

Abstract
The human T cell leukemia virus type 1 (HTLV-1) is a pathogenic retrovirus that persists as a provirus in the genome of infected cells and can lead to adult T cell leukemia (ATL). Worldwide, more than 10 million people are infected and approximately 5% of these individuals will develop ATL, a highly aggressive cancer that is currently incurable. In the last years, genome editing tools have emerged as promising antiviral agents. In this proof-of-concept study, we use substrate-linked directed evolution (SLiDE) to engineer Cre-derived site-specific recombinases to excise the HTLV-1 proviral genome from infected cells. We identified a conserved loxP-like sequence (loxHTLV) present in the long terminal repeats of the majority of virus isolates. After 181 cycles of SLiDE, we isolated a designer-recombinase (designated RecHTLV), which efficiently recombines the loxHTLV sequence in bacteria and human cells with high specificity. Expression of RecHTLV in human Jurkat T cells resulted in antiviral activity when challenged with an HTLV-1 infection. Moreover, expression of RecHTLV in chronically infected SP cells led to the excision of HTLV-1 proviral DNA. Our data suggest that recombinase-mediated excision of the HTLV-1 provirus represents a promising approach to reduce proviral load in HTLV-1-infected individuals, potentially preventing the development of HTLV-1-associated diseases.
Keywords: HTLV-1, adult T cell leukemia, ATL, HTLV-1-associated myelopathy/tropical spastic paraparesis, HAM/TSP, gene therapy, genome engineering, site-specific recombinases, molecular directed evolution, Cre-lox, retrovirus
Graphical abstract
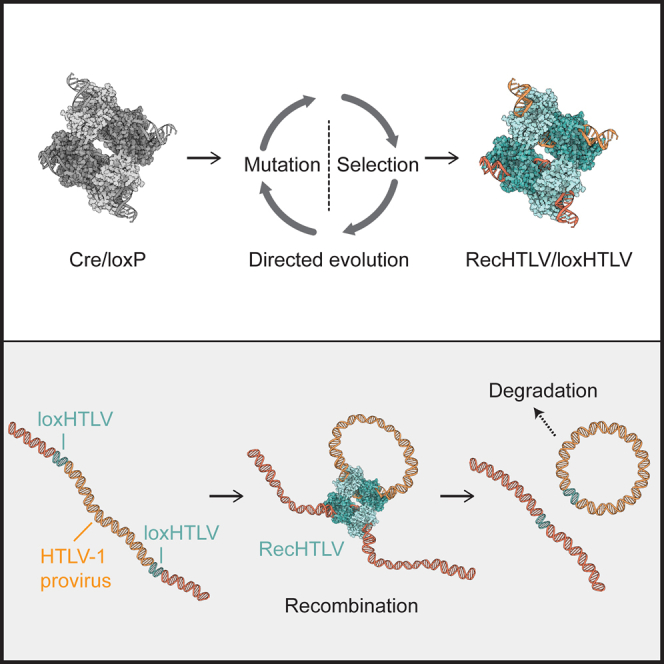
Directed molecular evolution is used to generate the designer-recombinase RecHTLV that can efficiently and specifically recombine a conserved sequence present in the LTRs of HTLV-1. Expression of RecHTLV in HTLV-1-infected cells results in excision of the HTLV-1 provirus in a precise and seamless fashion.
Introduction
Retroviral infections remain a challenge for modern medicine, mainly because there are still no effective therapies to cure them. The permanent integration of the proviral DNA into the host cell genome results in lifelong infection. For this reason, from the over 37 million human immunodeficiency virus (HIV)-infected people worldwide, the ones with access to therapy are dependent on lifelong drug treatments that suppress the virus but cannot cure the patients from the infection (UNAIDS 2021 estimates). A promising approach to solve this problem is the use of programmable DNA nucleases to compromise the virus. While some nuclease strategies aim to target the viral entry receptors of host cells,1,2,3,4,5 others focus on directly disrupting the genetic material of the virus.6,7,8 The use of designer-nucleases to treat HIV-1 infections has been possible because the integrated proviral DNA serves as a targetable substrate for genome scissors such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or CRISPR-Cas9.6,7,8,9,10 However, nucleases induce double-strand breaks and rely on the cellular DNA repair machinery, which processes them in an unpredictable manner. Indeed, the use of nucleases to target viral sequences leads to the rapid formation of resistant clones, potentially compromising the utility of this approach.11,12,13,14 Another class of genome editing tools, namely site-specific recombinases (SSRs), circumvent this problem, as they do not rely on host DNA repair mechanisms and edit DNA in a seamless manner.15 However, adapting SSRs to recombine custom-chosen sequences poses challenges. Nevertheless, directed evolution methods have proven useful to redirect the specificity of recombinases to recognize new target sequences.16,17,18 In fact, designer-recombinases have been developed as powerful tools for the removal of the HIV-1 provirus, with high efficiency and precision.19,20,21 Importantly, these designer-recombinases have demonstrated their anti-retroviral activity without the emergence of resistant clones in humanized mouse models, indicating that they have considerable therapeutic potential.19,20
Targeting infections using genome editing tools is not limited to the HIV virus and similar approaches could potentially be utilized for treating other human retroviral infections. Human T cell leukemia virus type 1 (HTLV-1) is a human-pathogenic retrovirus that causes an aggressive neoplasia, of adult T cell leukemia (ATL), in about 5% of carriers.22,23,24 Furthermore, in a smaller percentage of infected individuals, an inflammatory neurological disease called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is observed.25 Approximately 10 million people are infected with HTLV-1 around the world with a high percentage of them in endemic areas that include, among others, Japan, Central Australia, South America, the Caribbean, and Sub-Saharan Africa.24,26 The lack of effective therapies for ATL and HAM/TSP denote the need for new treatment approaches. Viral transmission occurs mostly through cell-to cell spread at tight cell contacts or via cellular protrusions (reviewed in Gross and Thoma-Kress27,28 and Sarkis et al.27,28). However, viral amplification occurs mainly by clonal expansion, although infectious spread persists during chronic infection.29 Thus, HTLV-1 has a remarkably stable genome, and there is little sequence variation between individuals and even within cells of an infected person.30,31 Nevertheless, defective proviruses are present not only in patients but also in asymptomatic HTLV-1 carriers.32 The HTLV-1 proviral genome is approximately 9 kb and it is flanked by 5′ and 3′ long terminal repeats (LTRs) that are identical in sequence.33 The genetic sequence stability of the virus and the fact that the provirus integrates in the genome of infected cells make HTLV-1 a suitable candidate for treatment with gene editing tools. Indeed, different groups have already attempted the disruption of the virus using nucleases: Tanaka et al. used zinc finger nucleases to disrupt the LTR promoter and Nakagawa et al. used CRISPR-Cas9 to target the Hbz gene, which led to a reduction in cell proliferation in three different ATL cell lines.34,35 However, with the use of nucleases it is likely that, as for HIV-1, resistant HTLV-1 clones will emerge. Here, we use directed molecular evolution to develop a Cre-based designer-recombinase able to recognize loxHTLV, a highly conserved sequence present in the LTRs of HTLV-1. Our results provide proof of concept that evolved recombinases can be generated to precisely excise the HTLV-1 provirus from infected cells.
Results
Target site selection
The target sites for Cre-like recombinases typically consist of an 8 bp spacer sequence flanked by 13 bp inverted repeats, which can harbor some positions of asymmetry.15,20,21 To identify a suitable recombinase target site, we applied the SeLOX algorithm36 to a collection of 949 LTR sequences deposited in the HTLV-1 Molecular Epidemiology Database.37 From these data, we identified the most conserved 34-mers, restricting the results to sequences present in at least 80% of all the deposited LTRs. After discarding sequences containing mononucleotide repeats of five identical bases or more, we obtained a set of eight candidate target sites. To avoid possible unwanted recombination on human off-target sites, we searched for all occurrences of sequences resembling the candidate target sites (reference assembly GRCh38 from December 2013). We allowed up to two mismatches per half-site and disregarded mismatches in the 8 bp spacer regions. As a result, we discarded five candidates with potential off-target risk. To select the most suitable of the three remaining target sites, we compared sequences of their half-sites with target sites of previously evolved recombinase libraries.16,20,21,38,39 We nominated the target site with the lowest number of mismatches against one of the previous target sites and named it loxHTLV (Figure 1A). The final target site is found at nucleotide positions from 317 to 350, and from 8,597 to 8,630 (GenBank entry AB513134.1). loxHTLV is highly conserved and found without mismatches in 86% of the sequences from the HTLV-1 Molecular Epidemiology Database (Figure S1A). Because loxHTLV is located in the LTR sequences of HTLV-1, flanking the genome of the retrovirus, recombination of these sites would result in the excision of the complete proviral coding sequences from infected cells (Figure S1B).
Figure 1.

Directed evolution of RecHTLV
(A) Alignment of the loxHTLV (RecHTLV) target sequence to loxP (Cre), loxLTR (Tre), and loxBTR (Brec1). Mismatches to the loxP sequence are marked in red and asymmetries within the sequences are underlined. The spacers are shown in gray. (B) Scheme of the RecHTLV evolution process. The activity of the recombinase libraries from the first and last cycle in each subsite are shown as images of the agarose gels, where the upper bands correspond to the non-recombined plasmids and are illustrated with two triangles. The smaller faster migrating bands indicate recombined plasmids (shown here as one triangle). The number of evolution cycles performed in each subsite is marked between the two agarose gels. The triangles below indicate the reduction of L-arabinose (L-ara) levels during evolution for each target site.
Directed evolution of HTLV-1 targeting recombinases
To develop a designer-recombinase able to recombine the loxHTLV site, we performed substrate-linked directed evolution (SLiDE) (materials and methods).16,20,38,39 SLiDE is performed in E. coli and links the excision activity of recombinases to the plasmid that encodes them. In each evolution cycle, the recombinases that successfully excise the target site were selected and amplified by PCR thus introducing variability in the library. Amplified recombinases were then inserted back into the evolution plasmid (pEVO) to allow successive adaptation (Figure S1C).
In total, we performed 181 evolution cycles via 7 subsites to obtain a library able to efficiently recombine the loxHTLV sequence at low recombinase expression levels, indicating that clones with high activity had been selected during SLiDE (Figures 1B, S1C, and S1D).
