

Abstract
The function and significance of RAS proteins in cancer have been widely studied for decades. In 2013, the National Cancer Institute established the RAS Initiative to explore innovative approaches for attacking the proteins encoded by mutant forms of RAS genes and to create effective therapies for RAS-driven cancers. This initiative spurred researchers to develop novel approaches and to discover small molecules targeting this protein that was at one time termed “undruggable.” More recently, advanced efforts in RAS degraders including PROTACs, linker-based degraders, and direct proteolysis degraders have been explored as novel strategies to target RAS for cancer treatment. These RAS degraders present new opportunities for RAS therapies and may prove fruitful in understanding basic cell biology. Novel delivery strategies will further enhance the efficacy of these therapeutics. In this review, we summarize recent efforts to develop RAS degraders, including PROTACs and E3 adaptor and ligase fusions as cancer therapies. This review also details the direct RAS protease degrader, RAS/RAP1-specific endopeptidase that directly and specifically cleaves RAS.
Keywords: RAS, degraders, PROTACs, RRSP
Graphical abstract
RAS degraders present new opportunities for RAS therapies that target not only KRAS mutants, but wild-type, RAS which may prove fruitful in understanding basic cell biology and reach a broader population of cancer patients. Novel delivery strategies will further enhance the efficacy of these therapeutics.
Introduction
The RAS family of genes including Kirsten rat sarcoma viral oncogene homolog (KRAS), neuroblastoma rat sarcoma viral oncogene homolog (NRAS), and Harvey rat sarcoma viral oncogene homolog (HRAS) encodes four proteins: KRAS4A, KRAS4B, NRAS, and HRAS.1 The RAS proteins exhibit 82%–90% overall amino acid sequence identity with variations in the C-terminal hypervariable region and allosteric lobe.1,2 The biological functions of the RAS isoforms vary based on membrane localization and effector use, which is controlled in part by post-translational modifications, including farnesylation, prenylation, methylation, and palmitoylation.3 RAS proteins are guanosine triphosphatases (GTPases) with GTP occupancy regulated by cell-surface receptors, which control the activity of guanine nucleotide exchange factors to activate RAS and GTPase activating proteins that stimulate GTP hydrolysis to inactivate RAS. In the active state, RAS GTPases stimulate cell proliferation and survival through the control of cytoplasmic signaling cascades.1,4 In addition, RAS is implicated in cell motility, polarity and morphology, differentiation, synaptic transmission, cell-cycle arrest and senescence, cytoskeletal rearrangements and pinocytosis, cytokinesis, and chemotaxis.5 Considering the important role RAS proteins play in cell growth and survival, constitutively activating mutations to these genes can drive oncogenesis.4
Between 20% and 30% of all human cancers exhibit mutations in the RAS genes. This makes them some of the most frequently mutated genes in cancer.6 Tumor cells express all three genes and mutations in any one gene can cause malignant transformation in cells and animal models.7 Historically, HRAS has been the most studied of the RAS genes,7 although KRAS is the most frequently mutated isoform (85%), followed by NRAS (11%) and HRAS (4%).6 Of the missense gain-of-function mutations in all three RAS genes, 98% occur at one of three mutational hotspots: glycine-12, glycine-13, or glutamine-61, although 130 different missense mutations have been identified in tumors.1,8 Each of these mutations produces distinct structural and biochemical effects on cellular signaling, leading to different clinical outcomes.1 RAS mutations are considered cancer drivers, but remain important in long-term tumor survival,9 making RAS an attractive therapeutic target for all cancers, including those with wild-type RAS.
Despite the high prevalence of RAS mutations in cancers, targeting RAS therapeutically has proven difficult. RAS has previously been termed “undruggable” because it lacks well defined binding sites and high concentrations of cellular GTP outcompete small molecules at its GTP-binding site.4,10 The existence of four RAS proteins presents another challenge; these RAS isoforms are differentially expressed depending on the cell type, and cell lines vary considerably in how dependent they are on RAS signaling for growth and survival. In addition, KRAS is transcribed as two splice variants KRAS4A and KRAS4B, which produce proteins with distinct biological functions and post-translational modifications.7 Researchers, however, have not been deterred in developing therapies against this elusive target. Here, we summarize the current landscape of RAS therapeutics, with an emphasis on the growing field of RAS degraders.
Small molecule inhibitors of RAS
Most therapeutic approaches have focused on the discovery of small molecules that target mutant KRAS. Here, we briefly explore the different inhibitors that have progressed into clinical and preclinical studies, as these have been extensively reviewed elsewhere.11,12,13,14,15,16,17,18,19,20,21,22 Table 1 summarizes the state of selected KRAS small molecule inhibitors.
Table 1.
Summary of small molecules targeting KRAS
| Selected small molecules targeting mutant KRAS | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Method of delivery | RAS Target | Concentration | In vitro studies | In vivo studies | Clinical trials | Ref(s). |
| AMG510 (Sotorasib or LUMA-KRAS) | Small molecule, His95 groove binder | KRAS G12C | 0.010–0.123 μM in 22 cell lines that had heterozygous or homozygous KRASG12C expression | in KRASG12C cell lines, NCI-H358 and MIA PaCa-2, AMG 510 almost completely inhibited p-ERK (IC50 ≈ 0.03 μM) after a 2-h treatment | 200 mg/kg AMG 510 resulted in inhibition of p-ERK in MIA PaCa-2 T2 and NCI-H358 tumors | Anticancer activity in 129 patients with locally advanced KRAS G12C-mutant solid tumors, Fast Track designation and undergoing a phase III study | 23,24,25,26 |
| MRTX849 (Adagrasib) | Small molecule, covalent KRAS G12C inhibitor | KRAS G12C | inhibited cell growth of KRASG12C-mutant cell lines with IC50 values ranging between 0.2 and 1,042 nM in the 3-D format | inhibited pERK; Thr202/Tyr204 ERK1, pS6; Ser235/236, and DUSP6, around 4.7 nM IC50s in KRASG12C-mutant H358 lung and MIA PaCa-2 | Tumor regression in 17 of 26 (65%) KRASG12C-positive cell line- and patient-derived xenograft models from multiple tumor types at a maximum tolerable dose between 30 and 100 mg/kg/day | ORR was 43%, the DCR was 80% in 112 KRAS G12C NSCLC patients, received Breakthrough Therapy designation, undergoing phase III study | 27,28,29,30 |
| RG6330/GDC-6036 | Small molecule, selective KRAS G12C inhibitor | KRAS G12C | IC50 of <0.01 μM | EC50 of 2 nM in K-RAS G12C-alkylation HCC1171 cells | Tumor growth inhibition in multiple KRAS G12C-positive cell lines and in xenograft mouse models | Recruiting patients for a phase Ia/Ib dose-escalation and dose-expansion study | 31,32,33 |
| D-1553 | Small molecule, selective KRAS G12C inhibitor | KRAS G12C | ND | anti-tumor activity across a panel of cancer cell lines including lung, pancreatic and colorectal cancers with KRAS-G12C mutation | Highly potent in vivo in various cell line-derived xenograft tumor models with KRAS-G12C mutation | Undergoing phase I/II open label study | 34,35 |
| BI 1829311 | KRASG12C selective small molecule inhibitor | KRAS G12C | ND | In a KRASG12C NSCLC cell line panel, downregulates DUSP6 and CCND1, and p-ERK | Daily oral dose of 60 mg/kg in a panel of lung and colon, mouse models showed comparable efficacy to AMG 510 and MRTX849 | Recruiting patients for phase Ia/Ib, open-label, multicenter dose-escalation, and expansion study | 36,37 |
| JAB-21822 | Covalent KRAS G12C inhibitor | KRAS G12C | <10–5000 nM in Ba/F3 cell lines bearing G12C mutation or secondary mutations | Cell growth inhibition in a variety of G12C mutant cancer cell lines | 50-100% tumor growth inhibition in CDX (10 mg/kg PO daily) or PDX (100 mg/kg PO daily) mouse models bearing G12C mutations | Undergoing phase I/II study, in the 800 mg daily cohort, ORR = 50% and DCR = 100% with 4 non-confirmed PR | 38,39,40 |
| JDQ443 | Small molecule covalent KRAS G12C inhibitor (GDP-bound) | KRAS G12C | 0.02 μM in KRAS G12C-mutated NCI-H358 cells, | Currently optimizing compound potency, reduced cell proliferation and cRAF recruitment in NCI-H2122/NCI-H1437 and Ba/F3 KRAS mutants | 30–100 mg/kg reduced tumor growth in multiple tumor xenograft and CDX models | Undergoing multiple clinical studies in G12C mutant NSCLC, CRC, and other patients | 41,42,43,44,45,46 |
| MK-1084 | KRAS G12C inhibitor | KRAS G12C | ND | ND | ND | Phase I study alone and in combination with pembrolizumab in NSCLC KRAS G12C patients | 47 |
| K20 | inhibitor of KRAS G12C (GTP bound) | KRAS G12C | IC50 of 0.78 μM in H358 cells and 1.55 μM H23 mutant G12C cells | 2.5 μM inhibited colony formation, induced apoptosis and reduced p-ERK levels in H23 and H358 cells | Tumor growth inhibition of 41% at 35 mg/kg and reduced p-ERK levels | NA | 48 |
| ARS-1620 | atropisomeric selective KRASG12C inhibitor (GDP-bound) | KRAS G12C | IC50 < 0.3 μM across a panel of cancer cell lines harboring either KRAS p.G12C (H358, MIA-PaCa2, and LU65) | in 3 G12C mutant cell lines, 150 nM reduced cell viability and p-ERK expression, also tested in 2D and 3D systems | Tumor growth inhibition at 200 mg/kg daily in MiaPaCa2 and PDX mutant G12C models | NA | 49 |
