

Abstract
IL-17-producing antigen-specific human T cells elicit potent antitumor activity in mice. Yet, refinement of this approach is needed to position it for clinical use. While activation signal strength regulates IL-17 production by CD4+ T cells, the degree to which T cell antigen receptor (TCR) and costimulation signal strength influences Th17 immunity remains unknown. We discovered that decreasing TCR/costimulation signal strength by incremental reduction of αCD3/costimulation beads progressively altered Th17 phenotype. Moreover, Th17 cells stimulated with αCD3/inducible costimulator (ICOS) beads produced more IL-17A, IFNγ, IL-2, and IL-22 than those stimulated with αCD3/CD28 beads. Compared with Th17 cells stimulated with the standard, strong signal strength (three beads per T cell), Th17 cells propagated with 30-fold fewer αCD3/ICOS beads were less reliant on glucose and favored the central carbon pathway for bioenergetics, marked by abundant intracellular phosphoenolpyruvate (PEP). Importantly, Th17 cells stimulated with weak αCD3/ICOS beads and redirected with a chimeric antigen receptor that recognizes mesothelin were more effective at clearing human mesothelioma. Less effective CAR Th17 cells generated with high αCD3/ICOS beads were rescued by overexpressing phosphoenolpyruvate carboxykinase 1 (PCK1), a PEP regulator. Thus, Th17 therapy can be improved by using fewer activation beads during manufacturing, a finding that is cost effective and directly translatable to patients.
Keywords: CAR-T, cellular therapy, Th17, inducible costimulator, ICOS, TCR signal strength
Graphical abstract
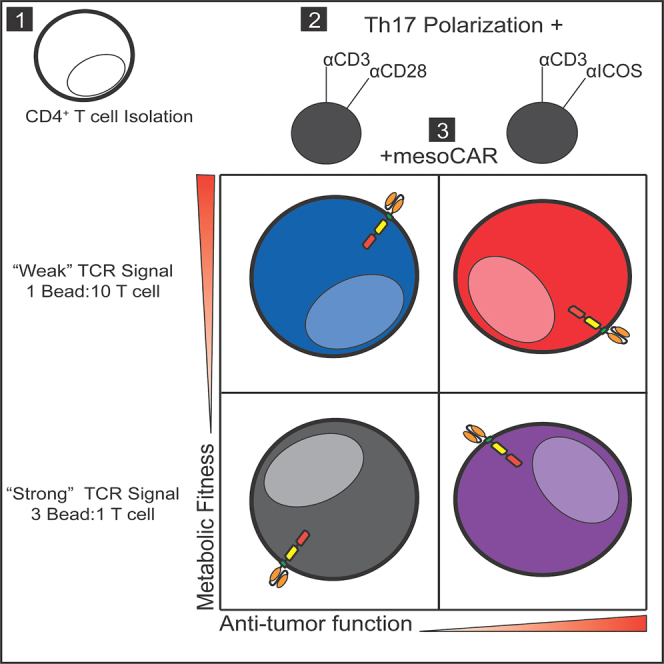
Wyatt and colleagues discovered that human Th17 CAR-T cells generated with lower TCR activation strength and ICOS costimulation have improved function and metabolic fitness and can regress solid tumors to a greater extent than cells generated with higher activation signal strength, resulting in a cost-saving approach to CAR-T cell manufacturing.
Introduction
T cells genetically redirected to express a chimeric or T cell antigen receptor (CAR, TCR) can elicit potent efficacy in a subset of patients with tumors harboring the antigenic target.1,2,3 While CD19-CAR T cell products have demonstrated success for individuals with B cell malignancies, CAR-based therapy approaches have been less effective at treating patients with solid tumors.4 The reason adoptive T cell transfer (ACT) therapy fails for patients with solid tumors is multifactorial, including inefficient trafficking of T cells into the tumor,5 inability to overcome the oppressive tumor microenvironment,6 and low persistence and response maintenance against aggressive malignancies.4,7
One potential strategy to overcome limitations of ACT therapy is to improve the quality of T cells generated ex vivo. Currently, standard FDA-approved ACT protocols recommend logarithmic growth of CAR T cells, which involves the selection of CD3+ T cells from the patient’s blood and expansion with artificial antigen-presenting cells (aAPCs). These aAPCs often consist of a magnetic bead coated with αCD3 and αCD28 antibodies to mediate TCR stimulation and costimulation, respectively. Three αCD3/CD28 beads are used per T cell in the culture to ensure logarithmic activation and propagation.8,9 As part of these protocols, T cells are genetically engineered to encode an antigen receptor and expanded in the presence of cytokines, such as IL-2, for 2 weeks before reinfusion into the lymphodepleted patient.
We posited that cell therapy against solid tumors can be advanced by generating human T helper (Th) 17 cell products in a less differentiated state. Th17 cells are a helper CD4+ T cell subset originally reported as pro-inflammatory and important in mediating defense against self-tissue attack and infections via their production of IL-17.10,11 We and others reported that Th17 subsets elicit potent efficacy against solid tumors in aggressive, pre-clinical tumor models, compared with other helper subsets, including Th1 or Th2 cells.12,13,14,15,16 Subsequent investigations revealed that Th17 cells possess hallmarks of stemness, marked by enhanced self-renewal, function, and proliferative potential, which make them particularly attractive as a novel ACT approach.14,17,18 Yet, the ideal way to expand antitumor Th17 cells for ACT without losing these characteristics remains elusive and a significant barrier to their implementation beyond the pre-clinical setting.
Prior work has demonstrated that lowering activation signal strength generates T cells with a younger memory phenotype and higher functionality.19,20,21 We hypothesized that reducing the initial human Th17 activation strength by using fewer αCD3 beads coated with either αCD28 or α-inducible costimulator (αICOS) in the culture would improve their antitumor activity, even when the CD8+ CAR T cells they are coinfused with are separately activated with the standard 3:1 αCD3/CD28 bead-to-T cell ratio condition. As signal strength was weakened, fewer Th17 cells differentiated into a full effector memory phenotype. Weak ICOS-costimulated Th17 cells maintained a more naive memory phenotype and cosecreted multiple cytokines. Th17 cells stimulated with the standard, strong signal strength relied more on glycolysis for growth and energy, while those stimulated with weak ICOS signal strength had enhanced mitochondrial bioenergetics. Global metabolomic analysis of these cells identified unique metabolites associated with the central carbon pathway and gluconeogenesis that are elevated in Th17 cells generated with weak signal strength ICOS bead preparations. In vivo, weak ICOS-costimulated Th17 cells redirected with a mesothelin-specific CAR and cotransfused with standard αCD3/CD28 strong-activated CD8+ mesoCAR T cells persisted and effectively controlled the growth of mesothelioma tumors. To understand the importance of the observed metabolic differences between ICOS strong and ICOS weak Th17 cells, we overexpressed phosphoenolpyruvate carboxykinase 1 (PCK1), a key enzymatic regulator of gluconeogenesis, in ICOS strong mesoCAR Th17 cells, which greatly improved their antitumor activity. Our work reveals that modulating metabolic pathways in Th17 cells is possible by simply using fewer beads in a culture, which results in potentiated CAR T cell immunity against solid tumors. These data enable further refinement of ex vivo expansion protocols for human Th17 cells that will accelerate the path to future clinical application.