Selection of RecHTLV recombinase clone
To enrich for recombinases that do not have adverse effects when expressed in mammalian cells, the final library was inserted into a lentiviral vector that allowed the expression of the recombinases from a tetracycline-inducible promoter (Figure 2A). Following infection of HeLa cells at low multiplicity of infection to ensure that most cells express a single recombinase clone, the library was expressed by the addition of doxycycline to the medium for 7 days, ensuring that expression of the recombinase was well tolerated in the cells (Figure 2A).39 The coding sequences of well-tolerated recombinases were then PCR amplified from genomic DNA and inserted into the pEVO vector to evaluate recombination activity in E. coli. Recombinase expression was induced by the addition of 10 μg/mL L-arabinose and the excision activity was tested for 96 clones using a PCR assay (Figure S2A; materials and methods). From the 96 clones, 44 showed recombination activity as indicated by the appearance of the recombination-specific band. Sanger sequencing of the clones revealed that some of them shared the same sequence (data not shown). Therefore, we selected the 29 unique active clones for further evaluation in mammalian cell culture. The selected recombinases were inserted into a mammalian expression vector and separately co-transfected into HeLa cells together with a fluorescent reporter plasmid to evaluate their activity on the loxHTLV site in mammalian cells (Figure S2B). The clone G9, hereafter referred to as RecHTLV, showed the highest activity in this assay and was hence selected for further evaluation as a candidate HTLV-1 recombinase (Figure S2B). To confirm that the initial screen for tolerability in HeLa cells was successful, we inserted the RecHTLV coding sequence into the tetracycline-inducible lentiviral system described above in which the expression of the recombinases is driven by a tetracycline-inducible promoter and generated HeLa-derived cell lines (Figure 2A). Because these vectors also express GFP, transduced cells can be followed over time. Upon the continuous addition of 100 ng/mL of doxycycline in the medium we observed that the percentage of GFP+ cells dropped only mildly for the cell lines expressing RecHTLV, similar to the GFP-only control (Figure S2C). These results confirmed the success of the initial screen and that RecHTLV does not affect cellular growth in this system. Therefore, RecHTLV is a well-tolerated candidate HTLV-1 recombinase.
Figure 2.

Screening for an active and specific HTLV-1 recombinase
(A) Overview of the process for the selection of clones. In the upper part, a scheme of the construct used for expression of the library is shown: expression of the recombinase fused to a nuclear localization signal (NLS-Rec) is regulated by a tetracycline-inducible promoter (TRE3G), which is bound by Tet-On 3G (Tet-On advanced transactivator protein) upon doxycycline treatment; bicistronic expression of Tet-On 3G and a GFP cassette is driven by an elongation factor 1 alpha (EF1a) promoter and separated by a self-cleaving 2A peptide (P2A). The lower part shows the process followed for selection of candidate clones: the recombinase library was cloned into a lentiviral vector and, after lentivirus production, HeLa cells were infected with the recombinase-containing lentiviral particles. After 7 days of expression, the recombinases were extracted by PCR and cloned in the pEVO bacterial plasmid for sequencing and evaluation of activity on loxHTLV. Furthermore, the clones were tested in the cell culture for activity on the loxHTLV target site. (B) Top: agarose gels for the plasmid activity assay showing recombination efficiency of Cre, Brec1, and RecHTLV on their respective target sites in bacteria. The performed test digest results in a smaller fragment for recombined (one triangle) and a larger fragment for non-recombined plasmids (two triangles). The recombinases were tested at 0, 1, 10, 50, 100, and 200 μg/mL L-arabinose as shown above. Quantification of recombination is shown below the gels as a fraction of 1 (1 fully recombined, 0 not recombined). Bottom: line graph showing the quantification of recombination at the different levels of arabinose for Cre, Brec1, and RecHTLV. Parts of the figure were created with BioRender.com.
Mutations acquired during evolution
To get an insight into the mutations that were acquired during evolution, we deep sequenced the final evolution library using PacBio long-read sequencing technology. We observed that through evolution, the library had acquired dominating mutations in 35 residues compared with Cre (L5Q, V7L, N10S, P12S, P15L, V23A, K25Q, D29V, M30T, R34H, S38P, K57E, Y77H, A84T, K86N, Q90K, G93A, Q94E, M97T, S108G, K122R, I166V, A175S, K244R, N245Y, A248V, A249V, R259H, L261M, E262Q, E266G, T268A, P307A, N317T, I320S) (Figure S3A). As described previously, many of these mutations are residues that have direct contact or proximity to the target site DNA, indicating a putative function of these residues in modulating the selectivity toward the new target site (Figure S3A).16,20,21 We next sequenced the selected RecHTLV clone using Sanger sequencing and observed that it differs from Cre in 46 residues (N3K, V7L, N10D, P12S, P15L, V16A, A18V, V23A, S38P, K43E, K57E, L58S, Y77H, A84T, K86N, I88V, Q90K, G93A, Q94E, M97T, S108G, S114T, K122R, Q144R, Q156L, I166V, A175S, T206A, S226T, V227A, K244R, N245Y, A248V, A249V, P250S, Q255R, R259H, L261M, E262Q, E266G, A267T, T268A, M299L, P307A, N317R, I320S). This is the highest number of changes observed in a Cre-derived recombinase to date, possibly due to the marked differences of loxHTLV when compared with loxP.
RecHTLV contains most of the mutations that emerged through the library evolution (Figures S3A and S3B), including mutations previously seen in other Cre-based designer-recombinases (Y77H, S108G, I166V, A175S, I320S), indicating a possible general role of these residues in protein stability.20,40 We also observed that RecHTLV has several changes in the first 20 amino acids (positions 3, 7, 10, 12, 15, 16, and 18). The role of the N terminus of Cre has been debated because wild-type Cre can efficiently recombine loxP even when the first 20 amino acids are removed, but it has recently been reported that for evolved Cre-type recombinases these amino acid changes contribute to the stability of the protein.41,42,43 Hence, the amino acid changes in the first 20 residues might also contribute to the protein stability of RecHTLV.
One of the hotspots of mutations for RecHTLV is in helix D, particularly at residues on positions 84–94, which have been shown in Cre to be important for recognizing the nucleotide positions close to the loxP spacer (Figure S3B). Interestingly, some of these changes were previously seen in the Brec1 recombinase.20 Nonetheless most of the mutations acquired are unique to RecHTLV possibly conferring specificity to the nucleotide changes in loxHTLV differing from the loxBTR target site (Figure S3B). Another mutation hotspot occurs in helix J, which makes direct contact with the major groove of the DNA, and has been shown to be involved in recognition of the nucleotide positions 8, 9, 10–25, 26, and 27 of the target site (Figure S3B).44,45,46 The loxHTLV target sequence is very different at these nucleotide positions when compared with loxP or other lox-like targets and we consistently observe unique changes in this region of the protein, including residues 261 and 267, which were never mutated in any other designer-recombinase investigated thus far (Figure S3B).20,21,38,39
Taken together, these data provide additional insight into the recognition of evolved recombinases to their target site, possibly contributing to efforts for a more rational engineering of Cre-based recombinases toward novel target sites.47,48,49
RecHTLV specifically recombines loxHTLV in E. coli
To characterize RecHTLV in more detail, we tested its recombination activity at different expression levels in E. coli. The pEVO vector allows for testing the activity of recombinase clones in bacteria at different expression levels by modulating the concentration of L-arabinose added to the growth medium. We expressed RecHTLV using 0, 1, 10, 50, 100, and 200 μg/mL of L-arabinose and loaded the isolated plasmid DNA after digestion with diagnostic restriction enzymes on agarose gels. As expected, we observed an increase of intensity of the band specific for the recombined form of the plasmid, indicating that the loxHTLV target sites were recombined in an L-arabinose dose-dependent fashion (Figure 2B). We compared the recombination activity of Cre on loxP and recombination of loxBTR by Brec1, a recombinase with potent antiretroviral activity in HIV-1-infected cells (Figure 2B).20 We found that at low levels of expression (0, 1 μg/mL of L-arabinose) Cre recombined loxP proficiently, whereas Brec1 and RecHTLV had less than 10% recombination activity on their respective target sites at this level of induction. However, when expressed at higher levels, RecHTLV efficiently recombined the loxHTLV sites (>80% recombination), displaying similar activity as Brec1 on loxBTR. The difference in activity can be explained by different factors including DNA-binding affinity or protein stability. Overall, RecHTLV shows slightly lower activity compared with Cre on loxP, but similar recombination activity to Brec1 on loxBTR.
RecHTLV recombines loxHTLV efficiently in HeLa cells
To test the activity of RecHTLV in mammalian cells, we constructed a mammalian reporter vector in which a three SV40 poly(A) cassette was flanked by two loxHTLV sites preventing the expression of the mCherry gene located downstream. Upon recombination of the loxHTLV sites, the poly(A) cassette is excised allowing the expression of mCherry driven by the CAG promoter (Figure 3A). To express the recombinases (RecHTLV or Cre as control), we used a plasmid that drives the expression from a cytomegalovirus (CMV) promoter and contained an IRES sequence, allowing linked expression of GFP and therefore enabling to control for transfection efficiencies of the plasmids (Figure 3A). When both plasmids were co-transfected in HeLa cells, we observed mCherry-positive cells only when RecHTLV was co-transfected with the reporter, whereas no mCherry-positive cells were detected in the Cre-transfected cells (Figures 3B, 3C, and S4).
Figure 3.

Activity of RecHTLV in human cells in transient reporter assay
(A) Schematic representation of the reporter and expression constructs used for assessing RecHTLV activity in HeLa cells. The reporter construct pCAGGS-lox-pA-lox-mCherry consists of 3 poly(A) (pA) sequences flanked by two loxHTLV sites which prevent the expression of mCherry driven by a CAG promoter. Expression of mCherry is only possible upon recombination of the loxHTLV sites. The IRES (internal ribosomal entry site) bicistronic expression construct allows simultaneous expression of the recombinase fused to a nuclear localization signal (NLS-Rec) and a GFP cassette driven by a CMV promoter. (B) Representative images showing the fluorescence-based recombination reporter assay. HeLa cells were co-transfected with the indicated recombinase and the reporter construct. Only upon the expression of RecHTLV we observed expression of mCherry. Scale bars, 200 μm. (C) Left: representative dot plots obtained by flow cytometry of HeLa cells transfected with the loxHTLV reporter construct and Cre or RecHTLV expression constructs. On the right side, quantification of the recombination efficiency measured as the ratio of mCherry+ cells and frequency of transfected cells (GFP+). The bar shows the mean ± SD of five independent experiments (∗∗∗p < 0.001, unpaired two-sided t test).
RecHTLV recombines loxHTLV in a genomic context
To test whether RecHTLV can recombine the loxHTLV sequence in a genomic context, we generated a HeLa reporter cell line where two loxHTLV target sites flank a puromycin cassette followed by an out-of-frame mCherry cassette. Upon recombination, the puromycin cassette is excised, allowing for the expression of mCherry driven by a spleen focus-forming virus promoter (Figure 4A). To express the recombinases, we transiently transfected a pIRES vector driving expression of the recombinase in addition to GFP to be able to track the transfection efficiency. Forty-eight hours post transfection we detected over 40% mCherry-positive cells transfected with RecHTLV (Figure 4B), signifying that RecHTLV can recombine the loxHTLV sequences in a genomic context. Importantly, expression of Cre recombinase in this reporter cell line did not lead to appreciable numbers of mCherry-positive cells (Figure 4B). To confirm the correct excision of the puromycin cassette, we extracted genomic DNA from the transfected cells. PCR assays with primers that flank the loxHTLV sites produced a band pattern consistent with the correct excision via the loxHTLV sites (Figure 4C). Furthermore, sequencing of the PCR band confirmed the exact nucleotide sequence expected from recombinase-mediated excision (Figure 4D). We conclude that RecHTLV is active in human cells and efficiently excises sequences flanked by two loxHTLV sites from the genome.
Figure 4.