| RM-018 | KRAS G12C “tricomplex” inhibitor (GTP bound) | KRAS G12C | ND | attenuated both RAS-MAPK signaling and cell viability in cancer cell lines bearing KRASG12Cmutations | Dose-dependent tumor regression in the NCI-H358 KRASG12CNSCLC xenograft mouse model | NA | 50,51 |
| SML-8–73-1, SML-10-70-1 | small molecule, GTP-competitive inhibitor of K-RAS | KRAS G12C | 26.6–100 mM | attenuated Akt and Erk phosphorylation at a concentration of 100 mM, antiproliferative effects in A549, H23, and H358 cells | NA | NA | 52,53 |
| HRS-4642 | Small molecule that targets KRAS G12D | KRAS G12D | ND | ND | ND | Phase I study to evaluate the safety, tolerability, and pharmacokinetics in patients with advanced solid tumors with KRAS G12D mutations | 54,55 |
| BI-2852 | KRAS inhibitor for the switch I/II pocket | KRAS G12D | binds to KRASG12Dwith a KD of 740 nM | inhibits GTP-KRASG12D binding to SOS1, CRAF, and PI3Kα with an IC50 of 490, 770, and 500 nM pERK modulation and antiproliferative effects in NCI-H358 | NA | NA | 56,57,58,59 |
| MRTX1133 | noncovalent KRAS G12D inhibitor for the switch II pocket | KRAS G12D | 0.2 pM | inhibited ERK phosphorylation in the AGS cell line with an IC50 of 2 nM in a 2D viability assay, the IC50 of MRTX1133 was 6 nM against the same cell line, with 500-fold higher selectivity against MKN1 cells |
Antitumor activity with 94% growth inhibition observed at 3 mg/kg twice daily (i.p.) and tumor regressions of −62% and −73% observed at 10 and 30 mg/kg twice daily, in KRASG12D mutant Panc 04.03 cell line | NA | 60 |
| RMC-6236 | RAS-MULTI(ON) inhibitor | RAS mutants | ND | ND | ND | Phase I clinical evaluation for patients with KRAS G12A, G12D, G12R, G12S, or G12V mutations | 55,61,62 |
| KRA-533 | small molecule KRAS agonist, binds the GTP/GDP-binding pocket of KRAS | KRAS mutants | 10 μM | 10 μM induced cell death in mutant KRAS cell lines A549, H157, Calu-1, and H292 | 7.5, mg/kg/day of KRA-533 i.p. for 28 days suppressed tumor growth in a dose-dependent manner in A549 xenografts | NA | 63 |
| THZ835 | small molecule, binds GDP and GTP-bound KRAS, disrupts the KRAS–CRAF interaction | KRAS G12D | low μM range | reduced pERK and pAKT levels, increased p21 and p27 levels and reduced CDK2/4/6 and cyclin D1 expression cells went through G1 cell-cycle arrest and apoptosis |
Mouse xenograft models of pancreatic cancer exhibited reduced tumor growth but mice experienced weight loss, suggesting the potential of off-target effects | NA | 64 |
| KAL-21404358 | small molecule allosteric ligand against P110 site | KRAS G12D | KD of 100 μM | impaired the interaction of K-RASG12Dwith B-Raf and disrupted the RAF-MEK-ERK and PI3K-AKT signaling pathways | NA | NA | 65 |
| 12VC1 | monobody, noncovalent inhibitor | KRAS G12V and G12C | expressed intracellularly alone or fused to VHL | inhibits ERK activation and the proliferation of RAS-driven cancer cell lines | H23 cells expressing VHL-12VC1 were significantly smaller than control tumors | NA | 66 |
| NS1 (aHRAS) | monobody, inhibits RAS-mediated signaling through targeting the α4–α5 surface | KRAS and HRAS | expressed intracellularly alone or fused to VHL | inhibited growth factor signaling and oncogenic H-RAS- and K-RAS-mediated signaling and transformation | NA | NA | 3,67 |
| Compound 3144 | multivalent small-molecule, pan-RAS inhibitor | KRAS HRAS NRAS |
Kd of 4.7/17/6.6/3.7 μM for KRAS G12D/KRAS wt/HRAS/NRAS, respectively | lethality in cells partially dependent on expression of RAS proteins | Displays anti-tumor activity in breast and pancreatic xenografts | NA | 68,69 |
| DCAI (dichloro-2-methyl-3-aminoethyl-indole) | competitive inhibitor that blocks the RAS-SOScat interaction | KRAS, KRAS mutant | 15.8 ± 0.4 μM | blocks the recruitment of the cRaf RBD-CRD domain to the cytoplasmic membrane, effect on cell viability ND | NA | NA | 70 |
Two G12C inhibitors, JNJ-74699157 and LY3499446, are not included as these compounds failed clinical trials.71,72
Revolution Medicines is also developing RMC-6291, a KRASG12C inhibitor, RMC-9805, a KRASG12D inhibitor and RMC-8839, a KRASG13Cinhibitor.
Jacobio Pharma is developing small molecule inhibitors: JAB-23400 (KRAS (multi)) and JAB-22000 (KRAS G12D).
Bridge Bio is performing preclinical studies for multiple compounds including the KRAS inhibitor BBP-454 and PI3Ka:RAS breakers.
CRC, colorectal cancer; DCR, disease control rate; ND, not disclosed; NA, information is not available; ORR, objective response rate; PI3K, phosphatidylinositol 3-kinase.
Mutant KRAS-specific small molecule inhibitors function by binding to the outer surface of KRAS in the expanded pocket of the switch II region.4,73 These include covalent inhibitors that specifically bind only the G12C mutant KRAS protein found in 14% of non-small cell lung cancers (NSCLC), 5% of colorectal cancers, and 2% of pancreatic cancers.74 The two most advanced small molecules in this class are AMG510 (sotorasib) and MRTX849 (adagrasib). In May 2021, the U.S. Food and Drug Administration (FDA) approved sotorasib for adult patients with KRAS G12C mutant locally advanced or metastatic NSCLC.23,24,75 Both a phase III multicenter, randomized, open label, active-controlled study (NCT04303780) and expanded access protocol (NCT04667234) are currently underway.23,24,25,26,75,76,77,78 For MRTX849, results have just been released on the KRYSTAL-1 study showing a 43% objective response rate in patients with KRAS G12C NSCLC. MRTX849 has now received a Breakthrough Therapy Designation status for previously treated patients with KRAS G12C NSCLC and is currently being reviewed by the FDA for accelerated approval. Many other KRAS G12C inhibitors are currently undergoing clinical trials (see Table 1).49,79,80 Of note, two covalent G12C inhibitors, JNJ-74699157 and LY34994446, have recently failed clinical trials because of off-target toxicities.23,71,72,81,82
Progress has also been made targeting the G12D mutant of KRAS, which is present in 51% of pancreatic, 40% of colorectal, and 17% of lung adenocarcinomas.22,83,84 These KRAS G12D inhibitors are currently all in preclinical development and include MRTX1133, BIKRASG12D1–3, KRA-533, and TH-Z835 (Table 1 and Figure 1). Notably, HRS-4642, a small molecule that targets G12D KRAS, is now entering phase I clinical trials (NCT05533463),54,55 although supporting pre-clinical data are not publically available.
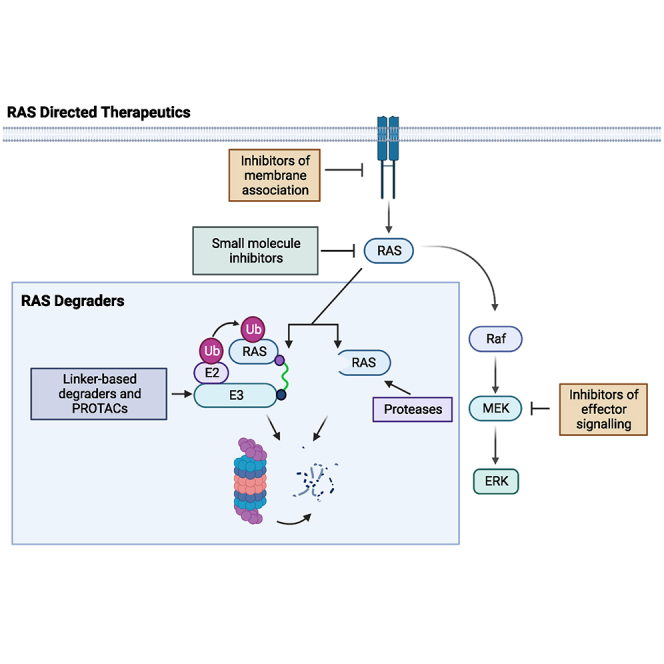
Figure 1.

Schematic of selected RAS targeted therapeutics
Current RAS targeted therapeutics that are in pre-clinical and clinical development are grouped by mechanism of action. Only those that specifically target RAS are listed. See Table 1 for more details on in vitro, in vivo, and clinical studies. Figure created with Biorender.com.
Other strategies have been explored for targeting multiple KRAS mutants locked in the “on” state. Revolution Medicines is developing multiple RAS-targeting molecules. One of these molecules, RMC-6236, is currently undergoing a phase I clinical evaluation for the treatment of patients with KRAS G12A, G12D, G12R, G12S, or G12V mutations. RMC-6236 is a RAS-MULTI(ON) inhibitor, selective for the active RAS(ON) form of both wild-type and mutant variants of the canonical RAS isoforms (HRAS, NRAS, and KRAS) (NCT05379985).55,61,62
Finally, approaches to inhibit RAS signaling more broadly include targeting indirect mechanisms,85 such as preventing interaction of RAS with its effectors,81,86 including the serine/threonine-protein kinases BRAF65 and RAF1,21,81,87,88,89,90 cyclophilin A,91 and guanine nucleotide exchange factor SOS1.1,23,56,70 The importance of RAS binding to the lipid membrane23 and appropriate membrane localization for RAS signaling have also been targeted therapeutically by inhibiting the delta subunit of cGMP phosphodiesterase92,93 or inhibiting palmitoylation (Figure 1).94,95
Overall, there has been substantial success with the discovery and preclinical and clinical development of RAS-directed small molecule therapeutics, which has culminated in improved patient outcomes. However, limitations to these therapies are important to consider. KRAS-independent tumors develop receptor tyrosine kinase dependency and signaling rebound kinetics84 of downstream proteins including ribosomal S6 kinase, mammalian target of rapamycin, rapidly accelerated fibrosarcoma kinases (RAF), YES-associated protein 1, and phosphatidylinositol 3-kinase.1,7,96,97 Alteration of these proteins and reactivation of RAS/MAPK signaling can result in outgrowth of resistant tumors.84 Tumor resistance to KRAS inhibitors can also arise from mutations that result in loss of the tumor suppressor protein neurofibromin 1 or activation of another RAS isoform.23,98,99,100 Resistance may also occur by promoting GDP and GTP exchange of RAS and indirect effects on the tumor microenvironment.101,102,103,104 Another caveat is that some of these compounds do not readily enter cells, inhibiting their therapeutic potential. Finally, the current dominant approach of targeting the somewhat rare G12C- and G12D-mutant KRAS limits the number of cancers treatable by targeting RAS.74,83 Indeed, 70% of cancers have a non-modified RAS or are heterozygous. Thus, there is a need for alternatives to small molecule targeting of mutant KRAS to expand the number of RAS-driven cancers that can be clinically treated, regardless of the association of RAS with GTP or GDP or its mutant status.
