

Abstract
Gene therapy with an adeno-associated virus serotype 8 (AAV8) vector (AAV8-LSPhGAA) could eliminate the need for enzyme replacement therapy (ERT) by creating a liver depot for acid α-glucosidase (GAA) production. We report initial safety and bioactivity of the first dose (1.6 × 1012 vector genomes/kg) cohort (n = 3) in a 52-week open-label, single-dose, dose-escalation study (NCT03533673) in patients with late-onset Pompe disease (LOPD). Subjects discontinued biweekly ERT after week 26 based on the detection of elevated serum GAA activity and the absence of clinically significant declines per protocol. Prednisone (60 mg/day) was administered as immunoprophylaxis through week 4, followed by an 11-week taper. All subjects demonstrated sustained serum GAA activities from 101% to 235% of baseline trough activity 2 weeks following the preceding ERT dose. There were no treatment-related serious adverse events. No subject had anti-capsid T cell responses that decreased transgene expression. Muscle biopsy at week 24 revealed unchanged muscle glycogen content in two of three subjects. At week 52, muscle GAA activity for the cohort was significantly increased (p < 0.05). Overall, these initial data support the safety and bioactivity of AAV8-LSPhGAA, the safety of withdrawing ERT, successful immunoprophylaxis, and justify continued clinical development of AAV8-LSPhGAA therapy in Pompe disease.
Keywords: adeno-associated virus, clinical trial, gene therapy, liver-specific, acid α-glucosidase, acid maltase, glycogen storage disease type II
Graphical abstract
Koeberl and colleagues report that gene therapy with an AAV8 vector could eliminate the need for enzyme replacement therapy in Pompe disease. We report a clinical trial of AAV8 gene therapy in patients with late-onset Pompe disease, which increased GAA activity for 52 weeks. This study suggests that gene therapy can be safe and effective.
Introduction
Pompe disease, with an incidence of approximately one in every 16,000 births,1 is caused by mutations in the GAA gene coding for the enzyme acid α-glucosidase (GAA) that leads to lysosomal glycogen accumulation in many organs, especially striated and smooth muscles, and is often fatal.2,3,4 A spectrum of severity has been defined based upon the amount residual GAA activity present, ranging from severe infantile-onset Pompe disease (IOPD) due to complete GAA deficiency that causes cardiomyopathy, to less severe late-onset Pompe disease (LOPD) that causes primarily skeletal and respiratory muscle weakness. Treatment of patients diagnosed with IOPD, in the form of enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA), has reversed cardiomyopathy, improved muscle function, and increased life expectancy. Although ERT has prolonged survival in the majority of patients with IOPD, long-term sequelae have been reported including persistent skeletal muscle weakness, ptosis, gait difficulties and in some instances the need for ventilator support.5,6 Many of these poor responders formed high, sustained anti-rhGAA IgG antibody titers (HSAT) that occurred in up to one-third of patients.5,6 In contrast, only a few adult patients with LOPD have formed HSAT during ERT that reduced efficacy.7,8 Even in patients with a good response to ERT, however, residual motor weakness (neck flexor weakness, dorsiflexor weakness, myopathic facies, ptosis and strabismus) has been observed.9,10,11 Furthermore, the benefits of ERT gradually wane over time as documented in patients with LOPD who had decreased 6-min walk test (6MWT) distance and forced vital capacity (FVC) performance after several years of treatment.12
Multiple preclinical experiments have demonstrated the ability of liver-based gene therapy with wild-type human GAA to secrete GAA into the bloodstream.13,14,15,16 This liver-based gene therapy with unmodified, wild-type GAA is conceptually similar to ERT with rhGAA using receptor-mediated uptake into muscle; however, continuous production of GAA that is secreted from a liver depot offers potential advantages.17 Liver depot gene therapy induced immune tolerance to rhGAA, prevented HSAT formation during ERT in GAA-knock out (KO) mice, and decreased mortality from hypersensitivity associated with ERT. Furthermore, ERT was efficacious in GAA-KO mice only in the setting of immune tolerance to GAA following AAV vector administration.14 The vector, AAV8-LSPhGAA, contained a liver-specific regulatory cassette that induced immune tolerance by expressing GAA exclusively in the liver and by activating regulatory T cells, which has consistently induced immune tolerance to human GAA in preclinical experiments.13,14,15,16 Importantly, AAV8-LSPhGAA corrected GAA deficiency in the heart and skeletal muscle at a minimum effective dose feasible for early phase clinical trials, confirming its potential as a stand-alone therapy to replace ERT.18 A GLP toxicology and biodistribution study demonstrated safety for AAV8-LSPhGAA at a dose of 1.6 × 1013 vg/kg in GAA-KO mice,19 which was 80-fold higher than the minimum effective dose.18,20
While the obvious strategy to correct GAA deficiency in Pompe disease muscle would appear to be muscle-targeted gene therapy, liver-targeted gene therapy has been more efficient due in part to the high tropism of AAV vectors for the liver. As demonstrated in the GAA-KO mouse model, liver-targeted gene therapy has achieved the generalized correction of muscle using liver-specific expression and secretion from a liver depot for GAA production with lower vector doses. For example, muscle-targeted gene therapy in GAA-KO mice with an AAV vector containing a highly active muscle-specific regulatory cassette required >10-fold higher doses, in comparison with the liver depot strategy.13,21,22 Furthermore, the efficacy of muscle-based GAA expression was blunted by anti-GAA antibody responses.21
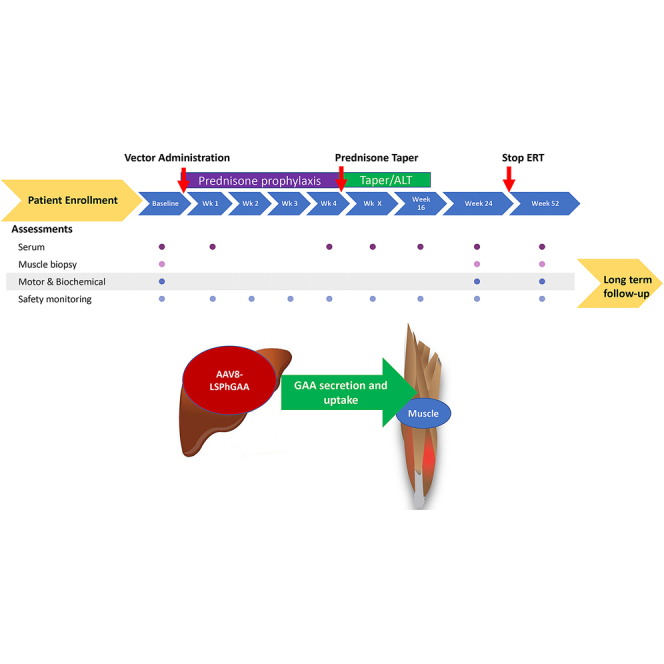
This is the initial report of the evaluation of safety and bioactivity for the first (low-dose) cohort (n = 3) in a 52-week, open-label, single-dose, dose-escalation study (NCT03533673) in patients with LOPD (Figure 1). The primary study objectives were to evaluate the safety as assessed by the incidence of adverse events (AEs), serious AEs (SAEs), and clinical laboratory abnormalities and bioactivity as assessed by serum GAA activity, and muscle GAA activity and glycogen content of AAV8-LSPhGAA in adult subjects. Secondary objectives included evaluating the effects of treatment on motor function by assessing 6MWT and on pulmonary function by assessing upright FVC.