Results
Lowering signal strength improves human Th17 cell polyfunctionality
Th17 subsets are more potent against solid tumors compared with other helper subsets, including Th1 or Th2 cells.13,14,22 We theorized that expanding human Th17 cells with fewer beads would generate lymphocytes with a more functional phenotype, marked by their capacity to secrete multiple cytokines at once. To test our hypothesis that reducing TCR/costimulation activation signal strength augments Th17 cell functionality and memory phenotype, we serially diluted the number of activation beads added to human Th17 cells to as low as 30-fold less than that used in the clinic, to mimic reduced antigen signaling. Specifically, CD4+ T cells were freshly isolated from peripheral blood mononuclear cells (PBMCs) of deidentified healthy human donors and cytokine-polarized toward a Th17 phenotype while being activated with αCD3 magnetic beads coated with either αCD28 or αICOS agonists, as depicted in Figure 1A. Th17-polarized cells were expanded in the presence of IL-2 (100 IU/mL, added 2 days post-polarization) for 10 days, and then the end-product cells were assayed for their capacity to secrete cytokines after phorbol myristate acetate (PMA) and ionomycin restimulation. We found that, as signal strength decreased, IFNγ and IL-17A production increased gradually (Figure 1B). Th17 cells expanded with the fewest beads (1:10 bead:T cell ratio) had the greatest percentage of cells that expressed both cytokines, and we observed that this polyfunctionality was most pronounced in cells activated with αCD3/ICOS beads (Figure 1C). Note that Th17 cell numbers in cultures were held constant (∼0.8 million/mL in a 24-well plate), while the αCD3/costimulatory bead numbers were altered as a rheostat for activation strength. Our data reveal that decreasing the activator beads profoundly bolsters Th17 polyfunctionality.
Figure 1.

Decreasing activation signal strength results in increased functionality of Th17 cells
(A) CD4+ T cells from normal human donors were isolated and expanded with either CD3/CD28 or CD3/ICOS artificial APC beads and Th17 polarizing cytokines. (B) Percentages of IFNγ and IL-17A cytokine-producing Th17 cells expanded for 9 days after activation with either CD3/CD28 (top) or CD3/ICOS (bottom) beads at indicated bead:T cell ratios, as assessed by flow cytometry. (C) Average percentage of IFNγ+IL-17A+Th17 cells (±SEM) at day 10 post-activation, n = 3. (D) Average percentages (±SEM) of IL-17A, IL-22, IFNγ, and IL-2 cytokine-producing Th17 cells activated with a range of αCD3/αICOS bead:T cell ratios as determined by flow cytometry. Shaded area indicates the range of observed optimal cytokine production containing the three highest averages for each cytokine (n = 6 healthy donors). Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparisons test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.001).
To uncover the ideal number of beads needed to optimize IL-17, IFNγ, IL-22, and IL-2 production by ICOS-costimulated Th17 cultures, cells were activated with a more dynamic titration range of activator beads (αCD3/ICOS) and then assessed for their functional profile (Figure 1D). Cytokine production of each peaked in T cells stimulated at an intermediate T cell-to-bead ratio. The range of the highest values containing the top three averages for each overlapped at one ratio, 1:10.7 bead:T cell, although the only significant differences observed were within IFNγ at the 1:10.7 to 1:42.7 bead:T cell conditions compared with the 3:1 bead:T cell condition (p < 0.05). Conversely, Th17 cells activated with strong signal strength using the standard ratio (3:1 bead:T cell) produced significantly lower levels of all cytokines tested except for IL-22, where one of the healthy donors analyzed had 43% IL-22+ cells at this ratio, while the other five donors had an average of 5% IL-22+ under this condition. Regardless, the intersection of highest cytokine production for all four cytokines analyzed occurs at ∼1:10 bead:T cell. At lower signal strengths of ∼1:43 bead:T cell and below, cytokine production gradually tapered, albeit the production of any four cytokines remained higher than that of those expanded with high-signal-strength cells (i.e., ∼3:1–1:1 bead:T cell). Overall, our data reveal an optimal bead-to-T cell ratio in which fewer beads can be used to manufacture Th17 cell products with enhanced multifunctionality.
Lowering activation signal strength slows Th17 cell division but sustains naive memory phenotypes
Current ACT protocols require expansion of large numbers of T cells before they are infused back into patients. Proliferation assays using carboxyfluoroscein succinimidyl ester (CFSE) were done to test if reducing the number of beads added to Th17 cells would affect their expansion and yield by culturing them with either a “Weak” (1 bead per 10 T cells) or a “Strong” bead stimulus protocol (the standard 3 beads per T cell). In Th17 cultures activated with αCD3/CD28 beads, we found that both the Strong and the Weak bead per T cell groups divided within 2 days (Figure 2A). By day 4, all cells in the CD28 Strong and CD28 Weak groups divided at least once. One week later, most cells in both groups had divided. In contrast, αCD3/ICOS-stimulated Th17 cells lagged in proliferation compared with those stimulated with αCD3/CD28 beads at the same ratios. In fact, by day 7 post-activation, a small percentage of ICOS Weak cells did not divide. These data suggest a difference in ICOS and CD28 signaling cues in promoting T cell proliferation, potentially due to the constitutive presence of CD28 on the T cell surface, while ICOS upregulates after TCR stimulation.23 However, the expansion rates and ultimate cell yield were not significantly different between Th17 cells undergoing either CD28 or ICOS costimulation, nor between the ICOS Strong and ICOS Weak conditions (Figure 2B). The CD28 Weak condition did significantly lower the yield of Th17 cells by day 10 post-activation, compared with the CD28 Strong condition (p = 0.0035).
Figure 2.

Lower activating signal strength programs an optimal T cell memory phenotype
(A) Cell division over 1 week of culture. CFSE-labeled cells were activated with either CD3/CD28 (black and blue) or CD3/ICOS (purple and red) beads at two different ratios: 3:1 bead:T cell and 1:10 bead:T cell. Cells from each culture were collected and fixed each day after activation, up to 7 days. After 7 days, all cells were analyzed via flow cytometry. (B) Cell growth rate was also observed. Cells from each culture were counted every other day, up to day 10 of culture (n = 3). (C–F) After 10 days of growth, expression of memory markers and coinhibitory receptors was analyzed by flow cytometry. Numbers in the fluorescence-activated cell sorting (FACS) plots represent percentage of positive lymphocytes. Bar graphs represent the mean ± SEM. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (n = 7, ∗p < 0.05, ∗∗p < 0.01).