RecHTLV recombines loxHTLV in a genomic reporter in HeLa cells
(A) Schematic representation of the reporter cell line and expression constructs used for assessing RecHTLV activity in HeLa cells in a genomic context. The reporter construct consists of a puromycin cassette flanked by two loxHTLV sites that prevent the expression of mCherry driven by a spleen focus-forming virus (SFFV) promoter. Expression of mCherry is only possible upon recombination of the loxHTLV sites. In the bicistronic recombinase expression construct, the IRES (internal ribosomal entry site) allows simultaneous expression of the recombinase fused to a nuclear localization signal (NLS-Rec) and a GFP cassette driven by a CMV promoter. (B) Left: representative dot plots obtained by flow cytometry of the loxHTLV reporter cells transfected with Cre or RecHTLV expression constructs. On the right side, quantification of the recombination efficiency measured as the ratio of mCherry+ cells and the total transfected cells (GFP+). The bar shows the mean ± SD of three independent experiments (∗∗∗∗p < 0.0001; ns, not significant, unpaired two-sided t test). (C) PCR performed on genomic DNA from transfected loxHTLV HeLa reporter cells. Left: schematic representation of the PCR where primers F and R are located in the flanking regions outside loxHTLV. The expected amplification product is 1.2 kb for non-recombined and 560 bp for the recombined reporter. Right: agarose gel of the PCR products from genomic DNA extracted from different samples: lane 1, HeLa cells; lane 2–4, reporter HeLa cells transfected with the indicated construct; lane 5, water control. The upper band on the gel shows unrecombined reporter (two triangles) while the lower one shows recombination (one triangle). (D) Chromatogram of the Sanger sequencing from the recombined PCR band shown in (C) from the RecHTLV-treated sample.
Experimental detection of potential off-target sites
To investigate the specificity of RecHTLV, we bioinformatically screened the human genome for potential off-target sites based on sequence similarity to loxHTLV. The three most closely related sites (Figure 5A) with five and six mismatches in the half-site sequences to the loxHTLV sequence, respectively, were tested experimentally in bacteria. We observed no detectable recombination on these three putative off-target sites, even at high recombinase expression levels, indicating that RecHTLV is rather specific and does not recombine human target sites with the closest sequence similarity to loxHTLV (Figure 5A).
Figure 5.

Off-target analysis of RecHTLV
(A) Top: alignment of the loxHTLV target sequence to the in-silico-predicted off-target sites: 1, 2, and 3. Off-target site 3 is asymmetric and is therefore shown as 3.1 and 3.2 in the alignment. Mismatches to the loxHTLV sequence are marked in red and spacers are shown in gray. Bottom: agarose gel showing the recombination activity of RecHTLV at 50 μg/mL L-arabinose for the on-target sequence (loxHTLV) and the three predicted off-targets in bacteria. Off-target sites 1 and 2 were inserted twice as excision substrates in the pEVO vector, whereas off-target site 3 consists of the 3.1 and the 3.2 sequences inserted as excision substrates. Below each lane, quantification of the recombination efficiency is shown. The upper unrecombined band is indicated with two triangles and the recombined band with one triangle. (B) Schematic representation of the plasmids used for testing the selected peaks from the ChIP-seq experiment in bacteria. From the selected peaks, 92 bp around the summit of the peak were cloned twice in the pEVO vector as excision substrates. (C) Bacterial plasmid-based for RecHTLV activity assay of the 10 peaks selected from the ChIP-seq binding sites. All peaks were tested at 200 μg/mL of L-arabinose. Quantification of the recombination is shown below each lane. (D) Dissecting peak 7 to find the RecHTLV recombinase recognition sequence. Top: sequence of the 92 bp tested in the plasmid assay with the indicated subdivisions tested: “left”, “middle”, “right” “lox”. Bottom: agarose gel from the bacterial test digest assay showing the recombination efficiencies of the different parts of peak 7. Only the lox-peak7 sequence shows recombination at 200 μg/mL L-arabinose. Quantification of the recombination is shown below each lane. The upper unrecombined band is indicated with two triangles and the recombined band with one triangle.
To experimentally identify putative RecHTLV off-targets in human cells with an unbiased approach, we used a chromatin immunoprecipitation followed by sequencing assay (ChIP-seq).39 To this aim, we fused RecHTLV to GFP and stably expressed the fusion protein in HeLa cells. We used a well-established anti-GFP antibody for the immunoprecipitation of the recombinase to enrich for genomic sequences bound by RecHTLV.39,50,51,52,53 After immunoprecipitation, the DNA fragments were deep sequenced using Illumina NGS sequencing. We identified 112 putative binding sites in the genome from which, based on the piled up reads when comparing the control and test samples, we selected 10 peaks for experimental testing in bacteria (Figure S5A). To investigate if the bound sequences can be recombined by RecHTLV, we inserted the 92 bp sequence found around the peak summit twice into the pEVO vector carrying RecHTLV as an excision substrate (Figure 5B). After induction of recombinase expression, 9 out of 10 constructs did not show any recombination-specific band (Figure 5C), confirming the high specificity of RecHTLV and validating that sequences bound by a designer-recombinase are not necessarily recombination substrates.39 However, one of the tested sites, namely peak 7 (Figure 5C), displayed a weak signal of the band expected from a recombination product. To find the exact 34 bp sequence that is recombined by RecHTLV, we split the 92 bp into three areas, called left, middle, and right, and inserted these sequences into the pEVO twice as excision substrates. We also tested in the same manner the sequence with the highest similarity to loxHTLV (lox-peak7) (Figures 5D and S5B). We observed that only the lox-peak7 sequence showed the recombination-specific band, thereby identifying exactly the 34 bp that are recognized by RecHTLV in the genomic context (Figure 5D). Despite being a potential off-target, the lox-peak7 sequence occurs only one time in the human genome.54 Hence, deleterious recombination events involving this sequence are highly unlikely and, because the spacer sequence differs from the spacer of loxHTLV, recombination between these two sites is implausible. This result demonstrates that RecHTLV has high specificity and that the ChIP-seq method is suitable for experimentally identifying putative genomic off-target sites of designer-recombinases.
RecHTLV recombines loxHTLV in the context of the full HTLV-1 LTR sequence
To test whether RecHTLV is able to recombine the loxHTLV sequences in the context of the full-length HTLV-1 LTR, we used the plasmid pHpX, which contains two full-size LTR sequences of HTLV-1 flanking the Tax protein coding sequence (Figure S6A).55 For expression of recombinases, we constructed a lentiviral plasmid in which the recombinase coding sequences are under control of the elongation factor-1 alpha (EF1a) promoter. Both plasmids were co-transfected into HEK293T or Jurkat T cell lines. For both cell lines we observed that the expression of the Tax protein was significantly reduced only when transfected with the plasmid expressing RecHTLV (Figures S6B and S6C), but not for the empty vector control (Puro, missing recombinase cassette) or Cre-transfected cells, indicating RecHTLV-mediated excision of the Tax expression cassette from the plasmid. To elucidate if the recombinases are also functional in the context of a full-length HTLV-1 proviral construct, we performed transient assays using a plasmid (X1MT) containing the entire genome of HTLV-1.56 Co-transfection together with the recombinase-expressing plasmids (Figure 6A) revealed that expression of Gag (detected with a Gag p19 antibody) was significantly reduced in the presence of RecHTLV compared with the empty vector (Puro) or Cre controls (Figure 6B). As an additional control, we performed the same assay using the HTLV-1 packaging plasmid pCMV-HT1M.57 In this case, expression of the viral proteins is driven by a CMV promoter while the 5′ LTR has been partially deleted, therefore lacking the loxHTLV site (Figure 6A). In contrast to the full-length proviral sequence, we observed for the 5′ LTR truncated provirus that expression of Gag was not reduced when RecHTLV was co-expressed, implying that RecHTLV-mediated reduction of Gag expression worked through recombination of the loxHTLV target sites (Figure 6C). We conclude that RecHTLV can recombine loxHTLV in the context of the full-length HTLV-1 genome.
Figure 6.

RecHTLV recombines loxHTLV in a viral context
(A) Scheme of the recombinase expression vector and the HTLV-1 proviral expression plasmids pCS-HTLV-X1MT and pCMV-HT1M. pCS-HTLV-X1MT contains two full-length LTRs while pCMV-HT1M has only one truncated 5′ LTR, therefore lacking one loxHTLV target site. In the bicistronic recombinase expression construct, the self-cleaving 2A peptide (P2A) allows simultaneous expression of the recombinase fused to a 3xFLAG peptide and a nuclear localization signal (NLS-Rec) and a GFP cassette driven by an EF1a. (B and C) Left: detection of Gag p55/p19 protein and FLAG-tagged recombinases 72 h after transient transfection of 293T cells with recombinase expression constructs Puro (control), Cre, and RecHTLV together with proviral expression plasmids (pCS-HTLV-X1MT, pCMV-HT1M). Hsp 90 served as control. Numbers indicate densitometric analysis of Gag (p55 + p19) protein, normalized to Hsp 90. Marked with red arrows are the expected sizes of p55 and p19 products. Blots were cut due to technical reasons. Right: densitometric analysis of Gag protein normalized to Hsp90. The bar shows the mean ± SD of three to four independent experiments (∗p < 0.05; ns, not significant, unpaired two-sided t test using the logarithm of the normalized values).
RecHTLV expression reduces HTLV-1 infection of Jurkat T cells
Next, we wanted to test if the expression of RecHTLV could reduce HTLV-1 infection in Jurkat T cells. To this end, we generated Jurkat cell lines that continuously express RecHTLV under control of the EF1a promoter, as well as control lines expressing Cre. Because HTLV-1 infection is dependent on close cell-cell contacts and hardly occurs from free viral particles,58,59,60,61 we co-cultured the recombinase-expressing Jurkat T cell lines with the chronically infected C91-PL cell line, which can produce infective viral particles (Figure S7A).60,62,63 To be able to differentiate Jurkat from C91-PL cells, we stained the Jurkat cell lines with 7-amino-4-chlormethylcumarin (CMAC) blue dye prior to co-culture (Figures 7A and 7B).64 As a marker of productive infection, we measured intracellular Tax protein expression by flow cytometry 5 days post co-culture. As a positive control, we applied the compound Raltegravir, which has been shown to inhibit HTLV-1 integrase activity and therefore blocks de novo infection of cells.65 As expected, Raltegravir reduced Tax levels of Jurkat T cells compared with the DMSO solvent control (Figures 7C and S7B), presumably because the drug prevented HTLV-1 integration into the Jurkat cell genome. Importantly, we observed a similar reduction of Tax expression in RecHTLV-expressing Jurkat T cells, while this reduction was not observed for cells expressing Cre (Figures 7B and 7C). These results indicate that expression of RecHTLV reduces stable integration of HTLV-1 in Jurkat T cells.
Figure 7.

RecHTLV expression reduces HTLV-1 infection of Jurkat T cells
(A) Schematic representation of the experimental work: chronically HTLV-1-infected C91-PL donor cells were co-cultured for 5 days with CMAC-Blue pre-stained Jurkat acceptor T cells, which constitutively express the RecHTLV recombinase or Cre as a control. Infected Jurkat T cells were detected by intracellular FACS staining of Tax protein. (B) Representative dot plots obtained by flow cytometry showing the gating strategy for co-culture of Jurkat acceptor cells (CMAC+) and C91-PL donor cells (CMAC–). The two populations are discriminated based on the CMAC staining and Tax expression is further detected in the distinct cell populations. (C) Quantification of the frequency of Tax+ cell in Jurkat (CMAC+) acceptor cells. Left: integrase inhibitor Raltegravir compared with the solvent control DMSO; Right: Jurkat cells expressing either Cre or RecHTLV. For statistical analysis, the fraction of C91-PL Tax+ cells in the co-culture was considered. Error bars show 95% confidence intervals (n = 8; ∗∗p < 0.01, ∗∗∗p < 0.001; linear mixed model followed by a t test). Parts of the figure were created with BioRender.com.