Figure 1.

Clinical laboratory testing for safety monitoring
Study design including summaries of eligibility and monitoring with study visits illustrated by a timeline. Laboratory testing was performed at each study visit to monitor for liver or muscle toxicity, including serum ALT, AST, GGT, and CK.
Results
Three subjects with LOPD were screened and enrolled in cohort 1 of the study at a dose of 1.6 × 1012 vg/kg (Figure S1). Eligible subjects had LOPD, no detectable anti-AAV8 neutralizing antibodies, >12 months stable ERT dosing, and ability to walk at least 100 m on the 6MWT. Subjects continued biweekly ERT until ERT withdrawal at week 26 based on the detection of quantifiable serum GAA activity from AAV8-LSPhGAA and the absence of clinically significant declines in FVC or 6MWT performance. The detection of serum GAA activity above baseline was attributed to transgene expression following vector administration, given that serum GAA was assayed at a trough level relative to ongoing ERT. Prednisone (60 mg/day) was administered as immunoprophylaxis through week 4, followed by an 11-week taper (5 mg/week). These subjects were typical for the Duke University Metabolic Clinic population, previously diagnosed with LOPD and having been treated with ERT for more than 2 years (Table 1). All three subjects were male and had decreased FVC consistent with eligibility criteria. Initial safety of AAV8-LSPhGAA was assessed by the incidence of vector-related AEs and SAEs. Related AEs included headaches, myalgias, and elevated transaminases and were elicited at phone and in-person research visits (Table 2). SAEs were unrelated to vector, and included pulmonary embolism and osteomyelitis in subject 001 (Table 3).
Table 1.
Initial demographics and baseline characteristics
| Patient ID | Sex | Race/Ethni-city | Age diag-nosed | Age enrolled | Wt. (kg) | Allele 1 | Allele 2 | ERT (mo) | FVC (% predict-ted) | 6MWT (m) | Serum GAA (nmol/ml/h) | Serum CK (U/L) | Muscle symptoms | Walking aids | Ventila-tion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 001 | M | White/non-Hispanic | 42 | 52 | 99.1 | c.-32-13T>G | c.2608C>T (R870X) | 125 | 50 | 320.6 | 165.6 | 1733 | Low back pain | None | BiPAP |
| 002 | M | White/non-Hispanic | 61 | 69 | 71 | c.-32-13T>G | c.1951_1982 delGGinsT | 109 | 75 | 400.1 | 189.2 | 208 | Back pain | Walking stick | BiPAP |
| 003 | M | White/non-Hispanic | 62 | 71 | 94 | c.-32-13T>G | c.1143delC | 113 | 69 | 462.5 | 251.5 | 290 | Back pain, right shoulder weakness | None | BiPAP |
Table 2.
Adverse events related to study drug
| Patient ID | Adverse event term | Outcome | Severity | Expectedness | Relationship to study drug | CTCAE grade | Action taken | Serious? |
|---|---|---|---|---|---|---|---|---|
| 101002 | Headache | Resolved | Mild | Unexpected | Possibly | Grade 1 | Not applicable | No |
| 101002 | Headache | Resolved | Moderate | Unexpected | Possibly | Grade 2 | Not applicable | No |
| 101001 | Myalgia | Resolved | Moderate | Unexpected | Possibly | Grade 2 | Not applicable | No |
| 101001 | ALT | Resolved | Mild | Unexpected | Possibly | Grade 1 | Not applicable | No |
| 101001 | AST | Resolved | Mild | Unexpected | Possibly | Grade 1 | Not applicable | No |
| 101001 | GGT | Resolved | Mild | Unexpected | Possibly | Grade 1 | Not applicable | No |
Table 3.
Serious adverse events
| Patient ID | Adverse event term | Outcome | Severity | Expectedness | Relationship to study drug | CTCAE grade | Action taken | Serious? |
|---|---|---|---|---|---|---|---|---|
| 101001 | Dehydrationa | Resolved | Severe | Unexpected | Not related | Grade 3 | None | Yes |
| 101001 | Pulmonary embolism | Resolved | Moderate | Unexpected | Not related | Grade 2 | Not applicable | Yes |
| 101001 | Osteomyelitis | Resolved | Severe | Unexpected | Not related | Grade 3 | Not applicable | Yes |
Prior to vector administration.
The risk for T cell-mediated anti-capsid immune response was assessed initially by safety laboratory assessments, including serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), γ-glutamyltransferase (GGT), and creatine kinase (CK) at each study visit and at weekly home visits from week 4 to week 16 (Figure S2). Subject 001 had elevated concentrations of all four of these biomarkers starting at week 28 that coincided with the onset and treatment of osteomyelitis, whereas subjects 002 and 003 had normal concentrations of all four biomarkers. The elevations for 001 were attributed to osteomyelitis and related drug therapy, as well as muscle damage given elevated CK. An initial elevation of CK and AST for subject 001, attributed to muscle damage, correlated with an illness with dehydration prior to vector administration (Figure S2 and Table 3). To further clarify whether T cell responses were the cause for liver toxicity as reflected by elevated ALT and GGT, the anti-AAV8 γ-interferon ELISPOT was assessed (Figure 2). Anti-AAV8 ELISPOT signals were only slightly elevated, up to 90 spot forming units (SFU) at week 16 (elevated: >40 SFU per million PBMCs and at least 3x the DMSO control). Serum GAA fluctuated, but remained increased above baseline without any downward trend at week 52 for subject 001, indicating sustained transduction and stable transgene expression with AAV8-LSPhGAA and a lack of significant destruction of transduced hepatocytes (Figure 2A). Thus, elevated transaminases were deemed by the principal investigator to be unrelated to vector administration. Elevated capsid ELISPOT signals (40–120 SPF) were observed in subjects 002 and 003 after week 15 in absence of any elevations of ALT, indicating a lack of associated hepatotoxicity (Figures 2B and 2C).