T cell differentiation and senescence are factors that limit the longevity of T cells used for ACT and hence its success. Thus, it is often a goal of novel manufacturing protocols to produce fewer differentiated T cell products for infusion. Because T cell expansion drives differentiation, we next assessed the memory profile of Th17 cells expanded with these different stimulation protocols, hypothesizing that higher signal strength (i.e., more beads to T cells) would foster greater effector memory differentiation. As expected, at day 10 post-activation, Strong, 3 beads per T cell, stimulatory conditions promoted the generation of differentiated cells, while Weak, 10 beads per T cell, signal strength supported more naive-like memory cells, as indicated by the percentages of CD45RA+CD45RO− cells (Figure 2C) and CD45RA+CD62L+ cells (Figure 2D). CCR7 and CD27, additional markers expressed on naive T cells, were also most elevated in Th17 cells given fewer beads (Figure 2E). Expression of T cell exhaustion and the coinhibitory markers CD39, Tim3, and PD-1 was elevated on Strong-stimulated cells (Figure 2F). Thus, we found that weaker signal strength-generated Th17 cells were ultimately those with a less differentiated memory phenotype.
Low signal strength improves Th17 mitochondrial function and reduces utilization of glucose
Mitochondrial health and metabolism are essential to T cell survival, function, and immunity.24,25,26,27 Consequentially, generating a metabolically fit T cell product is critical for durable antitumor immunity, as it enables resistance to the harsh and nutritionally restrictive tumor environment. Proliferation and T cell differentiation are tightly tied to the commitment to glycolysis,28 so we hypothesized that weaker signal strength promoted less glucose reliance in Th17 cells. We assayed the expression of glucose transporter 1 (GLUT1) and the cellular glucose uptake (using 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose [2-NBDG], a fluorescent glucose analog) in Th17 cells expanded with titrated αCD3 magnetic beads decorated with αCD28 or αICOS agonist antibodies at Strong (3 beads per T cell) and Weak (1 bead per 10 T cells) levels of signal strength over the course of in vitro expansion. GLUT1 was consistently elevated in the Strong CD28-activated Th17 cells, with a spike in expression on days 5–6 of culture (Figure 3A). All other activation treatments expressed less surface GLUT1 throughout culture compared with the Strong CD28-activated Th17 cells. Concomitantly, Th17 cells were measured for glucose uptake using 2-NBDG. Cells stimulated with a Weak signal strength consumed glucose at a slower rate than those cells activated at the Strong signal strength, regardless of ICOS or CD28 costimulatory signaling (Figure 3B). Th17 cells stimulated with the Strong concentration of αCD3/ICOS beads consumed less 2-NBDG than cells expanded with the Strong αCD3/CD28 beads after a week of expansion, suggesting that ICOS costimulation decreases T cell glucose utilization. Due to the observed differences in glucose reliance between the groups, we next probed the relative mitochondrial profile of Weak- vs. Strong-activated Th17 cells.
Figure 3.

Glucose uptake and mitochondrial dynamics are affected by signal strength and costimulatory signal
Flow cytometry was used to evaluate (A) the presence of the GLUT1 receptor on αCD3/CD28 or αCD3/ICOS activated cells as well as to assess (B) their ability to take up glucose using the fluorescent glucose reporter 2-NDGB over 7 days of culture. (C) At 10 days post-activation, markers of mitochondrial mass (MitoTracker deep red) and membrane potential (TMRM) were used to characterize the cells by flow cytometry. (D) The percentage of cells with low mitochondrial membrane potential is represented. (E) At the same time point, a Seahorse mitochondria stress test was used to calculate percentage spare respiratory capacity (SRC). For each assay, n = 4–6 healthy donors. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (∗p < 0.05). Data are represented as mean ± SEM.
T cells possessing mitochondria with low mitochondrial membrane potential (ΔѰm) retain enriched markers of stemness and are capable of enhanced tumor ablation in vivo.29 We hypothesized that weakening signal strength would increase the abundance of Th17 cells with a low ΔѰm signature, based on our findings regarding their memory and polyfunctionality. Th17 cells were profiled for their relative abundance of mitochondria, mitochondrial phenotype, and metabolic capacity. Mitochondrial mass (MitoTracker deep red; Thermo Fisher) and ΔѰm (tetramethyl rhodamine methyl ester [TMRM]) were analyzed by flow cytometry. As predicted, whether costimulated by αCD28 or αICOS agonists, lowering the activation signal strength expanded Th17 cells with more of their mitochondrial mass exhibiting lower ΔѰm (Figures 3C and 3D), potentially indicating that Weak signal strength preserves high mitochondrial function and mass. We theorized that the capacity for mitochondrial energy generation in Th17 cells would retain more spare respiratory capacity (SRC) when stressed following cell expansion with fewer activator beads. Using a Seahorse XF assay (Agilent), the percentage SRC increased when the activation signal strength was decreased, although not significantly (Figure 3E). Our data imply that mitochondria in Th17 cells activated with a weaker TCR signal strength and receiving ICOS costimulatory signaling are less differentiated, proliferate at lower rates, and are less reliant on glycolysis in vitro. Therefore, Th17 cells stimulated with 30-fold fewer activation beads have altered metabolic requirements compared with those expanded with more beads commonly used to generate clinical CAR products. Because of the marked decrease in glucose uptake, the ability to proliferate, and subtle changes in mitochondrial function, we hypothesized that these Th17 cultures expanded with a reduced signal strength and ICOS costimulatory signaling may be altering their metabolic needs to promote their survival and function, so we further probed the metabolic profile of these cells.
Gluconeogenesis is used by Weak TCR-activated/ICOS-costimulated Th17 cells
To interrogate the metabolic profile of human Th17 cells activated with a Strong vs. a Weak TCR signal, human CD4+ T cells from seven healthy PBMC donors were programmed toward a Th17 phenotype and then activated, or not, with a Strong (3 beads per T cell) or Weak (1 bead per 10 T cells) number of αCD3/ICOS beads for 4 days, to optimally capture the differences between treatments based on our metabolic phenotype time-course data (Figures 3A and 3B). Metabolomic analysis of the cells and corresponding media was done via mass spectrometry (Metabolon), and the fold changes in analyzed intracellular metabolites were calculated between treatment groups and grouped by metabolic pathway (Figure 4A). Increased glycolysis intermediates glucose-6-phosphate (G6P; p = 0.0001) and fructose-6-phosphate (F6P; p < 0.0001) were detected intracellularly in ICOS Strong-activated Th17 cells compared with ICOS Weak-activated Th17 cells (Figure 4B). Concordantly, Strong ICOS stimulation depleted glucose levels extracellularly (p < 0.0001) and increased lactate secretion (p < 0.0001) compared with Weak ICOS stimulation, suggesting high levels of glycolytic activity with that stimulation (Figure 4C). In contrast, Weak ICOS stimulation enriched isocitrate, phosphoenolpyruvate (PEP), and 3-phosphoglycerate intracellularly (p < 0.05; Figure 4B). Furthermore, Weak ICOS stimulation did not induce overt extracellular uptake of glucose, pyruvate, or glycerate (Figure 4C) or induce intracellular levels of G6P or F6P (Figure 4B), suggesting the direction of metabolic flux to favor that of gluconeogenesis instead of glycolysis.