RAS degraders
Targeted protein degradation (TPD) has become a major focus of research over the past 10 years. Publications including PROteolysis Targeting Chimeras (PROTACs) or linker-based degraders, also known as bioPROTACs,105 have gone from the single digits before 2015 to 450 publications in 2022.106 Because only 20%–25% of all protein targets are being pursued in drug discovery efforts, TPD has focused on undruggable targets with degraders of >42 distinct protein targets published.106 Unlike small molecules that bind to proteins to inhibit protein activity, degraders specifically target a protein inside cells for proteolysis using the physiologically normal process of disposing of old or damaged proteins, thus permanently removing the protein from the cellular pool. The targeted protein is first bound by the degrader molecule or protein and the bound molecule then recruits the cellular ubiquitination machinery to tag the protein for degradation by the 26S proteasome and recycling of its amino acids. Ubiquitination is performed by three enzymes, E1 (an activating enzyme), E2 (a conjugating enzyme), and E3 (a protein ligase).107 Some degraders forego the linker approach to the ubiquitination pathway and instead directly cleave the target to become inactive and ultimately removed by normal cellular protein turnover (Figure 2).
Figure 2.

Schematic of RAS degrader mechanism of action
(1) Linker-based degraders and PROTACs fuse a protein binding domain or chemical compound to a ligand for an E3 ligase. The degrader creates a ternary complex between the protein of interest, such as RAS, and the E3 ubiquitin machinery.10 E2 transfers ubiquitin to the protein of interest which allows it to be degraded by the 26S proteasome.107 (2) Direct proteolysis occurs where the protease binds to and cleaves the protein of interest, for example, RAS, and the protein is subsequently degraded. Figure created with Biorender.com.
To provide an alternative to targeting mutant RAS with small molecules, the recent development of RAS degraders has become a highly active and novel, therapeutic approach. As RAS is central in controlling cell proliferation, removal of RAS could be an effective strategy for the treatment of RAS-driven cancers. Degraders are in development that can treat all RAS isoforms (termed pan-RAS), as well as degraders specific for RAS isoforms and even for mutant KRAS. Current early stage investigation, pre-clinical, and clinical trials are underway for multiple classes of degraders, including PROTACs, linker-based degraders, and direct proteolysis strategies.
PROTACs
PROTACs are novel, selective chemical ligands that target a protein of interest for degradation.108 These molecules have one warhead with high specificity for the target with a second warhead that binds an E3 ligase adapter, thus bringing the protein of interest into close proximity of an E3 ligase substrate receptor protein, such as cereblon (CRBN). The targeted protein is ubiquitinated by E3 and thus re-directed to the 26S proteasome for degradation.10,109 The first instance of a PROTAC is when CRBN ligands were combined with an Src homology region 2 domain-containing phosphatase-2 (SHP2) inhibitor to promote SHP2 degradation.15
The first PROTAC targeting KRAS is compound 13 (also known as XY-4-88), which was created by fusing the KRAS G12C inhibitor ARS-1620 to CRBN. Although the compound permeates cells and recruits CRBN and KRAS G12C to a complex, the compound does not effectively degrade endogenous KRAS G12C.109 The PROTAC approach has been successful when fusing the G12C inhibitor MRTX849 (adagrasib) with a chemical ligand that recruits the von Hippel-Lindau (VHL) E3 ligase complex. This molecule LC-2 sustains endogenous degradation of KRAS G12C and decreased ERK signaling when tested in KRAS G12C cell lines.110 These compounds have yet to be tested in vivo.
In data from the AACR 2021 conference, Boehringer Ingelheim reported a panKRAS degrader and a KRAS-specific PROTAC.84,111 BIpanKRAS3 targets both KRAS and KRAS G12D. In a GP2d colon cancer cell line xenograft model, a 90 mg/kg dose of BIpanKRAS3 decreases pERK levels and nuclear protein Ki67 expression.84,111 In addition, 30-mg/kg doses reduce tumor progression in the GP2d model and a KRAS G13D HCT15 cell line xenograft model. The BIKRASdegrader1 degrades mutant KRAS and inhibits pERK expression and cell proliferation in GP5d cells. This PROTAC degrades KRAS in a panel of KRAS mutants, but has not as yet been tested in vivo.
The most advanced PROTAC-like molecule in development is ASP3082, a selective KRAS G12D degrader. This molecule binds both KRAS G12D and an undisclosed E3 ligase adapter. An abstract from the 34th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in October 2022 reports that ASP3082 degrades KRAS G12D in pancreatic cancer cells and exhibits dose-dependent reduction of pancreatic tumors after intravenous injection.112 A phase I study of this molecule began in June 2022 for patients with previously treated locally advanced or metastatic solid tumors with the KRAS G12D mutation (NCT05382559).113,114 See Table 2 for a summary of selected RAS PROTACs.
Table 2.
Summary of selected RAS degraders
| Compound | RAS Targets | Concentration | In vitro studies | In vivo studies | Ref(s). |
|---|---|---|---|---|---|
| RRSP | KRAS, NRAS, HRAS | 1-300 pM | Induces apoptosis or senescence in pancreatic, breast, and colon cancer | Tumor regression in pancreatic, breast, and colon xenografts | 115,116,117,118,119,120 |
| ASP3082 | PROTAC against KRAS G12D | NA | ND | In vivo results not disclosed but a phase I clinical trial began in June 2022 | 112,113,114 |
| RC-U | KRAS, NRAS, HRAS | transfection | Reduced RAS levels upon transfection in HEK293T cells | Transfection reduced tumor size | 121 |
| VHL-KRAS DARPin K19 | KRAS | transfection | Reduced KRAS levels in mutant KRAS cells | Doxycycline-inducible expression reduced tumor size | 122 |
| UBOX-Pan-RAS iDab | KRAS, NRAS, HRAS | transfection | Reduced RAS levels in mutant KRAS cells | Doxycycline-inducible expression reduced tumor size | 122 |
| BIpanKRAS3 | KRAS, KRAS G12D | 19–91 nM | Cleaved KRAS and reduced pERK levels in NCI-H2122 and SW837 cells | KRASG12/13D mutant CDX xenograft models a 30-mg/kg dose caused tumor regression | 84,111 |
| Compound 13 (XY-4-88) | KRAS G12C | >0.410 μM | Did not degrade endogenous RAS | NA | 109 |
| LC-2 | KRAS G12C | 0.25–0.76 μM | Degraded endogenous KRASG12C and ERK | NA | 110 |
| FKBP12F36V-KRASG12V + dTAG13 | KRAS G12V | transfection | Reduced KRAS G12V levels | NA | 123 |
| VHL-aHRAS AdPROM | KRAS, NRAS, HRAS | transfection | Reduced K,N,H-RAS in CRISPR-Cas9 HEK293 and U2OS cells | NA | 124 |
| DARPin K27-SPOP and others | various | plasmid and mRNA transfection | Several RAS degraders and E3 ligases reduced RAS expression and inhibited cell proliferation specific to each compound | NA | 74 |
| BIKRASdegrader1 | KRAS mutants | 2–116 nM | Degraded KRAS, reduced pERK levels and reduced cell proliferation | NA | 111 |
| Engineered subtilisin | KRAS | transfection | Cleaved eGFP-KRAS after doxycycline induction | NA | 125 |
ND-not disclosed, NA-information is not available
Linker-based degraders
Linker-based degraders, also known as bioPROTACs,105 are proteins rather than chemical ligands, but function similar to PROTACs. Linker-based degraders are generally plasmid-encoded fusion proteins that consist of a high-specificity target-binding protein domain linked to an E3 ubiquitin ligase or an E3 recruiting adapter. Upon transfection into cells, either the protein is ubiquitinated by the E3 ligase or the E3-recruiting adapter brings the corresponding E3/E2 complex to the vicinity of the target of interest and induces the transfer of polyubiquitin. In both cases, the targeted protein is tagged for proteasomal degradation (Figure 2).74,106,126,127,128
The first linker-based degrader that was specifically engineered to target RAS is the RCRAF−1-U-Box (RC-U) degrader.121 This expression construct expresses a fusion protein of the RAS binding (RBD) and cysteine-rich (CRD) domains of RAF1 with a U-Box family E3 ubiquitin ligase. Upon co-transfection, transient co-expression of a Flag-tagged mutant KRAS and RC-U reduces KRAS, HRAS, and NRAS protein levels and suppresses pancreatic cancer cell growth. For in vivo validation, PANC-1 xenograft tumors are injected with the expression plasmid in combination with in vivo-jetPEI (Polyplus transfection) reagent. When RC-U is transfected into tumors, RAS levels are reduced inside of the tumors and the rate of tumor growth is slowed.121
A distinct strategy to engineer degraders has taken advantage of the target specificity using antibody mimetics known as designed ankyrin repeats (DARPins). Expression plasmids have been generated to express the KRAS-specific DARPin K19 fused to the E3 adaptor ligand VHL under the control of a Tet-On promoter and H358 lung cancer cells have been stably transfected with these expression plasmids. In the presence of doxycycline to induce the promoter, the expressed KRAS degrader depletes only the KRAS isoform of RAS. Although the DARPin K19 does not specifically target mutant KRAS, cell proliferation is inhibited only in mutant KRAS cancer cells and the DARPin degrader has no effect on cells with wild-type KRAS or mutant NRAS or HRAS.122 In vivo experiments have been performed by injecting mutant KRAS H358 cancer cells stably expressing the RAS degraders into CD-1 nude mice. Upon doxycycline treatment to induce expression of the degrader in the grafted tumor cells, the degrader directly decreases RAS levels within the tumor cells and tumor regression is observed.122 Of note, no tumor decrease is observed in mouse xenografts of H1299 NRAS-mutant lung cancer cells, validating the specificity of the DARPin for KRAS over other RAS isoforms.