Figure 2.

Comparison of serum ALT and GAA with γ-interferon ELISPOT assay for anti-AAV8 T cell responses
ELISPOTs were uniformly collected through week 38, and thereafter were not obtained for subject 003 due to inability to travel related to the COVID19 pandemic. Each graph summarizes data for one subject: 001 (A), 002 (B), and 003 (C). The left y axis shows the ELISPOT in spot forming units (SFU), whereas the right y axis shows the scale for GAA (μmol/min/mL) and ALT (units/mL). The x axis shows study days corresponding to the data shown.
Bioactivity of AAV8-LSPhGAA was evaluated by detection of serum GAA activity, which was determined at the pharmacokinetic trough 2 weeks following the preceding ERT infusion (Figure 3). All subjects demonstrated sustained serum GAA activities from 101% to 235% of baseline activity. At week 24, all subjects met criteria for ERT withdrawal, which included GAA activity above baseline and lack of clinically meaningful decreases in the 6MWT and FVC. ERT was subsequently stopped at week 26 per protocol. The serum GAA at week 52 remained above baseline levels 26 weeks following withdrawal of ERT, which confirmed transgene expression and secretion of the transgene protein, GAA. Immune responses against GAA were not detected by anti-rhGAA quantification, which revealed stable or decreased antibody formation following vector administration (Figure S2). GAA activity was significantly increased in muscle biopsies obtained at week 52, in comparison with baseline muscle biopsy analysis that confirmed bioactivity of gene therapy (Figure 3B). Glycogen content of muscle was not significantly elevated relative to baseline, although one of three subjects had elevated muscle glycogen at week 52 above the normal range following treatment for osteomyelitis (subject 001; Figure 3C). Histology of the muscle biopsies revealed increased vacuoles and glycogen staining at week 52 only for 001 (Table S1), consistent with the observed increase in glycogen content (Figure 3C). Urinary Glc4, a biomarker for muscle glycogen accumulation, remained stable at week 52 despite the increase observed for subject 001 (Figure 3D).
Figure 3.

Bioactivity evaluated by quantification of biochemical testing
Samples were obtained 2 weeks following an infusion of ERT, when GAA activity from ERT was at a trough. (A) Serum GAA activity. (B) Muscle GAA activity. (C) Muscle glycogen content. (D) Urine Glc4. ∗ = p < 0.05; ns = not significant as determined by a paired t test for the comparison indicated by connecting lines. # indicates when ERT was discontinued at week 26. The dotted line box indicates the normal range, although no normal range has been established for the experimental endpoint of serum GAA.
Motor and pulmonary function testing relevant to LOPD was assessed to detect changes following vector administration. The 6MWT and upright FVC performance were not significantly different at week 52 from baseline (Figures 4A and 4B). The FVC done in the supine position, which is decreased relative to upright FVC in LOPD, was also stable at week 52 (Figure 4C). Similarly, patient-reported scales were unchanged from baseline at week 52 (Figures 4D–4F). Subsequently, all subjects met protocol-specified criteria to remain off ERT at weeks 52 and 104, although one subject voluntarily opted to resume ERT at week 97.
Figure 4.

Muscle function and patient-reported outcomes
Muscle function testing and patient-reported outcomes were assessed. (A) 6MWT, (B) FVC, (C) FVC in the supine position, (D) Fatigue Severity Scale, (E) PROMIS Fatigue Scale, and (F) Rasch-Built Pompe Specific Activity Scale. Statistical significance analyzed by paired t test, ns = not significant. # indicates that ERT was discontinued at week 26.
Discussion
This Phase I clinical trial of liver depot gene therapy for Pompe disease is the first report of systemic gene therapy to treat Pompe disease. A previous study evaluated the direct injection of the diaphragm with an AAV2 vector to correct GAA deficiency locally.23 We have demonstrated initial safety and partial bioactivity for AAV8-LSPhGAA at a dose of 1.6 × 1012 vg/kg in adult patients with LOPD. The lack of glycogen lowering suggested that despite the presence of increased GAA activity in skeletal muscle, the efficacy of gene therapy was not sufficient to replace ERT at this vector dose. The absence of any of the following all supported safety: dose-limiting toxicity based on laboratory and clinical parameters, including the absence of treatment-related SAEs, together with the observation that all subjects met the protocol-specified definition of clinical stability. Function of the AAV8-LSPhGAA vector in vivo was supported by the presence of consistently elevated serum GAA levels relative to trough baseline activities. Importantly, bioactivity was confirmed through the observations of elevated serum and muscle GAA activity above baseline at week 52, following the cessation of ERT at week 26. Furthermore, motor and pulmonary function remained stable for greater than 70 weeks following the withdrawal of ERT.