RecHTLV expression excises the integrated HTLV-1 provirus from infected cells
Finally, we tested if RecHTLV can excise the HTLV-1 provirus from a cell line isolated from an ATL patient. We used the chronically infected SP patient-derived cell line that has been shown to harbor four proviruses with known HTLV-1 integration sites, including two proviruses containing full-length LTRs.66 We first verified that our SP cells harbor these integrated proviruses, on chromosome 6 and 20, and that both contained the loxHTLV sequences in their respective LTRs (Figure S7C). We then transduced the SP cell line with a lentivirus that constitutively expresses the RecHTLV recombinase (SIN LV RecHTLV) or GFP as a negative control (SIN LV GFP) (Figure 8A). At day 9 or 10 after infection, we extracted genomic DNA from the infected cells and performed PCR reactions with primers flanking the integration site (Figures 8B and 8C). Consistent with the excision of the provirus, we could detect the genomic scar for both loci, as we obtained PCR bands of the expected size only in the cells expressing the RecHTLV recombinase (Figure 8D). Sequencing of the DNA fragments indeed confirmed the exact and precise removal of the provirus from these loci (Figure 8D). Hence, RecHTLV is capable of excising the HTLV-1 provirus from patient-derived cells.
Figure 8.

RecHTLV expression excises the integrated HTLV-1 provirus from infected cells
(A) Schematic representation of the used lentiviral expression constructs SIN LV RecHTLV and SIN LV GFP. The EF1a promoter drives the expression of the RecHTLV, the HTLV-1 specific recombinase fused to nuclear localization signal (NLS-Rec) or GFP, followed by a P2A peptide, which allows the expression of a puromycin resistance gene. (B) Experimental workflow: the HTLV-1-infected SP cell line was transduced with lentiviral vectors expressing RecHTLV or GFP. After 9–10 days, genomic DNA was extracted and a PCR was performed followed by Sanger sequencing to confirm the correct excision of the HTLV-1 provirus. (C) Scheme of the HTLV-1 provirus integrated into the genome of the SP cells. The black arrows indicate the position of the primers used, which bind in the genomic DNA of the host cell. Upon recombination on the loxHTLV sites by RecHTLV, the proviral genome will be excised leaving a “genomic scar” consisting of one LTR sequence. (D) Left: PCR on genomic DNA of recombinase-treated SP cells with primers as indicated in (B) for the proviruses in chromosomes 6 and 20. Marked with red arrows are the PCR products, which indicate excision of proviral DNA. MOI indicates multiplicity of infection of the recombinase or GFP constructs and d indicates the days post transduction. Right: representation of the sequencing strategy and Sanger sequencing results from the genomic scar on chromosomes 6 and 20 following RecHTLV expression. The red arrows indicate the extent of the Sanger sequencing reads, which confirm the correct excision product. Parts of the figure were created with BioRender.com.
Discussion
Up until today, HTLV-1 is an incurable infectious disease. However, the proviral integration in the host genome and the stability of viral sequences in infected cells make HTLV-1 a suitable target for gene editing tools. In this study, we developed a recombinase-based approach for removing the HTLV-1 provirus from infected cells. The RecHTLV recombinase is able to target and efficiently excise a sequence present in the HTLV-1 LTRs in bacteria and human cells. Sequencing of genomic DNA isolated from cells treated with RecHTLV revealed that the editing was successful and precise. Remarkably, RecHTLV harbors 46 mutations in its amino acid sequence when compared with Cre, likely due to the disparity between the loxHTLV target sequence and loxP, which demonstrates the flexibility to use designer-recombinases to target sequences, even when they only have remote similarity to the original loxP sequence.
To investigate potential side effects of RecHTLV, we employed an in-silico-based and an experimental-based off-target analysis. We could not detect activity of RecHTLV on the predicted potential off-target sequences, nominated based on sequence similarity to the loxHTLV target site. Furthermore, using an experimental unbiased approach, we showed that ChIP-seq can be used to find unspecific binding sites in the genome, with a sensitivity that allowed us to detect an off-target site showing less than 10% of recombination activity. Fortunately, this potential off-target sequence, lox-peak7, occurs in the genome only once, which alleviates the threat of undesired editing. Furthermore, recombination events between lox-peak7 and loxHTLV are highly unlikely since identical spacer sequences are required for recombination and the spacer sequences of lox-peak7 and loxHTLV are different.15 Nevertheless, to bring RecHTLV further toward potential therapy, additional efforts should be employed to ensure that the risk of off-target editing is brought down to a minimum.18,49,67 An extensive analysis of the identified peaks from the ChIP-seq experiment combined with whole-genome sequencing of treated cells may provide a broader overview of potential off-target sequences.39 If required, various approaches for enhancing specificity of evolved recombinases have been already described.20,39,67
To study the efficacy of recombinases in the context of HTLV-1, relevant cell line models are required. While it has been shown that HTLV-1 naturally infected T cells contain mostly a single integrated provirus, many cell lines used in the field harbor several copies of the provirus, accumulated due to de novo superinfection during passaging and culture, such as the extensively used MT-2 cell line.68,69 Furthermore, some of these copies are often truncated, thus obstructing the study of the virus as the biology no longer mirrors the situation of the cells in an infected individual. We found that RecHTLV excises HTLV-1 from the chronically infected SP cell line, which has been shown to contain four proviruses, including two proviruses containing full-length LTRs.66 However, for future studies, cellular models or animal models with higher resemblance to an infected patient should be systematically compared to analyze the effect of RecHTLV treatment on the expression of HTLV-1 viral proteins and evaluate its potential effect on cell proliferation and tumor development. For this aim, one could use some of the well-described cell lines in the field such as TL-oM1 or ED,70,71 which harbor only one copy of the HTLV-1 provirus, or work with primary material derived from infected patients and determine provirus sequence and integration site.32 Interestingly, the loxHTLV sequence is fully conserved in the LTRs of simian T cell leukemia virus (STLV-1), a retrovirus that causes similar pathologies in monkeys as HTLV-1 in humans and can be used as a model for ATL.72 Hence, it would be interesting to study the effects of RecHTLV on STLV-1 biology in naturally infected non-human primates, such as baboons or chimpanzees.73
Previous attempts at gene editing in HTLV-1 have focused on disrupting the expression of viral proteins, either by using zinc finger nucleases to target the LTR and thus disrupt its promoter function, or directly by using CRISPR-Cas9 on the viral Hbz gene.34,35 While the obtained results were impressive, the editing capacity of nucleases relies on the repair machinery of the treated cell. Therefore, the resulting sequence is unpredictable and escape mutants are likely to emerge, as has been documented for targeting HIV-1 with nucleases.11,12,13,14 Tanaka et al. aimed to target both LTRs and consequently remove the provirus of the infected cell using a similar approach to ours. While the efficiency of editing was high, they observed that most of the edited cells had indels in the LTRs.34 Therefore, also here, the appearance of indels after treatment could be detrimental since the edited virus could still be functional and therefore resistant to further treatment. In contrast, designer-recombinases offer predictability at nucleotide precision making them an excellent tool for excising the provirus from infected cells. Thus, RecHTLV could be used to reduce proviral load and improve the prognosis of HTLV-1 carriers with a latent infection, as high proviral load is associated with the development of HTLV-1-associated diseases such as HAM/TSP and ATL.74,75,76 However, the effectiveness of RecHTLV might be limited in patients already suffering from ATL since the HTLV-1 5′ LTR is frequently deleted or hypermethylated in ATL,77,78 which could hinder the accessibility of RecHTLV to the loxHTLV target sites.
Taken together, our data show that designer-recombinases are a promising tool to reverse HTLV-1 infections in human cells.
Materials and methods
All PCR primers used in this study are listed in Table S1.
Plasmids
The evolution plasmids pEVO-loxHTLV and all the pEVO-loxHTLV subsite plasmids, and the pEVO-lox containing the loxHTLV off-target sites, were generated using the Cold Fusion Cloning Kit (System Biosciences). The inserts were PCR amplified using the pEVO as a template and primers containing the new lox sites as an overhang (primers 3–18, 29–34). The backbone was prepared by digesting the pEVO vector with BglII restriction enzyme (NEB). The cold fusion reaction was performed following the manufacturer’s protocol.
The pCAGGS-lox-pA-lox-mCherry reporter plasmid used for the study of the activity of RecHTLV in HeLa cells was generated from the plasmids described by Lansing et al. (2020) and Lansing et al. (2022), originally derived from pCAG-loxPSTOPloxP-ZsGreen, a gift from Pawel Pelczar (Addgene plasmid no. 51269; http://n2t.net/addgene:51269; RRID:Addgene_51269).79 The target sites were introduced by PCR with overhang primers (primers 23–24) and were inserted in the pCAGGS reporter using SalI and EcoRI restriction digestion cloning.
The expression vector pIRES-NLS-eGFP was generated from the pIRESneo-Cre Vector as described by Lansing et al. (2020). The recombinases were inserted in the pIRES expression plasmid by restriction digest using XbaI and BsrGI.
The SVFF-loxHTLV-puro-mCherry reporter lentivirus plasmid (pLenti-loxHTLV-reporter) was generated from the bicistronic tagBFP/eGFP reporter obtained from D. Sürün80 and inserted in the plentiCRISPR v.2 lentiviral backbone, a gift from Feng Zhang (Addgene plasmid no. 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961).81 In brief, the eGFP was exchanged with an mCherry cassette using AgeI and XhoI (NEB). A puromycin cassette was flanked by the loxHTLV sites by PCR amplification using primers 27 and 28 and inserted by golden gate cloning with BsmBI.
The pLentiX-inducible system was constructed as described by Lansing et al. (2022), a DNA fragment for TRE3G-EF1a-Tet-ON 3G-P2A-eGFP was inserted into the lentiviral backbone of the plentiSAMv2, a gift from Feng Zhang (Addgene plasmid no. 75112; http://n2t.net/addgene:75112; RRID:Addgene_75112)82 using NheI and KpnI restriction enzymes (NEB). Recombinases were inserted in the pLentiX with restriction enzyme digestion and ligation using the BsrGI-HF and XbaI enzymes (NEB). For the ChIP-seq experiment, the pLentiX was further modified to fuse the recombinases to eGFP. In brief, the GFP cassette was exchanged for a puromycin cassette using HpaI restriction digest. Furthermore, the GFP cassette was inserted downstream of the XbaI restriction site. The recombinases were PCR amplified with a reverse primer which removes the stop codon from the coding sequence of the recombinases and cloned using BsrGI and XbaI.
The pLenti-EF1a-P2A-Puro was derived from the pLentiX vector by exchanging the TRE3G promoter with a minimal EF1a promoter using NheI and AscI. Furthermore, the Tet-On-3G coding sequence and the additional EF1a promoter was removed using XbaI and AgeI restriction enzymes. The resultant plasmid pLenti-EF1a-P2A-Puro allows the cloning of recombinases using BsrGI and XbaI restriction digest cloning.