Figure 4.

Changes in metabolic states of ICOS low vs. high activated T cells, compared with no activation, are revealed by metabolomics
(A) Metabolic analysis by mass spectrometry was performed on Th17 cells from healthy donors (n = 7) that were activated (or not, “None”) with αCD3/ICOS beads at the indicated bead per T cell ratio and cultured for 4 days before collection. Resulting values were normalized and fold changes were calculated and log2 transformed. Normalized values for selected metabolites measured intracellularly (B) and from theextracellular medium (C) were averaged. Statistical analysis was performed using paired t tests (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Data are represented as mean ± SEM.
Comparison of ICOS Weak- vs. ICOS Strong-activated Th17 intracellular metabolites revealed further differences in their use of the central carbon pathway, shown by increased abundance of metabolites involved in gluconeogenesis (PEP and 3-phosphoglycerate) as well as the pentose phosphate pathway (6-phosphogluconate), suggestive of active gluconeogenesis and shunting of carbon sources to the pentose phosphate pathway. An important rate-limiting step of gluconeogenesis is the formation of PEP from oxaloacetate (OAA) catalyzed by the enzyme PCK1.30 Importantly, we found that PEP was 14-fold higher in Weak ICOS-stimulated vs. Strong ICOS-stimulated Th17 cells (p = 0.0003; Figure 4B). This metabolic profile coupled with the proliferation, differentiation, and mitochondrial phenotype led us to hypothesize that Weak ICOS-stimulated cells may be more efficacious when infused into mice bearing a human solid tumor.
Weak ICOS signal strength augments Th17 cell engraftment and antitumor activity in vivo
Using an established human mesothelioma tumor model, we tested our hypothesis that decreasing the number of activation beads used to generate mesothelin-specific CAR-modified Th17 cells in vitro would improve their antitumor activity in vivo. To do so, bulk human CD4+ T cells were programmed toward a Th17 phenotype while being activated with one of four treatments, using either αCD3/CD28 or αCD3/ICOS beads at either a 3 bead per T cell (Strong) or 1 bead per 10 T cells (Weak) ratio. As shown in the diagram in Figure 5A, NOD/scid gamma chain knockout (NSG) mice bearing a human mesothelioma tumor were infused with the mesoCAR Th17 cell products. Human mesoCAR CD8+ T cells, expanded with αCD3/CD28 beads at the Strong signal strength (3 beads per T cell), were cotransferred into mice. As anticipated, tumor growth was only slightly and transiently halted in mice treated with CD28 Strong-activated Th17 cells compared with untreated animals. Tumor growth was controlled for 10 days post-cell infusion in mice treated with CD28 Weak-activated Th17 cells, after which their tumors rapidly expanded (Figure 5B). Mice infused with ICOS-costimulated Th17 cells showed superior antitumor responses compared with animals treated with CD28-costimulated Th17 cells, consistent with prior reports.31,32 Interestingly, ICOS Strong-activated Th17 cells prevented tumor growth for greater than 1 month until relapse, while ICOS Weak-activated Th17 cells imparted durable immunity, marked by sustained control of tumors post-ACT. This striking improvement in Th17 cell response was also reflected by the enhanced survival of ICOS Weak-activated Th17-treated mice (Figure 5C). Donor T cells (%CD45+) persisted in the blood of mice 2 weeks after infusion, revealing that ICOS Weak activation enhanced Th17 engraftment and persistence to the greatest extent compared with other groups (Figure 5D).
Figure 5.

ICOS low-activated Th17 cells persist in vivo and mediate a durable antitumor response
(A) Experimental schematic. Th17 cells activated with either αCD3/28 or αCD3/ICOS beads at a 3:1 (Strong) or 1:10 (Weak) bead:T cell ratio and transduced with the 4-1BB mesothelin CAR were expanded for 10 days prior to infusion into M108 tumor-bearing NSG mice. 4-1BB mesothelin CAR CD8 T cells from the same donor were coinfused. Tumor size was measured until mice reached the endpoint. (B) Tumor area (mm2) averages ± SEM as well as (C) survival to 200 mm2 tumor area are reported. (D) Blood from mice on day 7 post-ACT was processed and analyzed by FACS for presence of human CD45+ cells, n = 10 mice/group. (E) Th17 cells were activated with CD3/ICOS beads at a 3:1 (Strong) or 1:10 (Weak) bead:T cell ratio and transduced with the 4-1BB mesothelin CAR. A subset of ICOS Strong-activated cells was cotransduced with a PCK1 overexpression vector. Cells were expanded for 10 days prior to infusion into M108 tumor-bearing NSG mice (n = 5–11 mice/group). 4-1BB mesothelin CAR CD8 T cells from the same donor were coinfused. Tumor size was measured until mice reached the endpoint. Tumor area (mm2) averages ± SEM and (F) survival to 200 mm2 tumor area are reported. (G) Human CD45+ cells isolated from splenocytes obtained from mice 45 days post-ACT were activated overnight with K562-meso cells to assess reactivity potential, and the resulting IFNγ response was measured in the culture medium by ELISA, n = 3 mice/group. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test and survival log-rank Mantel-Cox test (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001). Data are represented as mean ± SEM.
