. doi: 10.1371/journal.pone.0196238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yarrington R.M., Verma S., Schwartz S., Trautman J.K., Carroll D. Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. USA. 2018;115:9351–9358. doi: 10.1073/pnas.1810062115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung C.-H., Allen A.G., Sullivan N.T., Atkins A., Nonnemacher M.R., Wigdahl B., Dampier W. Computational analysis concerning the impact of DNA accessibility on CRISPR-Cas9 cleavage efficiency. Mol. Ther. 2020;28:19–28. doi: 10.1016/j.ymthe.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciciani M., Demozzi M., Pedrazzoli E., Visentin E., Pezzè L., Signorini L.F., Blanco-Miguez A., Zolfo M., Asnicar F., Casini A., et al. Automated identification of sequence-tailored Cas9 proteins using massive metagenomic data. Nat. Commun. 2022;13:6474. doi: 10.1038/s41467-022-34213-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crudele J.M., Chamberlain J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018;9:3497. doi: 10.1038/s41467-018-05843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chew W.L., Tabebordbar M., Cheng J.K.W., Mali P., Wu E.Y., Ng A.H.M., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ensinck M., De Keersmaecker L., Heylen L., Ramalho A.S., Gijsbers R., Farré R., De Boeck K., Christ F., Debyser Z., Carlon M.S. Phenotyping of rare CFTR mutations reveals distinct trafficking and functional defects. Cells. 2020;9:754. doi: 10.3390/cells9030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anders C., Jinek M. In vitro enzymology of Cas9. Methods Enzymol. 2014;546:1–20. doi: 10.1016/B978-0-12-801185-0.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nobles C.L., Reddy S., Salas-McKee J., Liu X., June C.H., Melenhorst J.J., Davis M.M., Zhao Y., Bushman F.D. iGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol. 2019;20:14. doi: 10.1186/s13059-019-1625-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GUIDE-seq and targeted deep sequencing data have been deposited at BioProject (https://www.ncbi.nlm.nih.gov/bioproject/) under the accession number BioProject: PRJNA939956. All other relevant data are available from the authors upon request.