The coding sequence of RecHTLV was codon optimized for mammalian expression and synthesized as a fragment by Twist Bioscience (South San Francisco, CA). The codon-optimized RecHTLV was then cloned by digestion with BsrGI and XbaI restriction enzymes to the vectors used for viral experiments (pLenti-EF1a-P2A-Puro).
Furthermore, the following plasmids were used for transient transfection experiments: pcDNA3.1 (Life Technologies); the Tax-1 wild-type expression vector pHpX-Tax55; the full-length HTLV-1 proviral clone pCS-HTLV-X1MT56; the HTLV-1 packaging vector pCMV-HT1M harboring a deletion in the 5′ LTR, expressing all HTLV-1 gene products under the control of a CMV promoter.57
For delivery of RecHTLV in SP cells, an HIV-1-derived replication-incompetent lentiviral vector (SIN LV RecHTLV) was constructed that provides high safety levels due to a split packaging system and self-inactivating (SIN) vector design.83 The gene sequence encoding for RecHTLV and a P2A peptide puromycin resistance gene expression cassette was placed under the control of an EF1a promotor. For this purpose, the following sequences with homologous overhangs were amplified by PCR: the coding RecHTLV sequence with primers 63/64 from pLentiX-EF1a-flag-NLS-G9co-P2A-puro (insert 1); P2A puromycin resistance gene cassette with primers 65/66 from plasmid 1555 (insert 2); lentiviral plasmid backbone with primers 67/68 from plasmid 1539. For the lentiviral GFP control vector, insert 1 was replaced by GFP. GFP coding sequence was amplified using primers 69/70 from plasmid 1455 and primers 71/72 were used for the lentiviral vector backbone. The construction was carried out using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs) according to the manufacturer’s instructions.
Generation of recombinase libraries and evolution strategy
The initial library was generated by shuffling previously generated Cre-derived libraries. Substrate-linked protein evolution was performed as described previously16,20,21,38,39 by cloning the library of recombinases into the pEVO vector with the corresponding target sites (starting from the subsites to the final target site).
XL1 blue cells were transformed with the pEVO libraries and grown in 100 mL LB medium containing chloramphenicol (25 μg/mL) and the expression of the library was induced overnight by the addition of arabinose in the system. After 14–16 h, 10 mL of the culture was used for extraction of the plasmids using the GeneJET Plasmid Miniprep Kit. Plasmid DNA (500 ng) was digested using NdeI and AvrII (NEB) restriction enzymes and 25 ng of the digested DNA was used as template for PCR to amplify the active recombinases in the library. The PCR was performed using MyTaq polymerase (Bioline), which introduces mutations due to its lack of proof-reading activity (primers 1–2). The PCR product was digested with XbaI and BsrGI and the recombinase band (∼1 kb) was extracted from an agarose gel. The new insert was then cloned to the pEVO backbone starting therefore a new cycle of evolution.
Every fifth cycle of evolution, shuffling was performed. To this end, 25 ng of the NdeI and AvrII digested miniprep containing the recombinase library was PCR amplified using Herculase II Fusion Polymerase (Agilent) (primers 1–2). The PCR product was purified and sonicated using Covaris M220 to obtain fragments of about 200–300 bp (50 W peak incident power, 200 cycles/s, 150 s treatment time, and 20% duty factor). The fragments were reassembled by PCR, using the sonicated fragments as a template. This PCR was followed by a second reassembly PCR where the full-length recombinase was amplified using Herculase II Fusion polymerase (primers 19–20). The PCR was purified using the ISOLATE II PCR and Gel Kit (Bioline) following the manufacturer’s protocol and the elute was digested using XbaI and BsrGI-HF (NEB) and after gel purification the digested product was used as an insert for ligation into the pEVO plasmid to continue evolution.
Test digest for assessing the activity of libraries/recombinases
To assess the activity of the libraries during evolution, 500 ng of the miniprep DNA plasmid from the induced cultures was digested with XbaI and BsrGI-HF restriction enzymes (NEB) and 250 ng of the digestion was run in a 0.8% agarose gel stained with RedSafe (Intron Biotechnology). The same digestion was performed to test individual clones, using 500 ng of plasmid DNA extracted from 5 mL of induced culture.
Quantification of recombination from agarose gel
Recombination efficiency was quantified by the ratio of the non-recombined and the recombined band from the test digest of the pEVO plasmid. The gel images were obtained using an Infinity VX2-3026 transilluminator and the Infinity Capt software (Vilber) and the band intensities were calculated using the GelAnalyzer 19.1 (www.gelanalyzer.com) software by Istvan Lazar, Jr., PhD, and Istvan Lazar, Sr., PhD, CSc.
Deep sequencing of the recombinase libraries
To prepare the loxHTLV library for deep sequencing, 500 ng of the pEVO plasmid from the last cycle of evolution was digested with the restriction enzymes NdeI and AvrII (NEB) to select the active variants in the library. The digestion was further desalted with an MF-membrane and transformed into XL1-Blue E. coli competent cells (Agilent). The bacterial culture was grown for 14–16 h in 100 mL LB containing 25 μg/mL of chloramphenicol. The plasmid DNA was extracted using the GeneJet Plasmid Miniprep Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. From this plasmid DNA, 5 μg was digested with BsrGI-HF and XbaI (NEB) restriction enzymes and the 1,041 bp containing the recombinase library was enriched by twice binding of the pEVO backbone to the custom SPRI beads.84 The supernatant was cleaned with Ampure XP beads (Beckman Coulter) and the resulting DNA was quantified using a Qubit HS assay Kit on a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and sequenced by the CRTD Deep sequencing facility with a Sequel System 6.0 using the Pacbio HiFi method. Circular consensus sequencing data were generated with PacBio’s ccs v.3.4.1 and filtered to only keep sequences of length 1,034–1,200 bp with a minimum predicted accuracy of 99.997% (Phred score of ∼25). The data were converted to FASTA using SAMtools 1.11 and the coding sequences of the recombinases translated to amino acids using the protein2dna:bestfit alignment model of exonerate v.2.3.0. Furthermore, the sequences were filtered to guarantee that all start with a methionine and are 344 amino acids long with grep, sed, and awk. Further processing and analysis of the sequencing data were performed in R (v.4.1.0) using the dplyr, Sequence tools (https://github.com/ltschmitt/SequenceTools) and ggplot2 packages.
Cell culture
HeLa Kyoto (MPI-CBG, Dresden) cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) with 10% fetal bovine serum and 1% penicillin/streptomycin (Pen/Strep) (Thermo Fisher Scientific) at 37°C with 5% CO2 in a humidified incubator. HEK293T cells were kept in DMEM (Gibco, Life Technologies, Darmstadt, Germany) containing 10% fetal calf serum (FCS) (Capricorn Scientific, Ebsdorfergrund, Germany), L-glutamine (0.35 g/L), and Pen/Strep (0.12 g/L each). The CD4+ T cell line Jurkat85 was cultivated in RPMI 1640 (45%; Gibco, Life Technologies) and Panserin 401 medium (45%; PAN-Biotech, Aidenbach, Germany), supplemented with 10% FCS, L-glutamine, and Pen/Strep. The HTLV-1 in-vitro-transformed CD4+ T cell line C91-PL86 was cultured in RPMI 1640, containing 10% FCS, L-glutamine, and Pen/Strep. To confer puromycin resistance, C91-PL cells were transduced with pLenti-EF1a-P2A-Puro empty vector as described before87 and subsequently cultivated in C91-PL medium containing 1 μg/mL puromycin.
HTLV-1-infected cells (SP) (NIH AIDS Reagent Program, no. 3059) were cultured using RPMI 1640 (Lonza) containing 2.0 mM L-glutamine (PAN Biotech), 100 U/mL penicillin/100 mg/mL streptomycin (Merck), 10% FBS (PAN Biotech), and 250 U/mL human IL-2 (Biomol) at 37°C with 5% CO2 in a humidified incubator.
Screen in HeLa cells for tolerated recombinases
The final library was cloned in the pLentiX vector and the reporter loxHTLV cells were infected with the lentiviral particles containing the recombinases. Cells were transduced with a low multiplicity of infection to ensure that only one recombinase integrated in each cell. Expression of the library was induced during 7 days using 50 ng/mL doxycycline. The GFP+ cells were then sorted by flow cytometry and expanded for 5 days and then sorted again for GFP+mCherry+ to retrieve the active recombinases from the library. Approximately 3,500 cells were sorted in the second sorting round.
PCR to screen for active clones
Genomic DNA from the pooled sorted cells was extracted using the QIAamp DNA Blood Kit following the manufacturer’s protocol. Recombinases were then retrieved by PCR using the high-fidelity Herculase II Phusion DNA polymerase (Agilent) (primers 21–22) and the PCR product was cloned into the pEVO and the bacterial transformation was plated on a chloramphenicol plate. The next day, 96 colonies were picked into a deep-well 96-well plate and grown overnight using chloramphenicol LB containing 10 μg/mL L-arabinose. A colony PCR was performed using as a template 1 μL of the overnight culture and MyTaq polymerase (Bioline) (primers 82–84). The PCR products were loaded in a 2% agarose gel for analysis.
Cell culture plasmid transfection assays
The plasmid assay in cell culture to test the on-target activity of the recombinases was performed as follows: 15,000 HeLa cells/well were seeded in a 96-well plate. The next day, 100 ng of reporter plasmid and 150 ng of the pIRES expression plasmid were transfected using lipofectamine 2000 (0.3 μL per well). Forty-eight hours after transfection the cells well analyzed in a MacsQuant VYB (Miltenyi). After single-cell gating, recombination efficiency was calculated by dividing the percentage of double-positive cells (mCherry+GFP+) by the frequency of all GFP+ cells.
The test in the genomic reporter cells was performed as follows: 100,000 cells per well were seeded in a 24-well plate and transfected with 500 ng of pIRES expression plasmid using 1 μL of lipofectamine per well. Forty-eight hours post transfection half of the cells were analyzed for the expression of mCherry and GFP using a MacsQuant VYF flow cytometer (Miltenyi). Due to a low percentage of mCherry+ cells derived from the reporter cell line, only recombinase-expressing cells (GFP+) were considered to calculate recombination efficiency. The other half of the cells were used for genomic DNA extraction using the QIAamp DNA Blood Mini Kit (QIAGEN) following the manufacturer’s protocol. Genomic DNA (100 ng) was used as a template for PCR to detect recombination of the reporter (primers 25–26).
For other transfections using HEK293T cells, 24 h before transfection, 5 × 105 293T cells were seeded in 6-well plates. Transfection was conferred using GeneJuice transfection reagent (Merck Millipore, Darmstadt, Germany) according to the manufacturer’s protocol using a total amount of 2 μg DNA at a 1:1 ratio of the plasmids used.
Jurkat T cells were transfected as described previously,88 with minor modifications. In brief, 5 × 106 cells were electroporated with the Gene Pulser X Electroporation System (Bio-Rad) at 290 V and 1,500 μF, using a total amount of 50 μg DNA (30 μg pLenti-EF1a-P2A-Puro and 20 μg pHpX-Tax).