The magnitude of the circulating peripheral concentrations of secreted GAA that correlated with increased deposition of GAA in muscle tissue surpassed initial expectations, based upon data from preclinical studies. Given that the expected transduction of the human liver is approximately 20-fold lower than for mouse liver,24 the current dose of 1.6 × 1012 vg/kg translates to a dose for mice of 8 × 1010 vg/kg. When GAA-KO mice were treated with 8 × 1010 vg/kg of AAV8-LSPhGAA, biochemical correction of heart and diaphragm was observed without any associated changes in skeletal muscles, including the quadriceps. It is well established that the heart and diaphragm are more responsive to ERT or gene therapy than the skeletal muscle.14,25 The increased GAA activity in the quadriceps seen in this trial demonstrated the ability of liver depot gene therapy to correct skeletal muscle, which is resistant to ERT due to low expression of the cation independent mannose-6-phosphate receptor that takes up GAA in skeletal muscle.26 However, the demonstrated increase in GAA activity did not achieve glycogen content reduction in the quadriceps muscle beyond the level previously achieved with ERT in two of three subjects and the third subject had increased muscle glycogen. This discrepancy emphasizes that higher GAA activity will likely be needed to suppress muscle glycogen content over the long term, given that one of three subjects had increased muscle glycogen above the normal range at week 52. Muscle glycogen content is variable depending on the quality and the exact location of the biopsy; however, we previously reported statistically significant differences in muscle glycogen content following an experimental therapy, indicating that biochemical quantification of muscle glycogen content can be a robust endpoint in LOPD.27 It is possible that higher vector dosages planned for later cohorts will achieve higher serum GAA and muscle GAA activities. Alternatively, adjunctive therapies have been developed to increase the expression of the mannose-6-phosphate receptor in skeletal muscle and to improve biochemical correction from low-dose gene therapy, which could ameliorate the need for high vector dosages and the accompanying risks for vector-related toxicity.28,29,30 A Phase I clinical trial with clenbuterol revealed that adjunctive therapy with ERT could further decrease muscle glycogen content by up to 50%.27
An earlier study of gene therapy with an AAV8 vector in hemophilia B demonstrated the risk of T cell responses to the AAV8 capsid proteins at a dose of 2 × 1012 vg/kg, which was 25% higher than the dose for the current study. Four of six participants who received the 2 × 1012 vg/kg dose in that study developed ALT elevations at weeks 7–10, with concordant γ-interferon ELISPOT responses directed toward capsid proteins. In most patients, the administration of prednisone at onset of ALT increases prevented loss of transgene expression, although human factor IX concentrations were generally lower after the observation of transient increases in ALT. Based on these data, we designed our study to include prophylaxis with prednisone starting just prior to vector administration and continued at a stable dose for 4 weeks, followed by an 11-week taper, similar to the strategy used in the pivotal trial for AAV9 vector-mediated gene therapy for spinal muscular atrophy.31 The dose for our study was 80% of that used by Nathwani and colleagues32 in hemophilia B, but only one of three subjects developed mildly elevated ALT and ELISPOT signals (001, Figure 2) that resolved without further immune suppression and without an associated decrease in serum GAA, indicating that these elevations were less likely to be related to vector administration. However, the possibility that anti-AAV8 T cell responses were present in this subject cannot be excluded. This experience emphasizes the need for careful monitoring of AEs, such as osteomyelitis that occurred in subject 001, in order to determine relatedness to the study interventions. Of note, as a consequence of muscle damage, ALT is often elevated in LOPD,33 which decreases its reliability as a biomarker for anti-AAV8 T cell responses following vector administration. Thus, the initial immune prophylaxis with prednisone might have helped to preserve transgene expression in the current study despite the presence of anti-capsid T cell responses. GAA expression did not provoke higher anti-rhGAA responses, suggesting that liver depot gene therapy will avoid anti-GAA immunity that could interfere with efficacy.9,10,11
The motivation for developing a liver depot gene therapy for Pompe disease is to address an unmet need for advanced treatment of this condition. The current study was guided by the design of an earlier study of AAV8-mediated gene therapy for hemophilia B, which achieved long-term benefits including savings from decreased need for treatment with coagulation factor replacement,32 which has an annual cost of ∼$300,000 for coagulation factor IX replacement.32,34 In Pompe disease, the benefits of gene therapy might include (1) improved quality of life and savings due to the high costs of ERT in Pompe that is administered at least every 2 weeks – estimated at >$200,000 per year for ERT in Pompe disease35; (2) potentially inducing immune tolerance to GAA in CRIM-negative patients and CRIM-positive patients who form HSAT and fail to respond to ERT36,37; and (3) potentially improving the ability to treat Pompe disease very early following early detection by newborn screening by initiating pre-symptomatic treatment to prevent the complications of Pompe disease entirely.38 Several of these potential benefits would require the development of this treatment for IOPD, which would require additional clinical trials in pediatric patients. However, even a gene therapy limited to LOPD patients would provide treatment for up to 90% of patients detected by newborn screening,1 if methods become available to circumvent the effects of NAb against AAV8 that occur in up to half of patients at older ages.39
Neurologic complications have been reported in both infantile-onset, and more recently LOPD in association with accumulation of glycogen noted in the central (CNS) and peripheral nervous systems (PNS).40,41,42,43,44,45,46 Similarly, glycogen content is elevated in the CNS and PNS of GAA-KO mice,19,47,48,49,50 and has been correlated with behavioral deficits detected by systematic behavioral testing.44,51 In the latter study, an AAV vector cross-packaged as AAV-PHP.B corrected the CNS phenotype, when administered early in life to GAA-KO mice. Another study with an AAV8 vector encoding modified “secretable” GAA significantly decreased accumulated glycogen in the CNS of GAA-KO mice at the dose of 2 × 1012 vg/kg.42 An earlier study with an AAV8 vector encoding human GAA modified only by substitution of a human α1-antitrypsin signal peptide demonstrated significant reduction of brain glycogen at a dose of 1 × 1012 vg/kg, indicating that the wild-type GAA enzyme could penetrate the CNS following cleavage of the modified signal peptide and secretion into the bloodstream.47 However, neither of the two studies with an AAV8 vector confirmed the correction of behavioral abnormalities.47,48 A formal pharmacology-toxicology study of the AAV8-LSPhGAA vector used in this clinical trial revealed significantly decreased glycogen content in the CNS at the dose of 2 × 1013 vg/kg, as well as a lack of toxicity.19 Overall, these data suggest that AAV8 vector-mediated expression of GAA in the liver can correct the CNS involvement of Pompe disease in GAA-KO mice at higher vector dosages,19,47,48 which would require even higher dosages to achieve the same benefits in human patients.24
The current report is of a Phase I clinical trial for the initial low-dose cohort. The study has several limitations, including the lack of blinding, the lack of assessments for neurological (CNS/PNS) pathology or response to vector, the small number of subjects enrolled, which is typical for early phase clinical trials of gene therapy, and the fact that the study was not powered adequately to detect efficacy. One subject experienced SAEs unrelated to the study drug, which emphasized the risks of Pompe disease and the potential for disease-related complications in the context of a clinical trial. The risks from Pompe disease potentially may complicate participation in a clinical trial, and must be weighed against potential benefits that cannot be expected from participating in an early phase clinical trial. However, the lack of clinical progression following the cessation of ERT suggests that gene therapy might replace ERT for the treatment of LOPD, and justifies further clinical development of liver depot gene therapy for Pompe disease.