In a parallel study, the KRAS-specific DARPin degrader has been directly compared with a pan-RAS inhibitor created by fusion of a pan-RAS intracellular single domain antibody (iDAb) with U-BOX E3 ubiquitin ligase. In contrast with the specificity of the DARPin for KRAS, the pan-RAS degrader based on the iDAb decreases protein levels of KRAS, HRAS, and NRAS and inhibits cell proliferation upon doxycycline induction across eight different tumor cell lines. The pan-RAS inhibitor decreases the growth of both the KRAS mutant H358 xenografts, as well as the NRAS-mutant H1299 xenografts.122
A similar bio-degrader has been developed using the affinity-directed protein missile (AdPROM) system target degradation. These AdPROMs are likewise created by the fusion of E3 ligases or adapters with peptidic binders with high affinity for proteins of interest.124 To generate a RAS degrader, the engineered AdPROM has VHL fused to an HRAS/KRAS targeting monobody (aHRAS). When expression is induced in HEK293 and U2OS cells, the AdPROM decreases GFP-KRAS and H/N-RAS protein levels but has varying effects on cell proliferation, depending on the cell line.3 It is notable that the effectiveness of the RAS-AdPROM to degrade RAS in vivo by demonstration of tumor reduction has not as yet been determined.
In a broad-based study to determine the efficiency of 10 different E3 adaptors, speckle-type POZ protein (SPOP) was identified as the most efficient to degrade GFP-KRAS when fused to an anti-GFP binding protein. SPOP has next been tested after fusion to six different RBD proteins, including those tested by other groups: RAF RBD, RAF RBD+CRD, the monobody aHRAS (referred to in this paper as NS1), and DARPin K19.74 In addition, fusions have been created with DARPins K27 and K55, which have differential specificity for GDP and GTP-bound KRAS, respectively, and with R11.1.6, a RAS-specific combinatorial protein displayed in an SSo7d scaffold. All the fusions except for the DARPin K55 construct result in degradation of RAS when expressed in A549 lung cancer cells with the NS1 construct showing specificity for KRAS and HRAS, but not NRAS. This study suggests that RAS degraders can be tunable based on the ligand used to target RAS isoforms.74 However, the effectiveness of these SPOP fusions to degrade RAS in tumors resulting in tumor reduction has not yet been demonstrated.
Direct proteolysis degraders
Linker-based degraders and PROTACs are potent, versatile, and in theory could target any protein and be manufactured for tissue selectivity.106,127,128 Technically, it is tedious to create these chemical and biological linkers. Challenges include the discovery of the optimal RAS targeted ligand and pairing it with the best linker to the ubiquitination machinery. The final and perhaps most straightforward method of inhibiting RAS is through direct proteolysis, eliminating the process of bringing together multiple proteins in a complex. Direct proteolysis works by introducing a proteolytic enzyme into the cell that can cleave a target protein with high specificity, thus inhibiting its function.
One instance of engineered direct proteolysis against RAS has been completed by modification of the Bacillus protease subtilisin. Subtilisin is a canonical serine protease that was re-engineered to specifically cleave the conserved QEEYSAM amino acid sequence, which is found in the switch II region of RAS. In human cells with fluorescently tagged RAS (eGFP-KRAS), engineered subtilisin can self-activate, locate eGFP-KRAS at the plasma membrane, and cleave it as indicated by the presence of the eGFP fusion product and the precipitous disappearance of KRAS.125 In HEK293T cells, expression of the protease induced by doxycycline decreases eGFP-RAS expression.125 The function of RAS targeted subtilisin on endogenous levels of RAS is currently unknown and the effectiveness of this protein to reduce tumors has not as yet been evaluated.
Perhaps the most unique approach to a RAS degrader takes advantage of the multiple bacteria that produce protein toxins that have evolved over thousands of years to specifically target RAS. These, along with other bacterial effectors, could be harnessed for use as therapies.129,130 The most advanced and well studied RAS protease was first reported in 2014 and over the past 9 years has been developed into the most advanced proteinaceous RAS degrader in preclinical development.
The RAS/Rap1-specific endopeptidase (RRSP) is naturally produced by the bacterium Vibrio vulnificus. Originally termed domain of unknown function in the fifth position (DUF5), RRSP is now recognized as a highly potent cytotoxic effector domain from the multifunctional-autoprocessing repeats-in-toxin (MARTX) toxin.131 The X-ray structure of RRSP reveals that it is a three-domain protein with a membrane targeted domain and a catalytic domain in the same protease family as the eukaryotic Wnt-specific protease TIKI (Figure 3).132 RRSP as found in MARTX toxins has cytotoxic activity115 and blocks RAS-MAPK signaling by cleaving RAS and the closely related repressor activator protein 1 (RAP1) within the switch I region between the residues tyrosine-32 and aspartate-33.116 The enzyme is highly specific for the RAS/RAP1 switch I sequence, such that even the closely related RAS-like proto-oncogene A GTPase is not cleaved by RRSP. This activity is also found in other bacterial toxins, including Photorhabdus spp. insect pathogens and Aeromonas spp. fish pathogens.115 RRSP cleaves HRAS, NRAS, and KRAS as well as KRAS G12V, G12D, G12C, G13D, and Q61R mutants and is highly potent, with complete degradation of RAS occurring in the picomolar range.117,118 RRSP has activity against both GDP- and GTP-bound RAS, increasing its efficacy.117,118 The cleavage of RAS results in downstream loss of phosphorylated ERK, but has varying effects on cell proliferation based on the cell line. Colon carcinoma HCT116 and SW1463 cell lines are highly susceptible to RRSP and undergo apoptosis, while RRSP treatment of GP5d and SW620 colon cells induces G1 cell-cycle arrest. In fact, the predominant result of cleaving all RAS in the cell is to initiate CDK2 cell-cycle arrest. RRSP has also been linked to CDK1 cell-cycle arrest and rescues expression of the tumor suppressor p27.118 Further, RRSP from Photorhabdus luminescens inhibits cell proliferation, disturbs mitotic progression via CDK1, and increases the rate of HeLa cell death.133 Comprehensive analyses using both the NCI-60 cell line panel and a pancreatic cancer cell panel show that most tumor types are sensitive to RRSP, with those exhibiting RAS genomic abnormalities being the most sensitive to RRSP.118,119,120
Figure 3.

Functional domains within RRSP and RAS
(A) Schematic of RRSP from the Vibrio vulnificus MARTX toxin. The V. vulnificus MARTX is composed of six effector domains, including RRSP. The RRSP effector has three functional domains: C1, C2A, and C2B. C1 is the membrane localization domain and C2B contains the catalytic domain of RRSP. The function of the C2A domain is unknown. Within the C2B domain, four residues (denoted with asterisks) are critical for RAS/Rap1 cleavage. These include G3900, H3902, G3930, and H4030. If these residues are mutated catalytic activity is significantly reduced.115,117,132 (B) Schematic of the key functional domains of the RAS proteins. The RAS proteins (HRAS, NRAS, KRAS4A, and KRAS4B) have high sequence homology other than the C-terminal domain termed the hypervariable (HV) region. RAS proteins contain a P loop and Switch I/II regions.4,10 These three domains all are subject to the most common mutations found in RAS-driven cancers. The P loop and Switch II region each contain a GTP-binding domain and are the sites for the most frequent mutations G12/13 and Q61 (denoted with pink stars). The Switch I region contains the effector binding domain. RRSP cleaves RAS within this region between the residues Y32 and D33 (denoted with a yellow star), thus interfering with RAS downstream effector activation. Figure created with Biorender.com.
To demonstrate the function of RRSP in vivo, RRSP has been engineered as a chimeric toxin with RRSP fused to the translocation B fragment of diphtheria toxin (RRSP-DTB). DTB binds heparin-binding epidermal growth factor-like growth factor (HB-EGF) which is at least 1,000-fold less potent in cells expressing murine HB-EGF than in cells expressing human HB-EGF, making this an important system for testing the in vivo efficacy of RRSP.119 Intra-peritoneal administration of RRS-DTB at 0.1 mg/kg decreases total RAS expression in xenograft tumors. This results in tumor growth inhibition for wild-type and mutant KRAS triple-negative breast and in wild-type and KRAS-mutant colorectal xenografts,119 as well as a decrease in KRAS-mutant pancreatic patient-derived tumor xenografts.120 This protein is also stable for up to 16 h in the bloodstream of immunocompetent mice. Challenges remain for how to best deliver RRSP only to tumors. Loftis et al.134 have shown that RRSP fused to another toxin, anthrax toxin protective antigen, is specifically delivered to only cancer cells by targeting to epidermal growth factor receptor or carcinoembryonic antigen. In addition, RRSP-DTB has been redirected by changing the toxin to engage the IL-2 receptor.119 Thus, for the proteinaceous RAS degraders, the depth of data both in vitro and in vivo sets RRSP apart from other degraders as the most advanced in development.
Enhancing the delivery of RAS therapeutics
All of the RAS degrader strategies have the potential to have significant issues with delivery, as they require cytoplasmic exposure within the solid tumor for target engagement and efficacy. PROTACs have a high molecular weight because of the two-warhead design; in preclinical studies, some have demonstrated issues with membrane permeability, solubility, and pharmacokinetics.135 The cells are not taking up these molecules spontaneously. Because of these issues, many of these PROTACS have yet to be tested in vivo and may not show in vivo efficacy with exogenous addition or systemic injection. The success of ASP3082 in mouse xenograft studies and the start of a phase I clinical trial suggests that at least one PROTAC molecule has been developed to surmount the delivery barrier. However, the difficulty in development of these molecules may decrease the overall success to generate molecules beyond ASP3082 that can target pan-RAS or other mutant RAS proteins.
The delivery barrier is more profound for both the linker-based and directed proteolysis degraders, as these large biological proteins need to be translocated within the cells. The fusion RRSP-DTB demonstrates this barrier can be surmounted by using toxin-based cytosolic delivery, but other degraders in development have not yet been tested in tumor models. Further, although toxins can be used for the delivery of RRSP protein and potentially other protein degraders, these biologics must still be re-engineered for targeting against each type of cancer, which could ultimately limit clinical applications.