Lentivirus production
The lentiviral transfer plasmids were co-transfected using standard polyethylenimine transfection with the lentiviral gag/pol packaging plasmid (psPAX2, Addgene no. 12260) and the envelope plasmid VSV-G (pMD2.G, Addgene no. 12259) in a molar ratio of 3:1:1 in HEK293T cells. Forty-eight hours after transfection, the supernatant containing the viral particles was harvested and filtered (0.45 mm pore-size PVDF membrane filter, Millipore). The supernatants were either used directly to infect the desired target cells or stored at −80°C for later usage.
VSV-g pseudotyped lentiviral particles for transduction of SP cells were produced by transient cotransfection of HEK293T cells with the lentiviral plasmid and the respective packaging plasmids83 using TransitLT-1 as a transfection reagent according to the manufacturer’s protocol (Mirus Bio LLC). At 72 h post transfection, viral supernatants were collected and passed through 0.2 μm pore size filters to ensure removal of any viral aggregates. The viral supernatant was concentrated by ultracentrifugation through a 20% sucrose cushion and resuspended in RPMI medium. For the determination of the titer (IU/mL) HEK293T cells were transduced with various volumes of vector and, after 72 h, the genomic DNA was isolated and the titer was determined by ddPCR using primers 73, 74, and 75, and primers specific for the housekeeping gene ribonuclease P/MRP subunit P30 (RPP30) (PrimePCR ddPCR copy number assay: RPP30, Human, Bio-Rad). The supernatants were stored at −80°C for later usage.
Transduction and infection of cell lines
HeLa Kyoto cells or Jurkat cells were transduced with lentiviral particles generated from the corresponding lentiviral vectors. For the constructs containing a puromycin cassette, 72 h after transduction cells were selected with 2 μg/mL puromycin for 7 days.
SP and HEK293T cells were transduced with various amounts of vector in the presence of 5 μg/mL protamine sulfate (Merck) and spinoculated at 450 × g for 10 min at ambient temperature. After spinoculation cells were cultivated at 37°C and 5% CO2 until further use. SP cells were selected with 0.25 to 1 μg/mL puromycin for 7 days.
Toxicity assay
HeLa-derived cell lines were transduced to ∼46% RecHTLV and ∼89% empty control with a transgene expressing GFP and tetracycline-inducible recombinase system were cultured in tetracycline-free medium (Capricorn Scientific, FBS-TET-12A). A total of 15,000 cells/well were seeded in a flat-bottomed 96-well plate and continuously induced for 3, 6, or 9 days with 100 ng/mL of doxycycline. The percentage of GFP expressing cells was measured by flow cytometry using a MacsQuant VYB.
ChIP-seq and qPCR validation
The ChIP-seq experiment was performed using RecHTLV, RecHTLV-H289L (catalytic mutant), and another clone (E5 and E5-H289L). Recombinases were fused with eGFP and cloned in a modified version of the tetracycline-inducible pLentiX vector. HeLa cells were infected with the lentivirus and selected with 2 μg/mL of puromycin 72 h after transduction for 7 days. Cells were grown in 10-cm dishes and the expression of the fused recombinases with GFP was induced for 24 h with 100 ng/mL of doxycycline. Cells were inspected under the microscope for expression of nuclear GFP to confirm that the construct works. Cells were crosslinked with 1% formaldehyde for 10 min and chromatin extraction and shearing were performed using the truChIP Chromatin Shearing Kit (Covaris) following the manufacturer’s protocol for high cell number. Chromatin was sheared using a Covaris M220 sonicator. As input sample, for further qPCR validation, 1% of the sheared chromatin was separated and the rest of the sample was used for immunoprecipitation. The recombinase-protein fusion was immunoprecipitated using a goat GFP antibody (MPI-CBG antibody facility) and Protein G Sepharose beads (Protein G Sepharose 4 Fast Flow, GE Healthcare). Complexes were washed 1× with low salt immune complex wash buffer, 1× high salt wash buffer, 1× LiCl wash buffer, and 1× TE buffer and eluted with elution buffer (1% SDS, 0.1 M NaHCO3). Eluted samples and the input samples were treated with RNAse, reverse crosslinked, and further purified using the ISOLATE II PCR and Gel Kit (Bioline). The eluted DNA was sent to the CRTD Deep Sequencing Facility for library preparation and Illumina sequencing. Libraries for Illumina were prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina from 17 to 75 ng of ChIP DNA with 15 PCR amplification cycles and size selection using AMPure XP beads, and paired-end sequencing was performed on an Illumina HiSeq 2000. Sequencing reads were aligned to the GRCh38.p12 human genome assembly using STAR aligner89 using ChIP-seq analysis parameters. Peak calling was performed with Genrich (https://github.com/jsh58/Genrich) using the ENCODE blacklist (v.2).90 All manipulations and comparisons of genomic intervals were performed using BEDTools91 and visualizations of pile-up reads were generated with the UCSC Genome Browser92,93 directly from BAM files sorted by samtools command line tool.94
Recombination assay of ChIP-seq-selected peaks in bacteria
Ten peaks were selected for further recombination testing in bacteria in a plasmid-based assay. A total of 92 bp around the summit of the peaks was cloned twice in a modified version of the pEVO vector using BglII and PciI restriction sites (primers 35–62). The RecHTLV recombinase was cloned in the pEVO using BsrGI and XbaI restriction sites. After transformation in XL1blue E. coli the expression of the recombinase was induced using a concentration of 200 μg/mL L-arabinose. Plasmid extraction was performed and recombination was analyzed using test digestion (see above).
Western blot
Transfected cells were resuspended at the indicated time points after transfection (48 or 72 h) in lysis buffer (150 mM NaCl, 10 mM Tris-HCl [pH 7.0], 10 mM EDTA, 1% Triton X-100, 2 mM DTT, protease inhibitors leupeptin and aprotinin [20 μg/mL each] and 1 mM phenylmethylsulfonyl fluoride), subjected to repeated freeze-and-thaw cycles between −196°C (liquid nitrogen) and 30°C and additionally sonicated three times for 20 s. Equal amounts of proteins (40 μg) were boiled for 5 min at 95°C in sodium dodecyl sulfate (SDS) loading dye (10 mM Tris-HCl [pH 6.8], 10% glycerine, 2% SDS, 0.1% bromophenol blue, 5% β-mercaptoethanol). SDS-PAGE and immunoblot using Immobilon-FL PVDF (Merck Millipore, Billerica, MA) or nitrocellulose transfer membranes (Whatmann, Protran, Whatmann, Dassel, Germany) were performed using standard protocols. Proteins were detected with the following primary antibodies: mouse anti-Tax (derived from the hybridoma cell line 168B17-46-34, provided by B. Langton through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH),95 mouse monoclonal anti-HTLV-1 gag p19 (TP-7, ZeptoMetrix), mouse monoclonal anti-FLAG (M2, Sigma), mouse monoclonal anti-Hsp90 α/β (F-8, Santa Cruz Biotechnology), mouse monoclonal anti-GAPDH (sc-47724, Santa Cruz Biotechnology), and mouse monoclonal anti-α-tubulin (T9026, Sigma). Secondary antibodies were anti-mouse Alexa Fluor 647 (Life Technologies) or anti-mouse conjugated with horseradish peroxidase (GE Healthcare, Little Chalfont, UK). Fluorescence or chemiluminescence signals were detected using the Advanced Fluorescence Imager camera (ChemoStar, Intas Science Imaging, Göttingen, Germany). Densitometric analysis was performed with the advanced image data analyzer (AIDA) (v.4.22.034, Raytest Isotopenmessgeräte, Straubenhardt, Germany) to compare Tax or Gag expression levels, respectively.
Co-culture assay between chronically infected C91-PL-Puro donor cells and pre-stained Jurkat-pLenti-EF1a acceptor cells
C91-PL-Puro cells (1 × 106) were co-cultured with 1 × 106 Jurkat-pLenti-EF1a T cells for 5 days in a 1:1 mixture of C91-PL and Jurkat medium containing puromycin to maintain recombinase expression in Jurkat-pLenti-EF1a T cells. Co-cultures were split twice, at 24 and 72 h after the onset of the experiment, at a 1:2 ratio. Jurkat-pLenti-EF1a T cells have been pre-stained with the live cell dye CellTracker Blue CMAC (Thermo Fisher Scientific) as described earlier.64 As a control, pre-stained Jurkat-pLenti-EF1a T cells were pre-incubated with 10 μM of the integrase inhibitor Raltegravir for 24 h before initiating the co-culture. Raltegravir was replenished whenever fresh medium was added to the co-culture to keep a final concentration of 10 μM. Analogously, Jurkat-pLenti-EF1a T cells were treated with DMSO as solvent control. Cells were stained using mouse anti-Tax95 and anti-mouse Alexa Fluor 647-conjugated secondary antibodies (Life Technologies). In brief, cells were permeabilized with 0.3% Triton X-100, dispensed in FACS buffer (PBS/1% FCS/2 mM EDTA), for 10 min at room temperature. Staining with primary and secondary antibodies, diluted in FACS buffer, was conferred for 45 min at 37°C with three washing steps in between. Co-cultures were analyzed using the BD LSR II flow cytometer (BD Biosciences, San Jose, CA) and cells were discriminated by CMAC staining (Jurkat-pLenti-EF1a: CMAC-positive; C91-PL-Puro: CMAC-negative) and their different size (FSC/SSC). The percentage of Tax-positive cells within the Tax+ C91-PL donor and the CMAC-positive cells (Jurkat-pLenti-EF1a T cells) was examined to measure productive infection. For statistical analysis the coefficients were estimated by a linear mixed model followed by a t test using Satterthwaites’s method, provided by the lmerTest package for R.96 To measure background Tax+-stained cells in non-infected control, Tax+ Jurkat-pLenti-EF1a-Cre and Jurkat-pLenti-EF1a-RecHTLV were quantified (Figure S7B).
Verification of the HTLV-1 integration sites in SP cells
To verify the integration sites on chromosomes 6 and 20 and to demonstrate the integrity of the LTR of the proviruses in the SP cells published in Meissner et al.,66 the sequence regions were amplified with the following primers and verified by sanger sequencing using the same primers. For the verification of the integration on chromosome 6, primers 85/86 were used for the amplification of the 5ʹ LTR and primers 87/88 for the 3ʹ LTR. For the integration on chromosome 20, primers 89/90 were used for the 5ʹ LTR and primers 91/82 for the 3ʹ LTR.
Detection of the genomic scar in SP cells
Genomic DNA from cells transduced with RecHTLV or GFP control vector was isolated 9 or 10 days after transduction and the genomic scar was detected by PCR using the following primers. Primers 76 and 77 were used to detect the scar on chromosome 6. For detection of the scar on chromosome 20, primers 78/79 or 80/81 were used. The primers were designed to not be able to amplify the genomic site in the absence of the HTLV-1 provirus (one of the primers from each primer pair always binds partially to the LTR and partially to the genomic DNA sequence). The PCR fragment was visualized by agarose gel electrophoresis and verified by Sanger sequencing using the same primers.
Acknowledgments
We thank Lukas Theo Schmitt for preparing and sequencing the recombinase library. This work was supported by the European Union (ERC 742133, H2020 UPGRADE 825825], the Federal Ministry of Education and Research (BMBF) of the Federal Republic of Germany (Milk-TV, 01KI2023, to A.K.T.-K.), and by Deutsche Forschungsgemeinschaft (DFG) (TH2166/1-1, BU140/7-1). Furthermore, we thank the DRESDEN-concept Genome Center at the CMCB from the TU Dresden for performing the sequencing of the ChIP samples and the Flow Cytometry Core Facility of the CMCB Technology Platform at TU Dresden for their excellent support.