PCK1 overexpression rescues ineffective CAR T cell therapy
Murine tumor-specific T cells reprogrammed to produce more PEP via overexpression of PCK1, the rate-limiting enzyme of gluconeogenesis responsible for catalyzing the conversion of OAA into PEP, were previously reported to secrete more cytokines and lyse tumors more effectively in mice.33,34 Given the heightened PEP and other metabolites involved in gluconeogenesis detected in ICOS Weak-activated Th17 cells, we hypothesized that overexpression of PCK1 in less effective CAR T cells (i.e., ICOS Strong-activated Th17 cells) would bolster their in vivo ACT therapy potency to the extent mediated by ICOS Weak-activated Th17 cells. We cotransduced ICOS Strong-activated Th17 cells with a vector that contains PCK1 along with the mesoCAR vector and then infused them into M108-tumor-bearing mice along with CD28 Strong-activated CD8+ T cells. Compared with traditional ICOS Strong-activated mesoCAR Th17 cells, the PCK1-overexpressed (OE) ICOS Strong-activated mesoCAR Th17 cells were able to control tumor growth to a significantly greater extent, and this outcome was similar to that achieved with ICOS Weak-activated mesoCAR Th17 cells (Figure 5E). This result was also reflected in the survival of mice given ACT therapy (Figure 5F). A control group, in which mice received PCK1-OE ICOS Strong-activated Th17 cells that were not cotransduced with the mesoCAR vector, but still received the CD8+ mesoCAR cells, demonstrated no control over the tumor, highlighting the important contribution of the antigen specificity of the Th17 cells. Additional investigation revealed that hCD45+ cells isolated from the spleens of ICOS Strong-treated mice were unable to respond efficiently to antigen restimulation, as the production of IFNγ by these cells was not significantly higher than in cells from mice treated with non-transduced control ICOS Strong + PCK1-OE cells (Figure 5G). In comparison, cells from ICOS Weak-activated Th17-treated mice were able to produce more IFNγ when reactivated with antigen ex vivo compared with ICOS Strong-activated Th17 cells (p < 0.0001). Finally, we found that Strong-activated Th17 cells overexpressing PCK1 produced significantly more IFNγ than ICOS Strong-activated cells alone in vivo, although not as much IFNγ as mediated by ICOS Weak-activated Th17 cells. Taken together, our data implicate the alternative usages of the central carbon pathway by ICOS Weak-activated Th17 cells, leading to their superior response.
Discussion
T cells can be manipulated in various ways to yield an optimal antitumor cell product for therapy. Th17 cells have particularly demonstrated powerful antitumor prowess12,13,14,16; yet the optimal way to expand them has not been determined. In the present study, we found that the combination of lower TCR signaling and ICOS costimulation generated a far superior CAR Th17 cell product, with enhanced in vitro polyfunctionality, a stem-like memory phenotype, and improved antitumor activity. At a mechanistic level, this superior phenotype was mediated by a metabolic switch to oxidative phosphorylation. The concept of decreasing TCR signal strength to improve ACT has been reported previously, whereby decreasing the length of costimulation along with the addition of the cytokine IL-21 expanded stem-like memory T cells.19 Similarly, expanding bulk T cells with an equal bead-to-T cell ratio can promote optimal expansion while maintaining a younger memory phenotype.20 Further studies specifically focusing on Th17 cells found that either decreasing the length of time αCD3/28 beads are present in the Th17 culture or decreasing the number of αCD3/28 beads bolstered their capacity to secrete Th17-associated cytokines, and further elucidated that lowered αCD3/CD28 signal strength promoted Th17 cell responses through the binding of nuclear factor of activated T cells (NFAT) to the proximal region of the IL-17 promotor in low-stimulated cells only.21 However, modulating the strength of the TCR signal and the costimulatory effects of these cells’ ability to influence immunity against tumors was not tested.
ICOS costimulation, compared with CD28 costimulatory signaling, enhances Th17 cell generation and antitumor effectiveness.31,32 Herein, we are the first to correlate the concept of reduced TCR signal strength in concert with ICOS signaling to potentiate Th17 cells’ antitumor immunity. CD28 costimulatory signaling impairs Th17 function and development, which may impair their therapeutic potential.33 Pre-clinical studies of Th17 cells have characterized their advantages, including increased resistance to apoptosis and senescence and robust antitumor activity, compared with traditional bulk T cells, Th1 cells, or Th2 cells.13,14 By optimizing their production for use in CAR T cell therapy, we present a way to optimize this therapy for patients with solid tumors.
We found that decreasing the TCR activation signal modulated T cell metabolism in several ways. Metabolically fit Weak αCD3/ICOS-activated cells had lower mitochondrial membrane potential, greater SRC, and decreased glucose uptake, without any loss in cell yield. These data indicate an enhanced usage of oxidative phosphorylation (OXPHOS), which has been associated with long-lived memory T cells that have the capacity to produce energy under stressful metabolic conditions, as is often the case in hypoxic tumor microenvironments.24,25,34 In addition, OXPHOS has been implicated in bolstering Th17 pathogenicity by supporting mTOR and the BATF transcription factor, which lead to Th17 commitment over regulatory T cell (Treg) development in CD4+ T cells.35 In contrast, Strong αCD3/28-activated cells sustained high levels of glucose uptake, indicating that they may be more reliant on glucose and glycolysis than Weak-activated cells in vivo, which could be one detriment to their full functionality in the solid tumor microenvironment.
Weak stimulation of Th17 cells with a 1:10 αCD3/ICOS bead:T cell ratio promoted enhanced levels of metabolites involved in gluconeogenesis (PEP and 3-phosophoglycerate) and the pentose phosphate pathway (6-phosphogluconate). Our metabolite abundance data suggest that compared with ICOS high Th17 cells, which consume extracellular glucose and secrete lactate through glycolysis, ICOS low Th17 likely shunt glutamine or other amino acids toward isocitrate conversion to OAA and PEP through gluconeogenesis. The lack of abundance of G6P or F6P and the increase in 3-phosphoglycerate and 6-phosphogluconate suggest the flux of gluconeogenesis toward the pentose phosphate pathway to produce other carbon intermediates and the production of reducing potential, such as NADPH and reduced glutathione, to decrease levels of reactive oxygen species. Recent work has demonstrated that alternative fuels for the central carbon pathway such as inosine can be shunted through the pentose phosphate pathway as a substitute for glucose to improve T cell bioenergetics and tumor immunity.36 Thus, Weak ICOS stimulation could alter how T cells use gluconeogenesis for the synthesis of pentose phosphate pathway intermediates such as 6-phosphogluconate, which is a key intermediate for the generation of both carbon metabolite precursors and reducing potential molecules such as NADPH. Our data suggest that alternative usage of the central carbon pathway may be critical in the metabolic rewiring of ICOS Weak-activated Th17 cells and may contribute to their immune potency by providing them with an alternative pathway to supplement glucose in the tumor microenvironment. Our data complement other reports that PEP sustains NFAT and Ca2+ signaling, which results in enhanced effector functions in CD4+ T cells.37 By overexpressing the rate-limiting enzyme for PEP production and gluconeogenesis, PCK1, in cells stimulated with the ICOS Strong 3:1 bead:T cell ratio, we could partially rescue ineffective CAR T cell therapy, supporting our hypothesis that the observed differences in the metabolic phenotype of ICOS Weak cells are important to their improved response. Complementary work described upregulated PCK1 in memory CD8+ T cells, which supported the pentose phosphate pathway and reduced reactive oxygen species (ROS), which ultimately supported memory formation and maintenance.38 These data may explain why decreased TCR-stimulated cells have increased cytokine function while limiting glucose uptake, as NFAT has also been linked to IL-17 production in low-stimulated cells via its binding to the proximal region of the IL-17 promoter following its translocation to the nucleus.21 Furthermore, our Weak ICOS stimulation data and PCK1 overexpression data highlight an intriguing alternative rewiring of the central carbon pathway to increase gluconeogenesis and the pentose phosphate pathway to improve the metabolic capacity of Th17 cells. Although the mitochondrial bioenergetic changes observed with Weak ICOS stimulation were subtle compared with Weak CD28 stimulation, this alternative usage of gluconeogenesis seems to be enough to promote their potent antitumor response. Our data support recent reports showing that supplementation of alternative carbon sources, such as inosine, methionine, or L-arginine, can be sufficient to bolster T cell responses against solid tumors.36,39
We present a novel method to produce human CAR T cells by combining a weak TCR activation signal with ICOS costimulation to polarized Th17 cells. This finding has important clinical implications, not only to enhance antitumor response, but also to lower the cost of this expensive therapy by drastically reducing the number of antibody-coated beads necessary to generate the cell product with no impact on cell yield. An unknown concern may be that these potent cells may induce associated toxicities, such as cytokine release syndrome (CRS), in CAR-treated patients, as has occurred in other potent CAR T cell therapies,40 although we did not observe any evidence of this in our mouse models. Future studies will address whether combining ICOS Weak-activated Th17s with a similar activation strategy for the CD8+ CAR T cells to be coinfused into the patient is helpful or necessary, although any improvement may be only incremental to an already significantly improved response. Based on these studies, we believe this expansion method could improve the cell product and thus the outcome of patients with solid tumor malignancies, leading to long-term, durable responses.