A new frontier in RAS therapeutics may be through targeting and delivery with mRNA. In the wake of the success of mRNA vaccines during the coronavirus disease 2019 (COVID-19) pandemic, these therapies have become attractive targets for cancer treatments. Emerging evidence suggests KRAS mutant cancers are attractive targets for immune-based treatments in pancreatic and other cancers. Advances to target KRAS-driven cancer with mRNA-encoded proteins are already significantly advanced. The mRNA vaccine, mRNA-5671, which encodes neoepitopes for common KRAS mutations (G12C, G12D, G12V, and G13D) is delivered via a lipid nanoparticle. A phase I clinical trial (NCT03948763) in patients with NSCLC, colorectal cancer, and pancreatic adenocarcinoma has closed enrollment in 2022.84,136,137 RAS has also been targeted via AZD4785, a constrained ethyl-containing therapeutic antisense oligonucleotide. AZD4785 is complementary to a sequence in the 3′-UTR of KRAS mRNA. AZD4785 targets both the mutant and wild-type KRAS isoforms for ribonuclease H-mediated degradation without any delivery agent. In vivo, AZD4785 results in tumor growth inhibition in NCI-H358 KRAS mutant lung cancer xenografts, LXFA 983 patient-derived xenograft models, and several additional tumor models.6 However, in a clinical trial (NCT031018390) AZD4785 did not reduce KRAS levels, although work is ongoing to enhance its efficacy.23 In addition, a KRAS G12D mutant-selective small interfering RNA, siG12D LODER, has shown antitumor activity in mouse models of pancreatic cancer. In a phase I trial in combination with chemotherapy in 12 pancreatic cancer patients, 2 patients are shown to have a pathological response and 10 achieved stable disease (NCT01188785).138 A phase II trial in patients with KRAS G12D pancreatic cancer is currently underway (NCT01676259).23,139 KRAS G12D-directed small interfering RNA is currently being evaluated in pancreatic cancer6 and a short hairpin RNA against c-RAF in KRAS mutant lung cancers has led to partial tumor regression.140
Given these successes, the opportunity to pair RAS degrader strategies with mRNA delivery seems a promising approach. The linker-based RAS degrader comprised of DARPin K27 paired with SPOP is successfully expressed in cancer cells from a transfected mRNA and expression is correlated with a decrease in RAS levels.74 This is the first RAS degrader shown to work by a method beyond plasmid transfection or toxin-based protein delivery. The DARPin K27-SPOP mRNA, however, has not yet been tested for in vivo for tumor reduction.
As seen for BNT162b2, or mRNA-1273, to prevent COVID-19, the delivery of mRNA can be achieved through multiple types of nanoparticles.141 A variety of nanoparticle types have been created with compositions that alter their delivery properties, as discussed elsewhere.141,142,143 Most recently, an mRNA that expresses RRSP (referred to in the paper by the previous name DUF5) has been successfully delivered to colon cancer cells using a nanoparticle delivery system. The mRNA expressing RRSP degrades RAS in mutant HCT-116 (KRAS G13D) and H358 (KRAS G12C) cells. In vivo, RAS levels are decreased after the injection of RRSP mRNA-loaded nanoparticles in colorectal xenografts and lung adenocarcinoma, and the treatment inhibits tumor growth. The efficacy is comparable with the mutant KRAS inhibitor AMG510.144 Thus, a strategy is likely underway for implementation with other developed RAS degraders for in vivo applications and therapeutic development.
Impact on basic cell biology
The development of RAS degraders has the potential not only to expand therapeutic options, but also to expand our toolbox for studying RAS signaling pathways. RAS-less mouse embryonic fibroblasts145 and RNAi technologies146 are extensively used to study the impact of loss of RAS on cell biology and signaling. PROTACs and RRSP-DTB can similarly render cells RAS-less and have the advantage of being exogenously added inhibitors that can be used in a dose-dependent fashion. Linker-based degraders under control of the TET-on promoters also provide dose-dependent control of RAS levels. Several studies have been conducted that take advantage of these degraders to understand cell signaling.
A final unique iteration of linker-based degraders to decrease RAS expression is seen by the fusion of KRAS G12V to the linker FKBP12F36V (dTAG). In cells expressing this fusion protein, RAS expression is decreased after the treatment of cells with the chemical molecule dTAG13, which binds both FKBP12F36V and the CRBN E3 ligase. dTAG13 brings the RAS complex into close proximity of the ubiquitin machinery and, thus, decreases KRAS G12V expression and cell proliferation in NIH 3T3 mouse cells. The approach in combination with proteomics and transcriptomic profiling has been used to study the impact of KRAS degradation on signaling. This approach could conceptually also be employed in vivo with xenografts that stably express FKBP12F36V targeted against RAS. As proof of concept, a luciferase fusion to FKBP12F36V is degraded in vivo after the injection of dTAG13, but a similar study of the impact of RAS degradation has not yet been developed.123
Similarly, the AdPROM system is also valuable for studies of the impact of RAS depletion on cell signaling. A ligand-inducible AdPROM (L-AdPROM) created by addition of the PROTAC to FLAG-Halo-aHRAS expressing A549 cells also degrades RAS levels.147 Finally, as described above, RRSP-DTB can render cells RAS-less, and these cells have been used in phospho-proteomic profiling to demonstrate that depletion of all RAS proteins induces cell-cycle arrest while cell death by apoptosis or senescence varies by cell line.118 Thus, the development of RAS degraders could be excellent tools for many cell-based and in vivo basic science studies, even if the molecules are not ultimately forwarded for clinical development.
Conclusions
The field of RAS therapeutics has quickly expanded and presents great potential for future basic science and clinical applications. Specifically, the new field of RAS degraders alongside enhanced drug delivery technologies may benefit the 20%–30% of patients with RAS-driven cancers. Advancement of ASP3082 to clinical trials indicates that success is possible with chemical ligands being added to the small molecule repertoire of RAS-targeting therapeutics. Biologics are advancing through preclinical trials with the potential of tunable and also highly specific KRAS and pan-RAS degraders to be developed. In this class of protein biodegraders, RRSP is the only degrader that has shown in vivo efficacy across several different models, including breast, colon, and pancreatic mouse models, which is, thus far, the only degrader that reduced tumors by both exogenous protein addition and mRNA delivery. Indeed, investigating other bacterial proteases like RRSP presents untapped potential in terms of cancer therapeutics. RAS degraders present exciting potential to develop therapies that target not only KRAS mutants, but also wild-type RAS, which can expand our understanding of basic science and reach a broader population of cancer patients.
Acknowledgments
This work is funded by the Chicago Biomedical Consortium Accelerator Award (A-013 to K.J.F.S).
Author contributions
T.E.E. conducted the investigation, wrote the original draft of the manuscript, and prepared the figures and tables. T.E.E. and K.J.F.S. revised the subsequent drafts and approved the final version of the manuscript.
Declaration of interests
K.J.F.S. discloses that she holds a patent on use of RRSP as a cancer therapeutic (Patent # US10829752B2). K.J.F.S. has a significant interest in Situ Biosciences, a contract research organization that conducts research unrelated to this work.
References
- 1.Hobbs G.A., Der C.J., Rossman K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant K.L., Mancias J.D., Kimmelman A.C., Der C.J. KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Röth S., Macartney T.J., Konopacka A., Chan K.H., Zhou H., Queisser M.A., Sapkota G.P. Targeting endogenous K-RAS for degradation through the affinity-directed protein missile system. Cell Chem. Biol. 2020;27:1151–1163.e6. doi: 10.1016/j.chembiol.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGee J.H., Shim S.Y., Lee S.J., Swanson P.K., Jiang S.Y., Durney M.A., Verdine G.L. Exceptionally high-affinity Ras binders that remodel its effector domain. J. Biol. Chem. 2018;293:3265–3280. doi: 10.1074/jbc.M117.816348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkins A. MRC LMCB and Department of Physiology. University College London; 2000. The Function of Ras Proteins in Dictyostelium discoideum. [Google Scholar]
- 6.Ross S.J., Revenko A.S., Hanson L.L., Ellston R., Staniszewska A., Whalley N., Pandey S.K., Revill M., Rooney C., Buckett L.K., et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aal5253. [DOI] [PubMed] [Google Scholar]
- 7.McCormick F. Progress in targeting RAS with small molecule drugs. Biochem. J. 2019;476:365–374. doi: 10.1042/BCJ20170441. [DOI] [PubMed] [Google Scholar]
- 8.Muraoka S., Shima F., Araki M., Inoue T., Yoshimoto A., Ijiri Y., Seki N., Tamura A., Kumasaka T., Yamamoto M., Kataoka T. Crystal structures of the state 1 conformations of the GTP-bound H-Ras protein and its oncogenic G12V and Q61L mutants. FEBS Lett. 2012;586:1715–1718. doi: 10.1016/j.febslet.2012.04.058. [DOI] [PubMed] [Google Scholar]
- 9.Zeng M., Lu J., Li L., Feru F., Quan C., Gero T.W., Ficarro S.B., Xiong Y., Ambrogio C., Paranal R.M., et al. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem. Biol. 2017;24:1005–1016.e3. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 10.Martín-Acosta P., Xiao X. PROTACs to address the challenges facing small molecule inhibitors. Eur. J. Med. Chem. 2021;210 doi: 10.1016/j.ejmech.2020.112993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cucurull M., Notario L., Sanchez-Cespedes M., Hierro C., Estival A., Carcereny E., Saigí M. Targeting KRAS in lung cancer beyond KRAS G12C inhibitors: the immune regulatory role of KRAS and novel therapeutic strategies. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.793121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haider K., Sharma A., Yar M.S., Yakkala P.A., Shafi S., Kamal A. Novel approaches for the development of direct KRAS inhibitors: structural insights and drug design. Expert Opin. Drug Discov. 2022;17:247–257. doi: 10.1080/17460441.2022.2029842. [DOI] [PubMed] [Google Scholar]
- 13.Han Z., Zhou D., Wang J., Jiang B., Liu X. Reflections on drug resistance to KRAS(G12C) inhibitors and gene silencing/editing tools for targeting mutant KRAS in cancer treatment. Biochim. Biophys. Acta Rev. Cancer. 2022;1877 doi: 10.1016/j.bbcan.2022.188677. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Zhang J., Liu Q., Fan X.X., Leung E.L.H., Yao X.J., Liu L. Resistance looms for KRAS G12C inhibitors and rational tackling strategies. Pharmacol. Ther. 2022;229 doi: 10.1016/j.pharmthera.2021.108050. [DOI] [PubMed] [Google Scholar]
- 15.Hyun S., Shin D. Small-molecule inhibitors and degraders targeting KRAS-driven cancers. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naim N., Moukheiber S., Daou S., Kourie H.R. KRAS-G12C covalent inhibitors: a game changer in the scene of cancer therapies. Crit. Rev. Oncol. Hematol. 2021;168 doi: 10.1016/j.critrevonc.2021.103524. [DOI] [PubMed] [Google Scholar]
- 17.Nagasaka M., Potugari B., Nguyen A., Sukari A., Azmi A.S., Ou S.H.I. KRAS Inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 2021;101 doi: 10.1016/j.ctrv.2021.102309. [DOI] [PubMed] [Google Scholar]
- 18.Hamilton G., Plangger A. Cytotoxic activity of KRAS inhibitors in combination with chemotherapeutics. Expert Opin. Drug Metab. Toxicol. 2021;17:1065–1074. doi: 10.1080/17425255.2021.1965123. [DOI] [PubMed] [Google Scholar]
- 19.Yang A., Li M., Fang M. The research progress of direct KRAS G12C mutation inhibitors. Pathol. Oncol. Res. 2021;27 doi: 10.3389/pore.2021.631095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Indini A., Rijavec E., Ghidini M., Cortellini A., Grossi F. Targeting KRAS in solid tumors: current challenges and future opportunities of novel KRAS inhibitors. Pharmaceutics. 2021;13 doi: 10.3390/pharmaceutics13050653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H., Smaill J.B., Liu T., Ding K., Lu X. Small-molecule inhibitors directly targeting KRAS as anticancer therapeutics. J. Med. Chem. 2020;63:14404–14424. doi: 10.1021/acs.jmedchem.0c01312. [DOI] [PubMed] [Google Scholar]
- 22.Shetu S.A., Bandyopadhyay D. Small-molecule RAS inhibitors as anticancer agents: discovery, development, and mechanistic studies. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23073706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore A.R., Rosenberg S.C., McCormick F., Malek S. Author Correction: RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 2020;19:902. doi: 10.1038/s41573-020-0089-1. [DOI] [PubMed] [Google Scholar]
- 24.Amgen Study to Compare AMG 510 "Proposed INN Sotorasib" with Docetaxel in Non Small Cell Lung Cancer (NSCLC) (CodeBreak 200) 2020. https://ClinicalTrials.gov/show/NCT04303780
- 25.Amgen Expanded Access of Sotorasib. https://ClinicalTrials.gov/show/NCT04667234
- 26.Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C.G., Koppada N., et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 27.Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D.M., Sudhakar N., Bowcut V., Baer B.R., Ballard J.A., et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson I.C. Expanded Access of Adagrasib (MRTX849) in Patients With Advanced Solid Tumors Who Have a KRAS G12C Mutation. Identifier NCT05162443. 2021 https://ClinicalTrials.gov/show/NCT05162443 [Google Scholar]
- 29.Phase 3 Study of MRTX849 (Adagrasib) vs Docetaxel in Patients with Advanced Non-small Cell Lung Cancer with KRAS G12C Mutation. https://ClinicalTrials.gov/show/NCT04685135.