Author contributions
T.R.-R., J.K., F.L., P.M.S., and J.S. performed the directed evolution of the recombinase libraries. T.R.-R. performed the experiments for single-clone screening and off-target testing in bacteria and in reporter cell lines and analyzed the data. T.R.-R. performed the recombinase ChIP-seq experiment. M.P.-R. performed the bioinformatic analysis, including the Chip-seq data and the target site identification. S.M., N.B., and F.S. performed the experiments with the HTLV-1-infected cell lines. T.R.-R. and F.B. wrote the manuscript. F.B., A.K.T.-K., and J.H. contributed to the design and supervision of the study and critically revised the manuscript.
Declaration of interests
T.R.-R., N.B., and F.B. have filled a patent application based on work presented in the manuscript. N.B., J.H., and F.B. are co-founders of Provirex Genome Editing Therapies GmbH. T.R.-R., M.P.-R., F.L., and F.B. are co-founders of Seamless Therapeutics GmbH.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.03.014.
Supplemental information
Data availability
All original data are available from the authors upon request.
References
- 1.Ye L., Wang J., Beyer A.I., Teque F., Cradick T.J., Qi Z., Chang J.C., Bao G., Muench M.O., Yu J., et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA. 2014;111:9591–9596. doi: 10.1073/pnas.1407473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou P., Chen S., Wang S., Yu X., Chen Y., Jiang M., Zhuang K., Ho W., Hou W., Huang J., Guo D. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015;5:15577. doi: 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holt N., Wang J., Kim K., Friedman G., Wang X., Taupin V., Crooks G.M., Kohn D.B., Gregory P.D., Holmes M.C., Cannon P.M. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez E.E., Wang J., Miller J.C., Jouvenot Y., Kim K.A., Liu O., Wang N., Lee G., Bartsevich V.V., Lee Y.-L., et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilen C.B., Wang J., Tilton J.C., Miller J.C., Kim K.A., Rebar E.J., Sherrill-Mix S.A., Patro S.C., Secreto A.J., Jordan A.P.O., et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011;7:e1002020. doi: 10.1371/journal.ppat.1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ebina H., Misawa N., Kanemura Y., Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013;3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao H.-K., Gu Y., Diaz A., Marlett J., Takahashi Y., Li M., Suzuki K., Xu R., Hishida T., Chang C.-J., et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015;6:6413. doi: 10.1038/ncomms7413. [DOI] [PubMed] [Google Scholar]
- 8.Qu X., Wang P., Ding D., Li L., Wang H., Ma L., Zhou X., Liu S., Lin S., Wang X., et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41:7771–7782. doi: 10.1093/nar/gkt571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strong C.L., Guerra H.P., Mathew K.R., Roy N., Simpson L.R., Schiller M.R. Damaging the integrated HIV proviral DNA with TALENs. PLoS One. 2015;10:e0125652. doi: 10.1371/journal.pone.0125652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu W., Kaminski R., Yang F., Zhang Y., Cosentino L., Li F., Luo B., Alvarez-Carbonell D., Garcia-Mesa Y., Karn J., et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G., Zhao N., Berkhout B., Das A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. 2016;24:522–526. doi: 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z., Pan Q., Gendron P., Zhu W., Guo F., Cen S., Wainberg M.A., Liang C. CRISPR/Cas9-Derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016;15:481–489. doi: 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 13.Yoder K.E., Bundschuh R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci. Rep. 2016;6:29530. doi: 10.1038/srep29530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Silva Feelixge H.S., Stone D., Pietz H.L., Roychoudhury P., Greninger A.L., Schiffer J.T., Aubert M., Jerome K.R. Detection of treatment-resistant infectious HIV after genome-directed antiviral endonuclease therapy. Antivir. Res. 2016;126:90–98. doi: 10.1016/j.antiviral.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F. Cre recombinase and other tyrosine recombinases. Chem. Rev. 2016;116:12785–12820. doi: 10.1021/acs.chemrev.6b00077. [DOI] [PubMed] [Google Scholar]
- 16.Buchholz F., Stewart A.F. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol. 2001;19:1047–1052. doi: 10.1038/nbt1101-1047. [DOI] [PubMed] [Google Scholar]
- 17.Santoro S.W., Schultz P.G. Directed evolution of the site specificity of Cre recombinase. Proc. Natl. Acad. Sci. USA. 2002;99:4185–4190. doi: 10.1073/pnas.022039799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eroshenko N., Church G.M. Mutants of Cre recombinase with improved accuracy. Nat. Commun. 2013;4:2509. doi: 10.1038/ncomms3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauber I., Hofmann-Sieber H., Chemnitz J., Dubrau D., Chusainow J., Stucka R., Hartjen P., Schambach A., Ziegler P., Hackmann K., et al. Highly significant antiviral activity of HIV-1 LTR-specific tre-recombinase in humanized mice. PLoS Pathog. 2013;9:e1003587. doi: 10.1371/journal.ppat.1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpinski J., Hauber I., Chemnitz J., Schäfer C., Paszkowski-Rogacz M., Chakraborty D., Beschorner N., Hofmann-Sieber H., Lange U.C., Grundhoff A., et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016;34:401–409. doi: 10.1038/nbt.3467. [DOI] [PubMed] [Google Scholar]
- 21.Sarkar I., Hauber I., Hauber J., Buchholz F. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007;316:1912–1915. doi: 10.1126/science.1141453. [DOI] [PubMed] [Google Scholar]
- 22.Poiesz B.J., Ruscetti F.W., Gazdar A.F., Bunn P.A., Minna J.D., Gallo R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida M., Miyoshi I., Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA. 1982;79:2031–2035. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin F., Tagaya Y., Gallo R. Time to eradicate HTLV-1: an open letter to WHO. Lancet. 2018;391:1893–1894. doi: 10.1016/S0140-6736(18)30974-7. [DOI] [PubMed] [Google Scholar]
- 25.Osame M., Usuku K., Izumo S., Ijichi N., Amitani H., Igata A., Matsumoto M., Tara M. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;1:1031–1032. doi: 10.1016/s0140-6736(86)91298-5. [DOI] [PubMed] [Google Scholar]
- 26.Gessain A., Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol. 2012;3:388. doi: 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gross C., Thoma-Kress A.K. Molecular mechanisms of HTLV-1 cell-to-cell transmission. Viruses. 2016;8:74. doi: 10.3390/v8030074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkis S., Galli V., Moles R., Yurick D., Khoury G., Purcell D.F.J., Franchini G., Pise-Masison C.A. Role of HTLV-1 orf-I encoded proteins in viral transmission and persistence. Retrovirology. 2019;16:43. doi: 10.1186/s12977-019-0502-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laydon D.J., Sunkara V., Boelen L., Bangham C.R.M., Asquith B. The relative contributions of infectious and mitotic spread to HTLV-1 persistence. PLoS Comput. Biol. 2020;16:e1007470. doi: 10.1371/journal.pcbi.1007470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa Y., Kubota R., Tara M., Izumo S., Osame M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood. 2001;97:987–993. doi: 10.1182/blood.v97.4.987. [DOI] [PubMed] [Google Scholar]
- 31.Wattel E., Vartanian J.P., Pannetier C., Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J. Virol. 1995;69:2863–2868. doi: 10.1128/JVI.69.5.2863-2868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsuya H., Islam S., Tan B.J.Y., Ito J., Miyazato P., Matsuo M., Inada Y., Iwase S.C., Uchiyama Y., Hata H., et al. The nature of the HTLV-1 provirus in naturally infected individuals analyzed by the viral DNA-capture-seq approach. Cell Rep. 2019;29:724–735.e4. doi: 10.1016/j.celrep.2019.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Boxus M., Willems L. Mechanisms of HTLV-1 persistence and transformation. Br. J. Cancer. 2009;101:1497–1501. doi: 10.1038/sj.bjc.6605345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka A., Takeda S., Kariya R., Matsuda K., Urano E., Okada S., Komano J. A novel therapeutic molecule against HTLV-1 infection targeting provirus. Leukemia. 2013;27:1621–1627. doi: 10.1038/leu.2013.46. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa M., Shaffer A.L., Ceribelli M., Zhang M., Wright G., Huang D.W., Xiao W., Powell J., Petrus M.N., Yang Y., et al. Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell. 2018;34:286–297.e10. doi: 10.1016/j.ccell.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surendranath V., Chusainow J., Hauber J., Buchholz F., Habermann B.H. SeLOX--a locus of recombination site search tool for the detection and directed evolution of site-specific recombination systems. Nucleic Acids Res. 2010;38:W293–W298. doi: 10.1093/nar/gkq523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Araujo T.H.A., Souza-Brito L.I., Libin P., Deforche K., Edwards D., de Albuquerque-Junior A.E., Vandamme A.-M., Galvao-Castro B., Alcantara L.C.J. A public HTLV-1 molecular epidemiology database for sequence management and data mining. PLoS One. 2012;7:e42123. doi: 10.1371/journal.pone.0042123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lansing F., Paszkowski-Rogacz M., Schmitt L.T., Schneider P.M., Rojo Romanos T., Sonntag J., Buchholz F. A heterodimer of evolved designer-recombinases precisely excises a human genomic DNA locus. Nucleic Acids Res. 2020;48:472–485. doi: 10.1093/nar/gkz1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lansing F., Mukhametzyanova L., Rojo-Romanos T., Iwasawa K., Kimura M., Paszkowski-Rogacz M., Karpinski J., Grass T., Sonntag J., Schneider P.M., et al. Correction of a Factor VIII genomic inversion with designer-recombinases. Nat. Commun. 2022;13:422. doi: 10.1038/s41467-022-28080-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abi-Ghanem J., Samsonov S.A., Pisabarro M.T. Insights into the preferential order of strand exchange in the Cre/loxP recombinase system: impact of the DNA spacer flanking sequence and flexibility. J. Comput. Aided. Mol. Des. 2015;29:271–282. doi: 10.1007/s10822-014-9825-0. [DOI] [PubMed] [Google Scholar]
- 41.Guillén-Pingarrón C., Guillem-Gloria P.M., Soni A., Ruiz-Gómez G., Augsburg M., Buchholz F., Anselmi M., Pisabarro M.T. Conformational dynamics promotes disordered regions from function-dispensable to essential in evolved site-specific DNA recombinases. Comput. Struct. Biotechnol. J. 2022;20:989–1001. doi: 10.1016/j.csbj.2022.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rongrong L., Lixia W., Zhongping L. Effect of deletion mutation on the recombination activity of Cre recombinase. Acta Biochim. Pol. 2005;52:541–544. [PubMed] [Google Scholar]
- 43.Warren D., Laxmikanthan G., Landy A. A chimeric Cre recombinase with regulated directionality. Proc. Natl. Acad. Sci. USA. 2008;105:18278–18283. doi: 10.1073/pnas.0809949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo F., Gopaul D.N., van Duyne G.D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 45.Rüfer A.W., Sauer B. Non-contact positions impose site selectivity on Cre recombinase. Nucleic Acids Res. 2002;30:2764–2771. doi: 10.1093/nar/gkf399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S.T., Kim G.W., Lee Y.S., Park J.S. Characterization of Cre-loxP interaction in the major groove: hint for structural distortion of mutant Cre and possible strategy for HIV-1 therapy. J. Cell. Biochem. 2001;80:321–327. [PubMed] [Google Scholar]