Materials and methods
Isolation and expansion of human CAR Th17 cells
Peripheral blood buffy coats were obtained from deidentified healthy donors through the Oklahoma Blood Institute (Oklahoma City, OK). PBMCs were isolated via gradient separation using lymphocyte separation medium (Corning), and CD4+ and CD8+ T cells were further isolated with Dynabeads human CD4+ or CD8+ isolation kits (Thermo Fisher), following the manufacturer’s instructions. Cells were activated using Dynabeads M-280 Tosylactivated magnetic beads coated with αCD3 (clone OKT3; Biolegend) and either αCD28 (clone CD28.2; Biolegend) or αICOS (clone ISA-3; eBioscience). CD4+ cells were polarized toward a Th17 phenotype at the time of activation under the following conditions: IL-1β, 10 ng/mL; IL-6, 10 ng/mL; IL-23, 20 ng/mL; α-hIFN-γ, 5 μg/mL; and α-hIL-4, 5 μg/mL. Then they were cultured for up to 10 days in culture medium (CM; RPMI 1640 with L-glutamine containing 10% heat-inactivated FBS, 1% penicillin/streptomycin, 1% non-essential amino acids, 1% sodium pyruvate, 0.1% HEPES, and 0.1% β-mercaptoethanol) at 37°C, 5% CO2. IL-2 (100 IU/mL) and IL-23 (20 ng/mL) were added on day 2 of culture and any time the culture was split thereafter. Cells were debeaded on day 5 post-activation. CD8+ cells were activated with αCD3/28 beads (3 beads per T cell) and cultured in CM with IL-2 (100 IU/mL) for up to 10 days. T cells were transduced with lentivirus containing the second-generation mesothelin CAR with a CD3z signaling domain and a 4-1BB costimulatory domain (mesoCAR) vector as previously described.41
Flow cytometry
Cells were collected from culture and washed twice with fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% heat-inactivated FBS) and then stained with the appropriate extracellular antibodies (anti-CD45, clone H130, Biolegend; anti-CD3, clone HIT3a, Biolegend; anti-CD4, clone RPA-T4, Biolegend; anti-CD45RA, clone HI100, Biolegend; anti-CD45RO, clone UCHL1, Biolegend; anti-CD62L, clone DREG-56, Biolegend; anti-CCR7, clone G043H7, Biolegend; anti-CD27, clone LG.7F9, eBioscience; anti-CD39, clone A1, Biolegend; anti-Tim3, clone F38-2E2, Biolegend; anti-PD-1, clone EH12.2H7, Biolegend; anti-Glut1, clone 202915, R&D Systems) diluted in FACS buffer for 30 min at 4°C. For intracellular cytokine staining, cells were restimulated for 4 h with PMA/ionomycin and monensin at 37°C, 5% CO2 prior to FACS washes and extracellular antibody incubation. After extracellular staining, intracellular staining was performed in fixation and permeabilization buffers (Biolegend) and antibodies at a 1:200 dilution (anti-IFNγ, clone B27, Biolegend; anti-IL-17A, clone N49-653, BD Bioscience; anti-IL-22, clone 2G12A41, Biolegend; anti-IL-2, clone MQ1-17H12 BD Bioscience). Cells were washed and resuspended in FACS buffer and then analyzed on either an Accuri C6 flow cytometer (BD Biosciences) or a FACSVerse flow cytometer (BD Biosciences). CFSE (Thermo Fisher) staining was performed according to the manufacturer’s instructions. For mitochondrial stains, cells were resuspended in PBS containing 20 nM MitoTracker deep red (Thermo Fisher) and 250 nM TMRM (Thermo Fisher) for 30 min at 37°C prior to extracellular staining and flow cytometry. For glucose uptake, cells were resuspended in glucose-free medium containing 150 μM 2-NBDG (Thermo Fisher) and incubated for 45 min at 37°C.
Seahorse assay
Seahorse extracellular flux analyses were conducted using day 7 cells with the Seahorse XF (Agilent) reagents and XF24 or XF96 instrument, according to the manufacturer’s instructions. Briefly, the mitochondrial stress test injected oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP,500 nM), antimycin (2 μM), and rotenone (2 μM) while the instrument measured the oxygen consumption rate (OCR). Percentage SRC was calculated using the manufacturer’s spreadsheet macros.
Mice and tumor models
NSG mice were obtained from The Jackson Laboratory and maintained in the animal facilities of the University of Pennsylvania School of Medicine, in accordance with IACUC regulations. The human mesothelioma cell line M108 was cultured in E medium (RPMI 1640 containing L-glutamine with 0.5% human serum, 1× insulin-transferrin-ethanolamine-selenium (ITES), 10 mM HEPES, 0.5 mM sodium pyruvate, 0.1 mM non-essential amino acids, 1× Pen/Strep, 1 ng/mL recombinant human epithelial growth factor, 18 ng/mL hydrocortisone, and 0.1 nM triiodothyronine) for up to two passages. M108 cells (5 × 106) in a 1:1 mixture of PBS and Matrigel (Corning) were subcutaneously injected into NSG mice. Tumors were allowed to establish for 43 days, and then Th17 CAR T cells combined with CD8+ CAR T cells were adoptively transferred into tumor-bearing mice via tail vein injection. Prior to therapy, the mice were assigned randomly to groups based on tumor size, and L × W measurements via calipers were collected by personnel blinded to treatment group. Experimental endpoints were determined prior to study execution. Tumor control animal experiments were conducted over ∼60–70 days. Animals were excluded only if tumors were very small or immeasurable prior to therapy initiation. Outliers were reported. The tumor endpoint was established at 400 mm2.