- 30.Veluswamy R., Mack P.C., Houldsworth J., Elkhouly E., Hirsch F.R. KRAS G12C-mutant non-small cell lung cancer: biology, developmental therapeutics, and molecular testing. J. Mol. Diagn. 2021;23:507–520. doi: 10.1016/j.jmoldx.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Purkey H. Abstract ND11: discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. Cancer Res. 2022;82:ND11. doi: 10.1158/1538-7445.Am2022-nd11. [DOI] [Google Scholar]
- 32.MedChemExpress GDC-6036. https://file.medchemexpress.com/batch_PDF/HY-145928/GDC-6036-DataSheet-MedChemExpress.pdf
- 33.Malhotra S. & Xin J. (2020). Fused Ring Compounds (International Patent No. WO 2020/097537 A2). World Intellectual Property Organization International Bureau. https://patents.google.com/patent/WO2020097537A2/en.
- 34.InventisBio Co L., Sharp M., Corp D. Study to Evaluate D-1553 in Subjects with Solid Tumors. https://ClinicalTrials.gov/show/NCT04585035
- 35.Shi Z., Weng J., Fan X., Wang E., Zhu Q., Tao L., Han Z., Wang Z., Niu H., Jiang Y., et al. Abstract 932: discovery of D-1553, a novel and selective KRas-G12C Inhibitor with potent anti-tumor activity in a broad spectrum of tumor cell lines and xenograft models. Cancer Res. 2021;81:932. doi: 10.1158/1538-7445.Am2021-932. [DOI] [Google Scholar]
- 36.Savarese F., Gollner A., Rudolph D., Lipp J., Popow J., Hofmann M.H., Arnhof H., Rinnenthal J., Trapani F., Gmachl M., et al. Abstract 1271: in vitro and in vivo characterization of BI 1823911 - a novel KRASG12C selective small molecule inhibitor. Cancer Res. 2021;81:1271. doi: 10.1158/1538-7445.Am2021-1271. [DOI] [Google Scholar]
- 37.A Study to Test Different Doses of BI 1823911 Alone and Combined with Other Medicines in People with Different Types of Advanced Cancer with KRAS Mutation. https://ClinicalTrials.gov/show/NCT04973163
- 38.Li J., Zhao J., Cao B., Fang J., Li X., Wang M., Ba Y., Li X., Li Z., LIU Z., et al. A phase I/II study of first-in-human trial of JAB-21822 (KRAS G12C inhibitor) in advanced solid tumors. J. Clin. Oncol. 2022;40:3089. doi: 10.1200/JCO.2022.40.16_suppl.3089. [DOI] [Google Scholar]
- 39.JAB-21822 Activity in Adult Patients with Advanced Solid Tumors Harboring KRAS G12C Mutation. https://ClinicalTrials.gov/show/NCT05002270
- 40.Wang P., Zheng Q., Kang D., Sun X., Zhu S., Wang Y., Long W., Lin Y. 30P Investigation of KRAS G12C inhibitor JAB-21822 as a single agent and in combination with SHP2 inhibitor JAB-3312 in preclinical cancer models. Ann. Oncol. 2022;33:S1441. doi: 10.1016/j.annonc.2022.10.040. [DOI] [Google Scholar]
- 41.Study of JDQ443 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. https://ClinicalTrials.gov/show/NCT04699188 [DOI] [PMC free article] [PubMed]
- 42.Lorthiois E., Gerspacher M., Beyer K.S., Vaupel A., Leblanc C., Stringer R., Weiss A., Wilcken R., Guthy D.A., Lingel A., et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRAS(G12C) for the treatment of solid tumors. J. Med. Chem. 2022;65:16173–16203. doi: 10.1021/acs.jmedchem.2c01438. [DOI] [PubMed] [Google Scholar]
- 43.Weiss A., Lorthiois E., Barys L., Beyer K.S., Bomio-Confaglia C., Burks H., Chen X., Cui X., de Kanter R., Dharmarajan L., et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov. 2022;12:1500–1517. doi: 10.1158/2159-8290.cd-22-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Study of efficacy and safety of JDQ443 single-agent as first-line treatment for patients with locally advanced or metastatic KRAS G12C- mutated non-small cell lung cancer with a PD-L1 expression < 1% or a PD-L1 expression ≥ 1% and an STK11 Co-mutation. https://ClinicalTrials.gov/show/NCT05445843
- 45.Platform Study of JDQ443 in Combinations in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. https://ClinicalTrials.gov/show/NCT05358249
- 46.Study of JDQ443 in comparison with docetaxel in participants with locally advanced or metastatic KRAS G12C mutant non-small cell lung cancer. https://ClinicalTrials.gov/show/NCT05132075
- 47.A Study of MK-1084 as Monotherapy and in Combination with Pembrolizumab (MK-3475) in Participants with KRASG12C Mutant Advanced Solid Tumors (MK-1084-001) https://ClinicalTrials.gov/show/NCT05067283
- 48.Zhao H., Li L., Liu J., Mai R., Chen J., Chen J. Discovery of ARS-1620 analogs as KRas G12C inhibitors with high in vivo antitumor activity. Bioorg. Chem. 2022;121 doi: 10.1016/j.bioorg.2022.105652. [DOI] [PubMed] [Google Scholar]
- 49.Janes M.R., Zhang J., Li L.S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S.J., Darjania L., et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–589.e17. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 50.Schulze C.J., Bermingham A., Choy T.J., Cregg J.J., Kiss G., Marquez A., Reyes D., Saldajeno-Concar M., Weller C.E., Whalen D.M., et al. Abstract PR10: tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in vivo. Mol. Cancer Ther. 2019;18:PR10. doi: 10.1158/1535-7163.Targ-19-pr10. [DOI] [Google Scholar]
- 51.Nichols R., Schulze C., Bermingham A., Choy T., Cregg J., Kiss G., Marquez A., Reyes D., Saldajeno-Concar M., Weller C., et al. A06 tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in preclinical models of NSCLC. J. Thorac. Oncol. 2020;15:S13–S14. doi: 10.1016/j.jtho.2019.12.035. [DOI] [Google Scholar]
- 52.Hunter J.C., Gurbani D., Ficarro S.B., Carrasco M.A., Lim S.M., Choi H.G., Xie T., Marto J.A., Chen Z., Gray N.S., Westover K.D. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA. 2014;111:8895–8900. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim S.M., Westover K.D., Ficarro S.B., Harrison R.A., Choi H.G., Pacold M.E., Carrasco M., Hunter J., Kim N.D., Xie T., et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 2014;53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phase I Study of HRS-4642 in Patients with Advanced Solid Tumors Harboring KRAS G12D Mutation. https://ClinicalTrials.gov/show/NCT05533463
- 55.Ledford H. Cancer drugs are closing in on some of the deadliest mutations. Nature. 2022;610:620–622. doi: 10.1038/d41586-022-03392-2. [DOI] [PubMed] [Google Scholar]
- 56.Tran T.H., Alexander P., Dharmaiah S., Agamasu C., Nissley D.V., McCormick F., Esposito D., Simanshu D.K., Stephen A.G., Balius T.E. The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proc. Natl. Acad. Sci. USA. 2020;117:3363–3364. doi: 10.1073/pnas.1918164117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kessler D., Bergner A., Böttcher J., Fischer G., Döbel S., Hinkel M., Müllauer B., Weiss-Puxbaum A., McConnell D.B. Drugging all RAS isoforms with one pocket. Future Med. Chem. 2020;12:1911–1923. doi: 10.4155/fmc-2020-0221. [DOI] [PubMed] [Google Scholar]
- 58.Kessler D., Gmachl M., Mantoulidis A., Martin L.J., Zoephel A., Mayer M., Gollner A., Covini D., Fischer S., Gerstberger T., et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA. 2019;116:15823–15829. doi: 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haza K.Z., Martin H.L., Rao A., Turner A.L., Saunders S.E., Petersen B., Tiede C., Tipping K., Tang A.A., Ajayi M., et al. RAS-inhibiting biologics identify and probe druggable pockets including an SII-α3 allosteric site. Nat. Commun. 2021;12:4045. doi: 10.1038/s41467-021-24316-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X., Allen S., Blake J.F., Bowcut V., Briere D.M., Calinisan A., Dahlke J.R., Fell J.B., Fischer J.P., Gunn R.J., et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 2022;65:3123–3133. doi: 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- 61.Revolution Medicines, Inc.. Revolution Medicines Advances First RAS(ON) Inhibitor into Clinic, dosing first patient in phase 1/1b trial of RMC-6236. Globe NewsWire June 28,2022.