Materials and methods
Study design
This was a 52-week Phase I open-label study of AAV8-LSPhGAA in patients with LOPD. The study vector was manufactured per standard protocols at the Nationwide Children’s Center for Gene Therapy. The Duke Investigational Drug Service stored and dispensed the study vector, which was administered in the Duke Early Phase Research Unit. AAV8-LSPhGAA contains a liver-specific promoter (LSP) consisting of a thyroid hormone binding globulin promoter and two copies of an α1-microglobulin/bikunin enhancer upstream of a wild-type GAA cDNA without optimization or CpG depletion, which is upstream of a human growth hormone polyadenylation signal.13,14,15,16 Study vector was administered (1.6 × 1012 vg/kg body weight, infused by controlled intravenous infusion at a rate of 60 mL/h) approximately 3 days following a dose of ERT, and ERT continued every 2 weeks through week 24. Thereafter, study visits occurred approximately 2 weeks after the last dose of ERT, when the GAA concentration from ERT was at a trough level. At week 26, subjects had ERT suspended based on the following data relative to their screening values: (1) quantifiable GAA catalytic activity (i.e., endogenously expressed GAA activity as a result of dosing AAV8-LSPhGAA) in their serum at weeks 12, 16, and 24; and (2) no clinically significant declines in two or more parameters on two consecutive occasions (i.e., week 8 and week 24) in 6MWT, GSGC, QMFT, and FVC from pulmonary function testing performed in the upright position. Criteria for re-initiation of ERT were as follows: (1) serum GAA equivalent to or less than screening/baseline, and (2) significant decline on 6MWT and FVC performance on two consecutive visits, which included unscheduled visits at least 2 weeks apart. Significant 6MWT decline was defined as >30 m decrease from baseline; significant FVC decline was defined as >3% predicted performance decline from baseline. This study was approved by the Duke University Institutional Review Board, and written consent was obtained at study entry.
Patient selection
Inclusion criteria included the following: (1) Diagnosis of Pompe disease by blood or skin fibroblast GAA assay and two pathogenic variants in the GAA gene. (2) Age: Greater than or equal to 18 years at enrollment. (3) Ability to provide written informed consent. (4) FVC within the range of 30% to less than 90% (inclusive) of predicted in the upright position. (5) Ability to walk at least 100 m on the 6MWT (with assistive devices permitted). (6) Receiving ERT for at least 104 weeks (inclusive) immediately preceding screening and receiving a stable dose of ERT for the 52-week period immediately preceding dosing. (7) Subjects must use at least one acceptable birth control method throughout the study and for 6 months after dosing with ACTUS-101. Exclusion criteria included the following: (1) ELISA positive for neutralizing anti-AAV8 capsid IgG antibodies (NAb) with a titer >1:5. (2) Invasive ventilation required or noninvasive ventilation required while awake and upright. (3) FVC <20% of predicted (supine). (4) Clinically relevant illness within 2 weeks of enrollment including fever >38.2°C, vomiting more than once in 24 h, seizure, or other symptom deemed contraindicative to new therapy. (5) Any condition that would interfere with participation in the study as determined by the principal investigator. (6) Received any live vaccination 4 months prior to study day 1. (Subjects will also be prohibited from receiving any vaccination within the 4-month period after study day 1). (7) Pregnant or nursing mothers. (8) Anti-rhGAA IgG with sustained titer >1:12,800 for >6 months at time of enrollment. (9) Serology consistent with exposure to HIV, or serology consistent with active hepatitis A, B, or C infection. Any active liver disease. (10) GGT >1.2x upper limit of normal (ULN) adjusted for age and gender. (11) Bilirubin >1.2x ULN adjusted for age and gender (absent known history of Dubin-Johnson syndrome). (13) Active infection based upon clinical symptoms. (14) Having started respiratory muscle strength training in the last 6 months prior to study day 1 or having discontinued respiratory muscle strength training in the 6-month period preceding study day 1, or having started respiratory strength training more than 6 months prior to study day 1 and unwilling to continue for the first year of study participation. (15) Received an investigational drug or participated in another interventional study within 90 days of study day 1. (16) History of cardiomyopathy in first year of life.
Endpoints
The primary endpoint was safety of AAV8-LSPhGAA, including avoidance of the following stopping rules: if two subjects develop the same Grade 3 AE, or for any subject that develops a Grade 4 AE that in the opinion of the investigator is deemed related to the investigational product. Secondary endpoints included the 6MWT and PFTs. Biochemical endpoints included muscle GAA and glycogen content, and immune response as measured by anti-rhGAA antibody formation and anti-AAV8 and anti-GAA ELISPOT.
Clinical laboratory testing for monitoring safety
Clinical laboratory testing was performed by Duke Clinical Laboratories (Durham, NC). Antibodies against rhGAA were quantified by Sanofi Genzyme (Bridgewater, NJ).
NAb assay
NAb responses to the AAV8 capsid were analyzed using a cell-based AAV8 NAb assay described previously.52 The AAV8 vector used in this assay contains the gene encoding β-galactosidase (LacZ) driven by a cytomegalovirus (CMV) promoter (AAV8.CMV.LacZ.bGH) and was made by the Vector Core Laboratory at the University of Pennsylvania (Philadelphia, PA). The NAb titer values are reported as the reciprocal of the highest sample dilution that inhibits AAV8.CMV.LacZ.bGH transduction (β-gal expression) by ≥ 50%, compared with a naive mouse serum control. To determine the endpoint titer, samples were run in a dilution series of either 7 or 14 dilutions until a negative result was obtained. The highest concentration of sample tested in the assay was a 1:5 dilution. The variability of the assay is ± one 2-fold sample dilution. Validation controls included metrics for the average signal from the cell only wells, the average level of transduction obtained at every dilution tested, a human validation test sample with a known AAV8 NAb titer, and a rabbit polyclonal anti-AAV8 positive control serum sample.
Interferon-γ ELISPOT assay
T cell responses to AAV8 capsid as well as the hGAA transgene were analyzed by an interferon-γ ELISPOT assay described previously.52 AAV8 capsid and hGAA transgene peptide libraries were synthesized as 15-mers with a 10 amino acid overlap with the preceding peptides (Mimotopes, VIC, Australia). The resulting peptides were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 100 mg/mL. Peptide pools were aliquoted, stored at ≤ −60°C, validated, and used at a final concentration of approximately 2 μg/mL in the assay. The AAV8 capsid peptide library was grouped into three peptide pools of approximately 50 peptides each and the hGAA transgene peptide library was grouped into four peptide pools of approximately 50 peptides each. Data were imaged and analyzed on a CTL S6 Core Analyzer and the cutoff value for a positive response was >40 SFU per 1E+6 PBMCs and at least three times (3x) the medium (DMSO) control value. Wells stimulated with DMSO, peptide pools, CEF, and PHA contained 2E+5 cells per well with data presented as the average spot forming units (SFU) per million (1E+6) cells. A normal donor PBMC sample was tested on every assay plate and had to meet the above listed criteria for medium (DMSO), anti-CD3, and PMA + ION in order for the assay to be considered valid.