Metabolomic analysis
Th17 cells from seven normal healthy donors were activated with either Strong ICOS bead stimulation (3 beads per T cell) or Weak ICOS bead stimulation (1 bead per 10 T cells) or were left inactivated (no beads). Cells were cultured for 4 days and then harvested. The cells were removed from their cell medium by centrifugation and then frozen at −80°C along with the separated cell medium. Metabolon (Morrisville, NC) performed metabolomic analysis using mass spectrometry and bioinformatic analysis.
PCK1 overexpression and IFNγ ELISA
Th17 cells activated with high ICOS were cotransduced 72 h after bead activation, similar to previously described, with a lentivirus containing a PCK1 overexpression vector37,42 (coding sequence of GenBank: NM-002591) and a lentivirus containing the mesoCAR vector encoding 4-1BB and CD3z.41 These cells, along with ICOS Weak-activated Th17 cells and non-transduced ICOS standard-activated Th17 cells, were cultured and cotransferred with CD28 Strong-activated CD8+ Meso4-1BB/CD3z-CAR T cells into M108-tumor-bearing NSG mice at day 10 of culture as described above. On day 45 post-ACT, three mice per group were euthanized and spleens were harvested. hCD45+ cells were enriched using human CD45 MicroBeads (Miltenyi) according to the manufacturer’s instructions. Isolated cells were rested overnight and then cocultured with 100-Gy-irradiated mesothelin-expressing K562 cells for 24 h. Following this incubation period, cell culture supernatant was collected and assayed for IFNγ with a DuoSet ELISA kit (R&D Systems) according to the manufacturer’s instructions.
Statistical analysis
For in vitro experiments, a sample size of ≥3 was chosen. For in vivo experiments, a sample size of ≥5 was chosen. Statistical analysis was performed using GraphPad Prism software v.9 (GraphPad Software). Experiments comparing two groups were analyzed using a Student’s t test. For experiments comparing three or more groups, a one-way analysis of variance (ANOVA) was performed with a post-comparison of each group using Tukey’s multiple comparisons test. Graphs utilizing error bars display the mean as the center value, and the error bars indicate SEM. For tumor curve experiments, a one-way ANOVA with Tukey’s multiple comparisons test was performed at the final dates on which all mice from the compared groups remained alive. Survival curve analysis was performed using a log-rank Mantel-Cox test. For the metabolomics study, fold changes were calculated and log2 transformed and reported in the heatmap.
Data availability
All datasets generated and analyzed for this article are available from the corresponding author upon reasonable request.
Acknowledgments
This work was supported by NCI R50 CA233168 (M.M.W.), NCI R01 CA228406 and R21 CA266088 (G.B.L.), NCI R01 CA175061 and R01 CA208514 (C.M.P.), and V Foundation for Cancer Research Translational Award T2021-015 (C.M.P.). This work was supported in part by the Flow Cytometry and Cell Sorting Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). This work was supported in part by the Winship Cancer Institute Support Grant, Emory University (2P30 CA138292-09).
Author contributions
Conceptualization, M.M.W., L.W.H., M.H.N., L.R.N., and C.M.P.; data curation, M.M.W., L.W.H., M.H.N., L.R.N., A.R.M., G.O.R.R., A.S.S., A.M.R.R., H.M.K., G.B.L., and C.M.P.; formal analysis, M.M.W., M.H.N., G.B.L., and C.M.P.; investigation, M.M.W., L.W.H., M.H.N., L.R.N., A.R.M., G.O.R.R., A.S.S., A.M.R.R., H.M.K., and C.M.P.; methodology, M.M.W., L.W.H., M.H.N., L.R.N., and A.R.M.; visualization, M.M.W. and C.M.P.; writing – original draft, M.M.W. and C.M.P.; writing – review & editing, M.M.W., L.W.H., M.H.N., L.R.N., A.R.M., G.O.R.R., A.S.S., A.M.R.R., H.M.K., J.L.R., G.B.L., and C.M.P.; validation, G.O.R.R.; supervision, M.H.N., J.L.R., G.B.L., and C.M.P.; resources, J.L.R. and C.M.P.; funding acquisition, C.M.P.; project administration, C.M.P.; guarantor, C.M.P.
Declaration of interests
C.M.P. has a patent for the expansion of Tc17 and Th17 cells using the inducible costimulator agonist and inducible costimulator ligand-expressing artificial antigen-presenting cells and beads. C.M.P. is a cofounder of Ares Immunotherapy and has received research funding through a sponsored research agreement between the Medical University of South Carolina and Lycera Corporation, Thermo Fisher Scientific, and Obsidian Therapeutics and through a sponsored research agreement between Emory University and Vaccinex, Inc. G.B.L. has consulted for ProDa Biotech, LLC, and received compensation. G.B.L. has received research funding through a sponsored research agreement between Emory University and Merck and Co., Bristol-Myers Squibb, Boehringer-Ingelheim, and Vaccinex, Inc. J.L.R. is a founder and equity holder in Tmunity Therapeutics, which is now part of Kite, a Gilead Company. J.L.R. received research support from Tmunity Therapeutics.
Contributor Information
Megan M. Wyatt, Email: megan.meek.wyatt@emory.edu.
Chrystal M. Paulos, Email: cpaulos@emory.edu.