- 62.Evaluation of RMC-6236 in Subjects with Advanced Solid Tumors Harboring Specific Mutations in KRAS. https://ClinicalTrials.gov/show/NCT05379985
- 63.Xu K., Park D., Magis A.T., Zhang J., Zhou W., Sica G.L., Ramalingam S.S., Curran W.J., Deng X. Small molecule KRAS agonist for mutant KRAS cancer therapy. Mol. Cancer. 2019;18:85. doi: 10.1186/s12943-019-1012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mao Z., Xiao H., Shen P., Yang Y., Xue J., Yang Y., Shang Y., Zhang L., Li X., Zhang Y., et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8:5. doi: 10.1038/s41421-021-00368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng H., Zhang Y., Bos P.H., Chambers J.M., Dupont M.M., Stockwell B.R. K-Ras(G12D) has a potential allosteric small molecule binding site. Biochemistry. 2019;58:2542–2554. doi: 10.1021/acs.biochem.8b01300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teng K.W., Tsai S.T., Hattori T., Fedele C., Koide A., Yang C., Hou X., Zhang Y., Neel B.G., O'Bryan J.P., Koide S. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 2021;12:2656. doi: 10.1038/s41467-021-22969-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spencer-Smith R., Koide A., Zhou Y., Eguchi R.R., Sha F., Gajwani P., Santana D., Gupta A., Jacobs M., Herrero-Garcia E., et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017;13:62–68. doi: 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Welsch M.E., Kaplan A., Chambers J.M., Stokes M.E., Bos P.H., Zask A., Zhang Y., Sanchez-Martin M., Badgley M.A., Huang C.S., et al. Multivalent small-molecule pan-RAS inhibitors. Cell. 2017;168:878–889.e29. doi: 10.1016/j.cell.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Research Watch RAS can be targeted by a multivalent small-molecule inhibitor Cancer. Discov. 2017;7:350. doi: 10.1158/2159-8290.Cd-rw2017-042. [DOI] [Google Scholar]
- 70.Maurer T., Garrenton L.S., Oh A., Pitts K., Anderson D.J., Skelton N.J., Fauber B.P., Pan B., Malek S., Stokoe D., et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.A Study of LY3499446 in Participants With Advanced Solid Tumors With KRAS G12C Mutation. https://ClinicalTrials.gov/show/NCT04165031
- 72.First-in-Human Study of JNJ-74699157 in Participants With Tumors Harboring the KRAS G12C Mutation. 2019. https://ClinicalTrials.gov/show/NCT04006301
- 73.Punekar S.R., Velcheti V., Neel B.G., Wong K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022;19:637–655. doi: 10.1038/s41571-022-00671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim S., Khoo R., Juang Y.C., Gopal P., Zhang H., Yeo C., Peh K.M., Teo J., Ng S., Henry B., Partridge A.W. Exquisitely specific anti-KRAS biodegraders inform on the cellular prevalence of nucleotide-loaded states. ACS Cent. Sci. 2021;7:274–291. doi: 10.1021/acscentsci.0c01337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skoulidis F., Li B.T., Dy G.K., Price T.J., Falchook G.S., Wolf J., Italiano A., Schuler M., Borghaei H., Barlesi F., et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saiki A.Y., Gaida K., Rex K., Achanta P., Miguel T.S., Koppada N., Bagal D., Lanman B.A., Foti R.S., McCarter J.D., et al. Abstract 4484: discovery and in vitro characterization of AMG 510–a potent and selective covalent small-molecule inhibitor of KRASG12C. Cancer Res. 2019;79:4484. doi: 10.1158/1538-7445.Am2019-4484. [DOI] [Google Scholar]
- 78.Lanman B.A., Chen J.J., Liu L., Lopez P., Pickrell A.J., Reed A.B., Wang H.-L., Achanta P., Canon J., Erlanson D.A., et al. Abstract 4455: discovery of AMG 510, a first-in-human covalent inhibitor of KRASG12C for the treatment of solid tumors. Cancer Res. 2019;79:4455. doi: 10.1158/1538-7445.Am2019-4455. [DOI] [Google Scholar]
- 79.Shin Y., Jeong J.W., Wurz R.P., Achanta P., Arvedson T., Bartberger M.D., Campuzano I.D.G., Fucini R., Hansen S.K., Ingersoll J., et al. Discovery of N-(1-Acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as covalent inhibitors of KRAS(G12C) ACS Med. Chem. Lett. 2019;10:1302–1308. doi: 10.1021/acsmedchemlett.9b00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patricelli M.P., Janes M.R., Li L.S., Hansen R., Peters U., Kessler L.V., Chen Y., Kucharski J.M., Feng J., Ely T., et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 81.Chen K., Zhang Y., Qian L., Wang P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021;14:116. doi: 10.1186/s13045-021-01127-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J., Martin-Romano P., Cassier P., Johnson M., Haura E., Lenox L., Guo Y., Bandyopadhyay N., Russell M., Shearin E., et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist. 2022;27:536–e553. doi: 10.1093/oncolo/oyab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.AACR Project GENIE Consortium AACR project GENIE: powering precision medicine through an international Consortium. Cancer Discov. 2017;7:818–831. doi: 10.1158/2159-8290.Cd-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hofmann M.H., Gerlach D., Misale S., Petronczki M., Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov. 2022;12:924–937. doi: 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo W., Wu S., Liu J., Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shima F., Yoshikawa Y., Ye M., Araki M., Matsumoto S., Liao J., Hu L., Sugimoto T., Ijiri Y., Takeda A., et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA. 2013;110:8182–8187. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Z., Gao R., Hu Q., Peacock H., Peacock D.M., Dai S., Shokat K.M., Suga H. GTP-State-Selective cyclic peptide ligands of K-Ras(G12D) block its interaction with raf. ACS Cent. Sci. 2020;6:1753–1761. doi: 10.1021/acscentsci.0c00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ritt D.A., Abreu-Blanco M.T., Bindu L., Durrant D.E., Zhou M., Specht S.I., Stephen A.G., Holderfield M., Morrison D.K. Inhibition of ras/raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol. Cell. 2016;64:875–887. doi: 10.1016/j.molcel.2016.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yao Z., Gao Y., Su W., Yaeger R., Tao J., Na N., Zhang Y., Zhang C., Rymar A., Tao A., et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019;25:284–291. doi: 10.1038/s41591-018-0274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Athuluri-Divakar S.K., Vasquez-Del Carpio R., Dutta K., Baker S.J., Cosenza S.C., Basu I., Gupta Y.K., Reddy M.V.R., Ueno L., Hart J.R., et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell. 2016;165:643–655. doi: 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nichols R.J., Schulze C.J., Wang Z., Yang K., Jiang J., Whalen D.M., Hansen R., Garrenton L.S., Bermingham A., et al. American Association for Cancer Research Annual Meeting 2021; 2021. A Next Generation Tri-complex KRASG12C(ON) Inhibitor Directly Targets the Active, GTP-Bound State of Mutant RAS and May Overcome Resistance to KRASG12C(OFF) Inhibition. [Google Scholar]
- 92.Klein C.H., Truxius D.C., Vogel H.A., Harizanova J., Murarka S., Martín-Gago P., Bastiaens P.I.H. PDEdelta inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int. J. Cancer. 2019;144:767–776. doi: 10.1002/ijc.31859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leung E.L.H., Luo L.X., Liu Z.Q., Wong V.K.W., Lu L.L., Xie Y., Zhang N., Qu Y.Q., Fan X.X., Li Y., et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018;9:216. doi: 10.1038/s41419-017-0065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.A Phase 1, First-In-Human Study of Escalating Doses of Oral TVB-2640 in Patients with Solid Tumors. https://ClinicalTrials.gov/show/NCT02223247
- 95.Saha B., Nandi D. Farnesyltransferase inhibitors reduce Ras activation and ameliorate acetaminophen-induced liver injury in mice. Hepatology. 2009;50:1547–1557. doi: 10.1002/hep.23180. [DOI] [PubMed] [Google Scholar]
- 96.Kapoor A., Yao W., Ying H., Hua S., Liewen A., Wang Q., Zhong Y., Wu C.-J., Sadanandam A., Hu B., et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yuan T.L., Amzallag A., Bagni R., Yi M., Afghani S., Burgan W., Fer N., Strathern L.A., Powell K., Smith B., et al. Differential effector engagement by oncogenic KRAS. Cell Rep. 2018;22:1889–1902. doi: 10.1016/j.celrep.2018.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ryan M.B., Fece de la Cruz F., Phat S., Myers D.T., Wong E., Shahzade H.A., Hong C.B., Corcoran R.B. Vertical pathway inhibition overcomes adaptive Feedback resistance to KRASG12C inhibition. Clin. Cancer Res. 2020;26:1633–1643. doi: 10.1158/1078-0432.Ccr-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yaeger R., Solit D.B. Overcoming adaptive resistance to KRAS inhibitors through vertical pathway targeting. Clin. Cancer Res. 2020;26:1538–1540. doi: 10.1158/1078-0432.Ccr-19-4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ruess D.A., Heynen G.J., Ciecielski K.J., Ai J., Berninger A., Kabacaoglu D., Görgülü K., Dantes Z., Wörmann S.M., Diakopoulos K.N., et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018;24:954–960. doi: 10.1038/s41591-018-0024-8. [DOI] [PubMed] [Google Scholar]