Muscle function testing
Motor and pulmonary function was evaluated with the 6MWT and FVC, which are validated endpoints for Pompe disease.7 The 6MWT was performed as described.53 Pulmonary function tests were measured by electronic spirometer as described, including the forced expiratory volume in 1 s (FEV1), FVC, maximum expiratory pressure, and maximum inspiratory pressure.54 FEV1 and FVC were assessed in both the supine and upright positions to increase sensitivity for abnormalities detected in Pompe disease.55 All muscle function assessments were performed at screening, and at weeks 8, 24, 30, 38, and 52; and will be performed every 4 months in years 2 through 5.
Muscle biopsy
The impact of vector-produced GAA was analyzed by evaluating the biochemical correction of muscle in subjects with LOPD treated with ERT, both prior to and following vector administration. Subjects underwent a needle muscle biopsy of the quadriceps (vastus lateralis) at baseline, week 24, and week 52 visits by a neuromuscular specialist with expertise in Pompe disease. The needle muscle biopsy was performed under local anesthesia. The muscle biopsy was evaluated for biochemical correction by standard methods.56 Biopsy tissues were also processed for high-resolution light microscopy and stained for periodic acid-Schiff-positive glycogen.
Serum GAA assay
Serum samples isolated from standard serum separator tube were stored in a −80°C freezer with remote temperature monitoring system installed, until used for GAA activity determination as described.19 Briefly, serum samples were thawed on ice, while a 2.2-mM solution of 4-methyllumbelliferyl (4-MU)-α-D-glucoside (Sigma M9766) stock was boiled for 3 min and cooled at room temperature for more than 10 min. The reaction was set up in triplicate, consisting of 10 μL of serum and 20 μL of 4-MU substrate, in a 96-well plate (Corning, 3650). The reaction mixture was incubated at 37°C for 1 h, and was stopped by adding 130 μL of 0.5M sodium carbonate buffer (pH10.5). A standard curve (0–1,000 pmol/ μL of 4-MU; Sigma M1381) was used to measure the release of fluorescent 4-MU from individual reactions, using a TECAN microplate reader at 465-nm emission and 360-nm excitation. GAA activity measured in patients’ serum sample was reported by conversion of the artificial substrate 4-methylumbelliferyl (4-MU) α-D-glucoside to the fluorescent product umbelliferone (relative fluorescent units-RFUs) at acidic pH 4.3 using Magellan Software from TECAN. To calculate the GAA activity in milliliters, the released 4MU concentration was calculated based on the concentrations in the standard curve and activity was reported as nmol/h/mL.
Statistical analysis
Improvement in mean (±SD) values of each outcome at baseline and last study visit were evaluated using one-sided paired t tests. A p < 0.05 was considered statistically significant. All analyses were performed using Graphpad Prism (San Diego, CA).
Acknowledgments
The project described was supported by the National Center for Advancing Translational Sciences (NCATS) and by National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), through Grant Award Number U01AR071693, and by NCATS Award Number UL1TR001117 to the Duke Translational Medicine Institute/Duke CTSA, by the Alice and Y.-T. Chen Pediatric Genetics and Genomics Center, and by Asklepios Biopharmaceutical, Inc. We acknowledge project management by Dr. Jennifer E. Bond. Dr. Mai ElMallah was a co-investigator and was available for clinical decision making, when Dr. Smith was traveling. We acknowledge support of the study by Jian Dai, Songtao Li, and Ela Stefanescu. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author contributions
E.C.S.: Lead investigator; muscle biopsies; performed research; reviewed, edited, and approved the manuscript. S.H.: Designed and oversaw conduct of the study; data interpretation; reviewed, edited, and approved the manuscript. L.E.C.: Muscle testing; performed research; analyzed data; reviewed, edited, and approved the manuscript. S.-o.H.: Serum GAA assay; data analysis. J.C.: Supervised analysis of immune monitoring; reviewed, edited, and approved the manuscript. E.B.: Reviewed histology; reviewed, edited, and approved the manuscript. C.H. and T.S.: Performed data monitoring; analyzed data; reviewed, edited, and approved the manuscript. J.L.C.: Sub-investigator; Performed research; reviewed, edited, and approved the manuscript. D.B.: Supervised GAA activity and glycogen content measurement; data analysis; reviewed, edited, and approved the manuscript. P.S.K.: Clinical trial design; data interpretation; reviewed, edited, and approved the manuscript. D.D.K.: Designed the study; data interpretation; wrote the paper.
Declaration of interests
D.D.K. and P.S.K. have developed the technology that is being used in the study. If the technology is commercially successful in the future, the developers and Duke University may benefit financially. D.D.K. has served as a consultant for Sangamo Therapeutics and for Genzyme Sanofi, Amicus, and Vertex; has received grant support from Viking Therapeutics, Genzyme Sanofi, Roivant Rare Diseases, and Amicus; and has equity in Askbio, which is developing gene therapy for Pompe disease. E.C.S. received salary support for his role as PI on this study. S.H. is an employee of Askbio. L.E.C. has received honoraria from Genzyme Sanofi, has participated in research supported by Genzyme Sanofi, Valerion, Biomarin, and by Roivant Sciences; and is a member of the Pompe Registry North American Board of Advisors Genzyme Sanofi. D.B. has received research grant support and travel funds from Genzyme Sanofi, Baebies Inc., Biomarin, Alexion Inc., SOBI biopharma, and JCR biopharma. P.S.K. has received research/grant support from Genzyme Sanofi and Valerion Therapeutics. She received consulting fees and honoraria from Genzyme Sanofi, Amicus Therapeutics, and Vertex. She is a member of the Pompe and Gaucher Disease Registry Advisory Board for Genzyme Sanofi and on the Amicus Scientific advisory board.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.02.014.
Supplemental information
Data availability
Data from this study will be made available upon request.