References
- 1.Rosenberg S.A., Restifo N.P., Yang J.C., Morgan R.A., Dudley M.E. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restifo N.P., Dudley M.E., Rosenberg S.A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June C.H., O’Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 4.Knochelmann H.M., Smith A.S., Dwyer C.J., Wyatt M.M., Mehrotra S., Paulos C.M. CAR T cells in solid tumors: blueprints for building effective therapies. Front. Immunol. 2018;9:1740. doi: 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pule M.A., Savoldo B., Myers G.D., Rossig C., Russell H.V., Dotti G., Huls M.H., Liu E., Gee A.P., Mei Z., et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scarfò I., Maus M.V. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J. Immunother. Cancer. 2017;5:28. doi: 10.1186/s40425-017-0230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W., Sharma S.K., et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine B.L., Bernstein W.B., Connors M., Craighead N., Lindsten T., Thompson C.B., June C.H. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J. Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 9.Thomas A.K., Maus M.V., Shalaby W.S., June C.H., Riley J.L. A cell-based artificial antigen-presenting cell coated with anti-CD3 and CD28 antibodies enables rapid expansion and long-term growth of CD4 T lymphocytes. Clin. Immunol. 2002;105:259–272. doi: 10.1006/clim.2002.5277. [DOI] [PubMed] [Google Scholar]
- 10.Tesmer L.A., Lundy S.K., Sarkar S., Fox D.A. Th17 cells in human disease. Immunol. Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korn T., Bettelli E., Oukka M., Kuchroo V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 12.Muranski P., Boni A., Antony P.A., Cassard L., Irvine K.R., Kaiser A., Paulos C.M., Palmer D.C., Touloukian C.E., Ptak K., et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muranski P., Borman Z.A., Kerkar S.P., Klebanoff C.A., Ji Y., Sanchez-Perez L., Sukumar M., Reger R.N., Yu Z., Kern S.J., et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowers J.S., Nelson M.H., Majchrzak K., Bailey S.R., Rohrer B., Kaiser A.D., Atkinson C., Gattinoni L., Paulos C.M. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight. 2017;2 doi: 10.1172/jci.insight.90772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey S.R., Nelson M.H., Himes R.A., Li Z., Mehrotra S., Paulos C.M. Th17 cells in cancer: the ultimate identity crisis. Front. Immunol. 2014;5:276. doi: 10.3389/fimmu.2014.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majchrzak K., Nelson M.H., Bailey S.R., Bowers J.S., Yu X.-Z., Rubinstein M.P., Himes R.A., Paulos C.M. Exploiting IL-17-producing CD4+ and CD8+ T cells to improve cancer immunotherapy in the clinic. Cancer Immunol. Immunother. 2016;65:247–259. doi: 10.1007/s00262-016-1797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kryczek I., Zhao E., Liu Y., Wang Y., Vatan L., Szeliga W., Moyer J., Klimczak A., Lange A., Zou W. Human TH17 cells are long-lived effector memory cells. Sci. Transl. Med. 2011;3:104ra100. doi: 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez-Fernández C., Escribà-Garcia L., Vidal S., Sierra J., Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J. Transl. Med. 2016;14:214. doi: 10.1186/s12967-016-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y., Wu W., Wan T., Liu Y., Peng G., Chen Z., Zhu H. Impact of polyclonal anti-CD3/CD28-coated magnetic bead expansion methods on T cell proliferation, differentiation and function. Int. Immunopharmacol. 2013;15:129–137. doi: 10.1016/j.intimp.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 21.Purvis H.A., Stoop J.N., Mann J., Woods S., Kozijn A.E., Hambleton S., Robinson J.H., Isaacs J.D., Anderson A.E., Hilkens C.M.U. Low-strength T-cell activation promotes Th17 responses. Blood. 2010;116:4829–4837. doi: 10.1182/blood-2010-03-272153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson M.H., Knochelmann H.M., Bailey S.R., Huff L.W., Bowers J.S., Majchrzak-Kuligowska K., Wyatt M.M., Rubinstein M.P., Mehrotra S., Nishimura M.I., et al. Identification of human CD4+ T cell populations with distinct antitumor activity. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aba7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutloff A., Dittrich A.M., Beier K.C., Eljaschewitsch B., Kraft R., Anagnostopoulos I., Kroczek R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999;397:263–266. doi: 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 24.Pearce E.L., Walsh M.C., Cejas P.J., Harms G.M., Shen H., Wang L.-S., Jones R.G., Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Windt G.J.W., Everts B., Chang C.H., Curtis J.D., Freitas T.C., Amiel E., Pearce E.J., Pearce E.L., Pearce E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buck M.D., O’Sullivan D., Pearce E.L. T cell metabolism drives immunity. J. Exp. Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buck M.D., O'Sullivan D., Klein Geltink R.I., Curtis J.D., Chang C.H., Sanin D.E., Qiu J., Kretz O., Braas D., van der Windt G.J.W., et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sukumar M., Liu J., Ji Y., Subramanian M., Crompton J.G., Yu Z., Roychoudhuri R., Palmer D.C., Muranski P., Karoly E.D., et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sukumar M., Liu J., Mehta G.U., Patel S.J., Roychoudhuri R., Crompton J.G., Klebanoff C.A., Ji Y., Li P., Yu Z., et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakimi P., Johnson M.T., Yang J., Lepage D.F., Conlon R.A., Kalhan S.C., Reshef L., Tilghman S.M., Hanson R.W. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr. Metab. 2005;2:33. doi: 10.1186/1743-7075-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulos C.M., Carpenito C., Plesa G., Suhoski M.M., Varela-Rohena A., Golovina T.N., Carroll R.G., Riley J.L., June C.H. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci. Transl. Med. 2010;2:55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson M.H., Kundimi S., Bowers J.S., Rogers C.E., Huff L.W., Schwartz K.M., Thyagarajan K., Little E.C., Mehrotra S., Cole D.J., et al. The inducible costimulator augments Tc17 cell responses to self and tumor tissue. J. Immunol. 2015;194:1737–1747. doi: 10.4049/jimmunol.1401082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouguermouh S., Fortin G., Baba N., Rubio M., Sarfati M. CD28 co-stimulation down regulates Th17 development. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimeloe S., Mehling M., Frick C., Loeliger J., Bantug G.R., Sauder U., Fischer M., Belle R., Develioglu L., Tay S., et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J. Immunol. 2016;196:106–114. doi: 10.4049/jimmunol.1501766. [DOI] [PubMed] [Google Scholar]
- 35.Shin B., Benavides G.A., Geng J., Koralov S.B., Hu H., Darley-Usmar V.M., Harrington L.E. Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic Th17 and regulatory T cells. Cell Rep. 2020;30:1898–1909.e4. doi: 10.1016/j.celrep.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T., Gnanaprakasam J.N.R., Chen X., Kang S., Xu X., Sun H., Liu L., Rodgers H., Miller E., Cassel T.A., et al. Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat. Metab. 2020;2:635–647. doi: 10.1038/s42255-020-0219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho P.C., Bihuniak J.D., Macintyre A.N., Staron M., Liu X., Amezquita R., Tsui Y.C., Cui G., Micevic G., Perales J.C., et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma R., Ji T., Zhang H., Dong W., Chen X., Xu P., Chen D., Liang X., Yin X., Liu Y., et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 2018;20:21–27. doi: 10.1038/s41556-017-0002-2. [DOI] [PubMed] [Google Scholar]
- 39.Geiger R., Rieckmann J.C., Wolf T., Basso C., Feng Y., Fuhrer T., Kogadeeva M., Picotti P., Meissner F., Mann M., et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829–842.e13. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 2016;3 doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M., et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuo L., Xiang J., Pan X., Gao Q., Zhang G., Yang Y., Liang L., Xia J., Wang K., Tang N. PCK1 downregulation promotes TXNRD1 expression and hepatoma cell growth via the nrf2/keap1 pathway. Front. Oncol. 2018;8:611. doi: 10.3389/fonc.2018.00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated and analyzed for this article are available from the corresponding author upon reasonable request.