- 47.Schmitt L.T., Paszkowski-Rogacz M., Jug F., Buchholz F. Prediction of designer-recombinases for DNA editing with generative deep learning. Nat. Commun. 2022;13:7966. doi: 10.1038/s41467-022-35614-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soni A., Augsburg M., Buchholz F., Pisabarro M.T. Nearest-neighbor amino acids of specificity-determining residues influence the activity of engineered Cre-type recombinases. Sci. Rep. 2020;10:13985. doi: 10.1038/s41598-020-70867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abi-Ghanem J., Chusainow J., Karimova M., Spiegel C., Hofmann-Sieber H., Hauber J., Buchholz F., Pisabarro M.T. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucleic Acids Res. 2013;41:2394–2403. doi: 10.1093/nar/gks1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding L., Paszkowski-Rogacz M., Winzi M., Chakraborty D., Theis M., Singh S., Ciotta G., Poser I., Roguev A., Chu W.K., et al. Systems analyses reveal shared and diverse attributes of Oct4 regulation in pluripotent cells. Cell Syst. 2015;1:141–151. doi: 10.1016/j.cels.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding L., Paszkowski-Rogacz M., Mircetic J., Chakraborty D., Buchholz F. The Paf1 complex positively regulates enhancer activity in mouse embryonic stem cells. Life Sci. Alliance. 2021;4:e202000792. doi: 10.26508/lsa.202000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poser I., Sarov M., Hutchins J.R.A., Hériché J.K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A.W., et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F., et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 54.Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. Science. 2022;376:44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nerenberg M., Hinrichs S.H., Reynolds R.K., Khoury G., Jay G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science. 1987;237:1324–1329. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- 56.Derse D., Mikovits J., Ruscetti F. X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology. 1997;237:123–128. doi: 10.1006/viro.1997.8781. [DOI] [PubMed] [Google Scholar]
- 57.Derse D., Hill S.A., Lloyd P.A., Chung H., Morse B.A. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J. Virol. 2001;75:8461–8468. doi: 10.1128/jvi.75.18.8461-8468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan N., Gavalchin J., Paul B., Wells K.H., Lane M.J., Poiesz B.J. Infection of peripheral blood mononuclear cells and cell lines by cell-free human T-cell lymphoma/leukemia virus type I. J. Clin. Microbiol. 1992;30:905–910. doi: 10.1128/jcm.30.4.905-910.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pique C., Jones K.S. Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 2012;3:378. doi: 10.3389/fmicb.2012.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alais S., Mahieux R., Dutartre H. Viral source-independent high susceptibility of dendritic cells to human T-cell leukemia virus type 1 infection compared to that of T lymphocytes. J. Virol. 2015;89:10580–10590. doi: 10.1128/JVI.01799-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Igakura T., Stinchcombe J.C., Goon P.K.C., Taylor G.P., Weber J.N., Griffiths G.M., Tanaka Y., Osame M., Bangham C.R.M. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 2003;299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 62.Alais S., Dutartre H., Mahieux R. Quantitative analysis of human T-lymphotropic virus type 1 (HTLV-1) infection using Co-culture with Jurkat LTR-luciferase or Jurkat LTR-GFP reporter cells. Methods Mol. Biol. 2017;1582:47–55. doi: 10.1007/978-1-4939-6872-5_4. [DOI] [PubMed] [Google Scholar]
- 63.Popovic M., Lange-Wantzin G., Sarin P.S., Mann D., Gallo R.C. Transformation of human umbilical cord blood T cells by human T-cell leukemia/lymphoma virus. Proc. Natl. Acad. Sci. USA. 1983;80:5402–5406. doi: 10.1073/pnas.80.17.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donhauser N., Heym S., Thoma-Kress A.K. Quantitating the transfer of the HTLV-1 p8 protein between T-cells by flow cytometry. Front. Microbiol. 2018;9:400. doi: 10.3389/fmicb.2018.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seegulam M.E., Ratner L. Integrase inhibitors effective against human T-cell leukemia virus type 1. Antimicrob. Agents Chemother. 2011;55:2011–2017. doi: 10.1128/AAC.01413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meissner M.E., Mendonça L.M., Zhang W., Mansky L.M. Polymorphic nature of human T-cell leukemia virus type 1 particle cores as revealed through characterization of a chronically infected cell line. J. Virol. 2017;91 doi: 10.1128/JVI.00369-17. e00369003699-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoersten J., Ruiz-Gómez G., Lansing F., Rojo-Romanos T., Schmitt L.T., Sonntag J., Pisabarro M.T., Buchholz F. Pairing of single mutations yields obligate Cre-type site-specific recombinases. Nucleic Acids Res. 2022;50:1174–1186. doi: 10.1093/nar/gkab1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cook L.B., Rowan A.G., Melamed A., Taylor G.P., Bangham C.R.M. HTLV-1-infected T cells contain a single integrated provirus in natural infection. Blood. 2012;120:3488–3490. doi: 10.1182/blood-2012-07-445593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kobayashi N., Konishi H., Sabe H., Shigesada K., Noma T., Honjo T., Hatanaka M. Genomic structure of HTLV (human T-cell leukemia virus): detection of defective genome and its amplification in MT-2 cells. EMBO J. 1984;3:1339–1343. doi: 10.1002/j.1460-2075.1984.tb01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maeda M., Shimizu A., Ikuta K., Okamoto H., Kashihara M., Uchiyama T., Honjo T., Yodoi J. Origin of human T-lymphotrophic virus I-positive T cell lines in adult T cell leukemia. Analysis of T cell receptor gene rearrangement. J. Exp. Med. 1985;162:2169–2174. doi: 10.1084/jem.162.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuramitsu M., Okuma K., Yamagishi M., Yamochi T., Firouzi S., Momose H., Mizukami T., Takizawa K., Araki K., Sugamura K., et al. Identification of TL-Om1, an adult T-cell leukemia (ATL) cell line, as reference material for quantitative PCR for human T-lymphotropic virus 1. J. Clin. Microbiol. 2015;53:587–596. doi: 10.1128/JCM.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watanabe T., Seiki M., Tsujimoto H., Miyoshi I., Hayami M., Yoshida M. Sequence homology of the simian retrovirus genome with human T-cell leukemia virus type I. Virology. 1985;144:59–65. doi: 10.1016/0042-6822(85)90304-6. [DOI] [PubMed] [Google Scholar]
- 73.Jégado B., Kashanchi F., Dutartre H., Mahieux R. STLV-1 as a model for studying HTLV-1 infection. Retrovirology. 2019;16:41. doi: 10.1186/s12977-019-0503-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsuzaki T., Nakagawa M., Nagai M., Usuku K., Higuchi I., Arimura K., Kubota H., Izumo S., Akiba S., Osame M. HTLV-I proviral load correlates with progression of motor disability in HAM/TSP: analysis of 239 HAM/TSP patients including 64 patients followed up for 10 years. J. Neurovirol. 2001;7:228–234. doi: 10.1080/13550280152403272. [DOI] [PubMed] [Google Scholar]
- 75.Nagai M., Usuku K., Matsumoto W., Kodama D., Takenouchi N., Moritoyo T., Hashiguchi S., Ichinose M., Bangham C.R., Izumo S., Osame M. Analysis of HTLV-I proviral load in 202 HAM/TSP patients and 243 asymptomatic HTLV-I carriers: high proviral load strongly predisposes to HAM/TSP. J. Neurovirol. 1998;4:586–593. doi: 10.3109/13550289809114225. [DOI] [PubMed] [Google Scholar]
- 76.Okayama A., Stuver S., Matsuoka M., Ishizaki J., Tanaka G.-I., Kubuki Y., Mueller N., Hsieh C.-C., Tachibana N., Tsubouchi H. Role of HTLV-1 proviral DNA load and clonality in the development of adult T-cell leukemia/lymphoma in asymptomatic carriers. Int. J. Cancer. 2004;110:621–625. doi: 10.1002/ijc.20144. [DOI] [PubMed] [Google Scholar]
- 77.Takeda S., Maeda M., Morikawa S., Taniguchi Y., Yasunaga J.-I., Nosaka K., Tanaka Y., Matsuoka M. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer. 2004;109:559–567. doi: 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 78.Koiwa T., Hamano-Usami A., Ishida T., Okayama A., Yamaguchi K., Kamihira S., Watanabe T. 5’-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J. Virol. 2002;76:9389–9397. doi: 10.1128/jvi.76.18.9389-9397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hermann M., Stillhard P., Wildner H., Seruggia D., Kapp V., Sánchez-Iranzo H., Mercader N., Montoliu L., Zeilhofer H.U., Pelczar P. Binary recombinase systems for high-resolution conditional mutagenesis. Nucleic Acids Res. 2014;42:3894–3907. doi: 10.1093/nar/gkt1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sürün D., Schwäble J., Tomasovic A., Ehling R., Stein S., Kurrle N., von Melchner H., Schnütgen F. High efficiency gene correction in hematopoietic cells by donor-template-free CRISPR/Cas9 genome editing. Mol. Ther. Nucleic Acids. 2018;10:1–8. doi: 10.1016/j.omtn.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/JVI.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramawatar, Schwessinger B. DNA size selection (>3-4kb) and purification of DNA using an improved homemade SPRI beads solution. protocols.io. 2018 https://www.protocols.io/view/dna-size-selection-3-4kb-and-purification-of-dna-u-n7hdhj6 [Google Scholar]
- 85.Schneider U., Schwenk H.U., Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 86.Ho D.D., Rota T.R., Hirsch M.S. Infection of human endothelial cells by human T-lymphotropic virus type I. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7588–7590. doi: 10.1073/pnas.81.23.7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Millen S., Gross C., Donhauser N., Mann M.C., Péloponèse J.M., Jr., Thoma-Kress A.K. Collagen IV (COL4A1, COL4A2), a component of the viral biofilm, is induced by the HTLV-1 oncoprotein tax and impacts virus transmission. Front. Microbiol. 2019;10:2439. doi: 10.3389/fmicb.2019.02439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mann M.C., Strobel S., Fleckenstein B., Kress A.K. The transcription elongation factor ELL2 is specifically upregulated in HTLV-1-infected T-cells and is dependent on the viral oncoprotein Tax. Virology. 2014;464-465:98–110. doi: 10.1016/j.virol.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 89.Dobin A., Gingeras T.R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics. 2015;51:11-14.1–11-14.19. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Wang T., Nguyen N., Paten B., Zweig A.S., Karolchik D., Kent W.J. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014;30:1003–1005. doi: 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Langton B., Sliwkowski M., Tran K., Knapp S., Keitelmann E., Smith C., Wallingford S., Liu H., Ralston J., Brandis J. Development and characterization of monoclonal antibodies to the HTLV-I Tax (P40X) protein. Med. Virol. 1988;8:295. [Google Scholar]
- 96.Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest package: tests in linear mixed effects models. J. Stat. Soft. 2017;82:1–26. doi: 10.18637/jss.v082.i13. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data are available from the authors upon request.