References
- 1.Ficicioglu C., Ahrens-Nicklas R.C., Barch J., Cuddapah S.R., DiBoscio B.S., DiPerna J.C., Gordon P.L., Henderson N., Menello C., Luongo N., et al. Newborn screening for pompe disease: Pennsylvania experience. Int. J. Neonatal. Screen. 2020;6:89. doi: 10.3390/ijns6040089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elliott S., Buroker N., Cournoyer J.J., Potier A.M., Trometer J.D., Elbin C., Schermer M.J., Kantola J., Boyce A., Turecek F., et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016;118:304–309. doi: 10.1016/j.ymgme.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins P.V., Campbell C., Klug T., Rogers S., Raburn-Miller J., Kiesling J. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015;166:172–177. doi: 10.1016/j.jpeds.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani P.S., Hwu W.L., Mandel H., Nicolino M., Yong F., Corzo D., Infantile-Onset Pompe Disease Natural History Study Group A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 5.Messinger Y.H., Mendelsohn N.J., Rhead W., Dimmock D., Hershkovitz E., Champion M., Jones S.A., Olson R., White A., Wells C., et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet. Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banugaria S.G., Prater S.N., Patel T.T., Dearmey S.M., Milleson C., Sheets K.B., Bali D.S., Rehder C.W., Raiman J.A.J., Wang R.A., et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile Pompe disease: a step towards improving the efficacy of ERT. PLoS One. 2013;8:e67052. doi: 10.1371/journal.pone.0067052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Ploeg A.T., Clemens P.R., Corzo D., Escolar D.M., Florence J., Groeneveld G.J., Herson S., Kishnani P.S., Laforet P., Lake S.L., et al. A randomized study of alglucosidase alfa in late-onset Pompe's disease. N. Engl. J. Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 8.Patel T.T., Banugaria S.G., Case L.E., Wenninger S., Schoser B., Kishnani P.S. The impact of antibodies in late-onset Pompe disease: a case series and literature review. Mol. Genet. Metab. 2012;106:301–309. doi: 10.1016/j.ymgme.2012.04.027. [DOI] [PubMed] [Google Scholar]
- 9.Jones H.N., Muller C.W., Lin M., Banugaria S.G., Case L.E., Li J.S., O'Grady G., Heller J.H., Kishnani P.S. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25:277–283. doi: 10.1007/s00455-009-9252-x. [DOI] [PubMed] [Google Scholar]
- 10.Nicolino M., Byrne B., Wraith J.E., Leslie N., Mandel H., Freyer D.R., Arnold G.L., Pivnick E.K., Ottinger C.J., Robinson P.H., et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 11.Yanovitch T.L., Banugaria S.G., Proia A.D., Kishnani P.S. Clinical and histologic ocular findings in Pompe disease. J. Pediatr. Ophthalmol. Strabismus. 2010;47:34–40. doi: 10.3928/01913913-20100106-08. [DOI] [PubMed] [Google Scholar]
- 12.Schoser B., Stewart A., Kanters S., Hamed A., Jansen J., Chan K., Karamouzian M., Toscano A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J. Neurol. 2017;264:621–630. doi: 10.1007/s00415-016-8219-8. [DOI] [PubMed] [Google Scholar]
- 13.Franco L.M., Sun B., Yang X., Bird A., Zhang H., Schneider A., Brown T., Young S.P., Clay T.M., Amalfitano A., et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol. Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 14.Sun B., Bird A., Young S.P., Kishnani P.S., Chen Y.T., Koeberl D.D. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am. J. Hum. Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun B., Kulis M.D., Young S.P., Hobeika A.C., Li S., Bird A., Zhang H., Li Y., Clay T.M., Burks W., et al. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol. Ther. 2010;18:353–360. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler R.J., Bercury S.D., Fidler J., Zhao M.A., Foley J., Taksir T.V., Ryan S., Hodges B.L., Scheule R.K., Shihabuddin L.S., et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum. Gene Ther. 2008;19:609–621. doi: 10.1089/hum.2008.010. [DOI] [PubMed] [Google Scholar]
- 17.Bond J.E., Kishnani P.S., Koeberl D.D. Immunomodulatory, liver depot gene therapy for Pompe disease. Cell. Immunol. 2019;342:103737. doi: 10.1016/j.cellimm.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han S.O., Ronzitti G., Arnson B., Leborgne C., Li S., Mingozzi F., Koeberl D. Low-dose liver-targeted gene therapy for pompe disease enhances therapeutic efficacy of ERT via immune tolerance induction. Mol. Ther. Methods Clin. Dev. 2017;4:126–136. doi: 10.1016/j.omtm.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang G., Young S.P., Bali D., Hutt J., Li S., Benson J., Koeberl D.D. Assessment of toxicity and biodistribution of recombinant AAV8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol. Ther. Methods Clin. Dev. 2014;1:14018. doi: 10.1038/mtm.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han S.O., Gheorghiu D., Li S., Kang H.R., Koeberl D. Minimum effective dose to achieve biochemical correction with AAV vector-mediated gene therapy in mice with Pompe disease. Hum. Gene Ther. 2022;33:492–498. doi: 10.1089/hum.2021.252. [DOI] [PubMed] [Google Scholar]
- 21.Sun B., Young S.P., Li P., Di C., Brown T., Salva M.Z., Li S., Bird A., Yan Z., Auten R., et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol. Ther. 2008;16:1366–1371. doi: 10.1038/mt.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang P., Sun B., Osada T., Rodriguiz R., Yang X.Y., Luo X., Kemper A.R., Clay T.M., Koeberl D.D. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine Pompe disease. Hum. Gene Ther. 2012;23:460–472. doi: 10.1089/hum.2011.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith B.K., Collins S.W., Conlon T.J., Mah C.S., Lawson L.A., Martin A.D., Fuller D.D., Cleaver B.D., Clément N., Phillips D., et al. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum. Gene Ther. 2013;24:630–640. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vercauteren K., Hoffman B.E., Zolotukhin I., Keeler G.D., Xiao J.W., Basner-Tschakarjan E., High K.A., Ertl H.C., Rice C.M., Srivastava A., et al. Superior in vivo transduction of human hepatocytes using engineered AAV3 capsid. Mol. Ther. 2016;24:1042–1049. doi: 10.1038/mt.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raben N., Fukuda T., Gilbert A.L., de Jong D., Thurberg B.L., Mattaliano R.J., Meikle P., Hopwood J.J., Nagashima K., Nagaraju K., et al. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 26.Raben N., Danon M., Gilbert A.L., Dwivedi S., Collins B., Thurberg B.L., Mattaliano R.J., Nagaraju K., Plotz P.H. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 27.Koeberl D.D., Case L.E., Smith E.C., Walters C., Han S.O., Li Y., Chen W., Hornik C.P., Huffman K.M., Kraus W.E., et al. Correction of biochemical abnormalities and improved muscle function in a phase I/II clinical trial of clenbuterol in pompe disease. Mol. Ther. 2018;26:2304–2314. doi: 10.1016/j.ymthe.2018.