- 101.Hamarsheh S., Groß O., Brummer T., Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020;11:5439. doi: 10.1038/s41467-020-19288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Quandt J., Schlude C., Bartoschek M., Will R., Cid-Arregui A., Schölch S., Reissfelder C., Weitz J., Schneider M., Wiemann S., et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology. 2018;7 doi: 10.1080/2162402X.2018.1500671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsai Y.S., Woodcock M.G., Azam S.H., Thorne L.B., Kanchi K.L., Parker J.S., Vincent B.G., Pecot C.V. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J. Clin. Invest. 2022;132 doi: 10.1172/JCI155523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Maldegem F., Valand K., Cole M., Patel H., Angelova M., Rana S., Colliver E., Enfield K., Bah N., Kelly G., et al. Characterisation of tumour microenvironment remodelling following oncogene inhibition in preclinical studies with imaging mass cytometry. Nat. Commun. 2021;12:5906. doi: 10.1038/s41467-021-26214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lim S., Khoo R., Peh K.M., Teo J., Chang S.C., Ng S., Beilhartz G.L., Melnyk R.A., Johannes C.W., Brown C.J., et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA) Proc. Natl. Acad. Sci. USA. 2020;117:5791–5800. doi: 10.1073/pnas.1920251117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun X., Gao H., Yang Y., He M., Wu Y., Song Y., Tong Y., Rao Y. PROTACs: great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019;4:64. doi: 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zheng N., Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 2017;86:129–157. doi: 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- 108.Mullard A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021;20:247–250. doi: 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- 109.Zeng M., Xiong Y., Safaee N., Nowak R.P., Donovan K.A., Yuan C.J., Nabet B., Gero T.W., Feru F., Li L., et al. Exploring targeted degradation strategy for oncogenic KRAS(G12C) Cell Chem. Biol. 2020;27:19–31.e6. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 110.Bond M.J., Chu L., Nalawansha D.A., Li K., Crews C.M. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent. Sci. 2020;6:1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kraut N. Proceedings of the 112th Annual Meeting of the American Association for Cancer Research. 2021. Expanding the reach of precision oncology by drugging all KRAS mutants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nagashima T., Inamura K., Nishizono Y., Suzuki A., Tanaka H., Yoshinari T., Yamanaka Y. ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. Eur. J. Cancer. 2022;174:S30. doi: 10.1016/S0959-8049(22)00881-4. [DOI] [Google Scholar]
- 113.2022. A Study of ASP3082 in Adults with Previously Treated Solid Tumors.https://ClinicalTrials.gov/show/NCT05382559 [Google Scholar]
- 114.2022. Targeted Protein Degradation.https://www.astellas.com/en/innovation/tumor-directed-inhibition-research-unit [Google Scholar]
- 115.Antic I., Biancucci M., Satchell K.J.F. Cytotoxicity of the Vibrio vulnificus MARTX toxin effector DUF5 is linked to the C2A subdomain. Proteins. 2014;82:2643–2656. doi: 10.1002/prot.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Biancucci M., Rabideau A.E., Lu Z., Loftis A.R., Pentelute B.L., Satchell K.J.F. Substrate recognition of MARTX ras/rap1-specific endopeptidase. Biochemistry. 2017;56:2747–2757. doi: 10.1021/acs.biochem.7b00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Antic I., Biancucci M., Zhu Y., Gius D.R., Satchell K.J.F. Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat. Commun. 2015;6:7396. doi: 10.1038/ncomms8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stubbs C.K., Biancucci M., Vidimar V., Satchell K.J.F. RAS specific protease induces irreversible growth arrest via p27 in several KRAS mutant colorectal cancer cell lines. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-97422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vidimar V., Beilhartz G.L., Park M., Biancucci M., Kieffer M.B., Gius D.R., Melnyk R.A., Satchell K.J.F. An engineered chimeric toxin that cleaves activated mutant and wild-type RAS inhibits tumor growth. Proc. Natl. Acad. Sci. USA. 2020;117:16938–16948. doi: 10.1073/pnas.2000312117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vidimar V., Park M., Stubbs C.K., Ingram N.K., Qiang W., Zhang S., Gursel D., Melnyk R.A., Satchell K.J.F. Proteolytic pan-RAS cleavage leads to tumor regression in patient-derived pancreatic cancer xenografts. Mol. Cancer Ther. 2022;21:810–820. doi: 10.1158/1535-7163.MCT-21-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ma Y., Gu Y., Zhang Q., Han Y., Yu S., Lu Z., Chen J. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2013;12:286–294. doi: 10.1158/1535-7163.MCT-12-0650. [DOI] [PubMed] [Google Scholar]
- 122.Bery N., Miller A., Rabbitts T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020;11:3233. doi: 10.1038/s41467-020-17022-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A., et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fulcher L.J., Macartney T., Bozatzi P., Hornberger A., Rojas-Fernandez A., Sapkota G.P. An affinity-directed protein missile system for targeted proteolysis. Open Biol. 2016;6 doi: 10.1098/rsob.160255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen Y., Toth E.A., Ruan B., Choi E.J., Simmerman R., Chen Y., He Y., Wang R., Godoy-Ruiz R., King H., et al. Engineering subtilisin proteases that specifically degrade active. Commun. Biol. 2021;4:299. doi: 10.1038/s42003-021-01818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xi M., Chen Y., Yang H., Xu H., Du K., Wu C., Xu Y., Deng L., Luo X., Yu L., et al. Small molecule PROTACs in targeted therapy: an emerging strategy to induce protein degradation. Eur. J. Med. Chem. 2019;174:159–180. doi: 10.1016/j.ejmech.2019.04.036. [DOI] [PubMed] [Google Scholar]
- 127.Yao T., Xiao H., Wang H., Xu X. Recent advances in PROTACs for drug targeted protein research. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms231810328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Y., Jiang X., Feng F., Liu W., Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zeiser J., Gerhard R., Just I., Pich A. Substrate specificity of clostridial glucosylating toxins and their function on colonocytes analyzed by proteomics techniques. J. Proteome Res. 2013;12:1604–1618. doi: 10.1021/pr300973q. [DOI] [PubMed] [Google Scholar]
- 130.Simon N.C., Aktories K., Barbieri J.T. Novel bacterial ADP-ribosylating toxins: structure and function. Nat. Rev. Microbiol. 2014;12:599–611. doi: 10.1038/nrmicro3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Geissler B., Ahrens S., Satchell K.J.F. Plasma membrane association of three classes of bacterial toxins is mediated by a basic-hydrophobic motif. Cell. Microbiol. 2012;14:286–298. doi: 10.1111/j.1462-5822.2011.01718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Biancucci M., Minasov G., Banerjee A., Herrera A., Woida P.J., Kieffer M.B., Bindu L., Abreu-Blanco M., Anderson W.F., Gaponenko V., et al. The bacterial Ras/Rap1 site-specific endopeptidase RRSP cleaves Ras through an atypical mechanism to disrupt Ras-ERK signaling. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aat8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang X., Shen J., Jiang F., Jin Q. The Photorhabdus virulence cassettes RRSP-like effector interacts with cyclin-dependent kinase 1 and causes mitotic defects in mammalian cells. Front. Microbiol. 2020;11:366. doi: 10.3389/fmicb.2020.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loftis A.R., Santos M.S., Truex N.L., Biancucci M., Satchell K.J.F., Pentelute B.L. Anthrax protective antigen retargeted with single-chain variable fragments delivers enzymes to pancreatic cancer cells. Chembiochem. 2020;21:2772–2776. doi: 10.1002/cbic.202000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S., Wang J., Hamman B.D., Ishchenko A., Crews C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018;25:78–87.e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nagasaka M. ES28.04 emerging mechanisms to target KRAS directly. J. Thorac. Oncol. 2021;16:S96–S97. doi: 10.1016/j.jtho.2021.01.063. [DOI] [Google Scholar]
- 137.A study of mRNA-5671/V941 as monotherapy and in combination with pembrolizumab (V941-001) https://ClinicalTrials.gov/show/NCT03948763
- 138.Phase I - Escalating Dose Study of siG12D LODER (Local Drug EluteR) in Patients with Locally Advanced Adenocarcinoma of the Pancreas, and a Single Dose Study of siG12D LODER (Local Drug EluteR) in Patients with Non-operable Adenocarcinoma of the Pancreas. https://ClinicalTrials.gov/show/NCT01188785
- 139.A Phase 2 Study of siG12D LODER in Combination with Chemotherapy in Patients with Locally Advanced Pancreatic Cancer. https://ClinicalTrials.gov/show/NCT01676259
- 140.Sanclemente M., Francoz S., Esteban-Burgos L., Bousquet-Mur E., Djurec M., Lopez-Casas P.P., Hidalgo M., Guerra C., Drosten M., Musteanu M., Barbacid M. c-RAF ablation induces regression of advanced kras/trp53 mutant lung adenocarcinomas by a mechanism independent of MAPK signaling. Cancer Cell. 2018;33:217–228.e4. doi: 10.1016/j.ccell.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 141.Chatzikleanthous D., O'Hagan D.T., Adamo R. Lipid-based nanoparticles for delivery of vaccine adjuvants and antigens: toward multicomponent vaccines. Mol. Pharm. 2021;18:2867–2888. doi: 10.1021/acs.molpharmaceut.1c00447. [DOI] [PubMed] [Google Scholar]
- 142.Huang X., Kong N., Zhang X., Cao Y., Langer R., Tao W. The landscape of mRNA nanomedicine. Nat. Med. 2022;28:2273–2287. doi: 10.1038/s41591-022-02061-1. [DOI] [PubMed] [Google Scholar]
- 143.Mitchell M.J., Billingsley M.M., Haley R.M., Wechsler M.E., Peppas N.A., Langer R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021;20:101–124. doi: 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cai W., Luo T., Chen X., Mao L., Wang M. A Combinatorial Library of Biodegradable Lipid Nanoparticles Preferentially Deliver mRNA into Tumor Cells to Block Mutant RAS Signaling. Adv. Funct. Mater. 2022;32 doi: 10.1002/adfm.202204947. [DOI] [Google Scholar]
- 145.Lechuga C.G., Salmón M., Paniagua G., Guerra C., Barbacid M., Drosten M. RASless MEFs as a tool to study RAS-dependent and RAS-independent functions. Methods Mol. Biol. 2021;2262:335–346. doi: 10.1007/978-1-0716-1190-6_21. [DOI] [PubMed] [Google Scholar]
- 146.Kim D., Rossi J. RNAi mechanisms and applications. BioTechniques. 2008;44:613–616. doi: 10.2144/000112792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Simpson L.M., Macartney T.J., Nardin A., Fulcher L.J., Röth S., Testa A., Maniaci C., Ciulli A., Ganley I.G., Sapkota G.P. Inducible degradation of target proteins through a tractable affinity-directed protein missile system. Cell Chem. Biol. 2020;27:1164–1180.e5. doi: 10.1016/j.chembiol.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]