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S., Sun B., Nilsson M.I., Bird A., Tarnopolsky M.A., Thurberg B.L., Bali D., Koeberl D.D. Adjunctive beta2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J. 2013;27:34–44. doi: 10.1096/fj.12-207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han S.O., Gheorghiu D., Chang A., Mapatano S.H., Li S., Brooks E., Koeberl D. Efficacious androgen hormone administration in combination with AAV vector-mediated gene therapy in female mice with Pompe disease. Hum. Gene Ther. 2022;33:479–491. doi: 10.1089/hum.2021.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han S.O., Li S., Everitt J.I., Koeberl D.D. Salmeterol with liver depot gene therapy enhances the skeletal muscle response in murine Pompe disease. Hum. Gene Ther. 2019;30:855–864. doi: 10.1089/hum.2018.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 32.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huggins E., Holland M., Case L.E., Blount J., Landstrom A.P., Jones H.N., Kishnani P.S. Early clinical phenotype of late onset Pompe disease: lessons learned from newborn screening. Mol. Genet. Metab. 2022;135:179–185. doi: 10.1016/j.ymgme.2022.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Ponder K.P. Merry christmas for patients with hemophilia B. N. Engl. J. Med. 2011;365:2424–2425. doi: 10.1056/NEJMe1111138. [DOI] [PubMed] [Google Scholar]
- 35.Schoser B., Hahn A., James E., Gupta D., Gitlin M., Prasad S. A systematic review of the health economics of pompe disease. Pharmacoecon. Open. 2019;3:479–493. doi: 10.1007/s41669-019-0142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kishnani P.S., Goldenberg P.C., DeArmey S.L., Heller J., Benjamin D., Young S., Bali D., Smith S.A., Li J.S., Mandel H., et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banugaria S.G., Prater S.N., Ng Y.K., Kobori J.A., Finkel R.S., Ladda R.L., Chen Y.T., Rosenberg A.S., Kishnani P.S. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet. Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chien Y.H., Lee N.C., Thurberg B.L., Chiang S.C., Zhang X.K., Keutzer J., Huang A.C., Wu M.H., Huang P.H., Tsai F.J., et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–e1125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- 39.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus types 1, 2, 5, 6, 8 and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 40.Korlimarla A., Lim J.A., Kishnani P.S., Sun B. An emerging phenotype of central nervous system involvement in Pompe disease: from bench to bedside and beyond. Ann. Transl. Med. 2019;7:289. doi: 10.21037/atm.2019.04.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musumeci O., Marino S., Granata F., Morabito R., Bonanno L., Brizzi T., Lo Buono V., Corallo F., Longo M., Toscano A. Central nervous system involvement in late-onset Pompe disease: clues from neuroimaging and neuropsychological analysis. Eur. J. Neurol. 2019;26:442.e35. doi: 10.1111/ene.13835. [DOI] [PubMed] [Google Scholar]
- 42.Fuller D.D., Trejo-Lopez J.A., Yachnis A.T., Sunshine M.D., Rana S., Bindi V.E., Byrne B.J., Smith B.K. Case Studies in Neuroscience: neuropathology and diaphragm dysfunction in ventilatory failure from late-onset Pompe disease. J. Neurophysiol. 2021;126:351–360. doi: 10.1152/jn.00190.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr., Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hordeaux J., Dubreil L., Robveille C., Deniaud J., Pascal Q., Dequéant B., Pailloux J., Lagalice L., Ledevin M., Babarit C., et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol. Commun. 2017;5:66. doi: 10.1186/s40478-017-0464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrne B.J., Fuller D.D., Smith B.K., Clement N., Coleman K., Cleaver B., Vaught L., Falk D.J., McCall A., Corti M. Pompe disease gene therapy: neural manifestations require consideration of CNS directed therapy. Ann. Transl. Med. 2019;7:290. doi: 10.21037/atm.2019.05.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuller D.D., ElMallah M.K., Smith B.K., Corti M., Lawson L.A., Falk D.J., Byrne B.J. The respiratory neuromuscular system in Pompe disease. Respir. Physiol. Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun B., Zhang H., Benjamin D.K., Brown T., Bird A., Young S.P., McVie-Wylie A., Chen Y.T., Koeberl D.D. Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid alpha-glucosidase in glycogen storage disease type II. Mol. Ther. 2006;14:822–830. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puzzo F., Colella P., Biferi M.G., Bali D., Paulk N.K., Vidal P., Collaud F., Simon-Sola M., Charles S., Hardet R., et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 2017;9:eaam6375. doi: 10.1126/scitranslmed.aam6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner S.M.F., Hoyt A.K., ElMallah M.K., Falk D.J., Byrne B.J., Fuller D.D. Neuropathology in respiratory-related motoneurons in young Pompe (Gaa(-/-)) mice. Respir. Physiol. Neurobiol. 2016;227:48–55. doi: 10.1016/j.resp.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falk D.J., Todd A.G., Lee S., Soustek M.S., ElMallah M.K., Fuller D.D., Notterpek L., Byrne B.J. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum. Mol. Genet. 2015;24:625–636. doi: 10.1093/hmg/ddu476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim J.A., Yi H., Gao F., Raben N., Kishnani P.S., Sun B. Intravenous injection of an AAV-PHP.B vector encoding human acid alpha-glucosidase rescues both muscle and cns defects in murine Pompe disease. Mol. Ther. Methods Clin. Dev. 2019;12:233–245. doi: 10.1016/j.omtm.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calcedo R., Chichester J.A., Wilson J.M. Assessment of humoral, innate, and T-cell immune responses to adeno-associated virus vectors. Hum. Gene Ther. Methods. 2018;29:86–95. doi: 10.1089/hgtb.2018.038. [DOI] [PubMed] [Google Scholar]
- 53.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories ATS statement: guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002;166:111–117. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 54.Brooke M.H., Fenichel G.M., Griggs R.C., Mendell J.R., Moxley R., Miller J.P., Province M.A. Clinical investigation in Duchenne dystrophy: 2. Determination of the "power" of therapeutic trials based on the natural history. Muscle Nerve. 1983;6:91–103. doi: 10.1002/mus.880060204. [DOI] [PubMed] [Google Scholar]
- 55.van der Beek N.A.M., van Capelle C.I., van der Velden-van Etten K.I., Hop W.C.J., van den Berg B., Reuser A.J.J., van Doorn P.A., van der Ploeg A.T., Stam H. Rate of progression and predictive factors for pulmonary outcome in children and adults with Pompe disease. Mol. Genet. Metab. 2011;104:129–136. doi: 10.1016/j.ymgme.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 56.Koeberl D.D., Luo X., Sun B., McVie-Wylie A., Dai J., Li S., Banugaria S.G., Chen Y.T., Bali D.S. Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol. Genet. Metab. 2011;103:107–112. doi: 10.1016/j.ymgme.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from this study will be made available upon request.